

The prevalence of obesity, impaired glucose tolerance, and type 2 diabetes mellitus is escalating at an alarming rate. If current trends continue, 33% of adults in the United States may have diabetes by the year 2050,1 a prediction that underscores the need for new, effective, and safe strategies for prevention and treatment. Recently, Choi et al.2 reported findings that may enable the development of safer antidiabetic agents that target peroxisome-proliferator–activated receptor γ (PPARγ), a nuclear receptor and transcription factor that regulates the expression of hundreds of genes and has a fundamental role in adipocyte differentiation.
Synthetic activators of PPARγ, called thiazoli-dinediones, are one of the few classes of drugs that act primarily as effective insulin sensitizers, thereby reducing plasma insulin levels and, theoretically, the complications associated with hyperinsulinemia. The efficacy of thiazolidinediones was thought to result from altered transcriptional activity of PPARγ, although the glucose-lowering effects of different PPARγ ligands do not correspond with changes in the expression of genes regulated by PPARγ.3 Side effects of thiazolidinediones include weight gain, fluid retention, congestive heart failure, and bone fractures4 (Fig. 1). Since such side effects have become more evident, the use of these drugs has become controversial. Earlier this year, the Food and Drug Administration severely restricted the use of one of the most commonly prescribed thiazolidinediones, rosiglitazone, because of an associated increase in the rates of complications from cardiovascular causes.
Figure 1. Effects of Thiazolidinediones.
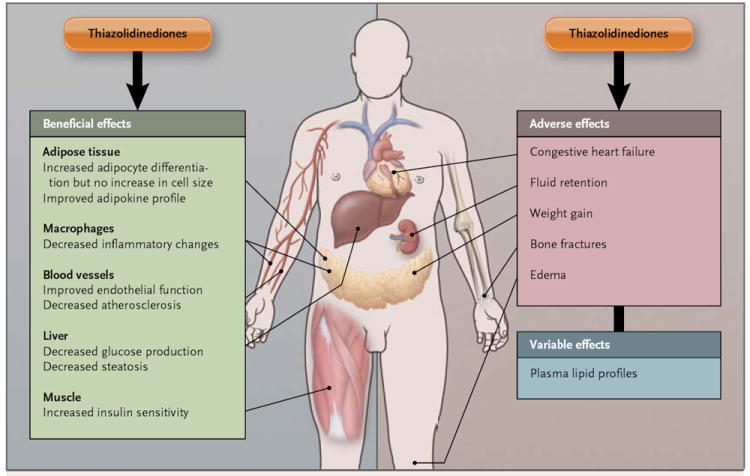
Thiazolidinediones have an array of beneficial and harmful effects on multiple tissues. Some of the effects are direct, whereas others are indirect. Adipocyte differentiation is thought to be beneficial for several reasons, including the production of favorable adipokines by small adipocytes and the provision of an expanded storage depot for lipids so that they are not deposited ectopically. Different thiazolidinediones have variable effects on plasma lipid profiles.4
The metabolic effects of thiazolidinediones are primarily the result of transcriptional regulation in adipose and other tissues (including cells such as macrophages) that affect whole-body insulin sensitivity (Fig. 1). Although the classic agonist effects of thiazolidinediones include the induction of an adipogenic gene program, the precise mechanisms of the insulin-sensitizing effects of these drugs are unknown. Adipocytes have an important role in metabolic regulation by storing lipids and hence reducing ectopic lipid accumulation in other tissues and by secreting hormones and cytokines that regulate systemic insulin sensitivity, inflammatory processes, and vascular biology, thereby affecting the risk of diabetes and cardiovascular disease.5 A better understanding of the actions of thiazolidinediones on adipocytes could provide insights for developing safer agents that target PPARγ and preserve the insulin-sensitizing effects while diminishing the deleterious side effects.
Choi et al. found that the serine residue at position 273 of PPARγ is phosphorylated by cyclin-dependent kinase 5 (CDK5), and its phosphorylation at this position alters its transcriptional activity. This results in the reduced expression of several genes that promote beneficial metabolic effects, most notably insulin sensitivity (Fig. 2). CDK5 is activated by proinflammatory cytokines that are elevated in the adipose tissue of obese subjects and in response to consuming a high-fat diet. This suggests that CDK5-altered PPARγ transcription to some degree attenuates the beneficial effects of adipose tissue on whole-body metabolism. This process could contribute to metabolic dysregulation in obesity.
Figure 2. A Comparison of PPARγ Activity in Lean and Obese Persons.

Regulation of the activity of peroxisome-proliferator–activated receptor γ (PPARγ) in the healthy, lean state (Panel A) differs from regulation in the obese, insulin-resistant state (Panel B). Obesity, a high-fat diet, and distinct types of lipids and fatty acids induce the production of inflammatory cytokines, which activate cyclin-dependent kinase 5 (CDK5). CDK5 phosphorylates PPARγ on serine residue 273 and induces a transcriptional program that is distinct from that of unphosphorylated PPARγ. The differences include both induction and repression of gene sets (represented by A, B, and C), as compared with transcriptional programs in adipose tissue in the nonobese or insulin-sensitive state. In addition to activating PPARγ, certain antidiabetic PPARγ ligands also block CDK5 phosphorylation. This effect may potentially restore the PPARγ transcription program to that of the healthy lean state or preferentially activate insulin-sensitizing gene programs.
Rosiglitazone not only activates PPARγ-controlled transcription but also inhibits CDK5 phosphorylation of PPARγ. This inhibition selectively restores the expression of adipocyte genes, such as adiponectin (ADIPOQ), which are important for the beneficial metabolic effects of adipose tissue. The antidiabetic activities of several non-thiazoli-dinedione PPARγ ligands correlate better with the inhibition of CDK5 phosphorylation of PPARγ than with the induction of adipogenesis. Provocatively, in a small group of persons with impaired glucose tolerance who were treated with rosiglitazone, the decrease in the phosphorylation of PPARγ on serine residue 273 correlated with the degree of improved insulin sensitivity.2 It will be important to determine whether the inhibition of CDK5-induced phosphorylation is a cause or result of the therapeutic effect of rosiglitazone treatment and whether such inhibition is necessary for the insulin-sensitizing effects of rosiglitazone.
These data contribute to the emerging picture that modifying the transcriptional machinery to fine-tune the expression of specific gene sets may allow for therapeutic efficacy and diminish side effects of drugs that target nuclear receptors. Similar goals have led to the development of selective agonists of the estrogen receptor, androgen receptor, and thyroid hormone receptor, although the mechanisms conferring specificity of these agonists differ for the various receptors. Array studies show overlapping but distinct profiles of gene expression associated with the use of different thiazolidinediones, and clinical studies show varying effects of individual thiazolidinediones on plasma lipoproteins,4,6 thus reinforcing the feasibility of developing selective PPARγ agonists.
The study by Choi et al. suggests a specific approach for developing selective PPARγ ligands: targeting CDK5 phosphorylation of PPARγ or the selective downstream effects of this phosphorylation (Fig. 2). Although it is too soon to conclude that CDK5 phosphorylation of PPARγ has a critical role in normal physiology or the patho-physiology of obesity and insulin resistance, these findings provide proof of concept that the transcriptional effects of PPARγ ligands can be separated from the effects underlying insulin sensitization. It will be important to determine whether the side effects and therapeutic effects can be separated. Would it be wise to develop more selective PPARγ ligands when other types of anti-diabetic drugs are in the pipeline? Quite possibly. Even in the absence of overt diabetes, insulin resistance confers an increased risk of cardiovascular disease, cancer, and shortened life span, and the vast majority of obese persons have insulin resistance and hence are at risk for these consequences. Safe insulin sensitizers could conceivably be used to prevent the development of diabetes and cardiovascular disease in persons with insulin resistance.
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
References
- 1.Boyle JP, Thompson TJ, Gregg EW, Barker LE, Williamson DF. Projection of the year 2050 burden of diabetes in the US adult population: dynamic modeling of incidence, mortality, and prediabetes prevalence. Popul Health Metr. 2010;8:29. doi: 10.1186/1478-7954-8-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choi JH, Banks AS, Estall JL, et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature. 2010;466:451–6. doi: 10.1038/nature09291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schopfer FJ, Cole MP, Groeger AL, et al. Covalent peroxisome proliferator-activated receptor γ adduction by nitro-fatty acids: selective ligand activity and anti-diabetic signaling actions. J Biol Chem. 2010;285:12321–33. doi: 10.1074/jbc.M109.091512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McGuire DK, Inzucchi S. New drugs for the treatment of diabetes mellitus. I. Thiazolidinediones and their evolving cardiovascular implications. Circulation. 2008;117:440–9. doi: 10.1161/CIRCULATIONAHA.107.704080. [DOI] [PubMed] [Google Scholar]
- 5.Galic S, Oakhill JS, Steinberg GR. Adipose tissue as an endocrine organ. Mol Cell Endocrinol. 2010;316:129–39. doi: 10.1016/j.mce.2009.08.018. [DOI] [PubMed] [Google Scholar]
- 6.DeSouza C, Fonseca V. Therapeutic targets to reduce cardiovascular disease in type 2 diabetes. Nat Rev Drug Discov. 2009;8:361–7. doi: 10.1038/nrd2872. [DOI] [PubMed] [Google Scholar]