

Abstract
The neurons in the central nervous system (CNS) with high O2 consumption and prolonged life span are chronically exposed to high levels of reactive oxygen species (ROS). Accumulation of ROS-induced genome damage in the form of oxidized bases and single-strand breaks (SSBs) as well as their defective or reduced repair in the brain has been implicated in the etiology of various neurological disorders including Alzheimer’s/Parkinson’s diseases (AD/PD). Although inactivating mutations in some DNA repair genes have been linked to hereditary neurodegenerative diseases, the underlying mechanisms of repair deficiencies for the sporadic diseases is not understood. The ROS-induced DNA damages are predominantly repaired via highly conserved and regulated base excision/SSB repair (BER/SSBR) pathway. We recently made an interesting discovery that transition metals iron (Fe) and copper (Cu) which accumulate excessively in the brains of AD, PD and other neurodegenerative diseases, act as a ‘double-edged sword’ by inducing genotoxic ROS and inhibiting DNA damage repair at the same time. These metals inhibit the base excision activity of NEIL family DNA glycosylases by oxidizing them, changing their structure, and inhibiting their binding to downstream repair proteins. Metal chelators and reducing agents partially reverse the inhibition, while curcumin with both chelating and reducing activities reverses the inhibition nearly completely. In this review, we have discussed the possible etiological linkage of BER/SSBR defects to neurodegenerative diseases and therapeutic potential of metal chelators in restoring DNA repair capacity.
Keywords: Alzheimer’s disease, Base excision repair, Curcumin, Metal chelators, Metal toxicity, Neurodegenerative diseases, Oxidative genome damage, Parkinson’s disease
INTRODUCTION
Both nuclear and mitochondrial genomes are susceptible to oxidative damage by reactive oxygen species (ROS) generated during cellular respiration or after exposure to various exogenous agents [1–3]. Ionizing radiation, commonly used in clinical diagnostics as well as therapy, also inflicts oxidative DNA damage both by direct hits and via ROS generated by radiolysis of water. The common ROS are O2•− (superoxide radical), OH• (hydroxyl radical) and H2O2 (hydrogen peroxide). High level of ROS-induced genome damage occurs in cells (e.g., neurons in brain) with high respiration rate. Furthermore, the long life span of postmitotic, differentiated neurons and lack of replication-coupled repair compared to cycling cells promote accumulation of unrepaired DNA damage over time [4]. It is thus not surprising that persistent oxidative genome damage has been implicated in several neurodegenerative diseases including AD [5, 6], PD [5, 7, 8] and Huntington’s disease (HD; [9, 10]) and amyotrophic lateral sclerosis (ALS; [11, 12]).
Furthermore, despite significant advancement in our understanding of the pathological and biochemical processes involved in neurodegenerative diseases, the etiology of progressive neuronal dysfunctions particularly in selective brain regions largely remains obscure. It was proposed that an imbalance in DNA damage and repair leading to accumulation of unrepaired damage initiates the neurodegenerative pathology. Decreased oxidative DNA repair capacity in AD and PD patients has been observed, but the levels of repair proteins was not found to be significantly affected [13, 14]. Mutations in some DNA repair proteins with decreased activity were implicated in the early-onset, inherited forms of neurological diseases, including those linked to protein kinase ATM (in ataxia telangiectasia), DNA helicases WRN (in Werner’s syndrome), and BLM (in Bloom syndrome), XPG (in xeroderma pigmentosum), CSB (in Cockayne’s syndrome) [15, 16]. However, the mechanism of repair defects observed in sporadic AD/PD was not understood until recently [17] when disorders were linked to accumulation of redox sensitive transition metals [e.g., iron (Fe) and copper (Cu)] in affected neurons that contribute to generation of ROS/other reactive species [18–20]. We showed that Fe and Cu not only induce DNA damage but also inhibit repair of oxidatively damaged DNA bases by NEIL1 and NEIL2 DNA glycosylases (DGs) [21]. The inhibition involves metal-induced oxidation as well as structural changes in NEILs and thus metal chelators and a reducing agent effectively reverse such inhibition. The major focus of this review is the etiological involvement of metal toxicity and oxidized genome damage repair in AD/PD and other neurodegenerative diseases.
OXIDIZED GENOME DAMAGE REPAIR IN HUMAN CELLS
ROS-induced genome damage constitutes the most pervasive insult to the genetic material in all aerobic organisms [3, 22]. It is generally estimated that more than 105 lesions are inflicted in the mammalian cell genome by endogenously generated ROS per cell per day [2]. We believe this to be gross underestimate due to continuous repair. ROS-induced DNA damage consist of several dozen oxidized bases (partial list in Table 1), oxidized abasic (AP) sites, oxidized sugar fragments (e.g., deoxyribonolactone, phosphoglycolate etc) and single-strand breaks (SSBs) with nonligatable termini [3, 22]. In addition, bi-stranded SSBs in close proximity may be converted into double-strand breaks (DSBs) [23]. While unrepaired DSBs are extremely lethal, oxidized base lesions can be mutagenic, cytotoxic or both [24]. The most common oxidation products of purines are 8-oxoguanine (8-oxoG), formamidopyrimidines (FapyG and FapyA), while the most common damage to pyrimidines are 5-OHU generated due to oxidative deamination of C, and thymine glycol [25]. Guanine is most sensitive to oxidation among the normal bases and 8-oxoG is commonly used as a marker of oxidative genome damage and oxidative stress [8]. Other oxidized base lesions that are not as common but highly mutagenic/toxic are 8-oxoA, 5-formylU, 5-OHC etc (Table 1). Furthermore, SSBs generated directly by reaction of deoxyribose with ROS or as intermediates during repair of oxidized bases contain various nonligatable termini including 3'phosphoglycolate, 3'phosphoglycoladehyde, 3'phosphate (3'P) or 5'OH, 5'phosphosugar derivative like deoxyribonolactone [26, 27]. All of these damages except DSBs are repaired by evolutionarily conserved base excision/SSB repair (BER/SSBR) pathways [3, 22]. DSBs are normally repaired by homologous recombination (HR) and non-homologous end-joining (NHEJ) pathways depending on the cell-cycle phases; NHEJ is active in all cell types while HR can function only in cycling cells during the S/G2 phase containing replicated genome [28]. However, recent studies have shown that SSBR proteins could also participate in DSBR via PARP-1-dependent alternative end-joining (ALT-EJ) pathway [29], which will not be discussed further in this review.
Table 1.
Major oxidative products of DNA bases and DGs primarily responsible for their excision in mammalian cells. Designations in red indicate oxidative modifications.
| Normal DNA base | Oxidized DNA bases | DGs responsible for excision |
|---|---|---|
1. Adenine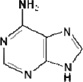
|
i) Formamidoadenine (FapyA)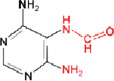
|
NEIL1 |
ii) 8-oxoadenine(8-oxoA)
|
OGG1(excises from 8-oxoA:C) Unknown DG for 8-oxoA:G pair | |
2. Guanine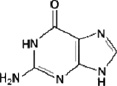
|
i) 8-oxoguanine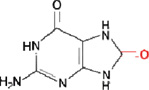
|
OGG1 (and NEIL1 in single-stranded DNA substrates) |
ii) Formamidoguanine (FapyG)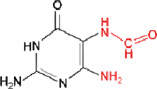
|
OGG1, NEIL1 | |
3.Cytosine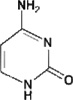
|
i) 5-hydroxycytosine (5-OHC)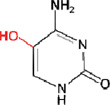
|
NEIL1, NEIL2, NTH1 |
ii) 5-hydroxyuracil (5-OHU)/5-formyluracil (5-formylU)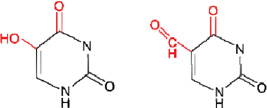
|
NEIL1, NEIL2, NTH1 NTH1 | |
4. Thymine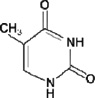
|
i) Thymine glycol
|
NTH1, NEIL1 |
ii) 5-hydroxy-5,6-dihydrothymine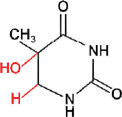
|
NTH1 NEIL1? | |
BER/SSBR, the most versatile among DNA repair processes, thus repairs majority of endogenous genome damage and involves four minimal steps in its simplest mode as described below and illustrated in Fig. 1.
Fig. 1. A schematic illustration of repair of ROS-induced DNA damage via BER/SSBR pathways.

ROS can induce oxidized bases (represented as a *), oxidized AP sites and SSBs with blocked termini. In addition, normal AP sites may be generated (by ROS) indirectly via deamination of C to form U which is removed by monofunctional UDG. After the base excision/AP lyase step in BER, the two pathways converge to common steps of cleaning blocked termini (in red), followed by gap-filling repair synthesis (in blue) and nick sealing. Divergent repair synthesis reactions with patch size of 1-nt (SN-BER) vs. 2–8 nts (LP-BER) are shown. Other details are given in the text.
Base lesion recognition, excision and cleavage of AP site
Repair of alkylated or abnormal bases is initiated by a monofunctional DG that recognizes and excises the damaged base generating an AP site [22]. Oxidized bases are excised by bifunctional DGs containing intrinsic AP lyase activity and using different chemistry from the monofunctional DGs as described below. Damage sensing is a critical step particularly for the oxidized base lesions that are similar in size to their normal counterparts to retain near normal base pairing and thus cause only minor perturbations in DNA double helix [30]. Thus lesion recognition, particularly in highly condensed chromatin poses a serious challenge. It should be mentioned that although DGs are involved in the recognition of base lesions owing to their specific affinity for a group of modified bases, there may be other signaling factors involved, an area which has not been extensively addressed.
Oxidized base-specific DGs discovered in human cells so far belong to two families: Nth and Nei, based on their prototypes in E. coli. The first discovered mammalian DG, 8-oxoguanine DNA glycosylase (OGG1), and NTH1 belong to the Nth family [31] and more recently discovered (by us and others) NEIL1 and NEIL2 belong to the Nei group [32–35]. Recently, a fifth member NEIL3 was added [36] to the list of DGs, but its functions have not been completely characterized. Another DG, MYH (ortholog of E. coli MutY) excises the normal base C from 8-oxoG•C pair in DNA [22].
The mechanism of base lesion excision involves extrahelical flipping of the damaged base into a lesion-specific recognition pocket in DGs [37, 38]. All DGs studied so far bind to the minor groove, kink DNA at the site of damage, and flip the lesion base out of the DNA major groove. Thus each DG is damage specific, and only those bases after nucleotide flipping that can be accommodated in the binding pocket provide the necessary contacts and orientation for their excision. As mentioned before, ROS produces more than 20 types of oxidized base lesions that are repaired by only four (or five) DGs in human cells; thus each DG acts on a specific set of base lesions. It is likely that plasticity of their catalytic pockets allows induced fit of diverse substrates. This could also explain why DGs are poor enzymes with low turnover, as a compromise for their promiscuity. However, recent studies have shown that in spite of broad substrate specificity, DGs have distinct preferential and back-up functions. We showed that NEILs are unique and distinct from OGG1/NTH1 in excising base lesions from single-stranded (ss) DNA sequences suggesting their preferential role during DNA replication and/or transcription [39, 40]. The preferred substrates of oxidized base-specific DGs are listed in Table 1.
Bifunctional DGs cleave the DNA strand at the AP site after the lesion base excision via β or βδ elimination. OGG1 and NTH1 carry out β elimination and generate 3'phospho-α,β-unsaturated aldehyde (3'PUA, also referred to as 3'dRP) and 5'P groups, while NEIL1 and NEIL2 catalyze βδ elimination and generate 3'P and 5'P termini [41]. The DG products are thus SSBs with blocked 3' termini due to concerted and sequential base excision and AP lyase reactions [41]. Further, AP sites induced by ROS or by the action of monofunctional DGs in human cells are primarily processed by AP endonuclease 1 (APE1) generating strand breaks with 3'OH and 5'deoxyribosephsopahte (5'dRP) end. The various types of SSBs directly induced by ROS or generated by the action of DGs or APE1 invariably possess nonligatable termini which are processed in the next step of BER.
Cleaning of blocked termini at the strand break
SSB end-processing, an essential step in BER/SSBR produces 3'OH/5'P termini required for subsequent gap-filling synthesis and ligation [22]. This is the most diverse step in BER/SSBR due to the multiple types of blocked termini.
End-processing in oxidized base/AP site repair: The two major end-processing enzymes involved in oxidized base repair are APE1 and polynucleotide kinase 3'phosphatase (PNKP) that remove 3'dRP (β elimination product) and 3'P (βδ elimination product), respectively, generating 3'OH [41]. Thus while APE1 is required for OGG1/NTH1-initiated BER, PNKP participates in NEIL-initiated BER. Further, 5'dRP residue (APE1’s product of AP site cleavage) is processed by DNA polymerase β (Polβ) in human cells which possesses 5'dRP lyase activity [42]. However, Polβ cannot remove if the 5'dRP residue is oxidized that may happen during oxidative stress [43]. Then the oxidized 5'dRP is displaced along with 2–8 nucleotides (nts) as a singlestranded flap and is removed by flap endonuclease I (FEN-1), as essential enzyme normally involved in the removal of Okazaki fragment primers during lagging strand DNA replication. A second flap endonuclease, DNA2, has been discovered in both nuclei and mitochondria of human cells that acts in concert with FEN-1 in processing long flap structures [44].
Other end-processing in SSBR: ROS-induced SSBs have blocked ends and several unique end-processing enzymes are involved in restoring these to conventional 3'OH/5'P moieties. The most common block at ROS induced SSB is a 3′ block mainly 3' phosphoglycolate or 3' phosphoglycolaldehyde which are generally processed by APE1. Tyrosylphosphodiesterase 1 (TDP1), another 3' end-processing enzyme in yeast and human cells, cleaves Top1(Tyr)-crosslinked to 3'P at the strand break generated by abortive topoisomerase 1 (Top1) reaction. The 3'P is then removed by PNKP [45–47], which also phosphorylates the 5'OH generated at an SSB. A unique type of 5' blocking groups is formed as intermediates during abortive DNA ligation, namely, adenylate linked via 5'•5' bond to the 5'P terminus at an SSB. Aprataxin releases 5'AMP to restore the 5' P terminus [48, 49].
Apart from these blocked termini-specific end-processing enzymes, various 3' exonucleases such as TREX1 and TREX2, ubiquitous in human tissues, are involved in editing or proof reading during repair/replication by DNA polymerases like Polβ lacking constitutive 3' exonuclease activity [50]. In addition, other DNA 3' and 5' exonucleases such as MRN complex (CtIP), EXO1, artemis are involved in end-trimming or end-resection to generate microhomology in DSBR [28], which is outside the scope of this review.
XRCC1 and PARP-1, two vital players in regulating BER/SSBR, act as accessory factors in end-processing activity [22, 41, 51]. While XRCC1 primarily acts a scaffold for recruiting BER proteins for excision or strand break repair, PARP acts a SSB sensor protein [22, 52]. XRCC1 has been shown to functionally interact with all end-processing enzymes, PNKP, APE1, aprataxin, TDP-1 and Polβ [53–56]. Reduced end-processing observed in XRCC1 deficient cells supports its role in end-processing [57]. PARP present in mammalian cells and absent in E. coli is activated by SSBs and transfers ADP-ribose moiety from NAD to a variety of proteins including itself to form poly(ADP-ribose) chains. PARP-1 and PARP-2 of the PARP superfamily have been shown to be important players in repair of SSBs both as a sensor and for recruiting repair proteins including end-processing enzymes to the strand break [51].
Interestingly, the majority of early BER/SSBR proteins in higher vertebrates (unlike their bacterial counterparts) possess a stretch of disordered peptide segment invariably at one of the termini or could serve sometimes as a linker bridging two domains, suggesting that such a common structural feature could be important for their functions like damage sensing, proteinprotein interactions, repair regulation via posttranslational modifications etc [30].
Gap-filling after nucleotide excision
Excision of oxidized base lesions and termini processing leaves a 1-nt gap at the damage site, which is filled with the appropriate nucleotide by a DNA polymerase. Depending on the repair patch size, two types of BER have been characterized: single-nt incorporation repair (SN-BER) involving replacement of the base lesion with the precursor base, and long-patch repair (LP-BER) involving repair patch size of 2–8 nts upstream of the lesion site, requiring FEN-1 (and possibly DNA2) to remove the displaced DNA flap.
Human cells express multiple DNA polymerases that function in BER/SSBR pathways. Polβ, ubiquitous in mammalian tissues, normally carries out SN-BER in non-dividing cells, although Polβ could also participate in LP-BER in co-ordination with FEN-1 [58]. LP-BER generally utilizes DNA replication machinery including replicative DNA pols, Polδ/ε, the sliding clamp PCNA, clamp loader replication factor-C (RF-C) and DNA ligase I (Lig I) and FEN-1 [59–61]. The choice of SN-BER vs. LP-BER is a complex issue that is yet to be completely understood. Initial studies suggested that the nature of the 5'-phosphoribose terminus (normal vs. oxidized) would be the deciding factor [62]. However, involvement of DNA replication proteins with LP-BER strongly suggests that LP-BER could be the preferred pathway during DNA replication, irrespective of the 5' terminal group.
Gap-filling or repair synthesis is also required for the repair of SSBs induced by ROS that oxidizes the sugar and cleaves the base. Thus either 3' or 5' terminus, or both may have blocking groups that have to be processed. In any case, repair synthesis (utilizing the undamaged strand as the template) is invariably required for repair of ROS-induced SSBs. If the strand cleavage is only due to inhibition of replication or generated by nucleases, a simple nick may be produced, repair of which requires only end-processing by PNKP and ligation. Any other end-processing involves loss of at least one base and thus requiring repair synthesis. Polβ could be the major DNA pol involved in SSBR, which would mostly carry out LP-BER. While Polβ and Polδ/ε are the major DNA pols involved in BER/SSBR, several other pols such as Polμ, Polλ and Polθ appear to be involved in DSBR via NHEJ or Alt-EJ [28, 29].
Nick sealing by DNA ligases
The sealing of the nick with 3'OH and 5'P termini by a DNA ligase to restore genomic integrity is the final step in BER/SSBR. DNA ligase IIIα (Lig IIIα) and Lig I are the two major DNA ligases in human cells; the former is generally associated with SN-BER and the latter in LP-BER although the distinction may not be absolute but preferential [22, 63].
A unique feature of mammalian BER/SSBR is that interactions among the repair proteins play a critical role in enhancing repair efficiency [40, 41, 59, 60]. We have shown that the early proteins in the BER pathway interact not only with the next protein, but also with distal downstream proteins. For example, NEIL1 or NEIL2 binarily interacts with Lig IIIα, the last enzyme in BER and most proteins in the intervening steps including Polβ, FEN-1 and XRCC1 [41, 60, 64]. We further showed that disrupting NEIL1/FEN-1 interaction significantly decreased the efficiency of LP-BER suggesting that such interactions are needed for optimum repair [60]. We propose that multiple interactions among BER/SSBR proteins forming larger repairosome complexes are vital for tight regulation of different repair sub-pathways [40].
MITOCHONDRIAL BER/SSBR
The mitochondria possess BER/SSBR activity for repairing oxidative (and alkylated) genome damage. Repair of oxidized bases and SSBs via SN- and LP-BER has been demonstrated for a number of cell types [65, 66]. The mitochondrial BER proteins are either identical to their nuclear counterparts or isoforms resulting from variant RNA splicing or are protease cleavage products. Among the DGs, unaltered OGG1, NTH1 and NEIL1 have been shown to localize in the mitochondria [67]. We showed that APE1 is also present in mammalian mitochondria [68]. Recent studies have shown the presence of TDP1 in mitochondria [69]. Furthermore, DNA polymerase γ (Polγ) is the only DNA polymerase in mammalian mitochondria and is thus essential for both mt genome replication and repair [70]. A splice variant of nuclear Lig III was shown to function in BER/SSBER in mitochondria [63].
BER/SSBR IN CENTRAL NERVOUS SYSTEM (CNS)
The CNS includes the brain and spinal cord consisting of multiple cell types including neurons and glial cells (astrocytes, oligodendrocytes and microglia). A distinct feature of neurons is that unlike the glial cells, they are terminally differentiated, postmitotic in which replication-associated DNA repair cannot occur [4]. However, recent studies have indicated that high levels of DNA damage in neurons could activate their S phase entry that either promotes DNA repair or more commonly apoptosis [71]. The major challenge for neurons is the repair of DNA damage during transcription [72]. Furthermore, the high metabolic activity and weak antioxidant defense system along with their long life span makes the neurons highly susceptible to oxidative overload [73]. Since the brain is generally protected from environmental genotoxins by the blood brain barrier, endogenous, ROS-induced genome damage is the most critical threat to neuronal cells which can carry out BER/SSBR [74, 75].
Among the DGs, OGG1 expression decreased during rat brain development (transition from non-differentiated to differentiated neurons), while NEIL1 and NEIL2 levels increased [14]. This is consistent with high transcriptional activity in brain [76] and NEILs’ role in transcription-associated repair [22, 59, 60, 77]. While NTH1 showed comparable expression and activity in brain, testes, liver and kidney, OGG1 activity was low in the brain relative to other tissues. PNKP and APE1 are generally highly expressed in the brain. Among DNA pols, Polβ is highly and ubiquitously expressed in brain regions, and appears to be the major DNA pol in neurons [78]. Similar to NTH1, Polβ activity decreases in the brain with age. Further, DNA replication/repair proteins FEN-1, Polδ/ε, PCNA, and Lig I levels are low in adult neurons, while XRCC1 and Lig IIIα are highly expressed in brain compared to other tissue types [14]. These studies suggest that neurons may lack LP-BER activity with SN-BER being the predominant mode of repair. However, such comparison among tissues/cell types may not provide insight into the repair capacity per se and the published studies only confirm the presence of BER/SSBR proteins in the neurons. Furthermore, the inconsistencies in the levels of these proteins in various reports could reflect disparities in methodology, animal strains and their age groups [14].
METAL TOXICITY, OXIDATIVE STRESS AND GENOME DAMAGE IN BRAIN LINKED TO NEURODEGENERATIVE DISEASES
Cellular dyshomeostasis of essential transition metals leading to abnormal free metal overload, oxidative stress and accumulation of oxidative genome damage in the brain are common features in the pathogenesis of most if not all neurodegenerative diseases. Specifically, excessive accumulation of Cu and Fe has been implicated in AD, PD, HD, hereditary ferritinopathy and Wilson’s disease (WD) [19, 20, 79, 80]. The mechanisms of neurotoxicity of essential transition metals are complex because of their essentiality need as micronutrients. In the normal tissue, the metal content is tightly regulated and the metals are released from storage proteins in response to metabolic need [81]. For example, in the normal brain the majority of Fe (∼70%) is sequestered in ferritin/transferrin and Cu in ceruloplasmin [82, 83]. However, neurodegenerative disorders often involve gradual, clinically silent accumulation of free Cu and Fe, followed by sudden onset of toxicity when the metal load exceeds the storage threshold. MRI imaging studies as well as postmortem evaluations of brain samples of patients afflicted with neurodegenerative disorders showed elevated Fe levels [20]. Close association between brain metal dyshomeostasis and the onset and/or progression of AD and PD has been established in a number of studies [84]. Recent studies have strongly implicated essential transition metals such as Cu, Fe, and Zn and nonessential metals such as Aluminum as key factors in the pathophysiology of AD and PD [85]. High levels of Cu (400 µM) and Zn (1 mM) were found in AD compared to that in the healthy brain (with 70 µM Cu and 350 µM Zn) [86]. Increasing accumulation of metals in AD brain during progression from moderate to severe AD was also observed [19, 87]. The levels of Cu(II), Fe(II) and Zn(II) were shown to increase in the earlyphase of AD, while the levels of trivalent Fe(III) and Al(III) increased mainly in frontal cortex and hippocampus in the late-phase of AD. Moreover, chronic occupational exposure to Fe and Cu has been shown to increase the risk of AD and PD [88]. It was also shown that unilateral injection of FeCl3 into the substantia nigra (affected region in PD) of adult rats caused substantial reduction of striatal dopamine (95%), supporting the hypothesis that iron initiates dopaminergic neurodegeneration in PD [89]. In general, a common trait of neurodegeneration appears to be (direct or indirect) perturbation in the homeostasis of Cu, Zn, Fe, Al etc, leading to their increased levels in affected cells. Decreased symptoms associated with administration of metal chelators in AD animal models provided compelling support for the involvement of metal toxicity in these diseases [90].
Fe and Cu can generate ROS causing DNA damage in brain cells [91–93], by producing hydroxyl and superoxide radicals via Fenton reaction as follows:
Fe(III) + H2O2→Fe(II) + HO2•+H+; Fe(II) + H2O2→Fe(III) + •OH + OH−
Cu(II) + H2O2→Cu(I) + HO2•+H+; Cu(I) + H2O2→Cu(II) + •OH + OH−
Metals can also cause DNA damage by direct binding. The metal-induced genotoxic damage was shown by the wide spectrum of damaged bases, oxidized AP sites as well as SSBs in cultured cells and animals exposed to pro-oxidant metals [94–96]. Accumulation of DNA damage in affected brain regions correlates with metal overload, but the mechanism of selective metal accumulation in affected regions vs. other regions is not understood.
Many studies have linked the increase in unrepaired oxidative DNA damage to neurodegenerative diseases [7, 8, 97]. Accumulation of 8-oxoG in distinct brain regions afflicted by neuronal dysfunction is a common finding in most neurological diseases. Further, the 8-oxoG level in cerebrospinal fluid of AD patients is nearly double of that in the control group. Other studies showed oxidative DNA damage as one of the early events in mild dementia [98]. Although most studies focused on changes in 8-oxoG, primarily because of the ease of its detection and quantitation, we predict similar increase in the levels of other oxidized bases as well as in these diseases. Increase in DNA SSBs with a small increase in DSBs was also observed in AD and PD brains [93]. Considering that ROS primarily induce genome damage in the brain, the DSBs might be the result of closely spaced bi-stranded SSBs and/or oxidized bases/AP sites during repair. Thus repair of these secondary DSBs which are the most toxic type of DNA damages is extremely critical for maintaining genomic integrity in the neurons.
As with nuclear DNA, oxidative damage to mitochondrial DNA has been linked to neuronal dysfunctions [99, 100]. Mitochondrial DNA is more vulnerable to oxidative damage than the nuclear genome, presumably due to its proximity to electron transport chain and to the lack of protective histones [101], particularly in the neurons because of their high O2 consumption. Thus increased levels of oxidized bases (e.g., 8-oxoG), AP sites and SSBs as well as mutations and deletions in the mitochondrial genome were observed in brain with aging, and in AD, PD, ALS, HD and other neurodegenerative diseases [102, 103]. The role of oxidative damage to mitochondrial DNA in neurodegenerative diseases was further supported by the fact that the steady state level of 8-oxoG in mitochondrial but not nuclear DNA inversely correlated with life span of brain cells [102, 104].
BER/SSBR DEFECTS IN NEURODEGENERATIVE DISEASES
As many as 200 neurological disorders with diverse etiologies and genetic characteristics have been reported so far in humans, many of which have been linked to inherited or acquired defect in one of the DNA repair pathways. Imbalance in genome damage and its repair has been proposed as a major reason for accumulation of unrepaired genome damage in neurodegenerative diseases, although the bases for such an imbalance were not understood until recently [4, 13, 105, 106]. Defective neuronal DNA repair resulting in genomic instability has been linked to neuronal dysfunctions. As shown in Table 2, mutations or altered expression of BER (e.g., OGG1, XRCC1; [107–109]), SSBR (e.g., TDP1, aprataxin, PNKP; [48, 54, 110]) and DSBR (e.g., ATM, NBS1) proteins have been observed in humans predisposed to various hereditary neurodegenerative diseases (see review by Caldecott, [52]). Deficiencies in OGG1 or APE1, combined with exposure to oxidative stress could lead to neurodegenerative symptoms. XRCC1 was indirectly linked to autosomal recessive spinocerebellar ataxias, which are characterized by various neurological symptoms. Further, defects in end-processing proteins aprataxin and TDP1 have been shown to be associated with ataxia with oculomotor apraxia1 and spinocerebellar ataxia with axonal neuropathy, respectively [54, 111, 112]. A recent study linked mutations in PNKP to autosomal recessive disease characterized by severe neurological abnormalities including microcephaly, early-onset, intractable seizures and developmental delay (denoted MCSZ; [110]).
Table 2.
Association of BER/SSBR protein defects with neurodegenerative diseases.
| Gene/Protein | Defect(s) | Disease connection |
|---|---|---|
| OGG1 | Ser326Cys polymorphism with reduced activity | Weakly associated with HD and ALS [107, 114, 115] |
| XRCC1 | Mutant variants with reduced binding to aprataxin/TDP1 | Autosomal recessive spinocerebellar ataxias [52, 111] |
| Arg399Gln polymorphism | ALS [117] | |
| Arg194Trp polymorphism | Late onset AD [108, 109] | |
| Aprataxin | Mutation/Deficiency | Ataxia with oculomotor apraxia1 [48, 111] |
| TDP1 | Inactivating mutations/Deficiency | Spinocerebellar ataxia with axonal neuropathy [112] |
| APE1 | Asp148Glu polymorphism with reduced repair activity | Increased ALS risk [116] |
| PNKP | Mutations | MCSZ (characterized by neurological abnormalities) [110] |
While compelling evidences exist linking deficiency in SSBR proteins to neurodegenerative disorders [113], there is controversy regarding similar association with the BER proteins. A common OGG1 Ser326Cys polymorphism with reduced enzyme activity and increased risk of various types of cancer is weakly associated with HD and ALS risk [114], but not with AD or PD [98, 107, 115]. Asp148Glu polymorphism in APE1 altering in the repair activity was associated with increased ALS risk [116]. Association between the XRCC1 Arg399Gln polymorphism and ALS risk was also reported [117]. Taken together, these studies suggest association of BER/SSBR gene polymorphisms with predisposition to neurodegenerative diseases.
Decreased capacity to repair oxidized DNA damage was also observed in brain cell extracts in sporadic neurodegenerative diseases constituting more than 80 % of most disease incidences, whose causes are not known. Weissman et al., [13] showed that significant BER deficiencies in brains of AD patients is due to limited DNA base damage processing by DGs and reduced repair synthesis by Polβ. Similarly, decreased mitochondrial Polγ activity in neurons affected with neurodegenerative diseases was observed [118], but a detailed picture of genome damage repair in neurodegenerative diseases is still lacking. Most of the difficulty in resolving these issues could be attributed to the fact that human brain genome damage/repair can only be studied in postmortem samples and difficulty in examining these processes in cell type (neurons, glial cells and astrocytes)-specific manner, which is very critical because of significant differences in damage tolerance in these cell types.
BER/SSBR INHIBITION BY PRO-OXIDANT METALS: A CASE OF DOUBLE JEOPARDY
Reduction in total BER capacity has not been correlated with the expression of BER enzymes in brain tissues from various neurodegenerative diseases which suggested additional mechanisms being involved in BER defects. Although, few early studies suggested inhibition of DNA repair activities by physiologically nonessential, heavy metals [119–122], the mechanism of such inhibition was not characterized. It was suggested that metals which accumulate in neurodegenerative brains could bind to DNA and interfere with damage scanning and detection by BER proteins, thus reducing the repair efficiency [121, 122].
Recently, we made an interesting discovery that Cu and Fe specifically inhibit the activities of NEIL1 and NEIL2 by forming stable complexes with these proteins [21]. We showed that Fe(II/III) and Cu(II) in physiologically relevant submicromolar concentration inhibit NEILs’ activity with 5-OHU or 8-oxoG-containing DNA substrates in vitro. Lack of similar inhibition of OGG1 with 8-oxoG substrates indicated the specificity of inhibition and suggested that the inhibition is due to metal binding to NEILs rather than to DNA. Furthermore, inhibition of NEIL-initiated total BER was due to NEIL1’s reduced interaction with Polβ and FEN-1 in presence of Fe(II). In-cell inhibition of NEILs by these metals was also observed based on the reduced repair activity of NEILs in extracts from SH-SY5Y neuroblastoma cells treated with FeSO4 or CuCl2. These studies, and evidence for increase in divalent Fe(II)/Cu(II) in the early phase of neurodegenerative diseases like AD, led us to propose that similar scenario could prevail in human neurodegenerative diseases.
We proposed that distinct mechanism/s could be involved in NEIL1 and NEIL2 inhibition by Fe/Cu [21]. NEIL2’s inhibition could at least partially involve displacement of the intrinsic Zn2+ in the Zn finger with Fe2+ or Cu2+. Earlier studies have shown the sensitivity of Zn-finger proteins such as bacterial Fpg and the mammalian XPA protein for heavy metals that could compete for the Zn finger centre [123, 124]. However, NEIL1 lacks the Zn finger motif but contains a Zn-less finger motif instead [125].
Many other BER/SSBR proteins are also inhibited by metals. Whiteside et al., showed that Cd and Cu inhibit both phosphatase and kinase activities of PNKP with human cell extracts and recombinant protein [126]. Heavy metals could delay SSB rejoining in mammalian cells [127, 128]. Another study revealed that elevated Fe levels caused reduction in FEN-1 and Lig III activities due to interference of repair protein binding to their DNA substrates [122].
Taken together, these studies showed that excessive Fe/Cu in neurodegenerative brains act as ‘double-edged sword’ by inducing ROS that damage the genome while also inhibiting damage repair (Fig. 2). Thus metal mediated genotoxicity could involve induction of oxidative genome damage via (i) free radical generation, and (ii) direct DNA binding; and inhibition of DNA repair via (i) inhibition of key BER/SSBR enzymes, and (ii) limiting protein-protein interactions among repair proteins.
Fig. 2. Metal genotoxicity in neurodegenerative diseases: A case of double jeopardy.

Iron and copper accumulating in human brain with neurodegenerative diseases induce oxidative genome damage via free radical generation and act as a ‘double-edged sword’ by also inhibiting damage repair by binding to the repair enzymes. Specific chelators and a reducing agent or curcumin allows repair to occur.
METAL CHELATORS TOGETHER WITH DTT/TCEP REVERSE BER INHIBITION
Metal chelators that sequester metal ions promoting their urinary and fecal excretion have limited therapeutic effectiveness in neurodegenerative diseases [129, 130]. Metal toxicity in AD/PD could involve dynamic (dys)homeostasis during disease progression. Hence any administration of metal chelators requires analysis of specific metal levels at the given time and universal chelators do not work [87]. Furthermore, choice of specific chelators with appropriate affinity and regulation of its dosage are very critical to prevent loss of essential metals much below the normal threshold. A comprehensive review of challenges associated with chelation therapy was published earlier [87].
We observed that metal chelators CaEDTA (a strong Cu chelator) only partially reversed Cu-induced inhibition of NEIL1, but produced nearly complete reversal in combination with the reducing agent DTT or TCEP suggesting that Cu oxidizes cysteine residues in NEIL1. Previous studies have shown oxidation of cysteines in proteins by Cu [131]. Inhibition of NEIL1 by H2O2 confirmed that oxidation could inhibit NEIL1. These studies suggested that metal chelators with reducing properties have therapeutic potential in restoring DNA repair capacity in neurodegenerative diseases.
Curcumin, a wonder molecule
Curcumin (1,7-bis[4-hydroxy 3-methoxy phenyl]-1,6-heptadiene-3,5-dione) is a common dietary pigment abundantly used in cuisines of several Asian countries and in traditional Indian medicine [132, 133]. Curcumin is one of the three curcuminoids in turmeric [87]. Beneficial effects of curcumin in human health have been known for decades, but its neuroprotective properties are gaining attention only recently. Early studies showed that curcumin protected PC12 cells against apoptosis induced by lethal dose of H2O2 and 1-methyl-4phenylpiriinium ions (MPP+)-induced apoptosis but the mechanism was not discovered [134]. Curcumin has both metal chelation [135] as well as reducing properties ([136]; see Fig. 3). The enolic hydroxyls in curcumin could form metal complexes [137]. Recent studies suggested that curcumin significantly reduces metal-induced neurotoxicity in rat hippocampal neurons [135]. It was proposed that the ability of curcumin to bind toxic metals and to form tight and inactive complexes could be a plausible pathway by which curcumin offers protection to the brain [138]. The interaction of curcumin with Cu and Fe reaches halfmaximum at approximately 3–12 µM Cu(II) and 2.5–5 µM Fe(II) [139]. Additionally, the antioxidant (free radical scavenger) and anti-inflammatory properties of curcumin along with its ability to cross blood brain barrier makes it a potent neuroprotectant [140, 141].
Fig. 3. Structure of curcumin (A), chemistry of metal chelation (B) and antioxidant properties (C).
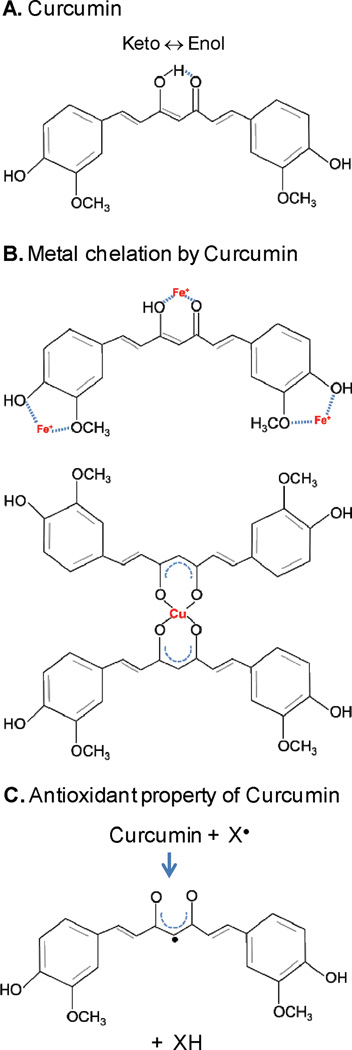
Curcumin exhibits keto-enol tautomerism in aqueous solution. Complexes of curcumin with iron (1:3) and copper (2:1 or 1:1, not shown) suggest its strong chelating activity. Curcumin also acts as a reducing agent as illustrated in (C).
We showed that curcumin could reverse both Fe(II/III) and Cu(II) mediated inhibition of NEILs’-initiated BER both in vitro and in SH-SY5Y cells [21]. Thus with its dual properties, curcumin could chelate Fe/Cu bound to NEILs and reverse NEIL1’s oxidation by Cu(II).
CONCLUSIONS AND FUTURE PERSPECTIVES
Elevated levels of unbuffered divalent transition metal ions, ROS and accumulation of oxidative genome damage coupled with reduction in their repair in the brain have been implicated in the etiology of most neurodegenerative diseases. One reason for targeted manifestation of oxidative stress in neurons may be due to limited DNA repair capacity of differentiated neurons lacking DNA replication. In addition, general reduction in DNA repair activity in the brain has been associated with the aging process. Furthermore, we and others have shown that inhibition of DNA repair processes by metal ions could also contribute to neuronal genotoxicity. One unexplored issue is how neuronal cells only in specific and localized brain regions are affected in different neurological diseases. Future studies should focus on the interplay of BER/SSBER components with specific etiologies associated with these diseases. Furthermore, as described before, a small but definite increase in DSBs (possibly generated from closely situated bi-stranded SSBs and/or base damages) occurs in neurodegenerative diseases; however their repair has not been investigated in the neurons. Future studies should also examine the deficiencies in other DNA repair processes and the inhibitory effect of diseaseslinked metal ions. Understanding how DNA repair deficiency occurs and affects the nervous system could provide a rational basis for therapies for ameliorating the genotoxicity in neurodegenerative diseases.
ACKNOWLEDGEMENTS
The review with limited focus was not meant to provide a comprehensive review of literature and many appropriate references could not be included for which the authors apologize. The research in the authors’ laboratory has been supported by USPHS grants, R01 CA81063, R01 CA53791 (S.M.) and American Parkinson’s Disease Association’s postdoctoral fellowship (M.L.H.). We thank Drs. Tapas Hazra, Luis Holthauzen (Department of Biochemistry and Molecular Biology), Istvan Boldogh (Department of Microbiology and Immunology) and Mitra lab members at UTMB for helpful discussion.
REFERENCES
- 1.Dawson TL, Gores GJ, Nieminen AL, Herman B, Lemasters JJ. Mitochondria as a source of reactive oxygen species during reductive stress in rat hepatocytes. Am J Physiol. 1993;264:C961–C967. doi: 10.1152/ajpcell.1993.264.4.C961. [DOI] [PubMed] [Google Scholar]
- 2.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 3.Mitra S, Izumi T, Boldogh I, Bhakat KK, Hill JW, Hazra TK. Choreography of oxidative damage repair in mammalian genomes. Free Radic Biol Med. 2002;33:15–28. doi: 10.1016/s0891-5849(02)00819-5. [DOI] [PubMed] [Google Scholar]
- 4.Hanawalt PC. Emerging links between premature ageing and defective DNA repair. Mech Ageing Dev. 2008;129:503–505. doi: 10.1016/j.mad.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dorszewska J, Florczak J, Rozycka A, Kempisty B, Jaroszewska-Kolecka J, Chojnacka K, Trzeciak WH, Kozubski W. Oxidative DNA damage and level of thiols as related to polymorphisms of MTHFR, MTR, MTHFD1 in Alzheimer's and Parkinson's diseases. Acta Neurobiol Exp (Wars) 2007;67:113–129. doi: 10.55782/ane-2007-1639. [DOI] [PubMed] [Google Scholar]
- 6.Lyras L, Cairns NJ, Jenner A, Jenner P, Halliwell B. An assessment of oxidative damage to proteins, lipids, and DNA in brain from patients with Alzheimer's disease. J Neurochem. 1997;68:2061–2069. doi: 10.1046/j.1471-4159.1997.68052061.x. [DOI] [PubMed] [Google Scholar]
- 7.Alam ZI, Jenner A, Daniel SE, Lees AJ, Cairns N, Marsden CD, Jenner P, Halliwell B. Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J Neurochem. 1997;69:1196–1203. doi: 10.1046/j.1471-4159.1997.69031196.x. [DOI] [PubMed] [Google Scholar]
- 8.Yasuhara T, Hara K, Sethi KD, Morgan JC, Borlongan CV. Increased 8-OHdG levels in the urine, serum, and substantia nigra of hemiparkinsonian rats. Brain Res. 2007;1133:49–52. doi: 10.1016/j.brainres.2006.11.072. [DOI] [PubMed] [Google Scholar]
- 9.Acevedo-Torres K, Berrios L, Rosario N, Dufault V, Skatchkov S, Eaton MJ, Torres-Ramos CA, Ayala-Torres S. Mitochondrial DNA damage is a hallmark of chemically induced and the R6/2 transgenic model of Huntington's disease. DNA Repair (Amst) 2009;8:126–136. doi: 10.1016/j.dnarep.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Polidori MC, Mecocci P, Browne SE, Senin U, Beal MF. Oxidative damage to mitochondrial DNA in Huntington's disease parietal cortex. Neurosci Lett. 1999;272:53–56. doi: 10.1016/s0304-3940(99)00578-9. [DOI] [PubMed] [Google Scholar]
- 11.Aguirre N, Beal MF, Matson WR, Bogdanov MB. Increased oxidative damage to DNA in an animal model of amyotrophic lateral sclerosis. Free Radic Res. 2005;39:383–388. doi: 10.1080/10715760400027979. [DOI] [PubMed] [Google Scholar]
- 12.Bogdanov M, Brown RH, Matson W, Smart R, Hayden D, O'Donnell H, Flint Beal M, Cudkowicz M. Increased oxidative damage to DNA in ALS patients. Free Radic Biol Med. 2000;29:652–658. doi: 10.1016/s0891-5849(00)00349-x. [DOI] [PubMed] [Google Scholar]
- 13.Weissman L, Jo DG, Sorensen MM, de Souza-Pinto NC, Markesbery WR, Mattson MP, Bohr VA. Defective DNA base excision repair in brain from individuals with Alzheimer's disease and amnestic mild cognitive impairment. Nucleic Acids Res. 2007;35:5545–5555. doi: 10.1093/nar/gkm605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilson DM, 3rd, McNeill DR. Base excision repair and the central nervous system. Neuroscience. 2007;145:1187–1200. doi: 10.1016/j.neuroscience.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 15.Rolig RL, McKinnon PJ. Linking DNA damage and neurodegeneration. Trends Neurosci. 2000;23:417–424. doi: 10.1016/s0166-2236(00)01625-8. [DOI] [PubMed] [Google Scholar]
- 16.Wilson DM, 3rd, Bohr VA. The mechanics of base excision repair, and its relationship to aging and disease. DNA Repair (Amst) 2007;6:544–559. doi: 10.1016/j.dnarep.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 17.Hart RW, Setlow RB. Correlation between deoxyribonucleic acid excision-repair and life-span in a number of mammalian species. Proc Natl Acad Sci U S A. 1974;71:2169–2173. doi: 10.1073/pnas.71.6.2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hegde ML, Shanmugavelu P, Vengamma B, Rao TS, Menon RB, Rao RV, Rao KS. Serum trace element levels and the complexity of inter-element relations in patients with Parkinson's disease. J Trace Elem Med Biol. 2004;18:163–171. doi: 10.1016/j.jtemb.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 19.Rao KSJ, Rao RV, Shanmugavelu P, Menon RB. Trace elements in Alzheimer's disease brain: A new hypothesis. Alz Rep. 1999;2:241–246. [Google Scholar]
- 20.Zecca L, Youdim MB, Riederer P, Connor JR, Crichton RR. Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci. 2004;5:863–873. doi: 10.1038/nrn1537. [DOI] [PubMed] [Google Scholar]
- 21.Hegde ML, Hegde PM, Holthauzen LM, Hazra TK, Rao KS, Mitra S. Specific Inhibition of NEIL-initiated repair of oxidized base damage in human genome by copper and iron: potential etiological linkage to neurodegenerative diseases. J Biol Chem. 2010;285:28812–28825. doi: 10.1074/jbc.M110.126664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008;18:27–47. doi: 10.1038/cr.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang N, Galick H, Wallace SS. Attempted base excision repair of ionizing radiation damage in human lymphoblastoid cells produces lethal and mutagenic double strand breaks. DNA Repair (Amst) 2004;3:1323–1334. doi: 10.1016/j.dnarep.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 24.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. 2. Washington: ASM Press; 2006. DNA Repair and Mutagenesis. [Google Scholar]
- 25.Slupphaug G, Kavli B, Krokan HE. The interacting pathways for prevention and repair of oxidative DNA damage. Mutat Res. 2003;531:231–251. doi: 10.1016/j.mrfmmm.2003.06.002. [DOI] [PubMed] [Google Scholar]
- 26.Chen Q, Marsh J, Ames B, Mossman B. Detection of 8-oxo-2'-deoxyguanosine, a marker of oxidative DNA damage, in culture medium from human mesothelial cells exposed to crocidolite asbestos. Carcinogenesis. 1996;17:2525–2527. doi: 10.1093/carcin/17.11.2525. [DOI] [PubMed] [Google Scholar]
- 27.Radak Z, Boldogh I. 8-Oxo-7,8-dihydroguanine: links to gene expression, aging, and defense against oxidative stress. Free Radic Biol Med. 2010;49:587–596. doi: 10.1016/j.freeradbiomed.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mahaney BL, Meek K, Lees-Miller SP. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem J. 2009;417:639–650. doi: 10.1042/BJ20080413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rassool FV, Tomkinson AE. Targeting abnormal DNA double strand break repair in cancer. Cell Mol Life Sci. 2010;67:3699–3710. doi: 10.1007/s00018-010-0493-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hegde ML, Hazra TK, Mitra S. Functions of disordered regions in mammalian early base excision repair proteins. Cell Mol Life Sci. 2010;67:3573–3587. doi: 10.1007/s00018-010-0485-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McCullough AK, Dodson ML, Lloyd RS. Initiation of base excision repair: glycosylase mechanisms and structures. Annu Rev Biochem. 1999;68:255–285. doi: 10.1146/annurev.biochem.68.1.255. [DOI] [PubMed] [Google Scholar]
- 32.Bandaru V, Sunkara S, Wallace SS, Bond JP. A novel human DNA glycosylase that removes oxidative DNA damage and is homologous to Escherichia coli endonuclease VIII. DNA Repair (Amst) 2002;1:517–529. doi: 10.1016/s1568-7864(02)00036-8. [DOI] [PubMed] [Google Scholar]
- 33.Hazra TK, Izumi T, Boldogh I, Imhoff B, Kow YW, Jaruga P, Dizdaroglu M, Mitra S. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc Natl Acad Sci U S A. 2002;99:3523–3528. doi: 10.1073/pnas.062053799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hazra TK, Izumi T, Kow YW, Mitra S. The discovery of a new family of mammalian enzymes for repair of oxidatively damaged DNA, and its physiological implications. Carcinogenesis. 2003;24:155–157. doi: 10.1093/carcin/24.2.155. [DOI] [PubMed] [Google Scholar]
- 35.Takao M, Kanno S, Shiromoto T, Hasegawa R, Ide H, Ikeda S, Sarker AH, Seki S, Xing JZ, Le XC, Weinfeld M, Kobayashi K, Miyazaki J, Muijtjens M, Hoeijmakers JH, van der Horst G, Yasui A. Novel nuclear and mitochondrial glycosylases revealed by disruption of the mouse Nth1 gene encoding an endonuclease III homolog for repair of thymine glycols. Embo J. 2002;21:3486–3493. doi: 10.1093/emboj/cdf350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu M, Bandaru V, Bond JP, Jaruga P, Zhao X, Christov PP, Burrows CJ, Rizzo CJ, Dizdaroglu M, Wallace SS. The mouse ortholog of NEIL3 is a functional DNA glycosylase in vitro and in vivo. Proc Natl Acad Sci U S A. 2010;107:4925–4930. doi: 10.1073/pnas.0908307107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parker JB, Bianchet MA, Krosky DJ, Friedman JI, Amzel LM, Stivers JT. Enzymatic capture of an extrahelical thymine in the search for uracil in DNA. Nature. 2007;449:433–437. doi: 10.1038/nature06131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Slupphaug G, Mol CD, Kavli B, Arvai AS, Krokan HE, Tainer JA. A nucleotide-flipping mechanism from the structure of human uracil-DNA glycosylase bound to DNA. Nature. 1996;384:87–92. doi: 10.1038/384087a0. [DOI] [PubMed] [Google Scholar]
- 39.Banerjee D, Mandal SM, Das A, Hegde ML, Das S, Bhakat KK, Boldogh I, Sarkar PS, Mitra S, Hazra TK. Preferential repair of oxidized base damage in the transcribed genes of mammalian cells. J Biol Chem. 2011 doi: 10.1074/jbc.M110.198796. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mitra S, Hegde ML, Theriot CA, Das A, Hegde PM, Hazra TK. Complexity in repair of oxidative genome damage and its regulation. Proceedings of Princess Takamatsu Symposium, Tokyo, Japan. 2009 [Google Scholar]
- 41.Wiederhold L, Leppard JB, Kedar P, Karimi-Busheri F, Rasouli-Nia A, Weinfeld M, Tomkinson AE, Izumi T, Prasad R, Wilson SH, Mitra S, Hazra TK. AP endonuclease-independent DNA base excision repair in human cells. Mol Cell. 2004;15:209–220. doi: 10.1016/j.molcel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 42.Sobol RW, Prasad R, Evenski A, Baker A, Yang XP, Horton JK, Wilson SH. The lyase activity of the DNA repair protein beta-polymerase protects from DNA-damage-induced cytotoxicity. Nature. 2000;405:807–810. doi: 10.1038/35015598. [DOI] [PubMed] [Google Scholar]
- 43.Frosina G, Fortini P, Rossi O, Carrozzino F, Raspaglio G, Cox LS, Lane DP, Abbondandolo A, Dogliotti E. Two pathways for base excision repair in mammalian cells. J Biol Chem. 1996;271:9573–9578. doi: 10.1074/jbc.271.16.9573. [DOI] [PubMed] [Google Scholar]
- 44.Duxin JP, Dao B, Martinsson P, Rajala N, Guittat L, Campbell JL, Spelbrink JN, Stewart SA. Human Dna2 is a nuclear and mitochondrial DNA maintenance protein. Mol Cell Biol. 2009;29:4274–4282. doi: 10.1128/MCB.01834-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.El-Khamisy SF, Saifi GM, Weinfeld M, Johansson F, Helleday T, Lupski JR, Caldecott KW. Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature. 2005;434:108–113. doi: 10.1038/nature03314. [DOI] [PubMed] [Google Scholar]
- 46.Yang SW, Burgin AB, Jr, Huizenga BN, Robertson CA, Yao KC, Nash HA. A eukaryotic enzyme that can disjoin dead-end covalent complexes between DNA and type I topoisomerases. Proc Natl Acad Sci U S A. 1996;93:11534–11539. doi: 10.1073/pnas.93.21.11534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pouliot JJ, Yao KC, Robertson CA, Nash HA. Yeast gene for a Tyr-DNA phosphodiesterase that repairs topoisomerase I complexes. Science. 1999;286:552–555. doi: 10.1126/science.286.5439.552. [DOI] [PubMed] [Google Scholar]
- 48.Ahel I, Rass U, El-Khamisy SF, Katyal S, Clements PM, McKinnon PJ, Caldecott KW, West SC. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature. 2006;443:713–716. doi: 10.1038/nature05164. [DOI] [PubMed] [Google Scholar]
- 49.Rass U, Ahel I, West SC. Actions of aprataxin in multiple DNA repair pathways. J Biol Chem. 2007;282:9469–9474. doi: 10.1074/jbc.M611489200. [DOI] [PubMed] [Google Scholar]
- 50.Mazur DJ, Perrino FW. Structure and expression of the TREX1 and TREX2 3' --> 5' exonuclease genes. J Biol Chem. 2001;276:14718–14727. doi: 10.1074/jbc.M010051200. [DOI] [PubMed] [Google Scholar]
- 51.Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 52.Caldecott KW. Single-strand break repair and genetic disease. Nat Rev Genet. 2008;9:619–631. doi: 10.1038/nrg2380. [DOI] [PubMed] [Google Scholar]
- 53.Dianova II, Sleeth KM, Allinson SL, Parsons JL, Breslin C, Caldecott KW, Dianov GL. XRCC1-DNA polymerase beta interaction is required for efficient base excision repair. Nucleic Acids Res. 2004;32:2550–2555. doi: 10.1093/nar/gkh567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.El-Khamisy SF, Hartsuiker E, Caldecott KW. TDP1 facilitates repair of ionizing radiation-induced DNA single-strand breaks. DNA Repair (Amst) 2007;6:1485–1495. doi: 10.1016/j.dnarep.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 55.Vidal AE, Boiteux S, Hickson ID, Radicella JP. XRCC1 coordinates the initial and late stages of DNA abasic site repair through protein-protein interactions. Embo J. 2001;20:6530–6539. doi: 10.1093/emboj/20.22.6530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Whitehouse CJ, Taylor RM, Thistlethwaite A, Zhang H, Karimi-Busheri F, Lasko DD, Weinfeld M, Caldecott KW. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell. 2001;104:107–117. doi: 10.1016/s0092-8674(01)00195-7. [DOI] [PubMed] [Google Scholar]
- 57.Lan L, Nakajima S, Oohata Y, Takao M, Okano S, Masutani M, Wilson SH, Yasui A. In situ analysis of repair processes for oxidative DNA damage in mammalian cells. Proc Natl Acad Sci U S A. 2004;101:13738–13743. doi: 10.1073/pnas.0406048101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu Y, Kao HI, Bambara RA. Flap endonuclease 1: a central component of DNA metabolism. Annu Rev Biochem. 2004;73:589–615. doi: 10.1146/annurev.biochem.73.012803.092453. [DOI] [PubMed] [Google Scholar]
- 59.Dou H, Theriot CA, Das A, Hegde ML, Matsumoto Y, Boldogh I, Hazra TK, Bhakat KK, Mitra S. Interaction of the human DNA glycosylase NEIL1 with proliferating cell nuclear antigen. The potential for replication-associated repair of oxidized bases in mammalian genomes. J Biol Chem. 2008;283:3130–3140. doi: 10.1074/jbc.M709186200. [DOI] [PubMed] [Google Scholar]
- 60.Hegde ML, Theriot CA, Das A, Hegde PM, Guo Z, Gary RK, Hazra TK, Shen B, Mitra S. Physical and functional interaction between human oxidized base-specific DNA glycosylase NEIL1 and flap endonuclease 1. J Biol Chem. 2008;283:27028–27037. doi: 10.1074/jbc.M802712200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Prasad R, Dianov GL, Bohr VA, Wilson SH. FEN1 stimulation of DNA polymerase beta mediates an excision step in mammalian long patch base excision repair. J Biol Chem. 2000;275:4460–4466. doi: 10.1074/jbc.275.6.4460. [DOI] [PubMed] [Google Scholar]
- 62.Matsumoto Y, Kim K. Excision of deoxyribose phosphate residues by DNA polymerase beta during DNA repair. Science. 1995;269:699–702. doi: 10.1126/science.7624801. [DOI] [PubMed] [Google Scholar]
- 63.Ellenberger T, Tomkinson AE. Eukaryotic DNA ligases: structural and functional insights. Annu Rev Biochem. 2008;77:313–338. doi: 10.1146/annurev.biochem.77.061306.123941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Das A, Wiederhold L, Leppard JB, Kedar P, Prasad R, Wang H, Boldogh I, Karimi-Busheri F, Weinfeld M, Tomkinson AE, Wilson SH, Mitra S, Hazra TK. NEIL2-initiated, APE-independent repair of oxidized bases in DNA: Evidence for a repair complex in human cells. DNA Repair (Amst) 2006;5:1439–1448. doi: 10.1016/j.dnarep.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bohr VA. Repair of oxidative DNA damage in nuclear and mitochondrial DNA, and some changes with aging in mammalian cells. Free Radic Biol Med. 2002;32:804–812. doi: 10.1016/s0891-5849(02)00787-6. [DOI] [PubMed] [Google Scholar]
- 66.Szczesny B, Tann AW, Longley MJ, Copeland WC, Mitra S. Long patch base excision repair in mammalian mitochondrial genomes. J Biol Chem. 2008;283:26349–26356. doi: 10.1074/jbc.M803491200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hu J, de Souza-Pinto NC, Haraguchi K, Hogue BA, Jaruga P, Greenberg MM, Dizdaroglu M, Bohr VA. Repair of formamidopyrimidines in DNA involves different glycosylases: role of the OGG1, NTH1, and NEIL1 enzymes. J Biol Chem. 2005;280:40544–40551. doi: 10.1074/jbc.M508772200. [DOI] [PubMed] [Google Scholar]
- 68.Chattopadhyay R, Wiederhold L, Szczesny B, Boldogh I, Hazra TK, Izumi T, Mitra S. Identification and characterization of mitochondrial abasic (AP)-endonuclease in mammalian cells. Nucleic Acids Res. 2006;34:2067–2076. doi: 10.1093/nar/gkl177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Das BB, Dexheimer TS, Maddali K, Pommier Y. Role of tyrosyl-DNA phosphodiesterase (TDP1) in mitochondria. Proc Natl Acad Sci U S A. 2010;107:19790–19795. doi: 10.1073/pnas.1009814107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Longley MJ, Prasad R, Srivastava DK, Wilson SH, Copeland WC. Identification of 5'-deoxyribose phosphate lyase activity in human DNA polymerase gamma and its role in mitochondrial base excision repair in vitro. Proc Natl Acad Sci U S A. 1998;95:12244–12248. doi: 10.1073/pnas.95.21.12244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kruman II. Why do neurons enter the cell cycle? Cell Cycle. 2004;3:769–773. [PubMed] [Google Scholar]
- 72.Nouspikel T, Hanawalt PC. DNA repair in terminally differentiated cells. DNA Repair (Amst) 2002;1:59–75. doi: 10.1016/s1568-7864(01)00005-2. [DOI] [PubMed] [Google Scholar]
- 73.Liu J, Head E, Gharib AM, Yuan W, Ingersoll RT, Hagen TM, Cotman CW, Ames BN. Memory loss in old rats is associated with brain mitochondrial decay and RNA/DNA oxidation: partial reversal by feeding acetyl-L-carnitine and/or R-alpha -lipoic acid. Proc Natl Acad Sci U S A. 2002;99:2356–2361. doi: 10.1073/pnas.261709299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen D, Cao G, Hastings T, Feng Y, Pei W, O'Horo C, Chen J. Age-dependent decline of DNA repair activity for oxidative lesions in rat brain mitochondria. J Neurochem. 2002;81:1273–1284. doi: 10.1046/j.1471-4159.2002.00916.x. [DOI] [PubMed] [Google Scholar]
- 75.Liu PK, Hsu CY, Dizdaroglu M, Floyd RA, Kow YW, Karakaya A, Rabow LE, Cui JK. Damage, repair, and mutagenesis in nuclear genes after mouse forebrain ischemia-reperfusion. J Neurosci. 1996;16:6795–6806. doi: 10.1523/JNEUROSCI.16-21-06795.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- 77.Dou H, Mitra S, Hazra TK. Repair of oxidized bases in DNA bubble structures by human DNA glycosylases NEIL1 and NEIL2. J Biol Chem. 2003;278:49679–49684. doi: 10.1074/jbc.M308658200. [DOI] [PubMed] [Google Scholar]
- 78.Wilson SH. Mammalian base excision repair and DNA polymerase beta. Mutat Res. 1998;407:203–215. doi: 10.1016/s0921-8777(98)00002-0. [DOI] [PubMed] [Google Scholar]
- 79.Campbell A, Smith MA, Sayre LM, Bondy SC, Perry G. Mechanisms by which metals promote events connected to neurodegenerative diseases. Brain Res Bull. 2001;55:125–132. doi: 10.1016/s0361-9230(01)00455-5. [DOI] [PubMed] [Google Scholar]
- 80.Zatta P, Lucchini R, van Rensburg SJ, Taylor A. The role of metals in neurodegenerative processes: aluminum, manganese, and zinc. Brain Res Bull. 2003;62:15–28. doi: 10.1016/s0361-9230(03)00182-5. [DOI] [PubMed] [Google Scholar]
- 81.Gaasch JA, Lockman PR, Geldenhuys WJ, Allen DD, Van der Schyf CJ. Brain iron toxicity: differential responses of astrocytes, neurons, and endothelial cells. Neurochem Res. 2007;32:1196–1208. doi: 10.1007/s11064-007-9290-4. [DOI] [PubMed] [Google Scholar]
- 82.Texel SJ, Xu X, Harris ZL. Ceruloplasmin in neurodegenerative diseases. Biochem Soc Trans. 2008;36:1277–1281. doi: 10.1042/BST0361277. [DOI] [PubMed] [Google Scholar]
- 83.Koeppen AH. A brief history of brain iron research. J Neurol Sci. 2003;207:95–97. doi: 10.1016/s0022-510x(02)00429-x. [DOI] [PubMed] [Google Scholar]
- 84.Cuajungco MP, Frederickson CJ, Bush AI. Amyloid-beta metal interaction and metal chelation. Subcell Biochem. 2005;38:235–254. doi: 10.1007/0-387-23226-5_12. [DOI] [PubMed] [Google Scholar]
- 85.Molina-Holgado F, Hider RC, Gaeta A, Williams R, Francis P. Metals ions and neurodegeneration. Biometals. 2007;20:639–654. doi: 10.1007/s10534-006-9033-z. [DOI] [PubMed] [Google Scholar]
- 86.Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR. Copper, iron and zinc in Alzheimer's disease senile plaques. J Neurol Sci. 1998;158:47–52. doi: 10.1016/s0022-510x(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 87.Hegde ML, Bharathi P, Suram A, Venugopal C, Jagannathan R, Poddar P, Srinivas P, Sambamurti K, Rao KJ, Scancar J, Messori L, Zecca L, Zatta P. Challenges associated with metal chelation therapy in Alzheimer's disease. J Alzheimers Dis. 2009;17:457–468. doi: 10.3233/JAD-2009-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gorell JM, Johnson CC, Rybicki BA, Peterson EL, Kortsha GX, Brown GG, Richardson RJ. Occupational exposure to manganese, copper, lead, iron, mercury and zinc and the risk of Parkinson's disease. Neurotoxicology. 1999;20:239–247. [PubMed] [Google Scholar]
- 89.Youdim MB, Ben-Shachar D, Riederer P. Iron in brain function and dysfunction with emphasis on Parkinson's disease. Eur Neurol. 1991;31(Suppl 1):34–40. doi: 10.1159/000116719. [DOI] [PubMed] [Google Scholar]
- 90.Kulkarni AP, Govender DA, Kotwal GJ, Kellaway LA. Modulation of Anxiety Behavior by Intranasally Administered Vaccinia Virus Complement Control Protein and Curcumin in a Mouse Model of Alzheimer's Disease. Curr Alzheimer Res. 2010 doi: 10.2174/156720511794604598. In Press. [DOI] [PubMed] [Google Scholar]
- 91.Berg D, Youdim MB. Role of iron in neurodegenerative disorders. Top Magn Reson Imaging. 2006;17:5–17. doi: 10.1097/01.rmr.0000245461.90406.ad. [DOI] [PubMed] [Google Scholar]
- 92.Gupta VB, Anitha S, Hegde ML, Zecca L, Garruto RM, Ravid R, Shankar SK, Stein R, Shanmugavelu P, Jagannatha Rao KS. Aluminium in Alzheimer's disease: are we still at a crossroad? Cell Mol Life Sci. 2005;62:143–158. doi: 10.1007/s00018-004-4317-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hegde ML, Gupta VB, Anitha M, Harikrishna T, Shankar SK, Muthane U, Subba Rao K, Jagannatha Rao KS. Studies on genomic DNA topology and stability in brain regions of Parkinson's disease. Arch Biochem Biophys. 2006;449:143–156. doi: 10.1016/j.abb.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 94.Hamilton-Koch W, Snyder RD, Lavelle JM. Metal-induced DNA damage and repair in human diploid fibroblasts and Chinese hamster ovary cells. Chem Biol Interact. 1986;59:17–28. doi: 10.1016/s0009-2797(86)80052-7. [DOI] [PubMed] [Google Scholar]
- 95.Robison SH, Cantoni O, Costa M. Analysis of metal-induced DNA lesions and DNA-repair replication in mammalian cells. Mutat Res. 1984;131:173–181. doi: 10.1016/0167-8817(84)90058-0. [DOI] [PubMed] [Google Scholar]
- 96.Snyder RD. Role of active oxygen species in metal-induced DNA strand breakage in human diploid fibroblasts. Mutat Res. 1988;193:237–246. doi: 10.1016/0167-8817(88)90034-x. [DOI] [PubMed] [Google Scholar]
- 97.Lovell MA, Markesbery WR. Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer's disease. Nucleic Acids Res. 2007;35:7497–7504. doi: 10.1093/nar/gkm821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Coppede F, Migliore L. DNA damage and repair in Alzheimer's disease. Curr Alzheimer Res. 2009;6:36–47. doi: 10.2174/156720509787313970. [DOI] [PubMed] [Google Scholar]
- 99.Deng X, Vidal R, Englander EW. Accumulation of oxidative DNA damage in brain mitochondria in mouse model of hereditary ferritinopathy. Neurosci Lett. 2010;479:44–48. doi: 10.1016/j.neulet.2010.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Martin LJ. Mitochondrial and Cell Death Mechanisms in Neurodegenerative Diseases. Pharmaceuticals (Basel) 2010;3:839–915. doi: 10.3390/ph3040839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ames BN, Shigenaga MK, Hagen TM. Oxidants, antioxidants, and the degenerative diseases of aging. Proc Natl Acad Sci U S A. 1993;90:7915–7922. doi: 10.1073/pnas.90.17.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Druzhyna NM, Wilson GL, LeDoux SP. Mitochondrial DNA repair in aging and disease. Mech Ageing Dev. 2008;129:383–390. doi: 10.1016/j.mad.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kang D, Hamasaki N. Alterations of mitochondrial DNA in common diseases and disease states: aging, neurodegeneration, heart failure, diabetes, and cancer. Curr Med Chem. 2005;12:429–441. doi: 10.2174/0929867053363081. [DOI] [PubMed] [Google Scholar]
- 104.Barja G, Herrero A. Oxidative damage to mitochondrial DNA is inversely related to maximum life span in the heart and brain of mammals. Faseb J. 2000;14:312–318. doi: 10.1096/fasebj.14.2.312. [DOI] [PubMed] [Google Scholar]
- 105.Obulesu M, Rao DM. DNA damage and impairment of DNA repair in Alzheimer's disease. Int J Neurosci. 2010;120:397–403. doi: 10.3109/00207450903411133. [DOI] [PubMed] [Google Scholar]
- 106.Subba Rao K. Mechanisms of disease: DNA repair defects and neurological disease. Nat Clin Pract Neurol. 2007;3:162–172. doi: 10.1038/ncpneuro0448. [DOI] [PubMed] [Google Scholar]
- 107.Coppede F, Ceravolo R, Migheli F, Fanucchi F, Frosini D, Siciliano G, Bonuccelli U, Migliore L. The hOGG1 Ser326Cys polymorphism is not associated with sporadic Parkinson's disease. Neurosci Lett. 2010;473:248–251. doi: 10.1016/j.neulet.2010.02.059. [DOI] [PubMed] [Google Scholar]
- 108.Dogru-Abbasoglu S, Aykac-Toker G, Hanagasi HA, Gurvit H, Emre M, Uysal M. The Arg194Trp polymorphism in DNA repair gene XRCC1 and the risk for sporadic late-onset Alzheimer's disease. Neurol Sci. 2007;28:31–34. doi: 10.1007/s10072-007-0744-x. [DOI] [PubMed] [Google Scholar]
- 109.Qian Y, Chen W, Wu J, Tao T, Bi L, Xu W, Qi H, Wang Y, Guo L. Association of polymorphism of DNA repair gene XRCC1 with sporadic late-onset Alzheimer's disease and age of onset in elderly Han Chinese. J Neurol Sci. 2010;295:62–65. doi: 10.1016/j.jns.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 110.Shen J, Gilmore EC, Marshall CA, Haddadin M, Reynolds JJ, Eyaid W, Bodell A, Barry B, Gleason D, Allen K, Ganesh VS, Chang BS, Grix A, Hill RS, Topcu M, Caldecott KW, Barkovich AJ, Walsh CA. Mutations in PNKP cause microcephaly, seizures and defects in DNA repair. Nat Genet. 2010;42:245–249. doi: 10.1038/ng.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rass U, Ahel I, West SC. Defective DNA repair and neurodegenerative disease. Cell. 2007;130:991–1004. doi: 10.1016/j.cell.2007.08.043. [DOI] [PubMed] [Google Scholar]
- 112.Hirano R, Interthal H, Huang C, Nakamura T, Deguchi K, Choi K, Bhattacharjee MB, Arimura K, Umehara F, Izumo S, Northrop JL, Salih MA, Inoue K, Armstrong DL, Champoux JJ, Takashima H, Boerkoel CF. Spinocerebellar ataxia with axonal neuropathy: consequence of a Tdp1 recessive neomorphic mutation? Embo J. 2007;26:4732–4743. doi: 10.1038/sj.emboj.7601885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Katyal S, McKinnon PJ. DNA strand breaks, neurodegeneration and aging in the brain. Mech Ageing Dev. 2008;129:483–491. doi: 10.1016/j.mad.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Coppede F, Mancuso M, Lo Gerfo A, Carlesi C, Piazza S, Rocchi A, Petrozzi L, Nesti C, Micheli D, Bacci A, Migliore L, Murri L, Siciliano G. Association of the hOGG1 Ser326Cys polymorphism with sporadic amyotrophic lateral sclerosis. Neurosci Lett. 2007;420:163–168. doi: 10.1016/j.neulet.2007.04.067. [DOI] [PubMed] [Google Scholar]
- 115.Coppede F, Mancuso M, Lo Gerfo A, Manca ML, Petrozzi L, Migliore L, Siciliano G, Murri L. A Ser326Cys polymorphism in the DNA repair gene hOGG1 is not associated with sporadic Alzheimer's disease. Neurosci Lett. 2007;414:282–285. doi: 10.1016/j.neulet.2006.12.035. [DOI] [PubMed] [Google Scholar]
- 116.Hayward C, Colville S, Swingler RJ, Brock DJ. Molecular genetic analysis of the APEX nuclease gene in amyotrophic lateral sclerosis. Neurology. 1999;52:1899–1901. doi: 10.1212/wnl.52.9.1899. [DOI] [PubMed] [Google Scholar]
- 117.Coppede F, Migheli F, Lo Gerfo A, Fabbrizi MR, Carlesi C, Mancuso M, Corti S, Mezzina N, del Bo R, Comi GP, Siciliano G, Migliore L. Association study between XRCC1 gene polymorphisms and sporadic amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2010;11:122–124. doi: 10.3109/17482960903220297. [DOI] [PubMed] [Google Scholar]
- 118.Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, Murphy MP, Sammut IA. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. Faseb J. 2005;19:1088–1095. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- 119.McNeill DR, Narayana A, Wong HK, Wilson DM., 3rd Inhibition of Ape1 nuclease activity by lead, iron, and cadmium. Environ Health Perspect. 2004;112:799–804. doi: 10.1289/ehp.7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zharkov DO, Rosenquist TA. Inactivation of mammalian 8-oxoguanine-DNA glycosylase by cadmium(II): implications for cadmium genotoxicity. DNA Repair (Amst) 2002;1:661–670. doi: 10.1016/s1568-7864(02)00074-5. [DOI] [PubMed] [Google Scholar]
- 121.Grin IR, Konorovsky PG, Nevinsky GA, Zharkov DO. Heavy metal ions affect the activity of DNA glycosylases of the fpg family. Biochemistry (Mosc) 2009;74:1253–1259. doi: 10.1134/s000629790911011x. [DOI] [PubMed] [Google Scholar]
- 122.Li H, Swiercz R, Englander EW. Elevated metals compromise repair of oxidative DNA damage via the base excision repair pathway: implications of pathologic iron overload in the brain on integrity of neuronal DNA. J Neurochem. 2009;110:1774–1783. doi: 10.1111/j.1471-4159.2009.06271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hartwig A, Asmuss M, Blessing H, Hoffmann S, Jahnke G, Khandelwal S, Pelzer A, Burkle A. Interference by toxic metal ions with zinc-dependent proteins involved in maintaining genomic stability. Food Chem Toxicol. 2002;40:1179–1184. doi: 10.1016/s0278-6915(02)00043-1. [DOI] [PubMed] [Google Scholar]
- 124.Asmuss M, Mullenders LH, Eker A, Hartwig A. Differential effects of toxic metal compounds on the activities of Fpg and XPA, two zinc finger proteins involved in DNA repair. Carcinogenesis. 2000;21:2097–2104. doi: 10.1093/carcin/21.11.2097. [DOI] [PubMed] [Google Scholar]
- 125.Doublie S, Bandaru V, Bond JP, Wallace SS. The crystal structure of human endonuclease VIII-like 1 (NEIL1) reveals a zincless finger motif required for glycosylase activity. Proc Natl Acad Sci U S A. 2004;101:10284–10289. doi: 10.1073/pnas.0402051101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Whiteside JR, Box CL, McMillan TJ, Allinson SL. Cadmium and copper inhibit both DNA repair activities of polynucleotide kinase. DNA Repair (Amst) 2010;9:83–89. doi: 10.1016/j.dnarep.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 127.De Boeck M, Lison D, Kirsch-Volders M. Evaluation of the in vitro direct and indirect genotoxic effects of cobalt compounds using the alkaline comet assay. Influence of interdonor and interexperimental variability. Carcinogenesis. 1998;19:2021–2029. doi: 10.1093/carcin/19.11.2021. [DOI] [PubMed] [Google Scholar]
- 128.Lynn S, Lai HT, Kao SM, Lai J, Jan KY. Cadmium inhibits DNA strand break rejoining in methyl methanesulfonate-treated CHO-K1 cells. Toxicol Appl Pharmacol. 1997;144:171–176. doi: 10.1006/taap.1997.8116. [DOI] [PubMed] [Google Scholar]
- 129.Carter DS, Harrison AJ, Falenski KW, Blair RE, DeLorenzo RJ. Long-term decrease in calbindin-D28K expression in the hippocampus of epileptic rats following pilocarpine-induced status epilepticus. Epilepsy Res. 2008;79:213–223. doi: 10.1016/j.eplepsyres.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Knudtson ML, Wyse DG, Galbraith PD, Brant R, Hildebrand K, Paterson D, Richardson D, Burkart C, Burgess E. Chelation therapy for ischemic heart disease: a randomized controlled trial. Jama. 2002;287:481–486. doi: 10.1001/jama.287.4.481. [DOI] [PubMed] [Google Scholar]
- 131.Jalilehvand F, Mah V, Leung BO, Mink J, Bernard GM, Hajba L. Cadmium(II) cysteine complexes in the solid state: a multispectroscopic study. Inorg Chem. 2009;48:4219–4230. doi: 10.1021/ic900145n. [DOI] [PubMed] [Google Scholar]
- 132.Sharma RA, Gescher AJ, Steward WP. Curcumin: the story so far. Eur J Cancer. 2005;41:1955–1968. doi: 10.1016/j.ejca.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 133.Aggarwal BB, Sung B. Pharmacological basis for the role of curcumin in chronic diseases: an age-old spice with modern targets. Trends Pharmacol Sci. 2009;30:85–94. doi: 10.1016/j.tips.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 134.Chen J, Tang XQ, Zhi JL, Cui Y, Yu HM, Tang EH, Sun SN, Feng JQ, Chen PX. Curcumin protects PC12 cells against 1-methyl-4-phenylpyridinium ion-induced apoptosis by bcl-2-mitochondria-ROS-iNOS pathway. Apoptosis. 2006;11:943–953. doi: 10.1007/s10495-006-6715-5. [DOI] [PubMed] [Google Scholar]
- 135.Daniel S, Limson JL, Dairam A, Watkins GM, Daya S. Through metal binding, curcumin protects against lead- and cadmium-induced lipid peroxidation in rat brain homogenates and against lead-induced tissue damage in rat brain. J Inorg Biochem. 2004;98:266–275. doi: 10.1016/j.jinorgbio.2003.10.014. [DOI] [PubMed] [Google Scholar]
- 136.Menon VP, Sudheer AR. Antioxidant and anti-inflammatory properties of curcumin. Adv Exp Med Biol. 2007;595:105–125. doi: 10.1007/978-0-387-46401-5_3. [DOI] [PubMed] [Google Scholar]
- 137.Barik A, Mishra B, Shen L, Mohan H, Kadam RM, Dutta S, Zhang HY, Priyadarsini KI. Evaluation of a new copper(II)-curcumin complex as superoxide dismutase mimic and its free radical reactions. Free Radic Biol Med. 2005;39:811–822. doi: 10.1016/j.freeradbiomed.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 138.Baum L, Ng A. Curcumin interaction with copper and iron suggests one possible mechanism of action in Alzheimer's disease animal models. J Alzheimers Dis. 2004;6:367–377. doi: 10.3233/jad-2004-6403. discussion 443–369. [DOI] [PubMed] [Google Scholar]
- 139.Barreto R, Kawakita S, Tsuchiya J, Minelli E, Pavasuthipaisit K, Helmy A, Marotta F. Metal-induced oxidative damage in cultured hepatocytes and hepatic lysosomal fraction: beneficial effect of a curcumin/absinthium compound. Chin J Dig Dis. 2005;6:31–36. doi: 10.1111/j.1443-9573.2005.00184.x. [DOI] [PubMed] [Google Scholar]
- 140.Cole GM, Teter B, Frautschy SA. Neuroprotective effects of curcumin. Adv Exp Med Biol. 2007;595:197–212. doi: 10.1007/978-0-387-46401-5_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, Cole GM. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005;280:5892–5901. doi: 10.1074/jbc.M404751200. [DOI] [PubMed] [Google Scholar]