

Abstract
Ambient ozone has a significant impact on human health. We have made considerable progress in understanding the fundamental mechanisms that regulate the biological response to ozone. It is increasingly clear that genes of innate immunity play a central role in both infectious and non-infectious lung disease. The biological response to ambient ozone provides a clinically relevant environmental exposure that allows us to better understand the role of innate immunity in non-infectious airways disease. In this brief review, we focus on: (1) specific cell types in the lung modified by ozone; (2) ozone and oxidative stress; (3) the relationship between genes of innate immunity and ozone; (4) the role of extracellular matrix in reactive airways disease; and (5) the effect of ozone on the adaptive immune system. We summarize recent advances in understanding the mechanisms that ozone contributes to environmental airways disease.
Keywords: ozone, oxidative stress, innate immunity, environment, surfactant, toll-like receptor, asthma, extracellular matrix, mindin, hyaluronan
HEALTH EFFECTS OF AMBIENT OZONE
Ozone is a reactive gas consisting of three oxygen atoms. Its highly oxidizing property defines ozone to be a harmful pollutant when existing in the lower atmosphere. Inhalation of ambient ozone is associated with adverse health consequences in vulnerable individuals and can lead to exacerbations of pre-existing respiratory diseases such as asthma and COPD. During days with high concentrations of ambient ozone, asthmatic patients have enhanced rates of emergency room visits and hospitalizations [1–2]. Adverse respiratory consequences are particularly prevalent in individuals participating in outdoor physical exercise[3], children [4–5], and elderly with predisposing cardiopulmonary diseases [6]. Typical ozone-induced pathophysiological manifestations in humans include immediate decrements in lung function, enhanced airways hyperreactivity, enhanced epithelial permeability, and inflammation [7]. Estimates support that for every 10 ppb increase in daily ozone there is an associated 1–4% increase in all-cause mortality [8–9]. However, evidence supports an ozone-induced biological response in human subjects to very low levels of ambient ozone, suggesting that adverse health consequences may exist at levels of ambient ozone below current regulatory standards[10–11]. Furthermore, ambient ozone levels are anticipated to rise with climate change [12] and rising surface temperatures are anticipated to enhance the biological consequences of ozone inhalation [13]. Based on predicted changes in climate by 2020, it is estimated that a 7.3% increase in ozone related asthma emergency room visits will occur in children [4]. Because of the ubiquitous exposure, the expected increase in ambient levels with climate change, and the clear health impact of ambient ozone, improved understanding of the molecular mechanisms that regulate the biological response to ozone are of high clinical significance.
MULTIPLE CELL-TYPES CONTRIBUTE TO THE RESPONSE TO OZONE
Inhalation of ambient ozone impacts numerous cell types in the lung and activates specific signaling cascades. Exposure to ozone elicits a variety of responses including: cellular damage, enhanced apoptosis, cytokine production, recruitment of inflammatory cells, and subsequent tissue repair. While many cells in the lung can be modified by inhaled ozone, the dominant cell types implicated in the acute response to ozone include: neutrophils, airway epithelia, and alveolar macrophages (Figure 1).
Figure 1. Ozone-induced oxidative stress results in epithelial injury and neutrophil inflammation.
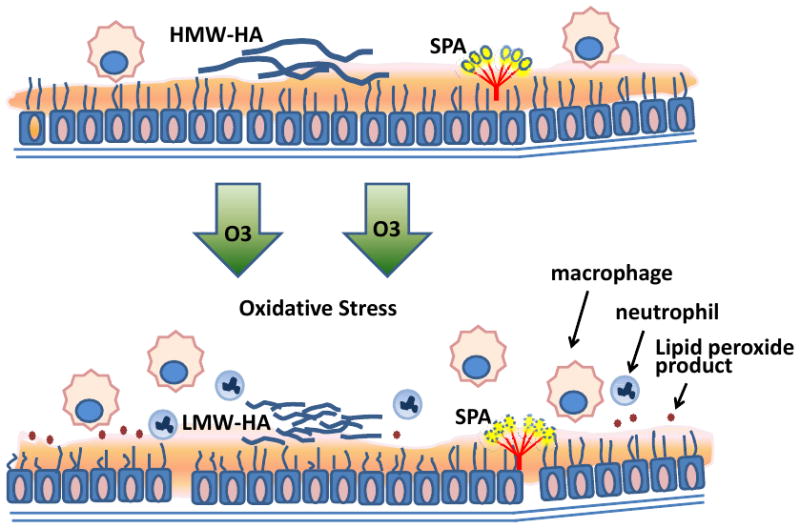
Ozone is a strong oxidant that oxidizes components in epithelial lining fluid such as SP-A and lipids producing toxic lipid peroxide products and protein adducts. The integrity of airway epithelium is impaired by ozone resulting in increased epithelial permeability. Ozone–induced oxidative stress induces fragmentation of extracellular matrix HMW-HA into LMW-HA and is associated with recruitment of neutrophils and macrophages into the airspace. (Abbreviation: HMW-HA, high molecular weight hyaluronan; LMWHA, low molecular weight hyaluronan.)
Neutrophils are frequently the first responder cells recruited into the sites of inflammation and tissue injury. Inhalation of ozone results in an early recruitment of neutrophils into the lung. Depending on the dose and duration of exposure, neutrophil inflammation is present 3–24 hours after inhalation [14–15]. Despite the presence of neutrophils in the lung after ozone, their functional impact in ozone-induced airways disease remains less clear. For example, airway hyperresponsiveness after inhalation to ozone has been demonstrated to be either dependent [16] or independent [17–18] of neutrophils. The reason for these discrepant effects of neutrophils on AHR may be due to the differences in experimental methodology. Independent of the role in AHR, recruited neutrophils can generate neutrophil elastase and matrix metalloproteinases that can contribute to airway injury [19]. Furthermore, exposure to ozone can impair neutrophil function that is associated with impaired pathogen clearance [20–21], a defect which may be related to apoptosis [22]. Together these observations support the notion that neutrophils are early responder cells that may contribute to ozone-induced lung injury and that exposure to ozone may impair the antibacterial function of neutrophils.
A critical aspect of host defense is dependent on intact barrier function, which in the lung is a principle function of epithelium. Airway epithelia are damaged after ozone inhalation resulting in loss of cilliary function [23], increased epithelial permeability [24–25], and defects in mucocilliary clearance [26]. In addition to barrier function, epithelia can also function as a central regulator of pulmonary immune responses [27–28]. After exposure to ozone, epithelia release cytokines and growth factors, such as interleukin 6 (IL-6), interleukin 8 (IL-8), and TNF-α that can result in recruitment of neutrophils and monocytes [29–32]. In addition, there is emerging evidence of cross-talk between epithelia and alveolar macrophages [33]. For example, airway epithelia-derived clara cell secretory protein (CCSP) regulate the response to ozone [34] and CCSP can attenuate macrophage-derived innate immune response [33]. Additionally, macrophages exposed to ozone can release IL-1α that further contributes to the epithelial-derived chemokine response [35]. Therefore, ozone exposure appears to cause a bi-directional communication between epithelia and alveolar macrophages. Comprehensive identification of molecules that serve as mediators between epithelia and alveolar macrophages in the biological response to ozone remains an area of considerable interest.
Lung macrophages are a dominant cell type in context of lung injury and repair. Macrophages contribute to the host response to ozone [36]. After inhalation of ozone, the number of macrophages in the lung is increased [37]. However, the regulatory role of macrophages remains incompletely elucidated. Challenges to understanding the role of macrophages in response to ozone are reflected in the fact that macrophages appear to have both pro-inflammatory and anti-inflammatory functions. Macrophage-derived mediators such as TNF-α, IL-1β, IL-6 and IL-8 contribute to the biological response to ozone and likely worsen injury and airway hyperresponsiveness [36, 38–40]. Despite the effect on injury and AHR, other macrophage functions include scavenger functions required for clearance of both apoptotic cells and oxidized lipids, which result in a reduction in inflammatory responses [41–42]. Given the divergent functions of macrophages, we reasoned that unique macrophage subsets were present in the lung with specific functions. Understanding macrophage heterogeneity and subpopulations in various organ systems has significantly expanded our understanding of this pleomorphic population of cells. One approach to understanding these divergent functions in the injured lung has been to understand the source of macrophages. Most macrophages are derived from monocytes in a manner either by recruitment from the blood in the setting of injury or by maturation from local intermediates. Much of this work in the lung has focused on the role of macrophages derived from monocytes in the setting of injury [43–44]. These inflammatory monocyte-derived macrophages have been termed exudate macrophages (ExMac) and are unique from resident alveolar macrophages by cell surface expression (ExMacs are CD11b+, whereas resident alveolar macrophages are CD11b−), morphology, and function [45–46]. We predicted that recruited ExMacs would be present in the lung after exposure to ozone and would account for the discrepancy in responses. To our surprise, ExMacs were not identified in the lung at 24hr after ozone exposure. Instead, we identified maturation of a lung resident macrophage, which were defined by specific cell surface markers by flow cytometry and unique gene expression from other lung macrophages. This population was dependent on CX3CR1 for its development. The loss of CX3CR1 was associated with worsened ozone induced AHR, cellular inflammation, cytokine production, and oxidant stress[47]. This work identified a unique macrophage subpopulation, which appears to protect the lung from ozone-induced pathology. Another approach to understanding divergent functions of macrophages has focused on understanding different phenotypes of macrophages which occur in the setting of injury and repair. This developed out of the observation that in vitro stimulation by different cytokines resulted in unique cellular programs. Classically activated macrophages (CAM) can be induced with stimulation by IFN-γ and LPS and are defined by expression of pro-inflammatory mediators such as IL-12 and iNOS. Alternatively activated macrophages (AAM) can be induced by stimulation by IL-4 and IL-13 and are classified based on expression of mannose receptor, arginase-1, Ym1 and FIZZ1[48]. Though in vitro stimulation of cytokines on macrophages clearly identifies unique cellular programing, how this works in vivo appears to be more complex with combinations of both CAM and AAM programs being evident after injury in several model systems. Recent work by Sunil et al. identified that markers of both classical and alternative activation of macrophages were present after ozone exposure in rats[49]. What is not clear from the present body of literature is how the identification of CAM and AAM in the setting of ozone exposure impact on the pathobiology. Also what remains unclear is how these phenotypic identifications relate to the known subsets of macrophages in the lung (ie alveolar macrophages, ExMacs and interstitial macrophages). Clearly given the central role of macrophages in the host response to ozone further work understanding the function of macrophage subpopulations and phenotypes will provide novel insight into fundamental mechanisms that regulate lung injury and repair.
OZONE AND OXIDANT STRESS
Oxidative stress is a conserved mechanism that contributes to numerous environmental lung injuries. Ozone, as a principle mediator of oxidative stress, is a clinically relevant model to understand the mechanisms that contribute to biological responses to oxidative stress. Oxidation products are either directly toxic and can cause injury to lung tissue or they can function as exogenous ligands via binding to cell surface receptors and thereby triggering intracellular inflammatory and/or apoptotic signaling pathways. Ozone-derived oxidative stress can also lead to changes in reactive oxygen species (ROS) in both the intracellular and extracellular compartments. While increased ROS in the setting of oxidant stress can directly induce cell and tissue injury, ROS can also modify innate immune signaling[50]. Ozone-induced oxidant stress modifies several known cell signaling mechanisms: activation of innate immune signaling pathways, upregulation of antioxidant genes, and enhanced release of damage-associated molecular pattern molecules (DAMPs). Tight regulation of oxidant stress is required to resolve tissue injury and maintain homeostasis.
Ozone exposure can directly lead to the development of oxidized intermediates, which are either formed in the epithelial lining fluid or on modified cell surface proteins and lipids (Figure 1). Ozone can result in either lipid peroxidation or lipid ozonation. Lipid peroxidation is a chain reaction between oxidant and polyunsaturated fatty acid, which severely damages the lipid structure of cell membrane [51]. By-products of lipid peroxidation such as 4-hydroxy-2-nonenal and isoprostanes are widely detected in the lung from ozone exposed humans [52–54] and animals [55–57]. 4-hydroxy-2-nonenal was found to induce cell death of alveolar macrophages [58], modulate airway remodeling [59], trigger intracellular ROS generation [60] and generate 4-hydroxy-2-nonenal-protein adducts [61]. Isoprostane can induce contraction of human bronchial smooth muscle in vitro [62] and induce airflow obstruction in guinea pigs [63]. Ozone exposure to unsaturated fatty acids can also result in lipid ozonation. Lipid ozonation products (LOPs) are generated by the oxidation of molecules in epithelial lining fluid and cell membranes in the lung by ozone. LOPs exposed ex vivo to bronchial epithelial cells leads to the activation of phospholipases A2, C, and D [64–65]. LOPs produced during ozone exposure also appear to direct macrophage and epithelial cell death, as well as induce pro-inflammatory cytokines production, such as IL-6 and IL-8 [65–66]. Oxidized or ozonized intermediates significantly contribute to the biological response to ozone.
Given the dominant role of oxidant stress in response to ozone, it is perhaps not surprising that antioxidant genes contribute to the biological response to ozone. NAD(P)H quinone oxidoreductase (NQO1), heme oxygenase-1 (HMOX1), glutathione-S-transferase isoforms M1 and P1 (GSTM1 and GSTP1) are critical detoxification enzymes in antioxidant defense. In human population studies, polymorphisms of NQO1 [67–68], HMOX1, GSTP1 [69], GSTM1 [68] are tightly associated with host susceptibility to ozone exposure although these associations appear to be complex. Unlike other antioxidant genes where impaired gene function results in decreased lung function after ozone inhalation, GSTM1-null polymorphisms alone had no association with ozone-related decrease in lung function [11, 68]. However, a combination of GSTM1-null and NQO1 major allele was significantly associated with ozone-induced FEV1 decrease [68]. In seeming contrast to human observations, glutathione (GSH) deficient mice were protected from ozone-induced lung injury, which was resultant from augmented expression of other antioxidant genes [70]. Additionally, ozone-exposed NQO1-deficient mice were protected from ozone-induced airway hyperreactivity and lung inflammation [71]. The level of ozone oxidation product F-isoprostane was lower in BALF from exposed NQO1-deficient mice, when compared with wild type control animals. However, it is recognized that NQO1 can have either pro-oxidant or antioxidant functions depending on the quinone substrate [72–74]. For example, in other non-infectious models of lung injury, NQO1 protects the lungs from oxidant-induced alveolar destruction [75]. In contrast, NQO1 appears necessary for the pro-oxidant response to ozone. Therefore, the response to ozone appears dependent on a combination of several antioxidant genes. Additionally, exposure to ozone can result in enhanced expression of many antioxidant genes.
SURFACTANT AND RESPONSE TO OZONE
Pulmonary surfactant, originally defined as a lipoprotein complex that aides in reducing surface tension at the air-liquid interface [76–77], has now been redefined to include multiple roles of surfactant proteins in lung host defense [78]. There are four known surfactant proteins: SP-A, SP-B, SP-C and SP-D. Of these, SP-A and SP-D are members of the collectin family of proteins, which have dual functions within the pulmonary innate immune system, acting either as an opsonin to enhance clearance of foreign materials [79] or as a modulator of ROS and pro-inflammatory cytokine production [80].
The surfactant film lining the airway and alveolus is exposed on a regular basis to a variety of oxidizing toxicants, including ozone. Excessive surfactant oxidation can lead to pulmonary complications including decreased lung compliance, impaired gas exchange and eventually airspace collapse. In addition, lipid ozonation products are thought to act as signal transduction messengers that relay response to ozone inhalation to other areas deeper in the lung [51, 81]. Both SP-A and SP-D directly function as effective inhibitors of lipid peroxidation and protect from oxidative cellular injury at physiologic concentrations [82]. Recent studies have found that alterations in SP-A function, either by ablation in the SP-A−/− animal or by reduction in activity by biochemical processes, renders mice more susceptible to the detrimental effects of ozone inhalation [83–84]. While less is known about the importance of SP-D in ozone-induced injury, decreased levels of functional SP-D correlate with increased inflammation after ozone exposure in mice, whereas enhanced SP-D production coincides with enhanced resolution of ozone-induced inflammation [85].
Interestingly, while surfactant proteins play protective roles in mediating the effects of oxidant-induced cellular damage, the function of these proteins themselves can also be impeded by oxidation [86]. Ozone oxidizes the methionine residues in SP-A and SP-B, which likely contributes to respiratory disorders and lung inflammation [87–88]. SP-A normally forms into an octadecamer structure (six trimeric subunits) by self-oligomerizing. Studies have shown that in vitro ozone exposure of SP-A results in an inability of SP-A to self-associate, to mediate lipid aggregation and to bind mannose [89]. Tubular myelin, a lipid transport system unique to the lungs, is formed exclusively by the combination of oligomerized SP-A and SP-B, along with phospholipids in the presence of calcium [90]. Therefore, it is not surprising that acute ozone stress leads to decreased tubular myelin organization, and as a result, perturbs the proportion of extracellular surfactant available for absorption into the surface film [91].
In addition to the involvement of SP-A in the biophysical activity of surfactant, several modulatory functions of SP-A in innate immunity are affected by ozone exposure. Ozone inhalation impairs the interactions between SP-A and either alveolar macrophages or the alveolar epithelia [89, 92]. Additionally, pathogen clearance via phagocytic mechanisms is impaired due to the decreased binding affinity of ozonated SP-A to LPS, bacteria, and viruses [93]. It was reported that SP-A exposed to ozone results in a diminished ability of SP-A to stimulate ROS production from macrophages under baseline conditions [92]. Seemingly in contrast, ozonated SP-A can additionally augment ability to modulate the respiratory burst of PMA stimulated macrophages [94]. While the precise mechanisms describing how ozonated SP-A functions differently from normal SP-A remain incompletely understood, studies strongly suggest that changes in SP-A structure due to ozonation affect multiple facets of SP-A including both physiological and immunological function [95].
Further understanding of the interaction between ozone and innate immunity has been elucidated by studies defining intrinsic connections between surfactant proteins and toll-like receptor 4 (tlr4). These interactions may account for the link between surfactant proteins and ozone-induced lung injury which occurs independent of oxidation. For example, both SP-A [96] and SP-D [97] can directly bind to tlr4/MD-2 complex. Such binding reduced the phosphorylation of tlr4 signaling proteins, such as MAPK family and AKT, thereby attenuating LPS derived immune responses in macrophage [98]. In contrast to this protective function, when SP-A is immunostimulatory, tlr4 is required for a complete inflammatory response [99]. Additionally, the expression of surfactant proteins can be regulated by ozone exposure through tlr4 signaling pathway [55]. Connor AJ, et al reported that the expression of pro-SP-C and SP-D was increased after ozone inhalation; reduced SP-C and SP-D expression in ozone-exposed tlr4-deficient animals was associated with reduced airway reactivity and lung inflammation. These findings suggest that surfactant proteins modulate ozone-host immune responses not only through their oxidization products, but also through interactions with other innate immune molecules.
Human SP-A is encoded by two functional genes, SP-A1 and SP-A2, of which several single nucleotide polymorphisms exist and are associated with acute and chronic lung diseases [100–102]. Variability in the susceptibility to ozone exposure has been described among individuals [32, 103–104]. It is possible that these differences are contributed by functional impairment of the different SP-A proteins (SP-A1 versus SP-A2) or by different combinations and/or proportions of SP-A1:SP-A2 and their respective genetic variants. Mikerov et al examined several of these possibilities and reported that in vitro ozone exposure reduces the ability of SP-A2 to aid in macrophage phagocytosis more than that of SP-A1, indicating that SP-A2 function may be more susceptible to ozone than SP-A1 [93]. This finding suggests that individuals bearingSP-A2 genetic polymorphisms may be more susceptible to the effects of oxidation and therefore ozone induced pathology.
TOLL-LIKERECEPTORS AND PULMONARY IMMUNE RESPONSE TO OZONE
The discovery of mammalian toll-like receptors (TLRs) [105] substantially advanced our understanding of innate immunity. TLRs were initially identified as pattern recognition receptors (PRR) that recognize pathogen-associated molecular patterns (PAMPs) [106]. Recent evidence support that PRRs additionally recognize damage-associated molecular patterns (DAMPs) including; hyaluronan (HA)[107], fibronectin [108], and fibrinogen [109–110]. We now recognize that TLRs and their downstream signaling pathways are central to the biological response to ambient ozone (Figure 2).
Figure 2. Ozone results in fragmentation of HMW-HA to LMW-HA that stimulates tlr4-dependent innate immune response.
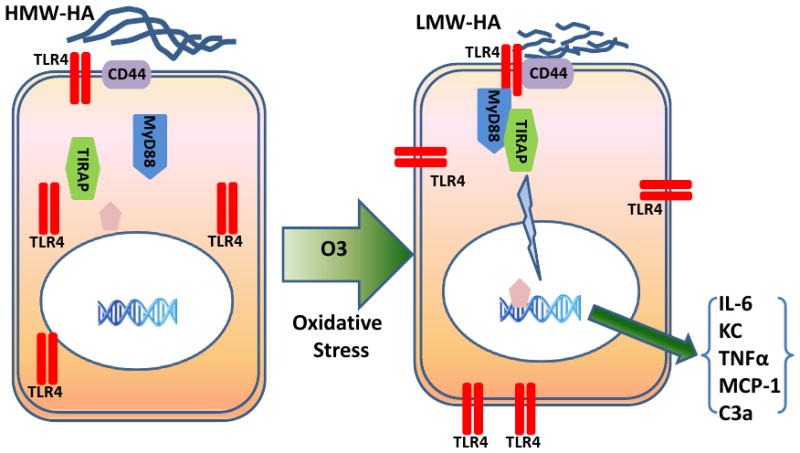
Ozone inhalation results in hyaluronan fragments with molecular weight lower than 300KD in the airspace, which is recognized to have immunostimmulatory properties. Both ozone and HA fragments can prime macrophage-derived innate immunity through trafficking oftlr4 to cell surface membrane. Hyaluronan interaction with the tlr4-cd44 complex on cell surface results in activation of the MyD88-tirap signaling pathway resulting in NF-κB activation and both the transcription of pro-inflammatory factors and the development of AHR.
Toll-like receptor 4 was initially discovered to be a susceptible gene for ozone-induced lung hyperpermeability [111–112]. Using genetically modified animals, we identified the role of tlr4 in ozone–induced AHR [113]. Further studies support that tlr4 signaling contributes to ozone-induced cytokine/chemokine production and neutrophil recruitment [114]. We now recognize that tlr4 signaling after exposure to ozone requires the intracellular adaptor proteins myeloid differentiation primary response gene 88 (MyD88) and toll-interleukin 1 receptor (TIR) domain containing adaptor protein (tirap)[115]. Tlr4 may additionally contribute to ozone-induced oxidative stress. Comparisons between ozone-exposed C3H/HeJ (tlr4 mutant) and C3H/HeOuJ mice reveal the levels of lipocalin 24p3 and 4-hydroxynonenal modified protein (markers for oxidative stress and lipid peroxidation) were significantly reduced in C3H/HeJ mice [55]. This observation suggests that tlr4 is required for ozone-induced oxidative stress, which is in contrast to previous observations suggesting an antioxidant role of tlr4 signaling in models of hyperoxia [116] and emphysema [117]. The precise mechanism that tlr4 signaling regulates oxidant stress remains an area of considerable interest. In addition to tlr4-MyD88-tirap signaling pathway, other TLRs including tlr2 may contribute to the response to ozone [114]. However, the current evidence strongly supports a central role of tlr4 in the biological response to ambient ozone.
Animal studies support the finding that tlr4 can regulate mRNA expression of numerous genes after ozone exposure, which has significant functional consequences. Prolonged or higher doses of ozone can enhance the expression of both tlr4 and MyD88 [111, 114, 118–119]. Ozone can also induce tlr4-dependent expression of heat-shock protein 70 (Hsp70) that contributes to pro-inflammatory signaling [119]. Macrophage receptor with collagenous structure (Marco) is increased on alveolar macrophages after ozone exposure in a manner dependent on tlr4 and functions to promote the uptake of surfactant-derived oxidation products in the epithelial lining fluid[41, 120]. Together, these findings support that tlr4 contributes to gene expression patterns after exposure to ozone with important functional consequences.
PRO-INFLAMMATORY FACTORS AND CELL SIGNALING
Previous studies have demonstrated that ozone exposure can induce the release of many pro-inflammatory factors, including: neutrophil elastase, fibronectin, prostaglandins, plasminogen activators, tissue factor, factor VIII, C3a fragment of complement, tumor necrosis factor (TNF)-α, IL-6, IL-8, IL-17, IL-1β, granulocyte macrophage-colony stimulating factor (GM-CSF), keratinocyte-derived chemokines (KC) and monocyte chemoattractant protein-1 (MCP-1)[30, 121–122]. Many of these factors are functionally required for the complete response to ambient ozone [123]. Additionally, many of these pro-inflammatory factors are recognized as downstream products of activated transcription factors required for innate immune response. For example, the p50 subunit of NF-κB is required for ozone-induced production of TNFα and nitric oxide [124]. Cho and colleagues built on this observation and identified that NF-κB and MAPK/AP-1 play key roles in ozone-induced lung inflammation and injury mediated through the TNF receptors [39]. It has been well documented that ozone exposure induces NF-κB activation in many cell types including alveolar macrophages and airway epithelial cells resulting to the production of cytokines and chemokines including; TNF-α, IL-6, and IL-8 [125–126].
EXTRACELLULAR MATRIX AND FUNCTIONAL RESPONSE TO OZONE
The extracellular matrix (ECM) refers to the structural components which comprise the space between tissue cells. Chronic exposure to ozone is known to impact lung structure [10, 127–128]. However, ECM can also contribute to immunological responses in the lung. In the setting of organ injury, degradation or fragmentation of the ECM can generate products that serve as extracellular ligands to TLR receptors that activate the innate immune system. Current evidence from our laboratory supports a role for extracellular matrix proteins including both mindin and hyaluronan (HA) in the functional response to ozone.
Mindin(spondin 2, SPON2) is an extracellular protein abundant in the spleen, lymph node, and the lung [129–130]. Mindin was initially identified as a necessary protein for an intact innate immune response to LPS and bacterial pathogens [129]. We now recognize that mindin additionally contributes to the response to antiviral host defense [131]and allergic airways disease [132–133]. In non-infectious lung injury caused by ozone inhalation, mindin-deficiency protected animals from augmented AHR and pro-inflammatory cytokine production [134]. Mindin contributes to cellular migration, dependent on integrin binding, in many cell types including: macrophages [130], neutrophils [129] and eosinophils [133]. However, the function extends beyond facilitating inflammatory cellular recruitment. After ozone exposure, bronchial rings derived from mindin-deficient mice demonstrated a reduced constriction in response to carbachol [134]. This finding suggests that mindin is required for airway smooth muscle contractility after exposure to ozone. However, the specific underlying mechanism remains unknown. Together these findings support that mindin contributes to innate immunity and inflammatory airways disease through divergent mechanisms. While previous data support a direct interaction between mindin and tlr4 [129], the functional consequences of this interaction in context of ozone remain incompletely defined.
Hyaluronan (HA) is an anionic non-sulfated glycosaminoglycan and a major component of ECM in the lung. The biological function of HA can either be protective or stimulatory depending on the size of the HA molecule [135]. We now recognize that HA can function as an endogenous ligand of tlr4[107]. Oxidative stress can degrade immune-suppressive high molecular weight HA into immunostimulatory low molecular weight HA [136–137]. Hyaluronan is functionally important in additional animal models of oxidative lung injury including; bleomycin [138] and asbestosis [139–140]. Clinically, HA is detected in airspace of patients with asthma [141–142] and COPD [143] implicating its importance in airways disease. Work from our lab demonstrates that hyaluronan fragments are increased in the lungs of mice after ozone exposure. Furthermore, we discovered that hyaluronan was both necessary and sufficient for ozone-induced airways hyperresponsiveness [144]. Interestingly, we were able to demonstrate that hyaluronan fragments function as endogenous ligands to tlr4 and contribute to reactive airways disease through a co-receptor complex with the dominant hyaluronan surface receptor CD44 [145]. We identified that hyaluronan signaling in the lung is dependent on the tlr4-MyD88-tirap signaling pathway, which results in both airway hyperresponsiveness and proinflammatory cytokines production [115]. Together, these observations support the finding that HA can function as an endogenous ligand to tlr4 to mediate ozone–induced reactive airways disease (Figure 2).
OZONE PRIMES PULMONARY INNATE AND ADAPTIVE IMMUNITY
In addition to the direct effects of ozone in the lung, it is important to recognize that exposure to ozone can modify the subsequent biological response to secondary challenges. Ozone inhalation enhances lung injury through modifying the function of both epithelial cells and macrophages. For example, oxidative stress decreases pathogen clearance by impairing anti-pathogen function of effector cells including; suppressing alveolar macrophage phagocytosis [93, 146–147], enhancing macrophage and neutrophil apoptosis [22], and increasing the susceptibility of human epithelial cells to H1N1 influenza infection [148]. While there are numerous mechanisms that could account for the effects of ozone on impaired antimicrobial host defense, we considered whether inhalation of ozone could directly modify subsequent innate immune response and either enhance or suppress the secondary response to pathogens.
To specifically determine whether ozone modifies subsequent innate immune responses, we exposed mice to ozone then subsequently challenged animals to the prototypic ligand to tlr4, bacterial lipopolysaccharide. We identified that pre-exposure to ozone dramatically enhanced innate immune response in the lung [22]. Ozone inhalation resulted in increased response to LPS including; lung injury, cytokine production, airway hyperresponsiveness, and cellular apoptosis. The enhanced response did not appear to be regulated though differences in gene expression, but rather through enhanced trafficking of tlr4 to the surface membrane of alveolar macrophages [22]. We now recognize that ozone results in fragmentation of extracellular matrix hyaluronan, which additionally contributes to ozone priming of the response to LPS [149]. Hyaluronan fragments prime alveolar macrophage for increased immune response to LPS through inducing tlr4 trafficking to surface lipid rafts on macrophages [149]. Previous observations in models of hemorrhagic shock [50] demonstrate that oxidative stress can alter tlr4 distribution on the cell surface. Recent work demonstrates that HA-induced intracellular ROS production contributes to trafficking of tlr4 to cell surface in both macrophage and dendritic cells [150]. Similar to observations in animal models, inhalation of ozone results in enhanced surface expression of TLR4 in airway macrophages derived from human subjects[151]. We would predict that enhanced surface expression of TLR4 would result in an enhanced response to LPS. The impact of primed innate immunity on health outcomes will certainly depend on the context of the host. Innate immunity is required for effective clearance of live bacterial pathogens, while uncontrolled response could result in enhanced lung injury or exacerbation of underlying airways disease. Understanding the environmental factors and fundamental mechanisms that regulate priming of innate immunity are of high clinical significance and will remain an area of continued investigation.
Previous work supports that exposure to ambient ozone is associated with an increase of asthma related hospitalizations [152–155]. It is well recognized that ozone can exacerbate existing allergic airways disease in human subjects [156–158]. Animal studies demonstrate that combined exposure to both ozone and to OVA challenges primes OVA-sensitized animals for an enhanced Th2 response [159]. These data suggest that ozone inhalation can modify adaptive immunity. Beyond modifying existing disease, ozone exposure has been shown to enhance sensitization to antigens. When rodents are co-exposed to OVA aerosol and ozone, then subsequently challenged to systemic OVA, there was a significant increase in the level of sensitization as supported by enhanced antigen-dependent fatal shock [160–161] and increased IgE-containing cells in the lung [162]. In monkeys, ozone exposure enhances the development of allergy to both house dust mite allergen [163] and inhaled platinum [164]. Cumulatively, these data suggest that ambient ozone promotes airway sensitization to airborne allergens through a previously unrecognized mechanism. Given our previous observations on the role of tlr4 in the biological response to ozone [113] and the role of tlr4 as an adjuvant [165], we sought to determine whether tlr4 contributes to the mechanism by which ozone can act as an adjuvant. We discovered that exposure to ozone during sensitization resulted in activation of dendritic cells in the airway in a tlr4-dependent manner resulting in enhanced allergic airways disease [166]. These data suggest that ozone can modulate adaptive immune response to allergen either during sensitization or after challenge to antigen. Ozone-induced priming of immunity could therefore have implications on lung injury, antibacterial host defense, and adaptive immunity.
CONCLUSIONS
Ambient ozone is a commonly encountered urban air pollutant with recognized adverse health effects. Current evidence supports a complex interaction between inhaled ozone and pulmonary innate immunity. Ozone can modify the function of many cell types in the lung required for intact innate immune response. Numerous genes of innate immunity are required for the complete response to ozone. We now recognize that components of the extracellular matrix contribute to activation of innate immunity in context of ozone inhalation and components of extracellular matrix can regulate intensity of subsequent innate immune signaling. Consistent with recognized cross-talk between the innate and adaptive immune systems, ozone activation of innate immunity can modify intensity of adaptive immunity. Given the effects of ozone inhalation on priming of innate immunity, analysis of the health effects of lower levels of ozone will require comprehensive immunological phenotyping for the response to secondary challenges. Improved understanding of the mechanisms that regulate host response to ambient ozone could provide insight into novel therapeutic approaches to reactive airways disease. Overall, current evidence support a complex relationship between ambient ozone and pulmonary innate immunity.
Acknowledgments
The authors appreciate funding support provided by the NIH (ES016126, AI081672, ES020426 and ES020350 to JWH) and training grants (HL105537 to RMT and HL111151 to JGL). The authors specifically appreciate support provided through the ONES Program at the NIEHS, which facilitated building a successful program focused on environmental lung disease.
References
- 1.Ji M, Cohan DS, Bell ML. Meta-analysis of the Association between Short-Term Exposure to Ambient Ozone and Respiratory Hospital Admissions. Environ Res Lett. 2011;6(2) doi: 10.1088/1748-9326/6/2/024006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Glad JA, Brink LL, Talbott EO, Lee PC, Xu X, Saul M, Rager J. The Relationship of Ambient Ozone and PM(2.5) Levels and Asthma Emergency Department Visits: Possible Influence of Gender and Ethnicity. Arch Environ Occup Health. 2012;67(2):103–108. doi: 10.1080/19338244.2011.598888. [DOI] [PubMed] [Google Scholar]
- 3.Carlisle AJ, Sharp NC. Exercise and outdoor ambient air pollution. Br J Sports Med. 2001;35(4):214–222. doi: 10.1136/bjsm.35.4.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sheffield PE, Knowlton K, Carr JL, Kinney PL. Modeling of regional climate change effects on ground-level ozone and childhood asthma. Am J Prev Med. 2011;41(3):251–257. doi: 10.1016/j.amepre.2011.04.017. quiz A253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Strickland MJ, Darrow LA, Klein M, Flanders WD, Sarnat JA, Waller LA, Sarnat SE, Mulholland JA, Tolbert PE. Short-term associations between ambient air pollutants and pediatric asthma emergency department visits. Am J Respir Crit Care Med. 2010;182(3):307–316. doi: 10.1164/rccm.200908-1201OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zanobetti A, Schwartz J. Ozone and survival in four cohorts with potentially predisposing diseases. Am J Respir Crit Care Med. 2011;184(7):836–841. doi: 10.1164/rccm.201102-0227OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Que LG, Stiles JV, Sundy JS, Foster WM. Pulmonary function, bronchial reactivity, and epithelial permeability are response phenotypes to ozone and develop differentially in healthy humans. J Appl Physiol. 2011;111(3):679–687. doi: 10.1152/japplphysiol.00337.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bell ML, McDermott A, Zeger SL, Samet JM, Dominici F. Ozone and short-term mortality in 95 US urban communities, 1987–2000. Jama. 2004;292(19):2372–2378. doi: 10.1001/jama.292.19.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jerrett M, Burnett RT, Pope CA, 3rd, Ito K, Thurston G, Krewski D, Shi Y, Calle E, Thun M. Long-term ozone exposure and mortality. N Engl J Med. 2009;360(11):1085–1095. doi: 10.1056/NEJMoa0803894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schelegle ES, Morales CA, Walby WF, Marion S, Allen RP. 6.6-hour inhalation of ozone concentrations from 60 to 87 parts per billion in healthy humans. Am J Respir Crit Care Med. 2009;180(3):265–272. doi: 10.1164/rccm.200809-1484OC. [DOI] [PubMed] [Google Scholar]
- 11.Kim CS, Alexis NE, Rappold AG, Kehrl H, Hazucha MJ, Lay JC, Schmitt MT, Case M, Devlin RB, Peden DB, Diaz-Sanchez D. Lung function and inflammatory responses in healthy young adults exposed to 0.06 ppm ozone for 6.6 hours. Am J Respir Crit Care Med. 2011;183(9):1215–1221. doi: 10.1164/rccm.201011-1813OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang HH, Zhou J, Fuentes M. Impact of climate change on ambient ozone level and mortality in southeastern United States. Int J Environ Res Public Health. 7(7):2866–2880. doi: 10.3390/ijerph7072866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Foster WM, Brown RH, Macri K, Mitchell CS. Bronchial reactivity of healthy subjects: 18–20 h postexposure to ozone. J Appl Physiol. 2000;89(5):1804–1810. doi: 10.1152/jappl.2000.89.5.1804. [DOI] [PubMed] [Google Scholar]
- 14.Williams AS, LS, Nath P, Khorasani NM, Bhavsar P, Issa R, Mitchell JA, Adcock IM, Chung KF. Role of TLR2, TLR4, and MyD88 in murine ozone-induced airway hyperresponsiveness and neutrophilia. J Appl Physiol. 2007;103(4):1189–1195. doi: 10.1152/japplphysiol.00172.2007. [DOI] [PubMed] [Google Scholar]
- 15.Johnston RA, MJ, Shore SA. CXCR2 is essential for maximal neutrophil recruitment and methacholine responsiveness after ozone exposure. Am J Physiol Lung Cell Mol Physiol. 2005;288(1):L61–67. doi: 10.1152/ajplung.00101.2004. [DOI] [PubMed] [Google Scholar]
- 16.O’Byrne PM, Walters EH, Gold BD, Aizawa HA, Fabbri LM, Alpert SE, Nadel JA, Holtzman MJ. Neutrophil depletion inhibits airway hyperresponsiveness induced by ozone exposure. Am Rev Respir Dis. 1984;130(2):214–219. doi: 10.1164/arrd.1984.130.2.214. [DOI] [PubMed] [Google Scholar]
- 17.Cooper PR, MA, Zhang J, Christmas P, Stark CM, Douaidy K, Mittelman MA, Soberman RJ, Blair IA, Panettieri RA. 20-HETE mediates ozone-induced, neutrophil-independent airway hyper-responsiveness in mice. PLoS One. 2010;5(4):e10235. doi: 10.1371/journal.pone.0010235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evans TW, BJ, Chung KF, Nadel JA, McDonald DM. Ozone-induced bronchial hyperresponsiveness in the rat is not accompanied by neutrophil influx or increased vascular permeability in the trachea. Am Rev Respir Dis. 1988;38(1):140–144. doi: 10.1164/ajrccm/138.1.140. [DOI] [PubMed] [Google Scholar]
- 19.Lazaar AL, SL, MacDonald AJ, Alexis NE, Chen C, Tal-Singer R. SB-656933, a novel CXCR2 selective antagonist, inhibits ex vivo neutrophil activation and ozone-induced airway inflammation in humans. Br J Clin Pharmacol. 2011;72(2):282–293. doi: 10.1111/j.1365-2125.2011.03968.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peterson ML, HS, Rummo N, House D. Effect of ozone on leukocyte function in exposed human subjects. Environ Res. 1978;15(3):485–493. doi: 10.1016/0013-9351(78)90127-5. [DOI] [PubMed] [Google Scholar]
- 21.Margalit M, AE, Attias D, Elstein D, Zimran A, Matzner Y. Effect of ozone on neutrophil function in vitro. Clin Lab Haematol. 2001;23(4):243–247. doi: 10.1046/j.1365-2257.2001.00401.x. [DOI] [PubMed] [Google Scholar]
- 22.Hollingsworth JW, Maruoka S, Li Z, Potts EN, Brass DM, Garantziotis S, Fong A, Foster WM, Schwartz DA. Ambient ozone primes pulmonary innate immunity in mice. J Immunol. 2007;179(7):4367–4375. doi: 10.4049/jimmunol.179.7.4367. [DOI] [PubMed] [Google Scholar]
- 23.Stephens RJ, Evans MJ, Sloan MF, Freeman G. A comprehensive ultrastructural study of pulmonary injury and repair in the rat resulting from exposures to less than one PPM ozone. Chest. 1974;65(Suppl):11S–13S. doi: 10.1378/chest.65.4_supplement.11s. [DOI] [PubMed] [Google Scholar]
- 24.Foster WM, Stetkiewicz PT. Regional clearance of solute from the respiratory epithelia: 18–20 h postexposure to ozone. J Appl Physiol. 1996;81(3):1143–1149. doi: 10.1152/jappl.1996.81.3.1143. [DOI] [PubMed] [Google Scholar]
- 25.Kehrl HR, Vincent LM, Kowalsky RJ, Horstman DH, O’Neil JJ, McCartney WH, Bromberg PA. Ozone exposure increases respiratory epithelial permeability in humans. Am Rev Respir Dis. 1987;135(5):1124–1128. doi: 10.1164/arrd.1987.135.5.1124. [DOI] [PubMed] [Google Scholar]
- 26.Foster WM, Costa DL, Langenback EG. Ozone exposure alters tracheobronchial mucociliary function in humans. J Appl Physiol. 1987;63(3):996–1002. doi: 10.1152/jappl.1987.63.3.996. [DOI] [PubMed] [Google Scholar]
- 27.Diamond G, Legarda D, Ryan LK. The innate immune response of the respiratory epithelium. Immunol Rev. 2000;173:27–38. doi: 10.1034/j.1600-065x.2000.917304.x. [DOI] [PubMed] [Google Scholar]
- 28.Leikauf GD, Simpson LG, Santrock J, Zhao Q, Abbinante-Nissen J, Zhou S, Driscoll KE. Airway epithelial cell responses to ozone injury. Environ Health Perspect. 1995;103(Suppl 2):91–95. doi: 10.1289/ehp.95103s291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nichols BG, Woods JS, Luchtel DL, Corral J, Koenig JQ. Effects of ozone exposure on nuclear factor-kappaB activation and tumor necrosis factor-alpha expression in human nasal epithelial cells. Toxicol Sci. 2001;60(2):356–362. doi: 10.1093/toxsci/60.2.356. [DOI] [PubMed] [Google Scholar]
- 30.Devlin RB, McKinnon KP, Noah T, Becker S, Koren HS. Ozone-induced release of cytokines and fibronectin by alveolar macrophages and airway epithelial cells. Am J Physiol. 1994;266(6 Pt 1):L612–619. doi: 10.1152/ajplung.1994.266.6.L612. [DOI] [PubMed] [Google Scholar]
- 31.Rusznak C, Devalia JL, Sapsford RJ, Davies RJ. Ozone-induced mediator release from human bronchial epithelial cells in vitro and the influence of nedocromil sodium. Eur Respir J. 1996;9(11):2298–2305. doi: 10.1183/09031936.96.09112298. [DOI] [PubMed] [Google Scholar]
- 32.Devlin RB, McDonnell WF, Mann R, Becker S, House DE, Schreinemachers D, Koren HS. Exposure of humans to ambient levels of ozone for 6.6 hours causes cellular and biochemical changes in the lung. Am J Respir Cell Mol Biol. 1991;4(1):72–81. doi: 10.1165/ajrcmb/4.1.72. [DOI] [PubMed] [Google Scholar]
- 33.Snyder JC, Reynolds SD, Hollingsworth JW, Li Z, Kaminski N, Stripp BR. Clara cells attenuate the inflammatory response through regulation of macrophage behavior. Am J Respir Cell Mol Biol. 2010;42(2):161–171. doi: 10.1165/rcmb.2008-0353OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnston CJ, Finkelstein JN, Oberdorster G, Reynolds SD, Stripp BR. Clara cell secretory protein-deficient mice differ from wild-type mice in inflammatory chemokine expression to oxygen and ozone, but not to endotoxin. Exp Lung Res. 1999;25(1):7–21. doi: 10.1080/019021499270394. [DOI] [PubMed] [Google Scholar]
- 35.Manzer R, Dinarello CA, McConville G, Mason RJ. Ozone exposure of macrophages induces an alveolar epithelial chemokine response through IL-1alpha. Am J Respir Cell Mol Biol. 2008;38(3):318–323. doi: 10.1165/rcmb.2007-0250OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pendino KJ, Meidhof TM, Heck DE, Laskin JD, Laskin DL. Inhibition of macrophages with gadolinium chloride abrogates ozone-induced pulmonary injury and inflammatory mediator production. Am J Respir Cell Mol Biol. 1995;13(2):125–132. doi: 10.1165/ajrcmb.13.2.7542894. [DOI] [PubMed] [Google Scholar]
- 37.Hotchkiss JA, Harkema JR, Kirkpatrick DT, Henderson RF. Response of rat alveolar macrophages to ozone: quantitative assessment of population size, morphology, and proliferation following acute exposure. Exp Lung Res. 1989;15(1):1–16. doi: 10.3109/01902148909069605. [DOI] [PubMed] [Google Scholar]
- 38.Arsalane K, Gosset P, Vanhee D, Voisin C, Hamid Q, Tonnel AB, Wallaert B. Ozone stimulates synthesis of inflammatory cytokines by alveolar macrophages in vitro. Am J Respir Cell Mol Biol. 1995;13(1):60–68. doi: 10.1165/ajrcmb.13.1.7598938. [DOI] [PubMed] [Google Scholar]
- 39.Cho HY, MD, Bauer AK, Kleeberger SR. Signal transduction pathways of tumor necrosis factor--mediated lung injury induced by ozone in mice. Am J Respir Crit Care Med. 2007;175(8):829–839. doi: 10.1164/rccm.200509-1527OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shore SA, I, Schwartzman N, Le Blanc B, Murthy GG, Doerschuk CM. Tumor necrosis factor receptor 2 contributes to ozone-induced airway hyperresponsiveness in mice. Am J Respir Crit Care Med. 2001;164(4):602–607. doi: 10.1164/ajrccm.164.4.2001016. [DOI] [PubMed] [Google Scholar]
- 41.Dahl M, Bauer AK, Arredouani M, Soininen R, Tryggvason K, Kleeberger SR, Kobzik L. Protection against inhaled oxidants through scavenging of oxidized lipids by macrophage receptors MARCO and SR-AI/II. J Clin Invest. 2007;117(3):757–764. doi: 10.1172/JCI29968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ishii Y, Hashimoto K, Nomura A, Sakamoto T, Uchida Y, Ohtsuka M, Hasegawa S, Sagai M. Elimination of neutrophils by apoptosis during the resolution of acute pulmonary inflammation in rats. Lung. 1998;176(2):89–98. doi: 10.1007/pl00007597. [DOI] [PubMed] [Google Scholar]
- 43.Maus U, Huwe J, Maus R, Seeger W, Lohmeyer J. Alveolar JE/MCP-1 and endotoxin synergize to provoke lung cytokine upregulation, sequential neutrophil and monocyte influx, and vascular leakage in mice. Am J Respir Crit Care Med. 2001;164(3):406–411. doi: 10.1164/ajrccm.164.3.2009055. [DOI] [PubMed] [Google Scholar]
- 44.Maus UA, Janzen S, Wall G, Srivastava M, Blackwell TS, Christman JW, Seeger W, Welte T, Lohmeyer J. Resident alveolar macrophages are replaced by recruited monocytes in response to endotoxin-induced lung inflammation. Am J Respir Cell Mol Biol. 2006;35(2):227–235. doi: 10.1165/rcmb.2005-0241OC. [DOI] [PubMed] [Google Scholar]
- 45.Lin KL, Suzuki Y, Nakano H, Ramsburg E, Gunn MD. CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality. J Immunol. 2008;180(4):2562–2572. doi: 10.4049/jimmunol.180.4.2562. [DOI] [PubMed] [Google Scholar]
- 46.Tighe RM, Liang J, Liu N, Jung Y, Jiang D, Gunn MD, Noble PW. Recruited exudative macrophages selectively produce CXCL10 after noninfectious lung injury. Am J Respir Cell Mol Biol. 2011;45(4):781–788. doi: 10.1165/rcmb.2010-0471OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tighe RM, Li Z, Potts EN, Frush S, Liu N, Gunn MD, Foster WM, Noble PW, Hollingsworth JW. Ozone Inhalation Promotes CX3CR1-Dependent Maturation of Resident Lung Macrophages That Limit Oxidative Stress and Inflammation. J Immunol. 2011 doi: 10.4049/jimmunol.1101312. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 48.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5(12):953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 49.Sunil VR, Patel-Vayas K, Shen J, Laskin JD, Laskin DL. Classical and alternative macrophage activation in the lung following ozone-induced oxidative stress. Toxicol Appl Pharmacol. 2012;263(2):195–202. doi: 10.1016/j.taap.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Powers KA, Szaszi K, Khadaroo RG, Tawadros PS, Marshall JC, Kapus A, Rotstein OD. Oxidative stress generated by hemorrhagic shock recruits Toll-like receptor 4 to the plasma membrane in macrophages. J Exp Med. 2006;203(8):1951–1961. doi: 10.1084/jem.20060943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pryor WA, Squadrito GL, Friedman M. The cascade mechanism to explain ozone toxicity: the role of lipid ozonation products. Free Radic Biol Med. 1995;19(6):935–941. doi: 10.1016/0891-5849(95)02033-7. [DOI] [PubMed] [Google Scholar]
- 52.Chen C, Arjomandi M, Balmes J, Tager I, Holland N. Effects of chronic and acute ozone exposure on lipid peroxidation and antioxidant capacity in healthy young adults. Environ Health Perspect. 2007;115(12):1732–1737. doi: 10.1289/ehp.10294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu L, Poon R, Chen L, Frescura AM, Montuschi P, Ciabattoni G, Wheeler A, Dales R. Acute effects of air pollution on pulmonary function, airway inflammation, and oxidative stress in asthmatic children. Environ Health Perspect. 2009;117(4):668–674. doi: 10.1289/ehp11813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alfaro MF, Walby WF, Adams WC, Schelegle ES. Breath condensate levels of 8-isoprostane and leukotriene B4 after ozone inhalation are greater in sensitive versus nonsensitive subjects. Exp Lung Res. 2007;33(3–4):115–133. doi: 10.1080/01902140701364367. [DOI] [PubMed] [Google Scholar]
- 55.Connor AJ, Laskin JD, Laskin DL. Ozone-induced lung injury and sterile inflammation. Role of toll-like receptor 4. Exp Mol Pathol. 2012;92(2):229–235. doi: 10.1016/j.yexmp.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tighe RM, Li Z, Potts EN, Frush S, Liu N, Gunn MD, Foster WM, Noble PW, Hollingsworth JW. Ozone inhalation promotes CX3CR1-dependent maturation of resident lung macrophages that limit oxidative stress and inflammation. J Immunol. 2011;187(9):4800–4808. doi: 10.4049/jimmunol.1101312. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 57.Long NC, Suh J, Morrow JD, Schiestl RH, Murthy GG, Brain JD, Frei B. Ozone causes lipid peroxidation but little antioxidant depletion in exercising and nonexercising hamsters. J Appl Physiol. 2001;91(4):1694–1700. doi: 10.1152/jappl.2001.91.4.1694. [DOI] [PubMed] [Google Scholar]
- 58.Li L, Hamilton RF, Jr, Kirichenko A, Holian A. 4-Hydroxynonenal-induced cell death in murine alveolar macrophages. Toxicol Appl Pharmacol. 1996;139(1):135–143. doi: 10.1006/taap.1996.0152. [DOI] [PubMed] [Google Scholar]
- 59.Tsukagoshi H, Kawata T, Shimizu Y, Ishizuka T, Dobashi K, Mori M. 4-Hydroxy-2-nonenal enhances fibronectin production by IMR-90 human lung fibroblasts partly via activation of epidermal growth factor receptor-linked extracellular signal-regulated kinase p44/42 pathway. Toxicol Appl Pharmacol. 2002;184(3):127–135. doi: 10.1006/taap.2002.9514. [DOI] [PubMed] [Google Scholar]
- 60.Uchida K, Shiraishi M, Naito Y, Torii Y, Nakamura Y, Osawa T. Activation of stress signaling pathways by the end product of lipid peroxidation. 4-hydroxy-2-nonenal is a potential inducer of intracellular peroxide production. J Biol Chem. 1999;274(4):2234–2242. doi: 10.1074/jbc.274.4.2234. [DOI] [PubMed] [Google Scholar]
- 61.Kirichenko A, Li L, Morandi MT, Holian A. 4-hydroxy-2-nonenal-protein adducts and apoptosis in murine lung cells after acute ozone exposure. Toxicol Appl Pharmacol. 1996;141(2):416–424. doi: 10.1006/taap.1996.0307. [DOI] [PubMed] [Google Scholar]
- 62.Kawikova I, Barnes PJ, Takahashi T, Tadjkarimi S, Yacoub MH, Belvisi MG. 8-Epi-PGF2 alpha, a novel noncyclooxygenase-derived prostaglandin, constricts airways in vitro. Am J Respir Crit Care Med. 1996;153(2):590–596. doi: 10.1164/ajrccm.153.2.8564103. [DOI] [PubMed] [Google Scholar]
- 63.Okazawa A, Kawikova I, Cui ZH, Skoogh BE, Lotvall J. 8-Epi-PGF2alpha induces airflow obstruction and airway plasma exudation in vivo. Am J Respir Crit Care Med. 1997;155(2):436–441. doi: 10.1164/ajrccm.155.2.9032175. [DOI] [PubMed] [Google Scholar]
- 64.Kafoury RM, Pryor WA, Squadrito GL, Salgo MG, Zou X, Friedman M. Lipid ozonation products activate phospholipases A2, C, and D. Toxicol Appl Pharmacol. 1998;150(2):338–349. doi: 10.1006/taap.1998.8418. [DOI] [PubMed] [Google Scholar]
- 65.Kafoury RM, Pryor WA, Squadrito GL, Salgo MG, Zou X, Friedman M. Induction of inflammatory mediators in human airway epithelial cells by lipid ozonation products. Am J Respir Crit Care Med. 1999;160(6):1934–1942. doi: 10.1164/ajrccm.160.6.9902025. [DOI] [PubMed] [Google Scholar]
- 66.Uhlson C, Harrison K, Allen CB, Ahmad S, White CW, Murphy RC. Oxidized phospholipids derived from ozone-treated lung surfactant extract reduce macrophage and epithelial cell viability. Chem Res Toxicol. 2002;15(7):896–906. doi: 10.1021/tx010183i. [DOI] [PubMed] [Google Scholar]
- 67.Bergamaschi E, De Palma G, Mozzoni P, Vanni S, Vettori MV, Broeckaert F, Bernard A, Mutti A. Polymorphism of quinone-metabolizing enzymes and susceptibility to ozone-induced acute effects. Am J Respir Crit Care Med. 2001;163(6):1426–1431. doi: 10.1164/ajrccm.163.6.2006056. [DOI] [PubMed] [Google Scholar]
- 68.Chen C, Arjomandi M, Tager IB, Holland N, Balmes JR. Effects of antioxidant enzyme polymorphisms on ozone-induced lung function changes. Eur Respir J. 2007;30(4):677–683. doi: 10.1183/09031936.00160806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Alexeeff SE, Litonjua AA, Wright RO, Baccarelli A, Suh H, Sparrow D, Vokonas PS, Schwartz J. Ozone exposure, antioxidant genes, and lung function in an elderly cohort: VA normative aging study. Occup Environ Med. 2008;65(11):736–742. doi: 10.1136/oem.2007.035253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Johansson E, Wesselkamper SC, Shertzer HG, Leikauf GD, Dalton TP, Chen Y. Glutathione deficient C57BL/6J mice are not sensitized to ozone-induced lung injury. Biochem Biophys Res Commun. 2010;396(2):407–412. doi: 10.1016/j.bbrc.2010.04.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Voynow JA, Fischer BM, Zheng S, Potts EN, Grover AR, Jaiswal AK, Ghio AJ, Foster WM. NAD(P)H quinone oxidoreductase 1 is essential for ozone-induced oxidative stress in mice and humans. Am J Respir Cell Mol Biol. 2009;41(1):107–113. doi: 10.1165/rcmb.2008-0381OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 72.Brar SS, Kennedy TP, Whorton AR, Sturrock AB, Huecksteadt TP, Ghio AJ, Hoidal JR. Reactive oxygen species from NAD(P)H:quinone oxidoreductase constitutively activate NF-kappaB in malignant melanoma cells. Am J Physiol Cell Physiol. 2001;280(3):C659–676. doi: 10.1152/ajpcell.2001.280.3.C659. [DOI] [PubMed] [Google Scholar]
- 73.Watanabe N, Forman HJ. Autoxidation of extracellular hydroquinones is a causative event for the cytotoxicity of menadione and DMNQ in A549-S cells. Arch Biochem Biophys. 2003;411(1):145–157. doi: 10.1016/s0003-9861(02)00716-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chan K, Kan YW. Nrf2 is essential for protection against acute pulmonary injury in mice. Proc Natl Acad Sci U S A. 1999;96(22):12731–12736. doi: 10.1073/pnas.96.22.12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Potts-Kant EN, Li Z, Tighe RM, Lindsey JY, Frush BW, Foster WM, Hollingsworth JW. NAD(P)H:quinone oxidoreductase 1 protects lungs from oxidant-induced emphysema in mice. Free Radic Biol Med. 2011;52(3):705–715. doi: 10.1016/j.freeradbiomed.2011.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 76.Pattle RE. Properties, function and origin of the alveolar lining layer. Nature. 1955;175(4469):1125–1126. doi: 10.1038/1751125b0. [DOI] [PubMed] [Google Scholar]
- 77.Clements JA. Surface tension of lung extracts. Proc Soc Exp Biol Med. 1957;95(1):170–172. doi: 10.3181/00379727-95-23156. [DOI] [PubMed] [Google Scholar]
- 78.Wright JR. Immunoregulatory functions of surfactant proteins. Nat Rev Immunol. 2005;5(1):58–68. doi: 10.1038/nri1528. [DOI] [PubMed] [Google Scholar]
- 79.van Iwaarden F, Welmers B, Verhoef J, Haagsman HP, van Golde LM. Pulmonary surfactant protein A enhances the host-defense mechanism of rat alveolar macrophages. Am J Respir Cell Mol Biol. 1990;2(1):91–98. doi: 10.1165/ajrcmb/2.1.91. [DOI] [PubMed] [Google Scholar]
- 80.Crouch E, Wright JR. Surfactant proteins a and d and pulmonary host defense. Annu Rev Physiol. 2001;63:521–554. doi: 10.1146/annurev.physiol.63.1.521. [DOI] [PubMed] [Google Scholar]
- 81.Leikauf GD, Zhao Q, Zhou S, Santrock J. Activation of eicosanoid metabolism in human airway epithelial cells by ozonolysis products of membrane fatty acids. Res Rep Health Eff Inst. 1995;(71):1–15. discussion 19–26. [PubMed] [Google Scholar]
- 82.Bridges JP, Davis HW, Damodarasamy M, Kuroki Y, Howles G, Hui DY, McCormack FX. Pulmonary surfactant proteins A and D are potent endogenous inhibitors of lipid peroxidation and oxidative cellular injury. J Biol Chem. 2000;275(49):38848–38855. doi: 10.1074/jbc.M005322200. [DOI] [PubMed] [Google Scholar]
- 83.Haque R, Umstead TM, Ponnuru P, Guo X, Hawgood S, Phelps DS, Floros J. Role of surfactant protein-A (SP-A) in lung injury in response to acute ozone exposure of SP-A deficient mice. Toxicol Appl Pharmacol. 2007;220(1):72–82. doi: 10.1016/j.taap.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mikerov AN, Haque R, Gan X, Guo X, Phelps DS, Floros J. Ablation of SP-A has a negative impact on the susceptibility of mice to Klebsiella pneumoniae infection after ozone exposure: sex differences. Respir Res. 2008;9:77. doi: 10.1186/1465-9921-9-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kierstein S, Poulain FR, Cao Y, Grous M, Mathias R, Kierstein G, Beers MF, Salmon M, Panettieri RA, Jr, Haczku A. Susceptibility to ozone-induced airway inflammation is associated with decreased levels of surfactant protein D. Respir Res. 2006;7:85. doi: 10.1186/1465-9921-7-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang G, Umstead TM, Phelps DS, Al-Mondhiry H, Floros J. The effect of ozone exposure on the ability of human surfactant protein a variants to stimulate cytokine production. Environ Health Perspect. 2002;110(1):79–84. doi: 10.1289/ehp.0211079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang G, Bates-Kenney SR, Tao JQ, Phelps DS, Floros J. Differences in biochemical properties and in biological function between human SP-A1 and SP-A2 variants, and the impact of ozone-induced oxidation. Biochemistry. 2004;43(14):4227–4239. doi: 10.1021/bi036023i. [DOI] [PubMed] [Google Scholar]
- 88.Manzanares D, Rodriguez-Capote K, Liu S, Haines T, Ramos Y, Zhao L, Doherty-Kirby A, Lajoie G, Possmayer F. Modification of tryptophan and methionine residues is implicated in the oxidative inactivation of surfactant protein B. Biochemistry. 2007;46(18):5604–5615. doi: 10.1021/bi062304p. [DOI] [PubMed] [Google Scholar]
- 89.Oosting RS, van Greevenbroek MM, Verhoef J, van Golde LM, Haagsman HP. Structural and functional changes of surfactant protein A induced by ozone. Am J Physiol. 1991;261(2 Pt 1):L77–83. doi: 10.1152/ajplung.1991.261.2.L77. [DOI] [PubMed] [Google Scholar]
- 90.Yu SH, Possmayer F. Role of bovine pulmonary surfactant-associated proteins in the surface-active property of phospholipid mixtures. Biochim Biophys Acta. 1990;1046(3):233–241. doi: 10.1016/0005-2760(90)90236-q. [DOI] [PubMed] [Google Scholar]
- 91.Balis JU, Paterson JF, Lundh JM, Haller EM, Shelley SA, Montgomery MR. Ozone stress initiates acute perturbations of secreted surfactant membranes. Am J Pathol. 1991;138(4):847–857. [PMC free article] [PubMed] [Google Scholar]
- 92.Oosting RS, Van Iwaarden JF, Van Bree L, Verhoef J, Van Golde LM, Haagsman HP. Exposure of surfactant protein A to ozone in vitro and in vivo impairs its interactions with alveolar cells. Am J Physiol. 1992;262(1 Pt 1):L63–68. doi: 10.1152/ajplung.1992.262.1.L63. [DOI] [PubMed] [Google Scholar]
- 93.Mikerov AN, Umstead TM, Gan X, Huang W, Guo X, Wang G, Phelps DS, Floros J. Impact of ozone exposure on the phagocytic activity of human surfactant protein A (SP-A) and SP-A variants. Am J Physiol Lung Cell Mol Physiol. 2008;294(1):L121–130. doi: 10.1152/ajplung.00288.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Su WY, Gordon T. Alterations in surfactant protein A after acute exposure to ozone. J Appl Physiol. 1996;80(5):1560–1567. doi: 10.1152/jappl.1996.80.5.1560. [DOI] [PubMed] [Google Scholar]
- 95.Muller B, Seifart C, Barth PJ. Effect of air pollutants on the pulmonary surfactant system. Eur J Clin Invest. 1998;28(9):762–777. doi: 10.1046/j.1365-2362.1998.00342.x. [DOI] [PubMed] [Google Scholar]
- 96.Yamazoe M, Nishitani C, Takahashi M, Katoh T, Ariki S, Shimizu T, Mitsuzawa H, Sawada K, Voelker DR, Takahashi H, Kuroki Y. Pulmonary surfactant protein D inhibits lipopolysaccharide (LPS)-induced inflammatory cell responses by altering LPS binding to its receptors. J Biol Chem. 2008;283(51):35878–35888. doi: 10.1074/jbc.M807268200. [DOI] [PubMed] [Google Scholar]
- 97.Yamada C, Sano H, Shimizu T, Mitsuzawa H, Nishitani C, Himi T, Kuroki Y. Surfactant protein A directly interacts with TLR4 and MD-2 and regulates inflammatory cellular response. Importance of supratrimeric oligomerization. J Biol Chem. 2006;281(31):21771–21780. doi: 10.1074/jbc.M513041200. [DOI] [PubMed] [Google Scholar]
- 98.Henning LN, Azad AK, Parsa KV, Crowther JE, Tridandapani S, Schlesinger LS. Pulmonary surfactant protein A regulates TLR expression and activity in human macrophages. J Immunol. 2008;180(12):7847–7858. doi: 10.4049/jimmunol.180.12.7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Guillot L, Balloy V, McCormack FX, Golenbock DT, Chignard M, Si-Tahar M. Cutting edge: the immunostimulatory activity of the lung surfactant protein-A involves Toll-like receptor 4. J Immunol. 2002;168(12):5989–5992. doi: 10.4049/jimmunol.168.12.5989. [DOI] [PubMed] [Google Scholar]
- 100.Floros J, Wang G, Mikerov AN. Genetic complexity of the human innate host defense molecules, surfactant protein A1 (SP-A1) and SP-A2--impact on function. Crit Rev Eukaryot Gene Expr. 2009;19(2):125–137. doi: 10.1615/critreveukargeneexpr.v19.i2.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Silveyra P, Floros J. Genetic variant associations of human SP-A and SP-D with acute and chronic lung injury. Front Biosci. 17:407–429. doi: 10.2741/3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Griese M, Birrer P, Demirsoy A. Pulmonary surfactant in cystic fibrosis. Eur Respir J. 1997;10(9):1983–1988. doi: 10.1183/09031936.97.10091983. [DOI] [PubMed] [Google Scholar]
- 103.Horstman DH, Folinsbee LJ, Ives PJ, Abdul-Salaam S, McDonnell WF. Ozone concentration and pulmonary response relationships for 6.6-hour exposures with five hours of moderate exercise to 0.08, 0.10, and 0.12 ppm. Am Rev Respir Dis. 1990;142(5):1158–1163. doi: 10.1164/ajrccm/142.5.1158. [DOI] [PubMed] [Google Scholar]
- 104.Kulle TJ, Sauder LR, Hebel JR, Chatham MD. Ozone response relationships in healthy nonsmokers. Am Rev Respir Dis. 1985;132(1):36–41. doi: 10.1164/arrd.1985.132.1.36. [DOI] [PubMed] [Google Scholar]
- 105.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282(5396):2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 106.Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11(4):443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 107.Taylor KR, Trowbridge JM, Rudisill JA, Termeer CC, Simon JC, Gallo RL. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J Biol Chem. 2004;279(17):17079–17084. doi: 10.1074/jbc.M310859200. [DOI] [PubMed] [Google Scholar]
- 108.Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, Chow JC, Strauss JF., 3rd The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem. 2001;276(13):10229–10233. doi: 10.1074/jbc.M100099200. [DOI] [PubMed] [Google Scholar]
- 109.Smiley ST, King JA, Hancock WW. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J Immunol. 2001;167(5):2887–2894. doi: 10.4049/jimmunol.167.5.2887. [DOI] [PubMed] [Google Scholar]
- 110.Kuhns DB, Priel DA, Gallin JI. Induction of human monocyte interleukin (IL)-8 by fibrinogen through the toll-like receptor pathway. Inflammation. 2007;30(5):178–188. doi: 10.1007/s10753-007-9035-1. [DOI] [PubMed] [Google Scholar]
- 111.Kleeberger SR, Reddy S, Zhang LY, Jedlicka AE. Genetic susceptibility to ozone-induced lung hyperpermeability: role of toll-like receptor 4. Am J Respir Cell Mol Biol. 2000;22(5):620–627. doi: 10.1165/ajrcmb.22.5.3912. [DOI] [PubMed] [Google Scholar]
- 112.Kleeberger SR, Reddy SP, Zhang LY, Cho HY, Jedlicka AE. Toll-like receptor 4 mediates ozone-induced murine lung hyperpermeability via inducible nitric oxide synthase. Am J Physiol Lung Cell Mol Physiol. 2001;280(2):L326–333. doi: 10.1152/ajplung.2001.280.2.L326. [DOI] [PubMed] [Google Scholar]
- 113.Hollingsworth JW, 2nd, Cook DN, Brass DM, Walker JK, Morgan DL, Foster WM, Schwartz DA. The role of Toll-like receptor 4 in environmental airway injury in mice. Am J Respir Crit Care Med. 2004;170(2):126–132. doi: 10.1164/rccm.200311-1499OC. [DOI] [PubMed] [Google Scholar]
- 114.Williams AS, Leung SY, Nath P, Khorasani NM, Bhavsar P, Issa R, Mitchell JA, Adcock IM, Chung KF. Role of TLR2, TLR4, and MyD88 in murine ozone-induced airway hyperresponsiveness and neutrophilia. J Appl Physiol. 2007;103(4):1189–1195. doi: 10.1152/japplphysiol.00172.2007. [DOI] [PubMed] [Google Scholar]
- 115.Li Z, Potts-Kant EN, Garantziotis S, Foster WM, Hollingsworth JW. Hyaluronan signaling during ozone-induced lung injury requires TLR4, MyD88, and TIRAP. PLoS One. 2011;6(11):e27137. doi: 10.1371/journal.pone.0027137. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 116.Qureshi ST, Zhang X, Aberg E, Bousette N, Giaid A, Shan P, Medzhitov RM, Lee PJ. Inducible activation of TLR4 confers resistance to hyperoxia-induced pulmonary apoptosis. J Immunol. 2006;176(8):4950–4958. doi: 10.4049/jimmunol.176.8.4950. [DOI] [PubMed] [Google Scholar]
- 117.Zhang X, Shan P, Jiang G, Cohn L, Lee PJ. Toll-like receptor 4 deficiency causes pulmonary emphysema. J Clin Invest. 2006;116(11):3050–3059. doi: 10.1172/JCI28139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Johnston CJ, Holm BA, Gelein R, Finkelstein JN. Postnatal lung development: immediate-early gene responses post ozone and LPS exposure. Inhal Toxicol. 2006;18(11):875–883. doi: 10.1080/08958370600822466. [DOI] [PubMed] [Google Scholar]
- 119.Bauer AK, Rondini EA, Hummel KA, Degraff LM, Walker C, Jedlicka AE, Kleeberger SR. Identification of candidate genes downstream of TLR4 signaling after ozone exposure in mice: a role for heat-shock protein 70. Environ Health Perspect. 2011;119(8):1091–1097. doi: 10.1289/ehp.1003326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Pulfer MK, Murphy RC. Formation of biologically active oxysterols during ozonolysis of cholesterol present in lung surfactant. J Biol Chem. 2004;279(25):26331–26338. doi: 10.1074/jbc.M403581200. [DOI] [PubMed] [Google Scholar]
- 121.Aris RM, CD, Hearne PQ, Kerr K, Finkbeiner WE, Balmes JR. Ozone-induced airway inflammation in human subjects as determined by airway lavage and biopsy. Am Rev Respir Dis. 1993;148(5):1363–1372. doi: 10.1164/ajrccm/148.5.1363. [DOI] [PubMed] [Google Scholar]
- 122.Koren HS, DR, Graham DE, Mann R, McGee MP, Horstman DH, Kozumbo WJ, Becker S, House DE, McDonnell WF, et al. Ozone-induced inflammation in the lower airways of human subjects. Am Rev Respir Dis. 1989;139(2):407–415. doi: 10.1164/ajrccm/139.2.407. [DOI] [PubMed] [Google Scholar]
- 123.Al-Hegelan M, Tighe RM, Castillo C, Hollingsworth JW. Ambient ozone and pulmonary innate immunity. Immunol Res. 49(1–3):173–191. doi: 10.1007/s12026-010-8180-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fakhrzadeh L, Laskin JD, Laskin DL. Ozone-induced production of nitric oxide and TNF-alpha and tissue injury are dependent on NF-kappaB p50. Am J Physiol Lung Cell Mol Physiol. 2004;287(2):L279–285. doi: 10.1152/ajplung.00348.2003. [DOI] [PubMed] [Google Scholar]
- 125.Garantziotis S, LZ, Potts EN, Lindsey JY, Stober VP, Polosukhin VV, Blackwell TS, Schwartz DA, Foster WM, Hollingsworth JW. TLR4 is necessary for hyaluronan-mediated airway hyperresponsiveness after ozone inhalation. Am J Respir Crit Care Med. 2010;181(7):666–675. doi: 10.1164/rccm.200903-0381OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 126.Wu W, DV, Diaz-Sanchez D, Samet JM, Kesic M, Dailey L, Zhang W, Jaspers I, Peden DB. GSTM1 modulation of IL-8 expression in human bronchial epithelial cells exposed to ozone. Free Radic Biol Med. 2011;51(2):522–529. doi: 10.1016/j.freeradbiomed.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Katre A, Ballinger C, Akhter H, Fanucchi M, Kim DK, Postlethwait E, Liu RM. Increased transforming growth factor beta 1 expression mediates ozone-induced airway fibrosis in mice. Inhal Toxicol. 2011;23(8):486–494. doi: 10.3109/08958378.2011.584919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Barr BC, Hyde DM, Plopper CG, Dungworth DL. A comparison of terminal airway remodeling in chronic daily versus episodic ozone exposure. Toxicol Appl Pharmacol. 1990;106(3):384–407. doi: 10.1016/0041-008x(90)90335-r. [DOI] [PubMed] [Google Scholar]
- 129.He YW, Li H, Zhang J, Hsu CL, Lin E, Zhang N, Guo J, Forbush KA, Bevan MJ. The extracellular matrix protein mindin is a pattern-recognition molecule for microbial pathogens. Nat Immunol. 2004;5(1):88–97. doi: 10.1038/ni1021. [DOI] [PubMed] [Google Scholar]
- 130.Jia W, Li H, He YW. The extracellular matrix protein mindin serves as an integrin ligand and is critical for inflammatory cell recruitment. Blood. 2005;106(12):3854–3859. doi: 10.1182/blood-2005-04-1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Jia W, Li H, He YW. Pattern recognition molecule mindin promotes intranasal clearance of influenza viruses. J Immunol. 2008;180(9):6255–6261. doi: 10.4049/jimmunol.180.9.6255. [DOI] [PubMed] [Google Scholar]
- 132.Li H, Oliver T, Jia W, He YW. Efficient dendritic cell priming of T lymphocytes depends on the extracellular matrix protein mindin. EMBO J. 2006;25(17):4097–4107. doi: 10.1038/sj.emboj.7601289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Li Z, Garantziotis S, Jia W, Potts EN, Lalani S, Liu Z, He YW, Foster WM, Hollingsworth JW. The extracellular matrix protein mindin regulates trafficking of murine eosinophils into the airspace. J Leukoc Biol. 2009;85(1):124–131. doi: 10.1189/jlb.0208135. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 134.Frush S, Li Z, Potts EN, Du W, Eu JP, Garantziotis S, He YW, Foster WM, Hollingsworth JW. The role of the extracellular matrix protein mindin in airway response to environmental airways injury. Environ Health Perspect. 2011;119(10):1403–1408. doi: 10.1289/ehp.1003339. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 135.McKee CM, Penno MB, Cowman M, Burdick MD, Strieter RM, Bao C, Noble PW. Hyaluronan (HA) fragments induce chemokine gene expression in alveolar macrophages. The role of HA size and CD44. J Clin Invest. 1996;98(10):2403–2413. doi: 10.1172/JCI119054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Monzon ME, Fregien N, Schmid N, Falcon NS, Campos M, Casalino-Matsuda SM, Forteza RM. Reactive oxygen species and hyaluronidase 2 regulate airway epithelial hyaluronan fragmentation. J Biol Chem. 2010;285(34):26126–26134. doi: 10.1074/jbc.M110.135194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bai KJ, Spicer AP, Mascarenhas MM, Yu L, Ochoa CD, Garg HG, Quinn DA. The role of hyaluronan synthase 3 in ventilator-induced lung injury. Am J Respir Crit Care Med. 2005;172(1):92–98. doi: 10.1164/rccm.200405-652OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, Homer RJ, Goldstein DR, Bucala R, Lee PJ, Medzhitov R, Noble PW. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11(11):1173–1179. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 139.Kliment CR, Tobolewski JM, Manni ML, Tan RJ, Enghild J, Oury TD. Extracellular superoxide dismutase protects against matrix degradation of heparan sulfate in the lung. Antioxid Redox Signal. 2008;10(2):261–268. doi: 10.1089/ars.2007.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cantin AM, Larivee P, Martel M, Begin R. Hyaluronan (hyaluronic acid) in lung lavage of asbestos-exposed humans and sheep. Lung. 1992;170(4):211–220. doi: 10.1007/BF00174118. [DOI] [PubMed] [Google Scholar]
- 141.Sahu S, Lynn WS. Hyaluronic acid in the pulmonary secretions of patients with asthma. Biochem J. 1978;173(2):565–568. doi: 10.1042/bj1730565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Liang J, Jiang D, Jung Y, Xie T, Ingram J, Church T, Degan S, Leonard M, Kraft M, Noble PW. Role of hyaluronan and hyaluronan-binding proteins in human asthma. J Allergy Clin Immunol. 2011;128(2):403–411. e403. doi: 10.1016/j.jaci.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Dentener MA, Vernooy JH, Hendriks S, Wouters EF. Enhanced levels of hyaluronan in lungs of patients with COPD: relationship with lung function and local inflammation. Thorax. 2005;60(2):114–119. doi: 10.1136/thx.2003.020842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Garantziotis S, Li Z, Potts EN, Kimata K, Zhuo L, Morgan DL, Savani RC, Noble PW, Foster WM, Schwartz DA, Hollingsworth JW. Hyaluronan mediates ozone-induced airway hyperresponsiveness in mice. J Biol Chem. 2009;284(17):11309–11317. doi: 10.1074/jbc.M802400200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 145.Garantziotis S, Li Z, Potts EN, Lindsey JY, Stober VP, Polosukhin VV, Blackwell TS, Schwartz DA, Foster WM, Hollingsworth JW. TLR4 is necessary for hyaluronan-mediated airway hyperresponsiveness after ozone inhalation. Am J Respir Crit Care Med. 2010;181(7):666–675. doi: 10.1164/rccm.200903-0381OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 146.Gilmour MI, Park P, Selgrade MK. Ozone-enhanced pulmonary infection with Streptococcus zooepidemicus in mice. The role of alveolar macrophage function and capsular virulence factors. Am Rev Respir Dis. 1993;147(3):753–760. doi: 10.1164/ajrccm/147.3.753. [DOI] [PubMed] [Google Scholar]
- 147.Valentine R. An in vitro system for exposure of lung cells to gases: effects of ozone on rat macrophages. J Toxicol Environ Health. 1985;16(1):115–126. doi: 10.1080/15287398509530723. [DOI] [PubMed] [Google Scholar]
- 148.Kesic MJ, Meyer M, Bauer R, Jaspers I. Exposure to ozone modulates human airway protease/antiprotease balance contributing to increased influenza A infection. PLoS One. 2012;7(4):e35108. doi: 10.1371/journal.pone.0035108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Li Z, Potts EN, Piantadosi CA, Foster WM, Hollingsworth JW. Hyaluronan fragments contribute to the ozone-primed immune response to lipopolysaccharide. J Immunol. 2010;185(11):6891–6898. doi: 10.4049/jimmunol.1000283. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 150.Kwon MJ, Han J, Kim BH, Lee YS, Kim TY. Superoxide dismutase 3 suppresses hyaluronic acid fragments mediated skin inflammation by inhibition of toll-like receptor 4 signaling pathway: superoxide dismutase 3 inhibits reactive oxygen species-induced trafficking of toll-like receptor 4 to lipid rafts. Antioxid Redox Signal. 2012;16(4):297–313. doi: 10.1089/ars.2011.4066. [DOI] [PubMed] [Google Scholar]
- 151.Hernandez ML, Lay JC, Harris B, Esther CR, Jr, Brickey WJ, Bromberg PA, Diaz-Sanchez D, Devlin RB, Kleeberger SR, Alexis NE, Peden DB. Atopic asthmatic subjects but not atopic subjects without asthma have enhanced inflammatory response to ozone. J Allergy Clin Immunol. 2010;126(3):537–544. e531. doi: 10.1016/j.jaci.2010.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Tenias JM, Ballester F, Rivera ML. Association between hospital emergency visits for asthma and air pollution in Valencia, Spain. Occup Environ Med. 1998;55(8):541–547. doi: 10.1136/oem.55.8.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Tolbert PE, Mulholland JA, MacIntosh DL, Xu F, Daniels D, Devine OJ, Carlin BP, Klein M, Dorley J, Butler AJ, Nordenberg DF, Frumkin H, Ryan PB, White MC. Air quality and pediatric emergency room visits for asthma in Atlanta, Georgia, USA. Am J Epidemiol. 2000;151(8):798–810. doi: 10.1093/oxfordjournals.aje.a010280. [DOI] [PubMed] [Google Scholar]
- 154.Jaffe DH, Singer ME, Rimm AA. Air pollution and emergency department visits for asthma among Ohio Medicaid recipients, 1991–1996. Environ Res. 2003;91(1):21–28. doi: 10.1016/s0013-9351(02)00004-x. [DOI] [PubMed] [Google Scholar]
- 155.Samoli E, Nastos PT, Paliatsos AG, Katsouyanni K, Priftis KN. Acute effects of air pollution on pediatric asthma exacerbation: evidence of association and effect modification. Environ Res. 2011;111(3):418–424. doi: 10.1016/j.envres.2011.01.014. [DOI] [PubMed] [Google Scholar]
- 156.Peden DB, Setzer RW, Jr, Devlin RB. Ozone exposure has both a priming effect on allergen-induced responses and an intrinsic inflammatory action in the nasal airways of perennially allergic asthmatics. Am J Respir Crit Care Med. 1995;151(5):1336–1345. doi: 10.1164/ajrccm.151.5.7735583. [DOI] [PubMed] [Google Scholar]
- 157.Ball BA, Folinsbee LJ, Peden DB, Kehrl HR. Allergen bronchoprovocation of patients with mild allergic asthma after ozone exposure. J Allergy Clin Immunol. 1996;98(3):563–572. doi: 10.1016/s0091-6749(96)70090-8. [DOI] [PubMed] [Google Scholar]
- 158.Peden DB, Boehlecke B, Horstman D, Devlin R. Prolonged acute exposure to 0.16 ppm ozone induces eosinophilic airway inflammation in asthmatic subjects with allergies. J Allergy Clin Immunol. 1997;100(6 Pt 1):802–808. doi: 10.1016/s0091-6749(97)70277-x. [DOI] [PubMed] [Google Scholar]
- 159.Neuhaus-Steinmetz U, Uffhausen F, Herz U, Renz H. Priming of allergic immune responses by repeated ozone exposure in mice. Am J Respir Cell Mol Biol. 2000;23(2):228–233. doi: 10.1165/ajrcmb.23.2.3898. [DOI] [PubMed] [Google Scholar]
- 160.Osebold JW, Gershwin LJ, Zee YC. Studies on the enhancement of allergic lung sensitization by inhalation of ozone and sulfuric acid aerosol. J Environ Pathol Toxicol. 1980;3(5–6):221–234. [PubMed] [Google Scholar]
- 161.Osebold JW, Zee YC, Gershwin LJ. Enhancement of allergic lung sensitization in mice by ozone inhalation. Proc Soc Exp Biol Med. 1988;188(3):259–264. doi: 10.3181/00379727-188-42733. [DOI] [PubMed] [Google Scholar]
- 162.Gershwin LJ, Osebold JW, Zee YC. Immunoglobulin E-containing cells in mouse lung following allergen inhalation and ozone exposure. Int Arch Allergy Appl Immunol. 1981;65(3):266–277. doi: 10.1159/000232766. [DOI] [PubMed] [Google Scholar]
- 163.Schelegle ES, Miller LA, Gershwin LJ, Fanucchi MV, Van Winkle LS, Gerriets JE, Walby WF, Mitchell V, Tarkington BK, Wong VJ, Baker GL, Pantle LM, Joad JP, Pinkerton KE, Wu R, Evans MJ, Hyde DM, Plopper CG. Repeated episodes of ozone inhalation amplifies the effects of allergen sensitization and inhalation on airway immune and structural development in Rhesus monkeys. Toxicol Appl Pharmacol. 2003;191(1):74–85. doi: 10.1016/s0041-008x(03)00218-7. [DOI] [PubMed] [Google Scholar]
- 164.Biagini RE, Moorman WJ, Lewis TR, Bernstein IL. Ozone enhancement of platinum asthma in a primate model. Am Rev Respir Dis. 1986;134(4):719–725. doi: 10.1164/arrd.1986.134.4.719. [DOI] [PubMed] [Google Scholar]
- 165.Eisenbarth SC, Piggott DA, Huleatt JW, Visintin I, Herrick CA, Bottomly K. Lipopolysaccharide-enhanced, toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J Exp Med. 2002;196(12):1645–1651. doi: 10.1084/jem.20021340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Hollingsworth JW, Free ME, Li Z, Andrews LN, Nakano H, Cook DN. Ozone activates pulmonary dendritic cells and promotes allergic sensitization through a Toll-like receptor 4-dependent mechanism. J Allergy Clin Immunol. 2010;125(5):1167–1170. doi: 10.1016/j.jaci.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]