

Abstract
Mutations in the BRCA1 gene substantially increase a woman's lifetime risk of breast cancer. However, there is great variation in this increase in risk with several genetic and non-genetic modifiers identified. The BRCA1 protein plays a central role in DNA repair, a mechanism that is particularly instrumental in safeguarding cells against tumorigenesis. We hypothesized that polymorphisms that alter the expression and/or function of BRCA1 carried on the wild-type (non-mutated) copy of the BRCA1 gene would modify the risk of breast cancer in carriers of BRCA1 mutations. A total of 9874 BRCA1 mutation carriers were available in the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA) for haplotype analyses of BRCA1. Women carrying the rare allele of single nucleotide polymorphism rs16942 on the wild-type copy of BRCA1 were at decreased risk of breast cancer (hazard ratio 0.86, 95% confidence interval 0.77–0.95, P = 0.003). Promoter in vitro assays of the major BRCA1 haplotypes showed that common polymorphisms in the regulatory region alter its activity and that this effect may be attributed to the differential binding affinity of nuclear proteins. In conclusion, variants on the wild-type copy of BRCA1 modify risk of breast cancer among carriers of BRCA1 mutations, possibly by altering the efficiency of BRCA1 transcription.
INTRODUCTION
Germline mutations in BRCA1 drastically increase breast cancer risk. They mainly consist of truncating mutations leading to loss of function of the mutant allele. In a meta-analysis of families with BRCA1 mutations detected through population-based studies, the risk of breast cancer by the age of 70 among BRCA1 mutation carriers was estimated to be 65% (1,2). This is lower than estimates based on families with multiple affected individuals (3). Furthermore, factors such as the age at diagnosis and the type of cancer in the index patient that led to the family ascertainment have been associated with differences in breast cancer risk among BRCA1 mutation carriers (1,2,4,5). The extent of breast cancer risk variability among mutation carriers in terms of a polygenic-modifying variance was estimated using segregation-analysis models (2,6). These observations led to the hypothesis that breast cancer risk among mutation carriers is modified by other genetic or environmental factors. In order to facilitate the analysis of genetic modifiers of risk for BRCA1 and BRCA2 mutation carriers across several studies, CIMBA (Consortium of Investigators of Modifiers of BRCA1/2) was established in 2005 (7), and since then multiple loci that modify risk among BRCA1/2 mutation carriers have been identified by this consortium (8–17).
The wild-type copy of BRCA1 is an interesting candidate for cancer risk modification given that the activity of the protein produced by the intact allele may influence cancer penetrance in individuals who have one inactivated BRCA1 copy, as it is the case for BRCA1 mutation carriers. Indeed, the BRCA1 protein plays a central role in DNA repair: it is not only essential for the repair of double-strand breaks by homologous recombination, but it is also required to signal the presence of these severe lesions to the cell (18). Yet, both environmental factors and normal biological activities constantly damage DNA through the course of an individual's lifetime. When normal repair processes fail and apoptosis does not occur, irreparable DNA damage in oncogenes and tumor suppressor genes may occur, ultimately leading to unregulated cell division and in turn to the formation of a tumor. Evidence that the amount and/or quality of the BRCA1 protein produced in cells might be important for keeping DNA integrity comes from several studies that have investigated cellular response to DNA damage in BRCA1 mutation carriers. High frequencies of micronuclei induction and chromosomal aberrations after exposure to mutagens have been reported (19–24), although not consistently (25–27). Furthermore, the observation that mRNA profiles are altered in normal breast epithelial cells heterozygous for mutations in BRCA1, including those of critical genes involved in BRCA signaling pathways, suggests that even a small alteration in the levels of BRCA1 may result in differential gene expression. It also indicates that BRCA1 haploinsufficiency is likely to be a driving mechanism leading to tumorigenesis in carriers (28) and, in turn, supports the hypothesis that individual BRCA1 variations may affect cancer risks in this population.
BRCA1 single nucleotide polymorphisms (SNPs) could exert their effect through two non-exclusive ways: missense polymorphisms could slightly modify BRCA1 protein function or stability, or SNPs could alter BRCA1 expression by acting on transcription, splicing or translation. It is not known as yet whether the few reported frequent BRCA1 missense polymorphisms alter BRCA1 function or stability. This is certainly not surprising given the technical challenges presented by the precise assessment of subtle changes in protein efficiency or stability, even more so in the case of BRCA1 because of its many described functions, large size and the poor quality of the antibodies directed against this protein. On the other hand, it is technically straightforward to accurately measure transcript levels in order to monitor gene expression and numerous such studies on BRCA1 have been published. In this regard, allelic imbalance of BRCA1 expression has been repeatedly reported, fueling the hypothesis that SNPs could influence BRCA1 transcription efficiency even if the mechanism(s) leading to the observed allelic imbalance is for the most part unknown (29–31).
Conflicting results concerning the associations between polymorphisms in BRCA1 and breast cancer risk in the general population have been reported (29,31–35). While three studies on Caucasian populations could not demonstrate any association (32,34,35), a more recent study of BRCA1 promoter polymorphisms identified four variants altering promoter activity which could affect susceptibility to breast cancer in the Chinese population (33). Furthermore, BRCA1 allelic imbalance has been shown to be associated with enhanced susceptibility to breast and/or ovarian cancer (29,31).
In 2003, we tested the hypothesis that polymorphisms in the wild-type copy of the BRCA1 gene could modify the risk of breast cancer among women with BRCA1 mutations, but the limited number of BRCA1 carriers that we were then able to genotype, and the less advanced state of knowledge of the patterns of human common genetic variation, prevented us from providing a convincing result (36).
With the establishment of CIMBA, we readdressed this question by determining common BRCA1 haplotypes and studying their effect in 9874 women with germline BRCA1 mutations. We also assessed the functional significance of the major BRCA1 promoter haplotypes.
RESULTS
In the present study, we genotyped SNPs tagging the BRCA1 region in BRCA1 mutation carriers and whenever possible reconstructed haplotypes in order to test the hypothesis that the wild-type copy of BRCA1, in these carriers, might modify cancer risk. As a second step, we studied the possibility that such an effect could be exerted through differential transcription efficiency.
Analysis of the association between wild-type BRCA1 genotypes and breast cancer risk in BRCA1 mutation carriers
While the BRCA1 gene is large (>80kb), only one block of linkage disequilibrium (LD) exists across the entire locus (Fig. 1 ), resulting in the occurrence of two major haplogroups tagged by the rs16942 SNP (see Materials and Methods). This tagging SNP was genotyped in 9874 BRCA1 mutation carriers available for these analyses when combining samples from 32 participating CIMBA centers (Table 1). Allele frequencies of rs16942 in our analysis were, on average, similar to those observed for other white populations (minor allele frequency ∼33%).
Figure 1.
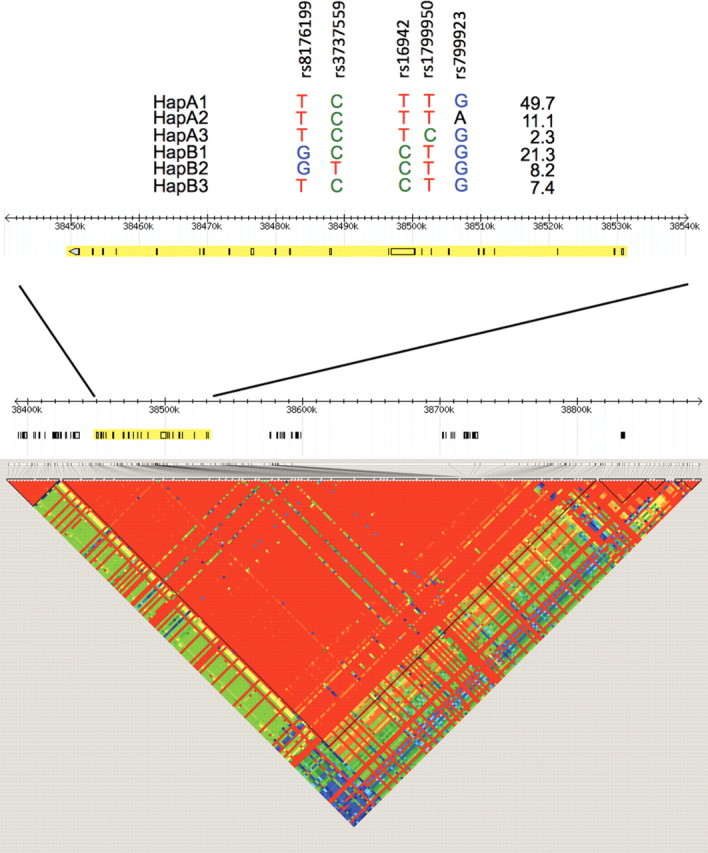
Haplotypes and LD structure of the BRCA1 gene. Region surrounding BRCA1 on chromosome 17 using HapMap Data Release 27 Phase II and III data, February 2009 build (accessed on 2 September 2010), on NCBI B36 assembly and dbSNP b126. SNPs used in haplotype analyses are shown, relative to their position in the BRCA1 gene.
Table 1.
Number of eligible BRCA1 mutation carriers by study group
| Study | Countrya | BRCA1 carriers | Genotyping platform |
|---|---|---|---|
| Breast Cancer Family Registry (BCFR) | USA/Australia/Canada | 592 | Taqman |
| Copenhagen Breast Cancer Study (CBCS) | Denmark | 162 | Taqman |
| Spanish National Cancer Centre (CNIO) | Spain | 156 | Taqman |
| CONsorzio Studi ITaliani sui Tumori Ereditari Alla Mammella (CONSIT TEAM) | Italy | 416 | Taqman |
| Deutsches Krebsforschungszentrum (DKFZ) | Germany | 160 | Taqman |
| HEreditary Breast and Ovarian study Netherlands (HEBON) | Netherlands | 773 | iPLEXb |
| Epidemiological Study of BRCA1 and BRCA2 Mutation Carriers (EMBRACE) | UK/Eire | 843 | iPLEXb |
| Fox Chase Cancer Centre (FCCC) | USA | 80 | iPLEXb |
| German Consortium of Hereditary Breast and Ovarian Cancer (GC-HBOC) | Germany | 916 | Taqman |
| Genetic Modifiers of cancer risk in BRCA1/2 mutation carriers (GEMO) | France/USA | 1100 | Taqman |
| Georgetown University (GEORGETOWN) | USA | 33 | iPLEXb |
| Gynecologic Oncology Group (GOG) | USA | 406 | Taqman |
| Hospital Clinico San Carlos (HCSC) | Spain | 116 | Taqman |
| Helsinki Breast Cancer Study (HEBCS) | Finland | 103 | iPLEXb |
| Institut Català d'Oncologia (ICO) | Spain | 113 | Taqman |
| Iceland Landspitali - University Hospital (ILUH) | Iceland | 6 | iPLEXb |
| Interdisciplinary Health Research International Team Breast Cancer Susceptibility (INHERIT BRCAs) | Canada (Quebec) | 73 | Taqman |
| Istituto Oncologico Veneto—Hereditary Breast Ovarian Cancer Study (IOVHBOCS) | Italy | 108 | Taqman |
| Kathleen Cuningham Foundation Consortium for research into Familial Breast cancer (kConFab) | Australia/New Zealand | 526 | iPLEXb |
| Mayo Clinic (MAYO) | USA | 218 | iPLEXb |
| Modifier Study of Quantitative Effects on Disease (MOD SQUAD) | Czech Republic/Belgium | 271 | Taqman |
| General Hospital Vienna (MUV) | Austria | 294 | iPLEXb |
| National Cancer Institute (NCI) | USA | 142 | Taqman |
| Ontario Cancer Genetics Network (OCGN) | Canada | 224 | Taqman |
| Ohio State University Clinical Cancer Center (OSU CCG) | USA | 80 | Taqman |
| Odense University Hospital (OUH) | Denmark | 263 | Taqman |
| Pisa Breast Cancer Study (PBCS) | Italy | 75 | iPLEXb |
| Sheba Medical Centre (SMC)—Tel Hashomer | Israel | 501 | Taqman |
| Swedish Breast Cancer Study (SWE-BRCA) | Sweden | 470 | iPLEXb |
| University of California Irvine (UCI) | USA | 193 | Taqman |
| UK and Gilda Radner Familial Ovarian Cancer Registries (UKGRFOCR) | UK/USA | 187 | Taqman |
| University of Pennsylvania (UPENN) | USA | 274 | iPLEXb |
| Total | 9874 |
aCountry of the clinic at which carriers are recruited.
bCentralized genotyping at Queensland Institute of Medical Research.
The results of association tests between rs16942 of the wild-type BRCA1 allele and breast cancer risk among BRCA1 mutation carriers are shown in Table 2. The analysis restricted to women homozygous for rs16942, in whom the phase of rs16942 with respect to their mutation was unambiguous (n = 5652), showed an inverse association between the C (minor) allele and breast cancer risk [hazard ratio (HR) 0.85, 95% confidence interval (CI) 0.74–0.96 P = 0.01]. Using familial information, we were able to phase an additional 1396 subjects (n = 7048), further refining the risk estimate to 0.86, 95% CI 0.77–0.95, P = 0.003. In these analyses, no heterogeneity was observed among centers (P = 0.94) (Fig. 2). HRs in analyses that excluded breast cancer cases diagnosed more than 1 year prior to the interview were similar to the overall results (HR 0.84, 95% CI 0.75–0.93). No differences in breast cancer risk between BRCA1 carriers of Class 1 (loss of function) or Class 2 (likely to generate potentially stable mutant protein) mutations were observed (Table 2).
Table 2.
Association between rs16942 genotypes on ‘wild-type’ allele of BRCA1 and breast cancer risk
| ‘T’ allele on wild-type (reference) |
‘C’ allele on wild-type |
|||||||
|---|---|---|---|---|---|---|---|---|
| Phasing method (see Materials and Methods) | Unaffected | Affected | Person-years | Unaffected | Affected | Person-years | HR (95% CI)a | P-value |
| rs16942 homozygotes | ||||||||
| Overall | 2065 | 2404 | 185 732 | 607 | 576 | 49 756 | 0.85 (0.74–0.96) | 0.01 |
| Family-based | ||||||||
| Overall | 2339 | 2608 | 206 355 | 1142 | 959 | 87 576 | 0.86 (0.77–0.95) | 0.003 |
| Class 1 mutations | 1636 | 1615 | 205 204 | 904 | 710 | 68 197 | 0.88 (0.79–0.99) | 0.04 |
| Class 2 mutations | 564 | 776 | 68 103 | 166 | 167 | 13 290 | 0.92 (0.71–1.20) | 0.55 |
aHRs and 95% CIs calculated using weighted Cox regressions with a robust sandwich estimator.
Figure 2.

Study-specific HRs between rs16942 genotypes on wild-type allele of BRCA1 and breast cancer risk. HRs were calculated in each specific study as described in Materials and Methods. The summary effect estimate for C allele carriers is 0.87 (0.78–0.97), p-heterogeneity 0.91 with a fixed effect model.
For some centers, additional SNPs that tag the six frequent haplotypes of the haplogroups A and B were genotyped. BRCA1 wild-type haplotypes could be inferred for a total of 1033 breast cancer cases and 1049 unaffected controls (Table 3, Fig. 1). Haplotype A2 showed an inverse association with breast cancer risk when compared with the referent haplotype A1 (HR 0.67, 95% CI 0.51–0.90, P = 0.007). Actually, all other haplotypes (A3, B1, B2 and B3) also had inverse, though weaker, associations with breast cancer risk.
Table 3.
Association between BRCA1 haplotypes on ‘wild-type’ allele of BRCA1 and breast cancer risk using family-based phasing (see Material and Methods)
| Group | Unaffected |
Affected |
HR (95% CI)a | P-value | ||
|---|---|---|---|---|---|---|
| Number | Person-years | Number | Person-years | |||
| HapA1 | 521 | 22 388 | 615 | 24 935 | 1.00 (Ref.) | |
| HapA2 | 116 | 5569 | 104 | 4306 | 0.67 (0.51–0.90) | 0.007 |
| HapA3 | 24 | 962 | 17 | 694 | 0.81 (0.41–1.61) | 0.37 |
| HapB1 | 224 | 9421 | 165 | 6673 | 0.77 (0.61–0.99) | 0.04 |
| HapB2 | 86 | 3686 | 69 | 2761 | 0.71 (0.50–1.02) | 0.07 |
| HapB3 | 78 | 3258 | 63 | 2663 | 0.80 (0.56–1.15) | 0.23 |
aHRs and 95% CIs calculated using Cox regressions.
Functional studies
More than 120 common SNPs (allele frequency >5%) have been detected across the BRCA1 LD block. Ten are located in the BRCA1 coding sequence, of which 7 are non-synonymous. The minor alleles of these seven missense SNPs are carried on the haplotype A2 (Asp693Asn—rs4986850), haplotype A3 (Gln356Arg—rs1799950 and Ser1040Asn—rs4986852) or haplogroup B (Pro871Leu—rs799917, Glu1038Gly—rs16941, Lys1183Arg—rs16942 and Ser1613Gly—rs1799966). These missense variants could potentially alter the function or the stability of the BRCA1 protein. However, none of them is located in a recognized functional domain of BRCA1 and they are predicted to be neutral by commonly used algorithms for assessing the functional effects of missense variants such as Align-GVGD and SIFT, mainly based on phylogenetic information and biochemical differences between the reference and variant amino acid.
On the other hand, differential allelic expression for BRCA1 has been reported in different studies, and it was hypothesized that variants in the promoter region could be involved in the regulation of this differential expression. We therefore chose to investigate the effect of SNPs located in the BRCA1 promoter region on transcription efficiency to try to explain the genetic effect revealed by our study.
Haplogroups A and B carry five frequent SNPs in the 2 kb region upstream BRCA1; the haplotype regions corresponding to the BRCA1 promoter were named thereafter pHapA and pHapB. As an initial approach to assess the functional significance of DNA variations in the promoter region, we performed transcriptional activity analyses using a gene-reporter assay in a HeLa cell line. As illustrated in Figure 3, differential transcriptional activity was observed between the two major haplotypes (27% decrease, P-value = 0.0011). These experiments were performed five times and the mean relative luciferase activity driven by pHapB was 20% lower than the levels driven by its counterpart pHapA, suggesting allele-specific differential promoter activity.
Figure 3.

Representative luciferase reporter assays of major BRCA1 haplotype constructs in HeLa cell line. Relative luciferase activity of constructs carrying BRCA1 promoter sequences corresponding to two haplotypes was measured following transient transfection into HeLa cells. The ratio of firefly:Renilla luciferase activity of each promoter construct was normalized against that of the promoter-less pGL3-Basic vector. The pGL3-SV40 vector containing the SV40 early promoter is used as a positive control. Promoter haplotype BRCA1-pHapA was used as reference against which pairwise comparisons were made. Significant differences between haplotype expressions are marked with asterisks (**P = 0.0011). The position of each SNP is given relative to the first base of the initiation codon.
To further investigate the effect of genetic variants of the BRCA1 promoter region on gene expression, we assessed the impact of these SNPs on DNA–protein binding capacity. Electrophoretic mobility shift assays (EMSAs) were performed for all identified SNPs using nuclear extracts from HeLa, MCF7 and MDA-MB-231 cell lines (Fig. 4). The EMSA assay performed on probes including polymorphism rs4793204 gave the most convincing results with clear-cut differential binding of nuclear proteins to probes carrying either the T or the C allele in all three cell lines. As illustrated in Figure 4A, probe-specific differential binding was observed for the complexes identified by solid arrows, which showed binding affinity for the rs4793204-T probe (Fig. 4, lane 2) but not for the rs4793204-C probe (Fig. 4, lane 8). Competition EMSAs performed in the presence of 50-fold molar excess of the unlabeled T probe confirmed binding specificity (Fig. 4, lane 3).
Figure 4.

Representative EMSAs illustrating allelic DNA–protein interactions in the promoter region of BRCA1. Labeled double-stranded oligonucleotide (ds-oligo) probes containing either allele of rs4793204 (A) and rs799908 (B) were incubated with nuclear extracts from HeLa, MCF7 and MDA-MB-231 cells. (A) Lanes 1–6 represent binding to the labeled ds-oligo containing the major allele; lanes 7–12, binding to the ds-oligo containing the minor allele. Lanes 1 and 7, negative control (no extracts). Lanes 3 and 10, competition with unlabeled allelic probes (specific competitors). Lanes 4 and 9, competition with mismatched unlabeled probes. Lanes 5 and 11, competition with a non-specific probe (non-specific competitor). Lanes 6 and 12, pre-incubation with anti-BRG1 antibody. (B) Lanes 1–5 represent binding to the labeled ds-oligo containing the major allele; lanes 6–10, binding to the ds-oligo containing the minor allele. Lanes 1 and 6, negative control (no extracts). Lanes 3 and 9, competition with unlabeled allelic probes (specific competitors). Lanes 4 and 8, competition with mismatched unlabeled probes. Lanes 5 and 10, competition with a non-specific probe (non-specific competitor).
In an attempt to identify the specific transcriptional factors responsible for this differential binding, in silico analyses were performed to assess the potential functional impact of SNP rs4793204 on predicted transcription factor binding sites (TFBSs). These analyses revealed that rs4793204 might alter the recognition/binding motifs of Brg-1, Oct-1 and Nkx-3.1. Further EMSAs were performed in the presence of antibodies raised against these transcription factors. Assays performed in the presence of anti-BRG1 antibodies revealed a supershifted band with the probe carrying the T allele in all cell lines tested (Fig. 4, lanes 6, dotted arrow). No supershift was observed when the assays were performed with antibodies raised against Oct-1 and Nkx-3.1 (data not shown). In the case of rs799908, several DNA–protein-specific complexes were observed with the rs799908-C probe using nuclear extracts from MCF7 and MDA-MB-231 cells, but not from HeLa cells, as illustrated in Figure 4B (solid arrows), suggesting cell-specific transcription factor binding. In silico analyses predicted that the minor allele of rs799908 altered the binding motif of transcription factors Elk-1, USF-1 and USF-2. However EMSAs performed in the presence of antibodies raised against any of these proteins did not yield supershifted bands, thus not allowing us to confirm that these transcription factors were responsible for the observed differential binding in these cell lines (data not shown).
The results we obtained for both of these SNPs suggest that the haplotype-specific differences observed in gene-reporter assays might involve differential binding of transcription factors in this promoter region. The other investigated SNPs located in the BRCA1 promoter region, rs11655505–rs799906–rs8176071, did not show significant allele-specific differential binding, as revealed by EMSA analyses using nuclear extracts from HeLa, MCF7 and MDA-MB-231 cells (data not shown).
DISCUSSION
The primary objective of this study was to test the hypothesis that polymorphisms of the non-mutated (wild-type) BRCA1 allele modify breast cancer risk among women who carry a BRCA1 mutation. We observed an association between a BRCA1 tag SNP, rs16942, and breast cancer risk, with women who carried the minor allele on their non-mutated BRCA1 copy having ∼14% decrease in risk (HR 0.86, 95% CI 0.77–0.95, P = 0.003). Perhaps not surprisingly, when phase was not taken into account, we observed weaker association between rs16942 and breast cancer risk using a per-allele test for trend (P = 0.02).
Characterizing the biological basis for this genetic effect is an important step towards further understanding of breast cancer susceptibility linked to BRCA1, and subsequent use in genetic counseling. Association studies, however, are limited in their ability to definitively identify causal variants due to correlation, or LD, between adjacent polymorphisms. This is particularly true for BRCA1 located within a 390 kb long block of LD. A constellation of more than 120 common SNPs exists across this LD block. Ten of these common SNPs are in the BRCA1 coding sequence, of which 7 are non-synonymous, potentially altering the function or the stability of the BRCA1 protein. However, the two most commonly used algorithms that evaluate the predicted pathogenicity of a missense variant revealed no potential effect. It should be noted that, where such an effect suspected, its assessment would prove difficult as only subtle function or stability impairment can be expected in this context. Another non-exclusive explanation for the genetic effect described here is that one or several SNPs alter(s) the level of expression of BRCA1, ultimately altering the amount of biologically active BRCA1. In favor of this hypothesis, previous studies have reported differential allelic expression of BRCA1 in lymphoblastoid cell lines, in B lymphocytes and in breast tissue (29–31). This BRCA1 differential allelic expression has been in some instances associated with breast (29) and ovarian (31) cancer susceptibility. Analysis of expression data available from the Genevar (GENe Expression VARiation) database (37) indicates that polymorphisms within the BRCA1 locus, including rs16942, were associated with BRCA1 expression. In lymphocytes, the most significant correlation coefficient between polymorphisms and BRCA1 expression (probe ILMN_1738027) was observed with rs16942, with a value of 0.41 (P = 2.2E−4). Some recent evidence that variation of expression levels is correlated with polymorphisms in the promoter region of BRCA1 (33) prompted us further to investigate the possibility that transcription efficiency could explain the genetic effect shown in this study.
We thus tried to assess the functional significance of five common SNPs present in the 2 kb BRCA1 promoter, using EMSA and in vitro transcriptional assays. Our results suggest that these polymorphisms can be involved in differential allelic expression. Indeed, EMSAs revealed that two of the five SNPs located in the promoter region showed allele-specific differential capacity of binding to nuclear proteins in HeLa cells and/or ER-positive (MCF7) and ER-negative (MDA-MB-231) breast cancer cell lines. For one of the SNPs, rs4793204, we observed in all cell lines studied a differential protein binding capacity to the BRG1 transcription factor, a subunit of the SWI/SNF chromatin remodeling complex previously shown to repress the BRCA1 promoter reporter activity (38). One can speculate that loss or decreased binding capacity of the minor C allele, carried on haplogroup B (and associated with decreased breast cancer risk for BRCA1 mutation carriers), to this transcription factor could lead to loss of repression resulting in an increase in the level of expression. Conversely, our gene-reporter assays show a tendency for the minor haplotype (corresponding to haplogroup B) to be expressed at lower levels than the major haplotype. However, it should be noted that only the proximal promoter region (2 kb upstream of the transcription start site) was used in these assays, as classically done and it is therefore possible that other regulatory elements or cis-regulatory modules present outside this region and potentially influencing the expression of this gene were missed. Indeed, regulatory elements can be located in far upstream and downstream regions still within the large block of LD surrounding BRCA1. Furthermore, given that BRCA1 allelic expression levels vary significantly between cell types (30), the results we obtained using luciferase reporter assays in HeLa cell line may be different in other cell types. Further studies will be needed to decipher the impact of putative regulatory SNPs in the complex transcriptional activation of the BRCA1 gene.
We genotyped four additional SNPs in 4050–4816 of the initial 9874 BRCA1 mutation carriers in order to define more precisely the haplotype(s) associated with modification of breast cancer risk. This analysis allowed us to define six haplotypes, three in haplogroup A and three in haplogroup B. It is interesting to note that all haplotypes carried by the wild-type allele were inversely associated with breast cancer risk when compared with the reference haplotype, haplotype A1. This implies that an allele(s) that alter(s) BRCA1 expression may actually be carried on haplotype A1 and not on the other haplotypes. However, caution must be used when interpreting our haplotype results, due to the reduction in sample size, which led to reduced power for these analyses. Indeed, an important limitation of the power of this study is that we restricted our analyses to homozygotes and heterozygotes that we were able to phase based on family information. Hence, although a total of 9874 BRCA1 mutation carriers were initially genotyped for rs16942, we were able to phase genotypes in 7048 individuals. For the haplotype analysis, this restriction had more serious consequence as haplotypes on the wild-type allele of BRCA1 could only be inferred in 2082 mutation carriers.
In addition to the haplotype analysis including four SNPs in LD with rs16942, a number of other related hypotheses have been evaluated, including the associations with different mutation types. None of the reported P-values has been adjusted for multiple testing as it is unclear what the correct type of adjustment would be in this context. However, both the observed P-values for the primary single SNP analysis and secondary haplotype analysis (P = 0.003 and 0.007, respectively) would survive a conservative Bonferroni correction (based on five and six tests, respectively). Furthermore, the functional evidence with respect to BRCA1 expression provides additional evidence for the association between this variant and breast cancer risk for BRCA1 mutation carriers.
In conclusion, we have shown an inverse association between minor polymorphic variants of the wild-type allele of the BRCA1 gene and breast cancer risk among women who carry a BRCA1 mutation. This association was limited to women who carry the variant allele of rs16942 on their wild-type (non-mutated) allele of BRCA1. This association is most likely due to the influence on BRCA1 expression of variants that are in LD with rs16942, which tags the two major haplotype groups present across the LD block of BRCA1. Some of these variants are likely to reside in the promoter, as we have shown here that polymorphisms located in the 2 kb promoter region of BRCA1 appear to be involved in differential allelic expression. However, it is reasonable to suspect that the polymorphisms we have examined, both in terms of association and functional testing, are not solely responsible for the genetic effect depicted here. Identification of true causal variants will provide important insight into the mechanisms by which BRCA1 exerts its tumor suppressor role in breast cancer.
MATERIALS AND METHODS
Ethics statement and study population
Eligible study subjects were women aged ≥18 years who carry a pathogenic mutation in BRCA1. Information on study subjects was submitted from 32 studies from 20 countries (Table 1). These women participated in clinical and research studies at the host institutions under institutional review board approved protocols. Data collected included year of birth, mutation description, family membership, ethnicity, country of residence, age at last follow-up, ages at diagnosis of breast and/or ovarian cancer, and information on bilateral prophylactic mastectomy. Mutations were included in the analysis if they were pathogenic according to generally recognized criteria.
To examine whether the effects of the SNPs are different in individuals carrying different types of mutations, we classified mutations according to their functional effect. Class 1 mutations (number of carriers = 7109) were defined as loss of function mutations expected to result in a reduced transcript or protein level because of nonsense-mediated mRNA decay (NMD) and/or degradation or instability of truncated proteins (39–42), translation re-initiation but no production of stable protein (43) or the absence of expression due to deletion of transcription regulatory regions. Class 2 mutations (number of carriers = 2085) comprised mutations likely to generate potentially stable mutant proteins that might have a dominant negative action, partially preserved normal function or loss of function. Class 2 mutations are missense substitutions and truncating mutations not triggering NMD (premature stop codon occurring in the last exon). A small proportion of mutations (number of carriers = 680) could not be categorized as belonging to Class 1 or Class 2.
Selection of haplotype tagging SNPs
To select a set of SNPs efficiently capturing common variation (tagSNPs) in the genomic region of BRCA1, we used data available from the HapMap project on CEPH trios (Utah-USA residents with ancestry from Northern and Western Europe) (http://www.hapmap.org). The 82 kb long BRCA1 gene is located within a 390 kb long block of LD that also comprises roughly 20 kb and 290 kb at its 5′ and 3′ ends, respectively. TagSNPs were selected using the ‘Haploview 4.0’ tool (http://www.broad.mit.edu/mpg/haploview/) (44). The ‘Tagger’ program was used to select a minimal set of tagSNPs such that all alleles to be captured (frequency >5%) were correlated at an r2 greater than 0.8 threshold (45). This resulted in the selection of five tagSNPs: rs16942, rs179950, rs799923, rs3737559 and rs8176199. The rs16942 SNP tags the two major haplogroups (further named haplogroups A and B). The combination of rs179950 and rs799923 tags haplotype A1, rs179950 tags haplotype A3 and rs799923 tags haplotype A2. rs3737559 tags haplotype B2 and rs8176199 tags haplotype B3, with the combination of these two SNPs tagging haplotype B1 (Fig. 1).
Genotyping and phasing
SNP rs16942 and minor haplotype tagging SNPs were genotyped using the 5′ nuclease assay (TaqMan) on the ABI 7900HT Sequence Detection System (Applied Biosystems) or using the iPLEX Mass Array platform. Additional SNPs that were genotyped varied by center (see details in Supplementary Material, Table S1). All centers included at least 2% of samples in duplicate, no template controls on every plate and a random mixture of samples of affected and unaffected mutation carriers on each plate. The minimum acceptable call rate was 95%. For each study, the genotype frequencies among unrelated carriers were consistent with the expected frequencies under the assumption of Hardy–Weinberg equilibrium.
A total of 9874 BRCA1 mutation carriers (5176 affected and 4698 unaffected) were available for these analyses. As our hypothesis was that the haplotype carried on the non-mutated BRCA1 allele would modify breast cancer risk, we initially restricted our analyses to carriers homozygous for rs16942 (4469 T/T homozygotes, 1183 C/C homozygotes). Since both rs16942 genotype and mutation status were available from multiple family members for some of the subjects, we next used this information to infer the phase of rs16942 alleles among heterozygotes with the specific mutation in each family. Specifically, we assumed that within each family, there was little probability of recombination between the mutation and rs16942. Therefore, if an rs16942 heterozygote and homozygote were observed within the same family, we assumed that the mutation was carried with the allele for which the family member was homozygous in that specific family. Polymorphisms rs179950, rs799923, rs3737559 and rs8176199 that define haplotypes within the major haplogroups tagged by rs16942 were genotyped in a subset of CIMBA centers (Supplementary Material, Table S1). Due to the complete LD between these SNPs, haplotypes were determined and phasing was carried out as for rs16942 described above.
In silico assessment of functional effects of missense SNPs
To predict potential functional impact of the BRCA1 missense SNPs, we used web-based algorithms with default settings: Align-GVGD (http://agvgd.iarc.fr/) (46) and SIFT (http://sift.jcvi.org/) (47).
Statistical analyses
To evaluate the association between wild-type BRCA1 genotype and breast cancer risk in BRCA1 mutation carriers, their phenotype was defined by their age at diagnosis of breast cancer or their age at last follow-up. For this purpose, individuals were censored at the age of the first of the following events: breast cancer diagnosis, ovarian cancer diagnosis, bilateral prophylactic mastectomy or last observation, and only carriers censored at breast cancer diagnosis were assumed to be affected. Risk reducing salpingo-oophorectomy was not considered in the analysis as it is not expected to be associated with the underlying SNP genotype (i.e. it is not a confounder).
Studying the associations with cancer risk for BRCA1 mutation carriers is complicated by the fact that mutation carriers in our study design are not randomly sampled with respect to their disease phenotype. Genetic testing is targeted at families with multiple affected individuals, and most genetic clinics tend to screen first young, affected family members. Therefore, the selection of mutation carriers is not random with respect to disease status or age at diagnosis. These study designs lead therefore to an oversampling of young affected mutation carriers. It has been shown in the past that under such study designs, standard cohort analysis (such as Cox regression, which assumes random sampling with respect to phenotype) yield biased estimates of the risk ratios. This can be illustrated by considering an individual affected at age t. In a standard analysis of a cohort study, the SNP genotype for the individual will be compared with those of all individuals at risk at age t. This analysis leads to consistent estimates of the HR. However, in the present design, mutation carriers are already selected on the basis of disease status (where affected individuals are over-sampled). If standard cohort analysis were applied to these data, it would lead to affected individuals at age t being compared with unaffected carriers selected on the basis of their future disease status. If the genotype is associated with the disease, the risk estimate will be biased to zero because too many affected individuals (in whom the at-risk genotype is overrepresented) are included in the comparison group. Simulation studies have shown that this effect can be quite marked.
To overcome this problem, a weighted cohort approach was previously proposed, under which affected and unaffected individuals are differentially weighted according to their age at diagnosis or last observation such that on the observed weighted age-specific BRCA1 breast cancer incidences in the study sample agree with established breast cancer incidences in mutation carriers (47,48). This method has been shown to provide risk ratio estimates which are close to unbiased (47,48). For analyses of rs16942, we estimated the log-HRs for CC genotypes using the TT homozygotes as the baseline category. Haplotype analyses used haplogroup A1 (the most common haplotype) as the baseline category. As some of the study participants had censoring events (bilateral mastectomy or breast cancer diagnosis) prior to study inclusion interview (887 unaffected, 228 affected), we also carried out analyses restricted to patients with censoring events less than 1 year prior to their study interview in order to exclude long-term survivors. We also performed analyses to examine whether SNP associations differed by type of BRCA1 mutations (class 1 and class 2 mutations). All analyses were stratified by study and country of residence. As sufficient detail regarding degree of family history was not available for all subjects, we were unable to take this into consideration in our analyses. In all instances, a robust variance approach was used to allow for the dependence between related carriers (49). Most statistical analyses were carried out in SAS v.9.1, with the exception of heterogeneity testing which used the rmeta package in R 2.10.1.
BRCA1 promoter polymorphisms
BRCA1 shares a well-characterized bi-directional promoter with its neighboring gene NBR2 in a 229 bp intergenic region. As is generally done in classical promoter studies, a 2 kb region upstream of the BRCA1 transcription start site was chosen (50,51) to assess the impact of upstream genetic variants on promoter activity. The 2067bp of the BRCA1 promoter region (chr17:41277361–41279427, GRCh37/hg19) was found in HapMap to contain five frequent polymorphisms that were confirmed on a population panel consisting of 40 unrelated individuals from five continental groups: rs4793204, rs799908, rs11655505, rs799906 and rs8176071. No other frequent SNPs were identified by sequencing. It should be noted that these five SNPs are in almost complete LD with rs16942 (r2 > 0.961). The haplotype regions corresponding to the BRCA1 promoter were named pHapA and pHapB.
In silico prediction of putative TFBSs
A computer-based search for putative transcription factor binding elements harbored by the BRCA1 promoter sequence corresponding to each of the two pHaps was performed using the MatInspector software (http://www.genomatix.de/online_help/help_matinspector/matinspector_help.htm) (52). Transcription factors that putatively bind to the sense strand sequence of the BRCA1 promoter in humans were identified, and those showing significantly altered predicted scores for any of the pHaps were selected for further analysis.
Cell culture
The human cervical carcinoma cell line HeLa was grown in EMEM (Wisent Bioproducts, St-Bruno, Québec, Canada) supplemented with 5% fetal bovine serum (FBS) and 1% penicillin–streptomycin. The human breast adenocarcinoma cell lines MCF7 and MDA-MB-231 were grown in DMEM/F12 (Wisent Bioproducts, St-Bruno, Québec, Canada) supplemented with 5% FBS, 1% penicillin–streptomycin and 10-9 m oestradiol (E2). All cells were grown at 37°C in a 5% CO2 incubator.
Electrophoretic mobility shift assay (EMSA)
EMSAs were performed on the regions of the five polymorphisms found in the BRCA1 promoter region. For each SNP, double-stranded oligonucleotide probes corresponding to the sequences surrounding the polymorphic site were 32P-radiolabeled and purified using MicroSpin G-25 columns. Binding experiments were conducted using nuclear protein extracts prepared from HeLa, MCF7 and MDA-MB-231 cell lines. Briefly, nuclear proteins from each cell line were quantified with the Bradford protein assay (Bio-Rad). Nuclear extracts (10 μg) were incubated with the various radiolabeled double-stranded DNA probes (35 fmol) in a buffer containing 50 mm Tris–HCl (pH 7.5), 5 mm MgCl2, 2.5 mm EDTA, 2.5 mm DTT, 250 mm NaCl, 0.25 μg/μl poly deoxyinosinate–deoxycytidylate and 20% glycerol, in a total volume of 10 μl for 20 min at room temperature. For competition experiments, 50-fold molar excess of the unlabeled probe oligonucleotide, the unlabeled corresponding mutant probe or a non-specific DNA probe was added before incubation. Supershift assays were performed in the presence of 1μl of antibodies (2 μg/μl). The variants tested and the corresponding antibodies were as follows: rs4793204: Brg-1, Oct-1, Nkx-3.1; rs799908: Elk-1, USF-1, USF-2; rs11655505: GATA-3, Oct-1; rs799906: ER; rs8176071: MEF2 (SantaCruz Biotechnology, Santa Cruz, CA, USA). Competitions and supershift experiments were carried out by pre-incubating nuclear extracts in binding buffer for 10 min at room temperature, followed by 20 min incubation at room temperature with the radiolabeled double-stranded DNA probes. Complexes were separated on a 6% non-denaturing polyacrylamide gel (acrylamide–bisacrylamide, 37.5:1) in 1× Tris-glycine-EDTA buffer (190 V at 4°C). After electrophoresis, gels were dried and subjected to autoradiographic analysis.
Promoter activity assay
Constructs
For the BRCA1 pHaps (A and B), genomic DNA samples from known homozygous or heterozygous individuals were polymerase chain reaction-amplified to obtain the 2067 bp haplotype-specific fragments that were then subcloned into the promoterless pGL3-Basic luciferase reporter vector (Promega, Madison, WI, USA). The following primers were used for amplification (5′-GGGGCCGCTCGAGACACAGAAGTTCTCCAAGTGC-3′, 5′-GGGGCCCAAGCTTCCCGTCCAGGAAGTCTCAG-3′) and detailed information on experimental conditions is available upon request. The resulting constructs were sequenced to confirm the presence of the expected polymorphic sites, amplified and then purified using QIAfilter Plasmid Kit (Qiagen Sciences, Maryland, USA) prior to transfection.
Transient transfection and luciferase reporter assay
HeLa cells were seeded in 24-well culture dishes at a density of 7 × 104cells/well for 24 h prior to transfection. Transient transfection was performed using ExGen 500 cationic polymer transfection reagent (MBI Fermentas Inc., Ontario, Canada) according to the supplier's protocol. Briefly, HeLa cells were co-transfected with 800 ng of pGL3-promoter haplotype-specific constructs encoding a modified firefly luciferase gene and 200 ng of pRL-null vector (Promega Corporation) encoding the Renilla luciferase gene as an internal standard. The promoterless pGL3-basic vector and pGL3-SV40 control vector, containing the SV40 early promoter, were used as negative and positive controls, respectively. Cells were harvested 24 h post-transfection and luciferase reporter gene activities measured with the Dual-Luciferase Reporter Assay System according to the manufacturer's instructions (Promega) in a MicroLumat Plus luminometer (EG&G Berthold). Promoter activities are expressed as a ratio of firefly luciferase to Renilla luciferase luminescence and are represented as the relative luciferase activity of four independent replicates (mean + standard deviation). The promoterless pGL3-basic vector was used to measure basal expression levels. Each experiment was performed five times. Pairs of haplotypes were compared by Student's unpaired t-test.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the European Community's Seventh Framework Programme under grant agreement no. 223175 (HEALTH-F2-2009-223175). D.G.C. received a grant from the INSERM/INCa. J.S. is Chairholder of the Canada Research Chair in Oncogenetics. This work was supported by the LIGUE CONTRE LE CANCER (S.M. and O.M.S.) and by the Canadian Institutes of Health Research for the ‘CIHR Team in Familial Risks of Breast Cancer’ program and by the Canadian Breast Cancer Research Alliance-grant #019511. D.S. holds the François-Karl Viau Chair in Pediatric Oncogenomics and is a scholar of the Fonds de la Recherche en Santé du Québec (FRSQ). A.C.A. is a CR-UK Senior Cancer Research Fellow, D.F.E. is CR-UK Principal Research Fellow and G.C.T. is a NHMRC Senior Principal Research Fellow.
Supplementary Material
ACKNOWLEDGEMENTS
Study-specific acknowledgments
Interdisciplinary Health Research International Team Breast Cancer Susceptibility (INHERIT BRCAs): We would like to thank Dr Martine Dumont for sample management and Martine Tranchant for skillful technical assistance.
National Cancer Institute (NCI): The research of Drs PL Mai and MH Greene was supported by the Intramural Research Program of the US National Cancer Institute, and by support services contracts NO2-CP-11019-50 and N02-CP-65504 with Westat, Inc., Rockville, MD.
Ontario Cancer Genetics Network (OCGN): We wish to thank Gord Glendon, Teresa Selander, Nayana Weerasooriya and members of the Ontario Cancer Genetics Network for their contributions to the study. The OCGN is supported by Cancer Care Ontario.
Swedish Breast Cancer Study (SWE-BRCA): SWE-BRCA collaborators: Per Karlsson, Margareta Nordling, Annika Bergman and Zakaria Einbeigi, Gothenburg, Sahlgrenska University Hospital; Marie Stenmark-Askmalm and Sigrun Liedgren, Linköping University Hospital; Åke Borg, Niklas Loman, Håkan Olsson, Ulf Kristoffersson, Maria Soller, Helena Jernström, Katja Harbst and Karin Henriksson, Lund University Hospital; Annika Lindblom, Brita Arver, Anna von Wachenfeldt, Annelie Liljegren, Gisela Barbany-Bustinza and Johanna Rantala, Stockholm, Karolinska University Hospital; Beatrice Melin, Henrik Grönberg, Eva-Lena Stattin and Monica Emanuelsson, Umeå University Hospital; Hans Ehrencrona, Richard Rosenquist Brandell and Niklas Dahl, Uppsala University Hospital.
Spanish National Cancer Centre (CNIO): This study was partially supported by Fundación Mutua Madrileña, Asociación Española Contra el Cáncer and the Spanish Ministry of Science and Innovation (FIS PI08 1120).
Deutsches Krebsforschungszentrum (DKFZ): The DKFZ study was supported by the DKFZ. We thank Diana Torres and Muhammad U. Rashid for providing DNA samples and supplying data. We thank Antje Seidel-Renkert for expert technical assistance.
The Hereditary Breast and Ovarian Cancer Research Group Netherlands (HEBON): HEBON Collaborating Centers: Coordinating center: Netherlands Cancer Institute, Amsterdam, NL: F.B.L. Hogervorst, S. Verhoef, M. Verheus, L.J. van't Veer, F.E. van Leeuwen, M.A. Rookus; Erasmus Medical Center, Rotterdam, NL: M. Collée, A.M.W. van den Ouweland, A. Jager, M.J. Hooning, M.M.A. Tilanus-Linthorst, C. Seynaeve; Leiden University Medical Center, NL, Leiden: C.J. van Asperen, J.T. Wijnen, M.P. Vreeswijk, R.A. Tollenaar, P. Devilee; Radboud University Nijmegen Medical Center, Nijmegen, NL: M.J. Ligtenberg, N. Hoogerbrugge; University Medical Center Utrecht, Utrecht, NL: M.G. Ausems, R.B. van der Luijt; Amsterdam Medical Center, NL: C.M. Aalfs, T.A. van Os; VU University Medical Center, Amsterdam, NL: J.J.P. Gille, Q. Waisfisz, H.E.J. Meijers-Heijboer; University Hospital Maastricht, Maastricht, NL: E.B. Gomez-Garcia, C.E. van Roozendaal, Marinus J. Blok, B. Caanen; University Medical Center Groningen University, NL: J.C. Oosterwijk, A.H. van der Hout, M.J. Mourits; The Netherlands Foundation for the Detection of Hereditary Tumours, Leiden, NL: H.F. Vasen. The HEBON study is supported by the Dutch Cancer Society grants NKI1998-1854, NKI2004-3088, NKI2007-3756 and the ZonMW grant 91109024.
Epidemiological study of BRCA1 & BRCA2 mutation carriers (EMBRACE): Douglas F. Easton is the PI of the study. EMBRACE Collaborating Centers are: Coordinating Centre, Cambridge: Susan Peock, Margaret Cook, Debra Frost, Radka Platte. North of Scotland Regional Genetics Service, Aberdeen: Zosia Miedzybrodzka, Helen Gregory. Northern Ireland Regional Genetics Service, Belfast: Patrick Morrison, Lisa Jeffers. West Midlands Regional Clinical Genetics Service, Birmingham: Trevor Cole, Kai-ren Ong, Jonathan Hoffman. South West Regional Genetics Service, Bristol: Alan Donaldson, Margaret James. East Anglian Regional Genetics Service, Cambridge: Joan Paterson, Sarah Downing, Amy Taylor. Medical Genetics Services for Wales, Cardiff: Alexandra Murray, Mark T. Rogers, Emma McCann. St James's Hospital, Dublin & National Centre for Medical Genetics, Dublin: M. John Kennedy, David Barton. South East of Scotland Regional Genetics Service, Edinburgh: Mary Porteous, Sarah Drummond. Peninsula Clinical Genetics Service, Exeter: Carole Brewer, Emma Kivuva, Anne Searle, Selina Goodman, Kathryn Hill. West of Scotland Regional Genetics Service, Glasgow: Rosemarie Davidson, Victoria Murday, Nicola Bradshaw, Lesley Snadden, Mark Longmuir, Catherine Watt, Sarah Gibson, Eshika Haque, Ed Tobias, Alexis Duncan. South East Thames Regional Genetics Service, Guy's Hospital London: Louise Izatt, Chris Jacobs, Caroline Langman, Anna Whaite. North West Thames Regional Genetics Service, Harrow: Huw Dorkins. Leicestershire Clinical Genetics Service, Leicester: Julian Barwell. Yorkshire Regional Genetics Service, Leeds: Julian Adlard, Carol Chu, Julie Miller. Merseyside & Cheshire Clinical Genetics Service, Liverpool: Ian Ellis, Catherine Houghton. Manchester Regional Genetics Service, Manchester: D Gareth Evans, Fiona Lalloo, Jane Taylor. North East Thames Regional Genetics Service, NE Thames, London: Lucy Side, Alison Male, Cheryl Berlin. Nottingham Centre for Medical Genetics, Nottingham: Jacqueline Eason, Rebecca Collier. Northern Clinical Genetics Service, Newcastle: Fiona Douglas, Oonagh Claber, Irene Jobson. Oxford Regional Genetics Service, Oxford: Lisa Walker, Diane McLeod, Dorothy Halliday, Sarah Durell, Barbara Stayner. The Institute of Cancer Research and Royal Marsden NHS Foundation Trust: Ros Eeles, Susan Shanley, Nazneen Rahman, Richard Houlston, Elizabeth Bancroft, Lucia D'Mello, Elizabeth Page, Audrey Ardern-Jones, Kelly Kohut, Jennifer Wiggins, Elena Castro, Anita Mitra, Lisa Robertson. North Trent Clinical Genetics Service, Sheffield: Jackie Cook, Oliver Quarrell, Cathryn Bardsley. South West Thames Regional Genetics Service, London: Shirley Hodgson, Sheila Goff, Glen Brice, Lizzie Winchester, Charlotte Eddy, Vishakha Tripathi, Virginia Attard. Wessex Clinical Genetics Service, Princess Anne Hospital, Southampton: Diana Eccles, Anneke Lucassen, Gillian Crawford, Donna McBride, Sarah Smalley. EMBRACE is supported by Cancer Research UK Grants C1287/A10118 and C1287/A11990. D. Gareth Evans and Fiona Lalloo are supported by an NIHR grant to the Biomedical Research Centre, Manchester. The Investigators at The Institute of Cancer Research and The Royal Marsden NHS Foundation Trust are supported by an NIHR grant to the Biomedical Research Centre at The Institute of Cancer Research and The Royal Marsden NHS Foundation Trust. Ros Eeles, Elizabeth Bancroft and Lucia D'Mello are also supported by Cancer Research UK Grant C5047/A8385.
Fox Chase Cancer Centre (FCCC): University of Kansas Medical Center (KUMC): A.K.G. was funded by R01CA140323, U01CA69631, 5U01CA113916, and the Eileen Stein Jacoby Fund.
Genetic Modifiers of cancer risk in BRCA1/2 mutation carriers (GEMO) study: Cancer Genetics Network ‘Groupe Géneétique et Cancer’, Fédération Nationale des Centres de Lutte Contre le Cancer, France. The study was supported by the Ligue National Contre le Cancer; Association for International Cancer Research Grant (AICR-07-0454); and the Association ‘Le cancer du sein, parlons-en!’ Award. We wish to thank all the GEMO collaborating groups for their contribution to this study. GEMO Collaborating Centers are: Coordinating Centres, Unité Mixte de Génétique Constitutionnelle des Cancers Fréquents, Centre Hospitalier Universitaire de Lyon/Centre Léon Bérard, & Equipe «Génétique du cancer du sein», Centre de Recherche en Cancérologie de Lyon, Lyon: Olga Sinilnikova, Laure Barjhoux, Carole Verny-Pierre, Sophie Giraud, Mélanie Léone, Sylvie Mazoyer; and Service de Génétique Oncologique, Institut Curie, Paris: Dominique Stoppa-Lyonnet, Marion Gauthier-Villars, Bruno Buecher, Claude Houdayer, Virginie Moncoutier, Muriel Belotti, Carole Tirapo, Antoine de Pauw. Institut Gustave Roussy, Villejuif: Brigitte Bressac-de-Paillerets, Audrey Remenieras, Véronique Byrde, Olivier Caron, Gilbert Lenoir. Centre Jean Perrin, Clermont–Ferrand: Yves-Jean Bignon, Nancy Uhrhammer. Centre Léon Bérard, Lyon: Christine Lasset, Valérie Bonadona. Centre François Baclesse, Caen: Agnès Hardouin, Pascaline Berthet. Institut Paoli Calmettes, Marseille: Hagay Sobol, Violaine Bourdon, Tetsuro Noguchi, François Eisinger. Groupe Hospitalier Pitié-Salpétrière, Paris: Florence Coulet, Chrystelle Colas, Florent Soubrier. CHU de Arnaud-de-Villeneuve, Montpellier: Isabelle Coupier, Pascal Pujol. Centre Oscar Lambret, Lille: Jean-Philippe Peyrat, Joëlle Fournier, Françoise Révillion, Philippe Vennin, Claude Adenis. Hôpital René Huguenin/Institut Curie, St Cloud: Etienne Rouleau, Rosette Lidereau, Liliane Demange, Catherine Nogues. Centre Paul Strauss, Strasbourg: Danièle Muller, Jean-Pierre Fricker. Institut Bergonié, Bordeaux: Michel Longy, Nicolas Sevenet. Institut Claudius Regaud, Toulouse: Christine Toulas, Rosine Guimbaud, Laurence Gladieff, Viviane Feillel. CHU de Grenoble: Dominique Leroux, Hélène Dreyfus, Christine Rebischung. CHU de Dijon: Fanny Coron, Laurence Faivre. CHU de St-Etienne: Fabienne Prieur, Marine Lebrun. Hôtel Dieu Centre Hospitalier, Chambéry: Sandra Fert Ferrer. Centre Antoine Lacassagne, Nice: Marc Frénay. CHU de Limoges: Laurence Vénat-Bouvet. CHU de Nantes: Capucine Delnatte. CHU Bretonneau, Tours: Isabelle Mortemousque. Creighton University, Omaha, USA: Henry T. Lynch, Carrie L. Snyder.
The Breast Cancer Family Registry (BCFR): BCFR is supported by the National Cancer Institute, National Institutes of Health under RFA-CA-06-503 and through cooperative agreements with members of the BCFR and Principal Investigators, including Cancer Care Ontario (U01 CA69467), Columbia University (U01 CA69398), Fox Chase Cancer Center (U01 CA69631), Huntsman Cancer Institute (U01 CA69446), Northern California Cancer Center (U01 CA69417), University of Melbourne (U01 CA69638) and the Georgetown University Informatics Support Center (RFP No. N02PC45022-46). Samples from the NCCC, FCCC and HCI were processed and distributed by the Coriell Institute for Medical Research. The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the BCFR, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government or the BCFR.
Copenhagen Breast Cancer Study (CBCS): We would like to thank Bent Ejlertsen, Mette K. Andersen, and Susanne Kjaergaard for clinical data. Moreover, we thank the NEYE Foundation for financial support.
Gynecologic Oncology Group (GOG): GOG's participation was supported through funding provided by both intramural (Clinical Genetics Branch, DCEG) and extramural (Community Oncology and Prevention Trials Program, COPTRG) NCI programs, as well as by GOG's Cancer Prevention and Control Committee.
Ohio State University Clinical Cancer Center (OSU CCG): We thank Leigha Senter and Kevin Sweet for accrual of study participants and Michelle O'Connor and Caroline Craven for database entry. The OSU Human Genetics Sample Bank processed DNA samples and the OSU CCC Nucleic Acids Shared Resource performed the Taqman Plate Reads. This work was supported by funds from the OSU Comprehensive Cancer Center.
Istituto Oncologico Veneto–Hereditary Breast Ovarian Cancer Study (IOVHBOCS): The study was supported by the Ministero dell'Università e della Ricerca (MIUR), Ministero della Salute (P.I.O. V and ‘Progetto Tumori Femminili’) and Alleanza Contro il Cancro.
UK and Gilda Radner Familial Ovarian Cancer Registries (UKGRFOCR): UKFOCR was supported by a project grant from CRUK to Paul Pharoah. We thank Paul Pharoah, Susan Ramus, Carole Pye, Patricia Harrington and Eva Wozniak for their contributions towards the UKFOCR. We'd like to acknowledge the Roswell Park Alliance Foundation for their continued support of the Gilda Radner Ovarian Family Cancer Registry. GRFOCR would like to acknowledge Lara Sucheston (Department of Cancer Prevention and Control) and Kunle Odunsi (Departments Gynecologic Oncology and Immunology).
The German Consortium of Hereditary Breast and Ovarian Cancer (GC-HBOC): GC-HBOC is supported by a grant of the German Cancer Aid (grant 107054). We thank Raymonda Varon-Mateeva (Center Berlin), Karin Kast (Center Dresden), Sabine Preisler-Adams (Center Münster), Helmut Deissler (Center Ulm), Ines Schönbuchner (Center Würzburg), Wolfram Heinritz (Center Leipzig) and Dieter Schäfer (Center Frankfurt) for providing samples and clinical data and Juliane Köhler for her excellent technical assistance.
Helsinki Breast Cancer Study (HEBCS): The HEBCS study has been financially supported by the Helsinki University Central Hospital Research Fund, Academy of Finland (132473), the Finnish Cancer Society and the Sigrid Juselius Foundation. HEBCS thanks Drs Kristiina Aittomäki, Carl Blomqvist and Tuomas Heikkinen and RN Irja Erkkilä for their help with the patient data and samples.
Kathleen Cuningham Foundation Consortium for research into Familial Breast cancer (kConFab): We wish to thank Heather Thorne, Eveline Niedermayr, all the kConFab research nurses and staff, the heads and staff of the Family Cancer Clinics and the Clinical Follow Up Study (funded by NHMRC grants 145684, 288704 and 454508) for their contributions to this resource, and the many families who contribute to kConFab. kConFab is supported by grants from the National Breast Cancer Foundation, the National Health and Medical Research Council (NHMRC) and by the Queensland Cancer Fund, the Cancer Councils of New South Wales, Victoria, Tasmania and South Australia, and the Cancer Foundation of Western Australia. A.B.S. and G.C.-T. are supported by a NHMRC Senior Research and Principal Research Fellowships, respectively.
University of California Irvine (UCI) (now the Beckman Research Institute of City of Hope): Genotyping at UCI was funded by grant NIH CA74415.
Mayo Clinic (MAYO): This work was supported by grants from the Breast Cancer Research Foundation (BCRF), Komen Foundation for the Cure, Department of Defense ovarian cancer research award (W81XWH-10-1-0341) and US National Cancer Institute, National Institutes of Health grant CA128978.
Georgetown University (GEORGETOWN): C.I. received support from the Familial Cancer Registry and the Tissue Culture Shared Registry at Georgetown University (NIH/NCI grant P30-CA051008), the Cancer Genetics Network (HHSN261200744000C), and Swing Fore the Cure.
University of Pennsylvania (UPENN): We acknowledge support by the Breast Cancer Research Foundation to K.L.N., by the Susan G. Komen Foundation to S.M.D., and by R01-CA083855 and R01-CA102776 to T.R.R. for the MAGIC consortium.
Hospital Clinico San Carlos (HCSC): The HCSC study was partially supported by Instituto de Salud Carlos III: RD06/0020/0021. We wish to thank Dr Miguel de la Hoya and Pedro Perez-Segura for their contribution to this study.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Antoniou A., Pharoah P.D., Narod S., Risch H.A., Eyfjord J.E., Hopper J.L., Loman N., Olsson H., Johannsson O., Borg A., et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am. J. Hum. Genet. 2003;72:1117–1130. doi: 10.1086/375033. doi:10.1086/375033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antoniou A.C., Cunningham A.P., Peto J., Evans D.G., Lalloo F., Narod S.A., Risch H.A., Eyfjord J.E., Hopper J.L., Southey M.C., et al. The BOADICEA model of genetic susceptibility to breast and ovarian cancers: updates and extensions. Br. J. Cancer. 2008;98:1457–1466. doi: 10.1038/sj.bjc.6604305. doi:10.1038/sj.bjc.6604305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ford D., Easton D.F., Bishop D.T., Narod S.A., Goldgar D.E. Risks of cancer in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Lancet. 1994;343:692–695. doi: 10.1016/s0140-6736(94)91578-4. doi:10.1016/S0140-6736(94)91578-4. [DOI] [PubMed] [Google Scholar]
- 4.Begg C.B., Haile R.W., Borg A., Malone K.E., Concannon P., Thomas D.C., Langholz B., Bernstein L., Olsen J.H., Lynch C.F., et al. Variation of breast cancer risk among BRCA1/2 carriers. JAMA. 2008;299:194–201. doi: 10.1001/jama.2007.55-a. doi:10.1001/jama.2007.55-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simchoni S., Friedman E., Kaufman B., Gershoni-Baruch R., Orr-Urtreger A., Kedar-Barnes I., Shiri-Sverdlov R., Dagan E., Tsabari S., Shohat M., et al. Familial clustering of site-specific cancer risks associated with BRCA1 and BRCA2 mutations in the Ashkenazi Jewish population. Proc. Natl Acad. Sci. USA. 2006;103:3770–3774. doi: 10.1073/pnas.0511301103. doi:10.1073/pnas.0511301103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Antoniou A.C., Pharoah P.D., McMullan G., Day N.E., Stratton M.R., Peto J., Ponder B.J., Easton D.F. A comprehensive model for familial breast cancer incorporating BRCA1, BRCA2 and other genes. Br. J. Cancer. 2002;86:76–83. doi: 10.1038/sj.bjc.6600008. doi:10.1038/sj.bjc.6600008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chenevix-Trench G., Milne R.L., Antoniou A.C., Couch F.J., Easton D.F., Goldgar D.E. An international initiative to identify genetic modifiers of cancer risk in BRCA1 and BRCA2 mutation carriers: the Consortium of Investigators of Modifiers of BRCA1 and BRCA2 (CIMBA) Breast Cancer Res. 2007;9:104. doi: 10.1186/bcr1670. doi:10.1186/bcr1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Antoniou A.C., Sinilnikova O.M., Simard J., Leone M., Dumont M., Neuhausen S.L., Struewing J.P., Stoppa-Lyonnet D., Barjhoux L., Hughes D.J., et al. RAD51 135G–>C modifies breast cancer risk among BRCA2 mutation carriers: results from a combined analysis of 19 studies. Am. J. Hum. Genet. 2007;81:1186–1200. doi: 10.1086/522611. doi:10.1086/522611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Antoniou A.C., Spurdle A.B., Sinilnikova O.M., Healey S., Pooley K.A., Schmutzler R.K., Versmold B., Engel C., Meindl A., Arnold N., et al. Common breast cancer-predisposition alleles are associated with breast cancer risk in BRCA1 and BRCA2 mutation carriers. Am. J. Hum. Genet. 2008;82:937–948. doi: 10.1016/j.ajhg.2008.02.008. doi:10.1016/j.ajhg.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Antoniou A.C., Sinilnikova O.M., McGuffog L., Healey S., Nevanlinna H., Heikkinen T., Simard J., Spurdle A.B., Beesley J., Chen X., et al. Common variants in LSP1, 2q35 and 8q24 and breast cancer risk for BRCA1 and BRCA2 mutation carriers. Hum. Mol. Genet. 2009;18:4442–4456. doi: 10.1093/hmg/ddp372. doi:10.1093/hmg/ddp372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Antoniou A.C., Wang X., Fredericksen Z.S., McGuffog L., Tarrell R., Sinilnikova O.M., Healey S., Morrison J., Kartsonaki C., Lesnick T., et al. A locus on 19p13 modifies risk of breast cancer in BRCA1 mutation carriers and is associated with hormone receptor-negative breast cancer in the general population. Nat. Genet. 2010;42:885–892. doi: 10.1038/ng.669. doi:10.1038/ng.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Antoniou A.C., Beesley J., McGuffog L., Sinilnikova O.M., Healey S., Neuhausen S.L., Ding Y.C., Rebbeck T.R., Weitzel J.N., Lynch H.T., et al. Common breast cancer susceptibility alleles and the risk of breast cancer for BRCA1 and BRCA2 mutation carriers: implications for risk prediction. Cancer Res. 2010;70:9742–9754. doi: 10.1158/0008-5472.CAN-10-1907. doi:10.1158/0008-5472.CAN-10-1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Antoniou A.C., Kartsonaki C., Sinilnikova O.M., Soucy P., McGuffog L., Healey S., Lee A., Peterlongo P., Manoukian S., Peissel B., et al. Common alleles at 6q25.1 and 1p11.2 are associated with breast cancer risk for BRCA1 and BRCA2 mutation carriers. Hum. Mol. Genet. 2011;20:3304–3321. doi: 10.1093/hmg/ddr226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Engel C., Versmold B., Wappenschmidt B., Simard J., Easton D.F., Peock S., Cook M., Oliver C., Frost D., Mayes R., et al. Association of the variants CASP8 D302H and CASP10 V410I with breast and ovarian cancer risk in BRCA1 and BRCA2 mutation carriers. Cancer Epidemiol. Biomarkers Prev. 2010;19:2859–2868. doi: 10.1158/1055-9965.EPI-10-0517. doi:10.1158/1055-9965.EPI-10-0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramus S.J., Kartsonaki C., Gayther S.A., Pharoah P.D., Sinilnikova O.M., Beesley J., Chen X., McGuffog L., Healey S., Couch F.J., et al. Genetic variation at 9p22.2 and ovarian cancer risk for BRCA1 and BRCA2 mutation carriers. J. Natl Cancer Inst. 2010;103:105–116. doi: 10.1093/jnci/djq494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walker L.C., Fredericksen Z.S., Wang X., Tarrell R., Pankratz V.S., Lindor N.M., Beesley J., Healey S., Chen X., Stoppa-Lyonnet D., et al. Evidence for SMAD3 as a modifier of breast cancer risk in BRCA2 mutation carriers. Breast Cancer Res. 2010;12:R102. doi: 10.1186/bcr2785. doi:10.1186/bcr2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X., Pankratz V.S., Fredericksen Z., Tarrell R., Karaus M., McGuffog L., Pharaoh P.D., Ponder B.A., Dunning A.M., Peock S., et al. Common variants associated with breast cancer in genome-wide association studies are modifiers of breast cancer risk in BRCA1 and BRCA2 mutation carriers. Hum. Mol. Genet. 2010;19:2886–2897. doi: 10.1093/hmg/ddq174. doi:10.1093/hmg/ddq174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murphy C.G., Moynahan M.E. BRCA gene structure and function in tumor suppression: a repair-centric perspective. Cancer J. 2010;16:39–47. doi: 10.1097/PPO.0b013e3181cf0204. doi:10.1097/PPO.0b013e3181cf0204. [DOI] [PubMed] [Google Scholar]
- 19.Baldeyron C., Jacquemin E., Smith J., Jacquemont C., De Oliveira I., Gad S., Feunteun J., Stoppa-Lyonnet D., Papadopoulo D. A single mutated BRCA1 allele leads to impaired fidelity of double strand break end-joining. Oncogene. 2002;21:1401–1410. doi: 10.1038/sj.onc.1205200. doi:10.1038/sj.onc.1205200. [DOI] [PubMed] [Google Scholar]
- 20.Febrer E., Mestres M., Caballin M.R., Barrios L., Ribas M., Gutierrez-Enriquez S., Alonso C., Cajal T., Francesc B.J. Mitotic delay in lymphocytes from BRCA1 heterozygotes unable to reduce the radiation-induced chromosomal damage. DNA Repair (Amst.) 2008;7:1907–1911. doi: 10.1016/j.dnarep.2008.08.001. doi:10.1016/j.dnarep.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 21.Rothfuss A., Schutz P., Bochum S., Volm T., Eberhardt E., Kreienberg R., Vogel W., Speit G. Induced micronucleus frequencies in peripheral lymphocytes as a screening test for carriers of a BRCA1 mutation in breast cancer families. Cancer Res. 2000;60:390–394. [PubMed] [Google Scholar]
- 22.Speit G., Trenz K. Chromosomal mutagen sensitivity associated with mutations in BRCA genes. Cytogenet. Genome Res. 2004;104:325–332. doi: 10.1159/000077511. doi:10.1159/000077511. [DOI] [PubMed] [Google Scholar]
- 23.Trenz K., Rothfuss A., Schutz P., Speit G. Mutagen sensitivity of peripheral blood from women carrying a BRCA1 or BRCA2 mutation. Mutat. Res. 2002;500:89–96. doi: 10.1016/s0027-5107(01)00300-1. [DOI] [PubMed] [Google Scholar]
- 24.Trenz K., Landgraf J., Speit G. Mutagen sensitivity of human lymphoblastoid cells with a BRCA1 mutation. Breast Cancer Res. Treat. 2003;78:69–79. doi: 10.1023/a:1022157528247. doi:10.1023/A:1022157528247. [DOI] [PubMed] [Google Scholar]
- 25.Baeyens A., Thierens H., Claes K., Poppe B., de Ridder R.L., Vral A. Chromosomal radiosensitivity in BRCA1 and BRCA2 mutation carriers. Int. J. Radiat. Biol. 2004;80:745–756. doi: 10.1080/09553000400017937. doi:10.1080/09553000400017937. [DOI] [PubMed] [Google Scholar]
- 26.Baria K., Warren C., Roberts S.A., West C.M., Scott D. Chromosomal radiosensitivity as a marker of predisposition to common cancers? Br. J. Cancer. 2001;84:892–896. doi: 10.1054/bjoc.2000.1701. doi:10.1054/bjoc.2000.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kotsopoulos J., Chen Z., Vallis K.A., Poll A., Ainsworth P., Narod S.A. DNA repair capacity as a possible biomarker of breast cancer risk in female BRCA1 mutation carriers. Br. J. Cancer. 2007;96:118–125. doi: 10.1038/sj.bjc.6603528. doi:10.1038/sj.bjc.6603528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bellacosa A., Godwin A.K., Peri S., Devarajan K., Caretti E., Vanderveer L., Bove B., Slater C., Zhou Y., Daly M., et al. Altered gene expression in morphologically normal epithelial cells from heterozygous carriers of BRCA1 or BRCA2 mutations. Cancer Prev. Res. (Phila) 2010;3:48–61. doi: 10.1158/1940-6207.CAPR-09-0078. doi:10.1158/1940-6207.CAPR-09-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen X., Weaver J., Bove B.A., Vanderveer L.A., Weil S.C., Miron A., Daly M.B., Godwin A.K. Allelic imbalance in BRCA1 and BRCA2 gene expression is associated with an increased breast cancer risk. Hum. Mol. Genet. 2008;17:1336–1348. doi: 10.1093/hmg/ddn022. doi:10.1093/hmg/ddn022. [DOI] [PubMed] [Google Scholar]
- 30.Maia A.T., Spiteri I., Lee A.J., O'Reilly M., Jones L., Caldas C., Ponder B.A. Extent of differential allelic expression of candidate breast cancer genes is similar in blood and breast. Breast Cancer Res. 2009;11:R88. doi: 10.1186/bcr2458. doi:10.1186/bcr2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen J., Medico L., Zhao H. Allelic imbalance in BRCA1 and BRCA2 gene expression and familial ovarian cancer. Cancer Epidemiol. Biomarkers Prev. 2011;20:50–56. doi: 10.1158/1055-9965.EPI-10-0720. doi:10.1158/1055-9965.EPI-10-0720. [DOI] [PubMed] [Google Scholar]
- 32.Baynes C., Healey C.S., Pooley K.A., Scollen S., Luben R.N., Thompson D.J., Pharoah P.D., Easton D.F., Ponder B.A., Dunning A.M. Common variants in the ATM, BRCA1, BRCA2, CHEK2 and TP53 cancer susceptibility genes are unlikely to increase breast cancer risk. Breast Cancer Res. 2007;9:R27. doi: 10.1186/bcr1669. doi:10.1186/bcr1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan K.Y., Liu W., Long J.R., Yip S.P., Chan S.Y., Shu X.O., Chua D.T., Cheung A.N., Ching J.C., Cai H., et al. Functional polymorphisms in the BRCA1 promoter influence transcription and are associated with decreased risk for breast cancer in Chinese women. J. Med. Genet. 2009;46:32–39. doi: 10.1136/jmg.2007.057174. doi:10.1136/jmg.2007.057174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cox D.G., Kraft P., Hankinson S.E., Hunter D.J. Haplotype analysis of common variants in the BRCA1 gene and risk of sporadic breast cancer. Breast Cancer Res. 2005;7:R171–R175. doi: 10.1186/bcr973. doi:10.1186/bcr973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Freedman M.L., Penney K.L., Stram D.O., Riley S., Kean-Cowdin R., Le M.L., Altshuler D., Haiman C.A. A haplotype-based case-control study of BRCA1 and sporadic breast cancer risk. Cancer Res. 2005;65:7516–7522. doi: 10.1158/0008-5472.CAN-05-0132. doi:10.1158/0008-5472.CAN-05-0132. [DOI] [PubMed] [Google Scholar]
- 36.Ginolhac S.M., Gad S., Corbex M., Bressac-de-Paillerets B., Chompret A., Bignon Y.J., Peyrat J.P., Fournier J., Lasset C., Giraud S., et al. BRCA1 wild-type allele modifies risk of ovarian cancer in carriers of BRCA1 germ-line mutations. Cancer Epidemiol. Biomarkers Prev. 2003;12:90–95. [PubMed] [Google Scholar]
- 37.Dimas A.S., Deutsch S., Stranger B.E., Montgomery S.B., Borel C., Attar-Cohen H., Ingle C., Beazley C., Gutierrez A.M., Sekowska M., et al. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science. 2009;325:1246–1250. doi: 10.1126/science.1174148. doi:10.1126/science.1174148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baker K.M., Wei G., Schaffner A.E., Ostrowski M.C. Ets-2 and components of mammalian SWI/SNF form a repressor complex that negatively regulates the BRCA1 promoter. J. Biol. Chem. 2003;278:17876–17884. doi: 10.1074/jbc.M209480200. doi:10.1074/jbc.M209480200. [DOI] [PubMed] [Google Scholar]
- 39.Anczukow O., Ware M.D., Buisson M., Zetoune A.B., Stoppa-Lyonnet D., Sinilnikova O.M., Mazoyer S. Does the nonsense-mediated mRNA decay mechanism prevent the synthesis of truncated BRCA1, CHK2, and p53 proteins? Hum. Mutat. 2008;29:65–73. doi: 10.1002/humu.20590. doi:10.1002/humu.20590. [DOI] [PubMed] [Google Scholar]
- 40.Mikaelsdottir E.K., Valgeirsdottir S., Eyfjord J.E., Rafnar T. The Icelandic founder mutation BRCA2 999del5: analysis of expression. Breast Cancer Res. 2004;6:R284–R290. doi: 10.1186/bcr785. doi:10.1186/bcr785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perrin-Vidoz L., Sinilnikova O.M., Stoppa-Lyonnet D., Lenoir G.M., Mazoyer S. The nonsense-mediated mRNA decay pathway triggers degradation of most BRCA1 mRNAs bearing premature termination codons. Hum. Mol. Genet. 2002;11:2805–2814. doi: 10.1093/hmg/11.23.2805. doi:10.1093/hmg/11.23.2805. [DOI] [PubMed] [Google Scholar]
- 42.Ware M.D., DeSilva D., Sinilnikova O.M., Stoppa-Lyonnet D., Tavtigian S.V., Mazoyer S. Does nonsense-mediated mRNA decay explain the ovarian cancer cluster region of the BRCA2 gene? Oncogene. 2006;25:323–328. doi: 10.1038/sj.onc.1209033. [DOI] [PubMed] [Google Scholar]
- 43.Buisson M., Anczukow O., Zetoune A.B., Ware M.D., Mazoyer S. The 185delAG mutation (c.68_69delAG) in the BRCA1 gene triggers translation reinitiation at a downstream AUG codon. Hum. Mutat. 2006;27:1024–1029. doi: 10.1002/humu.20384. doi:10.1002/humu.20384. [DOI] [PubMed] [Google Scholar]
- 44.Barrett J.C., Fry B., Maller J., Daly M.J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. doi:10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 45.de Bakker P.I., Yelensky R., Pe'er I., Gabriel S.B., Daly M.J., Altshuler D. Efficiency and power in genetic association studies. Nat. Genet. 2005;37:1217–1223. doi: 10.1038/ng1669. doi:10.1038/ng1669. [DOI] [PubMed] [Google Scholar]
- 46.Tavtigian S.V., Greenblatt M.S., Lesueur F., Byrnes G.B. In silico analysis of missense substitutions using sequence-alignment based methods. Hum. Mutat. 2008;29:1327–1336. doi: 10.1002/humu.20892. doi:10.1002/humu.20892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ng P.C., Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–874. doi: 10.1101/gr.176601. doi:10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Antoniou A.C., Goldgar D.E., Andrieu N., Chang-Claude J., Brohet R., Rookus M.A., Easton D.F. A weighted cohort approach for analysing factors modifying disease risks in carriers of high-risk susceptibility genes. Genet. Epidemiol. 2005;29:1–11. doi: 10.1002/gepi.20074. doi:10.1002/gepi.20074. [DOI] [PubMed] [Google Scholar]
- 49.Boos D.D. American Statistician. 1992;46:327–333. doi: 10.1198/tas.2011.10129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Labuda D., Labbe C., Langlois S., Lefebvre J.F., Freytag V., Moreau C., Sawicki J., Beaulieu P., Pastinen T., Hudson T.J., Sinnett D. Patterns of variation in DNA segments upstream of transcription start sites. Hum. Mutat. 2007;28:441–450. doi: 10.1002/humu.20463. doi:10.1002/humu.20463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sinnett D., Beaulieu P., Belanger H., Lefebvre J.F., Langlois S., Theberge M.C., Drouin S., Zotti C., Hudson T.J., Labuda D. Detection and characterization of DNA variants in the promoter regions of hundreds of human disease candidate genes. Genomics. 2006;87:704–710. doi: 10.1016/j.ygeno.2006.01.001. doi:10.1016/j.ygeno.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 52.Quandt K., Frech K., Karas H., Wingender E., Werner T. MatInd and MatInspector: new fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Res. 1995;23:4878–4884. doi: 10.1093/nar/23.23.4878. doi:10.1093/nar/23.23.4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.