


Abstract
Transcription from the commonly used GTP- initiating T7 class III promoter φ6.5 frequently produces heterogeneous RNA at both 3′ and 5′ ends. We demonstrate here that RNA transcripts from the T7 class II promoter φ2.5 have superior 5′ homogeneity over those from the φ6.5 promoter, with comparable total RNA yields. The overall homogeneity of RNA transcripts is improved to different degrees depending on RNA sequences, although transcription under φ2.5 does not affect the 3′ heterogeneity of RNA. In combination with 3′ RNA trimming by DNAzymes or ribozymes, this ATP- initiated transcription system based on the T7 φ2.5 promoter can provide excellent quality of RNA for applications requiring a high degree of RNA size homogeneity.
INTRODUCTION
In vitro transcription by T7 RNA polymerase (1) has become a standard procedure for the preparation of RNA in a variety of applications. The commonly used in vitro transcription system is derived from the T7 consensus promoter (same as the class III φ6.5 promoter) (2). Transcription under the promoter is initiated by GTP (1,2), and usually requires two or more consecutive guanosines (2) at the first few nucleotide positions of RNA for efficient transcription. Repeated abortive initiation may result in substantial concentrations of oligomers of guanosine. Subsequently, initiation by both GTP and its oligomers may produce significant 5′ heterogeneity of RNA (3–5). While standard RNA purification methods such as gel electrophoresis can be used to fractionate different sizes of RNA, it is difficult to remove 5′ RNA heterogeneity (4). We have recently developed an adenosine-initiating transcription system (6–8) based on the T7 class II promoter φ2.5 (2). In the present work, we compare the two T7 promoters and demonstrate that the T7 class II promoter produces superior 5′ homogeneity of RNA over the most commonly used T7 class III promoter. Such high quality RNA provided by in vitro transcription under the T7 φ2.5 promoter may be ideal for applications such as structural investigation by NMR (9–15) and X-ray crystallography (16–18).
MATERIALS AND METHODS
RNA preparation
Three DNA sequences were prepared using either the T7 class III promoter (φ6.5) or the class II promoter (φ 2.5) for a total of six separate DNA template molecules (Fig. 1 shows the top strand of each). Duplex DNA was prepared for each sequence by PCR amplification of two separate synthetic DNA oligomers (IDT). All DNA oligomers were purified by 8% denaturing PAGE prior to use. To ensure that the concentration for each template was the same, the concentration of each oligo was determined by UV and the same PCR mixture (excluding sequence-specific DNA oligomers) was used to prepare all dsDNA templates.
Figure 1.

DNA sequences used in transcription. Only the sense strands (top strands) are shown. The T7 promoter sequences are underlined and the initiating nucleotides are in bold. The grouped pair sequences are the same except for the promoter sequences and the initiating nucleotides. CA0 is Escherichia coli tRNAAla, and CA2 is the GC4-69 mutant of CA0 described previously (4). 35n is a RNA sequence used previously in our work (7,8).
Transcription was carried out under the following standard conditions: 1 mM each of ATP, UTP, GTP and CTP (Roche), 40 mM Tris (pH 8.0), 6 mM MgCl2, 2 mM spermidine, 0.01% Triton X-100, 5 mM DTT, and 5 U/µl T7 RNA polymerase. Double strand DNA concentration was 0.3 µM for all transcriptions. As an internal label, [α-32P]ATP (NEN) was added to the transcription mixture. All transcription reactions were made from the same stock solution of the above components to ensure identical conditions, and were allowed to proceed for 4 h at 37°C before product analysis by 8% low resolution denaturing PAGE. Transcription yields were determined by quantitation of the RNA bands by phosphorimaging followed by analysis by Molecular Analyst software (Bio-Rad).
DNAzyme trimming of 3′ end of RNA transcripts
For both the CA2 and CA0 sequences, a DNAzyme with the sequence TTGCATGCCAGGCTAGCTACAACGAGCAA GCGCTC was used to specifically cleave off a portion of the 3′ heterogeneous ends of the RNA transcripts, yielding a 3′ homogenous 34-nt RNA for the N band. Any 5′ heterogeneity would therefore appear as 35- and 36-nt bands for the 5′ N+1 and N+2 RNA transcripts. Similarly, the 35n RNA sequences were treated with a separate DNAzyme (TGATCGGCT AGGCTAGCTACAACGAGGCTGGCCGC) (7) to produce a 3′ homogenous 25-nt RNA for the N band. For DNAzyme treatment, RNA transcripts (1 µM) were incubated with 1 µM DNAzymes in 50 mM MgCl2 and 50 mM Tris (pH 8.0) for 1 h at 37°C, ethanol-precipitated and analyzed by 8% single nucleotide resolution denaturing PAGE.
RESULTS AND DISCUSSION
In vitro transcription usually produces heterogeneous sizes of RNA, mainly due to the non-templated addition of nucleotides at the 3′ end (7,8). In addition, RNA transcripts may also possess significant 5′ heterogeneity (3–5). Based on the mechanism of 5′ RNA heterogeneity generation by repeated initiation and abortive initiation, we reasoned that the adenosine-initiating T7 φ2.5 promoter (with the RNA sequence of 5′ AG…) (2,6–8) would produce a high degree of 5′ RNA homogeneity, in contrast to RNA transcripts prepared under the guanosine-initiating T7 φ6.5 promoter. In addition, our previous experiments have suggested that transcription under either the T7 φ2.5 promoter or φ6.5 promoter yields a similar amount of total RNA transcripts (6–8). Experiments below compare both the total RNA yields and 5′ RNA heterogeneity of different RNA sequences under the two T7 promoters.
The sense strand (top strand) DNA sequences (including the T7 promoter sequences and RNA-coding sequences) are listed in Figure 1. The RNA sequences labeled as CA2 and CA0 are identical to those described previously (4) for a direct comparison. The two sequences differ by a single nucleotide (G or C) at the fourth nucleotide position of RNA. In addition, a 35-nucleotide RNA sequence (35n), which was used in our previous work (7,8), was included to assess sequence variation effects. For all three RNA sequences, both the T7 φ6.5 promoter and the φ2.5 promoter were used to initiate transcriptions with GTP or ATP, respectively.
Total RNA yields of the six individual sequences (Fig. 1) under identical transcription conditions are shown in Figure 2 by a low resolution PAGE analysis (15 × 13 cm gel). To ensure equal concentrations of DNA templates, synthetic oligodeoxyribonucleotide sequences were gel-purified and quantitated by UV spectroscopy. The resulting DNA oligomers were then used for full-length double stranded DNA (dsDNA) template construction by PCR (one cycle). Relative dsDNA concentrations of each pair were checked by non-denaturing PAGE before use in transcription. For the three RNA sequences (CA2, CA0 and 35n), the average of four independent experiments showed that the φ2.5 promoter had an efficiency of 94 ± 19% that of the φ6.5 promoter. Considering the variations of the experiments, we conclude that the two T7 promoters have similar transcription initiation efficiencies, consistent with our previous results (6,7).
Figure 2.

Comparison of total transcription yields of RNA for the six sequences listed in Figure 1. For each RNA sequence, four independent experiments indicate that guanosine- and adenosine-initiated transcriptions lead to similar total RNA yields. The 8% denaturing gel was run at a low resolution (15 × 13 cm). Therefore, each transcription produced a single RNA band that contained heterogeneous RNA transcripts of similar sizes.
To examine the 5′ heterogeneity of RNA transcripts from the two different promoters, the commonly occurring 3′ RNA heterogeneity must first be eliminated. Otherwise, RNA size fractionation by PAGE cannot resolve the 3′ and 5′ RNA heterogeneities. RNA 3′ end trimming can be easily achieved through site-specific cleavage by the DNAzyme 10-23 (19), which was used in our earlier work (7). As shown in Figure 3A, there are nine different RNA species from transcription (assuming N, N+1 and N+2 at both 3′ and 5′ ends), with RNA sizes ranging from N to N+4. A DNAzyme can be designed to cleave the RNA at the site indicated by the arrow. After the DNAzyme treatment, all 3′ RNA heterogeneities are removed, reducing the original nine different RNA species to three 5′ heterogeneous sizes: 5′ N, 5′ N+1 and 5′ N+2, which can then be easily resolved by high resolution PAGE (single-nucleotide resolution).
Figure 3.
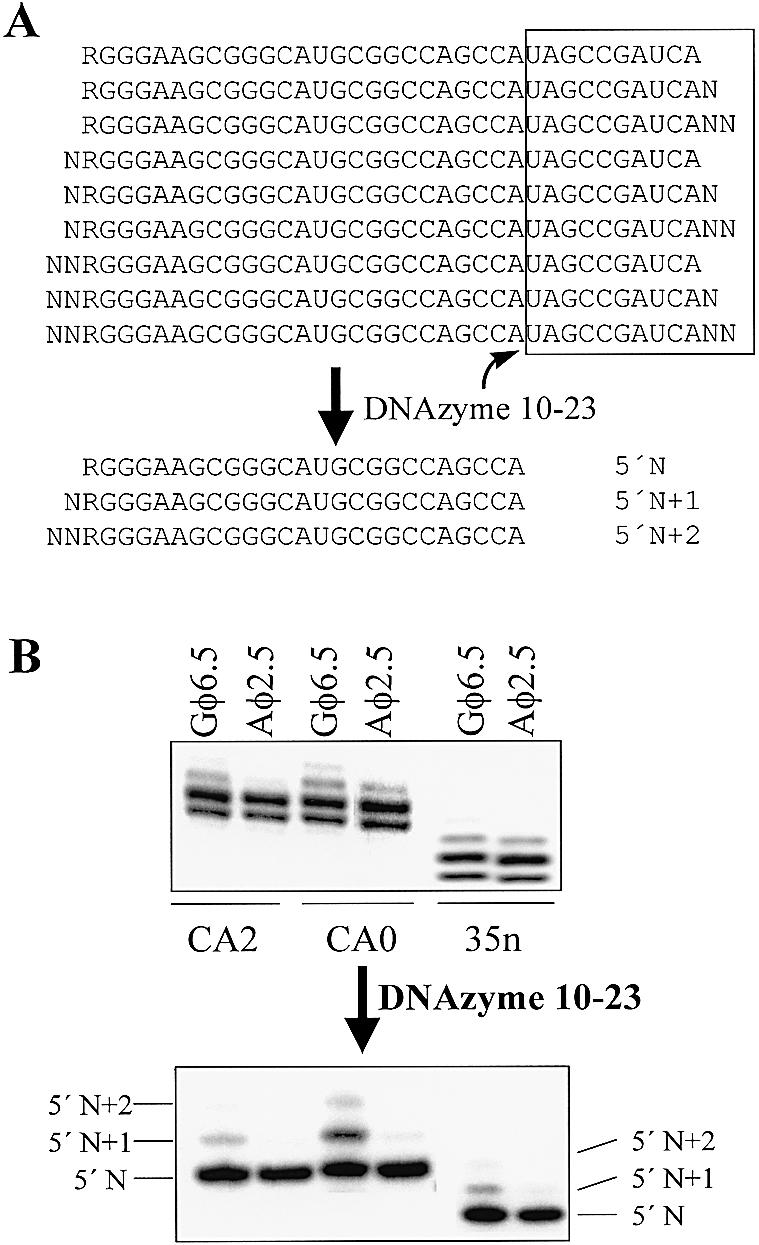

RNA size heterogeneity analysis by 8% high resolution PAGE (42 × 34 cm) and phosphorimaging of different RNA sequences prepared from the T7 φ2.5 and φ6.5 promoters. (A) Combination of RNA heterogeneities at both 3′ and 5′ ends and elimination of the 3′ heterogeneity by site-specific DNAzyme cleavage, which reduces the nine differently-sized RNA species to three 5′ heterogeneous RNA bands. (B) Resolution of RNA bands by single nucleotide resolution PAGE before and after DNAzyme treatment. The top panel shows the original RNA size-heterogeneities (both 3′ and 5′ ends) directly after transcription. The bottom panel indicates 5′ RNA heterogeneity of different sequences. It is noticeable that the φ2.5 promoter produces less heterogeneity even before the DNAzyme cleavage. DNAzyme trimming greatly improves the detection of 5′ RNA heterogeneity and reveals the superiority of the φ2.5 promoter over the φ6.5 promoter. (C) Radioactivity profiles from the bottom panel of (B). The major RNA bands at the right side of the figures are N band RNAs, while the two smaller peaks to the left are associated with the 5′ N+1 and 5′ N+2 bands, respectively. The solid line in each figure represents the result from the φ6.5 promoter with GTP-initiated transcription, and the dotted line shows the RNA from the φ2.5 promoter with ATP-initiated transcription. The percentages of radioactivity for different RNA bands prepared under the φ6.5 promoters are indicated on the figure. For all ATP-initiated transcriptions (dotted lines), the N band RNA is 94 ± 2%.
Figure 3B shows high-resolution gels (42 × 34 cm) before and after DNAzyme cleavage. Before the DNAzyme treatment, differences between the two promoters look small but clearly visible (Fig. 3B, top panel) (comparing lane 1 with 2, 3 with 4 and 5 with 6). It can be seen that the T7 φ6.5 promoter generates more RNA bands than the φ2.5 promoter does. Transcription under the φ2.5 promoter reduces the overall heterogeneity by 17, 30 and 10%, respectively, for the CA2, CA0 and 35nt RNA sequences relative to their corresponding G-initiated transcripts. The differences between the two promoters become more apparent after a small portion of the 3′ end was removed by a DNAzyme (Fig. 3B bottom panel). Comparing the two gels, it is evident that the φ2.5 promoter produces little 5′ heterogeneous RNA and the 3′ RNA heterogeneity contributes to the overall RNA size heterogeneity under the φ2.5 promoter. The degree of 3′ RNA heterogeneity depends on RNA sequences (7,8). On the other hand, the gels reveal that the φ6.5 promoter generates significant amount of both 3′ and 5′ heterogeneous RNA transcripts. Therefore, the combination of the two different heterogeneities at both ends can result in a high degree of overall RNA size heterogeneity.
Radioactivity profiles of the gel (bottom panel in Fig. 3B) can better illustrate different 5′ heterogeneities produced by the two promoters (Fig. 3C). As demonstrated previously (4), the degree of 5′ RNA heterogeneity under the GTP-initiating φ6.5 promoter is sequence-dependent. For CA2 and 35n RNA transcription under the φ6.5 promoter, 80–86% of total RNA transcripts are N band RNA, while 12–15% RNA has an additional 5′ G (5′ N+1 band) and 3–5% total RNA transcripts carry additional 5′ GG (5′ N+2 band). The degree of 5′ RNA heterogeneity of CA0 transcripts is significantly higher than those of CA2 and 35n under identical transcription conditions, with the 5′ N+1 and 5′ N+2 bands reaching 29 and 8%, respectively. These experiments not only confirm the previous findings on 5′ heterogeneous RNA transcription under the GTP-initiating φ6.5 promoter, but also agree quantitatively with those results (4).
Combined with our previous experiments (6–8), the present finding has demonstrated distinct advantages of the class II φ2.5 promoter (A-initiation) over the commonly used class III φ6.5 promoter (G-initiation). First, transcription under the φ2.5 promoter produces 5′ homogeneous RNA transcripts, while the φ6.5 promoter can lead to a significant degree of 5′ RNA heterogeneity (4,5). Although both systems may add non-templated nucleotides at the 3′ end of RNA depending on RNA sequences, such 3′ RNA heterogeneity can be easily eliminated by either DNAzymes (7,19) or ribozymes (20–23). The 3′ RNA heterogeneity can also be dramatically reduced by the introduction of C2′ methoxy groups (–OCH3) on the last two nucleotides of DNA templates (24,25). As a result, highly homogeneous RNA sizes can be prepared by in vitro transcription under the φ2.5 promoter, followed by 3′ end RNA trimming. On the other hand, it is difficult to eliminate the 5′ RNA heterogeneity. Such heterogeneity can lower RNA functional activities (4) and may cause problems in RNA structure, folding and mechanism investigation such as by NMR (9–15) and X-ray crystallography (16–18), where high purity of RNA is essential. Second, numerous adenosine analogs can be incorporated onto the 5′ ends of RNA transcripts. Adenosine itself may be used to prepare adenosine-initiated RNA with free 5′ hydroxyl groups, allowing easy 32P-labeling of the 5′ end by polynucleotide kinase. AMP can also be used for transcription initiation, and the resulting RNA transcripts may be directly used in RNA ligation by T4 RNA ligase. Further, two kinds of extensive modifications (at the either 5′ or N6 sites of adenosine) can be introduced without abolishing transcription initiation. A variety of functional groups including coenzymes, fluorophores and biotin can be used to label RNA at the specific 5′ end (7,8,26), providing functionalized RNA for biophysical, biochemical and biomedical applications. Third, an option for the 5′ end nucleotide (either A or G) of RNA is provided with the choice of T7 φ2.5 and φ6.5 promoters. In addition, the two transcription systems may use the same conditions, and give similar total RNA yields. Based on the above discussion, we recommend the use of the T7 φ2.5 promoter over the commonly used φ6.5 promoter for RNA transcription, if experiments are compatible with the 5′ end switch from a guanosine to an adenosine.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by an NSF grant MCB9974487 and a NASA grant NAG5-10668.
REFERENCES
- 1.Milligan J.F., Groebe,D.R., Witherell,G.W. and Uhlenbeck,O.C. (1987) Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res., 15, 8783–8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dunn J.J. and Studier,F.W. (1983) Complete nucleotide sequence of bacteriophage T7 DNA and the locations of T7 genetic elements. J. Mol. Biol., 166, 477–535. [DOI] [PubMed] [Google Scholar]
- 3.Nam S.C. and Kang,C.W. (1988) Transcription initiation site selection and abortive initiation cycling of phage SP6 RNA polymerase. J. Biol. Chem., 263, 18123–18127. [PubMed] [Google Scholar]
- 4.Pleiss J.A., Derrick,M.L. and Uhlenbeck,O.C. (1998) T7 RNA polymerase produces 5′ end heterogeneity during in vitro transcription from certain templates. RNA, 4, 1313–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Helm M., Brule,H., Giege,R. and Florentz,C. (1999) More mistakes by T7 RNA polymerase at the 5′ ends of in vitro-transcribed RNAs. RNA, 5, 618–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang F., Bugg,C.W. and Yarus,M. (2000) RNA-Catalyzed CoA, NAD and FAD synthesis from phosphopantetheine, NMN and FMN. Biochemistry, 39, 15548–15555. [DOI] [PubMed] [Google Scholar]
- 7.Huang F. (2003) Efficient incorporation of CoA, NAD and FAD into RNA by in vitro transcription. Nucleic Acids Res., 31, e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang F., Wang,G., Coleman,T.M. and Li,N. (2003) Synthesis of adenosine derivatives as transcription initiators and preparation of 5′ fluorescein- and biotin-labeled RNA through one-step in vitro transcription. RNA, 9, 1562–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Batey R.T., Inada,M., Kujawinski,E., Puglisi,J.D. and Williamson,J.R. (1992) Preparation of isotopically labeled ribonucleotides for multidimensional NMR spectroscopy of RNA. Nucleic Acids Res., 20, 4515–4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Puglisi J.D., Chen,L., Frankel,A.D. and Williamson,J.R. (1993) Role of RNA structure in arginine recognition of TAR RNA. Proc. Natl Acad. Sci. USA, 90, 3680–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Batey R.T., Battiste,J.L. and Williamson,J.R. (1995) Preparation of isotopically enriched RNAs for heteronuclear NMR. Methods Enzymol., 261, 300–322. [DOI] [PubMed] [Google Scholar]
- 12.Nikonowicz E.P. and Pardi,A. (1992) Three-dimensional heteronuclear NMR studies of RNA. Nature, 355, 184–186. [DOI] [PubMed] [Google Scholar]
- 13.Pardi A. (1995) Multidimensional heteronuclear NMR experiments for structure determination of isotopically labeled RNA. Methods Enzymol., 261, 350–380. [DOI] [PubMed] [Google Scholar]
- 14.Mollova E.T. and Pardi,A. (2000) NMR solution structure determination of RNAs. Curr. Opin. Struct. Biol., 10, 298–302. [DOI] [PubMed] [Google Scholar]
- 15.Furtig B., Richter,C., Wohnert,J. and Schwalbe,H. (2003) NMR spectroscopy of RNA. Chem. Biochem., 4, 936–962. [DOI] [PubMed] [Google Scholar]
- 16.Pley H.W., Flaherty,K.M. and McKay,D.B. (1994) Three-dimensional structure of a hammerhead ribozyme. Nature, 372, 68–74. [DOI] [PubMed] [Google Scholar]
- 17.Cate J.H., Gooding,A.R., Podell,E., Zhou,K., Golden,B.L., Kundrot,C.E., Cech,T.R. and Doudna,J.A. (1996) Crystal structure of a group I ribozyme domain: principles of RNA packing. Science, 273, 1678–1685. [DOI] [PubMed] [Google Scholar]
- 18.Golden B.L., Gooding,A.R., Podell,E.R. and Cech,T.R. (1998) A preorganized active site in the crystal structure of the Tetrahymena ribozyme. Science, 282, 259–264. [DOI] [PubMed] [Google Scholar]
- 19.Santoro S.W. and Joyce,G.F. (1997) A general purpose RNA-cleaving DNA enzyme. Proc. Natl Acad. Sci. USA, 94, 4262–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Price S.R., Ito,N., Oubridge,C., Avis,J.M. and Nagai,K. (1995) Crystallization of RNA-protein complexes. I. Methods for the large-scale preparation of RNA suitable for crystallographic studies. J. Mol. Biol., 249, 398–408. [DOI] [PubMed] [Google Scholar]
- 21.Ferre-D’Amare A.R. and Doudna,J.A. (1996) Use of cis- and trans-ribozymes to remove 5′ and 3′ heterogeneities from milligrams of in vitro transcribed RNA. Nucleic Acids Res., 24, 977–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schurer H., Lang,K., Schuster,J. and Morl,M. (2002) A universal method to produce in vitro transcripts with homogeneous 3′ ends. Nucleic Acids Res., 30, e56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walker S.C., Avis,J.M. and Conn,G.L. (2003) General plasmids for producing RNA in vitro transcripts with homogeneous ends. Nucleic Acids Res., 31, e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kao C., Zheng,M. and Rudisser,S. (1999) A simple and efficient method to reduce nontemplated nucleotide addition at the 3 terminus of RNAs transcribed by T7 RNA polymerase. RNA, 5, 1268–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kao C., Rudisser,S. and Zheng,M. (2001) A simple and efficient method to transcribe RNAs with reduced 3′ heterogeneity. Methods, 23, 201–205. [DOI] [PubMed] [Google Scholar]
- 26.Coleman T.M. and Huang,F. (2002) RNA-catalyzed thioester synthesis. Chem. Biol., 9, 1227–1236. [DOI] [PubMed] [Google Scholar]