

Abstract
Background and aims
Celiac disease (cd) is a common small intestinal inflammatory disorder that results from a breach of intestinal tolerance to dietary gluten proteins, driven by gluten-reactive effector T cells. We aimed to assess the pathogenic role of gluten-reactive effector T cells and to generate a model of gluten-induced enteropathy.
Methods
CD4+CD25− T cell fractions were adoptively transferred into lymphopenic mice, leading to “baseline” small intestinal inflammation.
Results
Rag1−/− recipients of gliadin-presensitized CD4+CD45RBlowCD25− T cells, but not CD4+CD45RBhigh naive T cells, gained less weight and suffered from more severe duodenitis when challenged with oral gluten than recipients on gluten-free diet, or recipients of control (ovalbumin)-presensitized T cells. This was accompanied by deterioration of mucosal histological features characteristic of cd, and increased Th1/Th17 cell polarization in the duodenum and the periphery. Interestingly, reintroduction of a gluten-free diet led to weight gain, improvement of histological duodenitis, and a decrease in duodenal IFNγ and IL-17 transcripts. Moreover, B cell-competent nude recipients of gliadin-presensitized T cells produced high levels of serum anti-gliadin IgA and IgG1/IgG2c only when challenged with oral gluten.
Conclusions
CD4+ T cell immunity to gluten leads to a breach of oral gluten tolerance and small intestinal pathology in lymphopenic mice, similar to human cd. This model will be useful for the study of cd pathogenesis, but also for testing novel non-dietary therapies for cd.
Keywords: Celiac disease, gluten-sensitive enteropathy, small intestine, duodenitis, enteritis, gliadin, gluten, memory T cells, regulatory T cells, CD4+CD45RBlowCD25− T cells, intestinal antigen, food protein, oral tolerance, mucosal immunology
Introduction
Celiac disease (cd) is a gluten-sensitive enteropathy that has a prevalence of 0.5–1% in most Western populations1,2. 90–95% of patients carry the human leukocyte antigen (HLA-) DQ2, and the remainder HLA-DQ83. Cd is triggered by exposure to oral gluten (the storage proteins of wheat and related cereals) and is characterized by a dysregulation of gluten-specific T cell responses. The presentation of gluten peptides on the cd-associated HLA class II molecules leads to activation of gluten-specific CD4+ T helper 1 (Th1) cells that are implicated in driving the adaptive immune response causing intestinal pathology3,4. Small intestinal lesions in cd are defined by mucosal infiltration with lymphocytes, crypt hyperplasia and villus atrophy1.
The exploration of cd pathogenesis and the design of novel therapeutics for cd have been hampered by the lack of a small animal model. Approaches using HLA-DQ2 or -DQ8 transgenic mice did not lead to small intestinal damage after oral gluten challenge5,6. Tolerance to intestinal antigens in general and food proteins in particular depends on regulatory T cell function7. Most of the current animal disease models of impaired regulatory T cell function are based on a deficiency of CD4+CD25+ T cells (Treg)8–10. Treg deficiency can lead to multiorgan inflammatory disease, with mucosal surfaces, primarily of the intestine, being most severely affected. This is explained by predominant recognition of microbial antigens at mucosal sites by disease-causing, dysregulated effector T cells11. Other studies have demonstrated that compensatory homeostatic T cell expansion -as occurs upon transfer of T cells into lymphopenic mice- facilitates both the induction of antigen-specific immunity and the spontaneous development of organ-specific autoimmune disease12,13. We therefore hypothesized that recognition of dietary gluten by transferred gliadin-sensitized CD4+CD25− T cells will drive duodenitis/enteritis in lymphopenic recipient mice. Here, we report that an adaptive T cell response to gluten exacerbates baseline proximal small bowel pathology in Rag1 (−/−) mice, demonstrating experimental abrogation of tolerance to oral gluten in vivo and resembling human cd.
Methods
Animals and treatments
Male C57BL/6 donor mice were maintained on a gluten-free standardized diet (AIN-76A, Research Diets) from birth. Mice were immunized at the tail base on day −28 (100μg antigen in CFA) and day −14 (50μg antigen in IFA) with gliadin or ovalbumin (all Sigma) and sacrificed on day 1. Rag1−/− (Jackson Laboratory) and nude (Taconic) recipient mice (all of C57BL/6 genetic background) were kept in autoclaved cages and changed to irradiated AIN-76A on day −7. On day 1, groups of weight-matched male Rag1−/− (7–9 weeks of age) or male nude (8–24 weeks of age) recipient mice were injected intraperitoneally with 4.5×105 fractionated splenic donor T cells9,10. Some groups of recipients were changed to a customized, irradiated diet (based on AIN-76A, Research Diets) containing 2.5g wheat gluten (Sigma) per kg. After sacrifice, blood was collected and samples of small bowel, colon, pancreas, lung, liver, kidney and skin were taken for histology. Approval for all procedures had been obtained from the institutional animal care and use committee (Protocol # 043-2005).
Preparation of T cell fractions for adoptive transfer experiments
Non-T cells were depleted from donor splenocyte suspensions using CD3+ T cell enrichment columns (R&D). CD4+CD45RBlowCD25−, CD4+CD45RBlowCD25+ or CD4+CD45RBhigh T cells were selected by flow cytometry9,10 using a FACSAria Sorter (BD Biosciences). PE anti-mouse CD45RB, PE-Cy5 anti-mouse CD4 and FITC anti-mouse CD25 (all BD Biosciences) were used for fluorescent labeling. Cell fractions were >97% pure on post-sort analysis.
Clinical and macroscopical observations and scores
At necropsy, the weight of the entire small bowel was recorded. Colitis activity was assessed clinically/macroscopically calculating the sum of 4 parameters: Hunching (0–1), wasting (0–1), colon thickening (mild, moderate, severe; 0–3), stool consistency (normal, soft, not formed, watery; 0–3); maximal score =8.
Histological analyses and scores; immunohistochemistry
Organ samples were paraffin-embedded and sections (6μm) stained with hematoxylin and eosin (H/E). In addition, small bowel sections were stained using rabbit anti-Ki-67 IgG (clone SP6, dilution 1:200, Lab Vision), biotinylated goat anti-rabbit IgG, avidin-biotin complex reagent, diaminobenzidine substrate (all Vector Laboratories) and hematoxylin for counterstaining. To assess the severity of duodenitis at representative, well-oriented sites of maximal damage, we adapted a histological composite score for ileitis14. The mean depth/mean height of 5 adjacent proliferative (“crypt”) and non-proliferative villus zones was measured at magnification 100x in sections stained for Ki-67 using SPOT advanced software (Diagnostic Instruments), assigning C/V (crypt/villus) ratio scores (higher scores reflecting increased severity): C/V ratio score 0 (V/C ratio >3), 0.5 (2.5–3.0), 1.0 (2.0–2.49), 1.5 (1.5–1.99), 2.0 (1–1.49), 2.5 (0.5–0.99), 3.0 (<0.5). Villus mononuclear cell infiltration was graded as follows (means of 5 independent measurements): Score 0 (villus lamina propria diameter <0.5xcrypt diameter), 1 (0.5–1x), 2 (1–2x), 3 (>2x). Basal infiltration with neutrophils was scored as follows: Score 0 (normal crypts), 1 (focal invasion of crypt epithelium by granulocytes, cryptitis)), 2 (1–3 crypt abscesses per duodenal section), 3 (>3 crypt abscesses). Each animal was assigned a composite duodenitis score by combining the three separate scores obtained in a blinded fashion (max. score 3+3+3=9). Colitis severity was assessed histologically as described previously15.
Cytokine ELISA
Splenocytes from individual mice were restimulated with 3μg/ml of anti-CD3 mAb (positive control, clone 145-2C11, eBioscience), 10μg/ml of gliadin or 10μg/ml ovalbumin (negative control; both from Sigma) in complete RPMI 1640. Cytokine concentrations (pg/ml) were measured in supernatants after 48h using IFNγ, IL-2, IL-4 and IL-10 ELISA kits (R&D, eBioscience). For statistical comparisons, cytokine concentrations in individual gliadin- or ovalbumin (negative control)-stimulated samples were referred to the internal standard of maximal stimulation (anti-CD3 mAb-activated splenocytes of the same mouse).
Quantitative RT-PCR
Samples from the small intestine (0.5cm segment, 2cm distal to the pylorus) were collected at sacrifice and snap-frozen for further analysis. Relative mRNA transcript levels were quantified using the LightCycler FastStart DNA Master Hybridization Probes kit on a LightCycler (all from Roche) applying the TaqMan principle as described previously16. TaqMan probes and primer sets are shown in supplementary Table I. mRNA transcript levels were quantified using second derivative maximums and internal calibration curves, normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA and expressed either in arbitrary units as relative changes (x-fold) over means for healthy C57BL6 controls, or as ratios.
Determination of serum antibodies to gliadin and tissue transglutaminase
As described previously17, 96-well plates were coated for ELISA with 1μg gliadin (Sigma) or recombinant mouse tissue transglutaminase (tTG) per well. Sera were assayed in serial dilutions. Horseradish peroxidase-labeled anti-mouse IgA, IgG (both Sigma), IgG1 (Serotec) or IgG2c (Bethyl Lab; dilutions 1:10,000–1:100,000) was followed by tetramethylbenzidine substrate (Sigma) and detection at 450nm.. Antibody titers were calculated according to the formula: (O.D. of sample − O.D. of blank) × serum dilution. Titers below 20 were considered negative.
Statistical analysis
Data were analyzed with Prism 5 software (GraphPad). For body weight data, p values (*p<0.05; **p<0.01; ***p<0.001) were calculated using repeated measure one-way ANOVA and Tukey’s multiple comparison tests or a paired t-test (2 group analysis). Small bowel weights were compared using unpaired t-tests. Non-parametric data from histological observations was analyzed using Mann-Whitney U tests. Cytokine and serum antibody data (ELISA) was compared using one-way ANOVA and Tukey’s multiple comparison tests between groups, and paired t-tests for comparisons between matched samples within the same group (cytokines). RT-PCR results were analyzed using unpaired t-tests and correlation analysis. In all graphs, error bars depict standard errors of the mean.
Results
Rag1−/− mice gain less weight when challenged with oral gluten after adoptive transfer of gliadin-presensitized T cells
C57BL/6 donor mice were immunized with gliadin (the ethanol-soluble fraction of gluten) or ovalbumin (control) in complete Freund’s adjuvant (CFA). Splenocytes were harvested from immunized donors and suspensions were enriched for CD3+ T cells. CD4+CD45RBlowCD25− memory T cells were selected by fluorescent activated cell sorting (supplementary Fig.S1a,b) and injected i.p. into T and B cell deficient Rag1−/− recipient mice. 3 groups of recipient mice were monitored for 8.5 weeks while maintained on a standardized gluten-free diet (gfd) or the same diet containing 2.5g gluten/kg, equivalent to an average challenge with ~12.5mg gluten/day18 (Fig. 1a). Strikingly, Rag1−/− mice that received adoptive T cell transfers from gliadin-sensitized donors and were gluten-challenged (gliadin/gluten group) gained less weight than mice maintained on a gfd (gliadin/gfd), or than mice that were challenged with oral gluten, but had received adoptive transfers from control-immunized donors (ovalbumin/gluten; (Fig. 1b)). This suggested that gliadin-priming of donor T cells led to exacerbation of intestinal inflammatory disease in Rag1−/− recipients challenged with oral gluten.
Fig. 1. Generation of a mouse model of gluten-sensitive enteropathy.


a) Splenic CD3+ T cell fractions from immunized donor mice were stained with fluorescent antibodies. Rag1−/− or nude mice were injected i.p. with 4.5×105 CD4+CD45RBlow CD25− T cells (Facs) from gliadin-immunized donors, or from ovalbumin (control)-immunized donors. After T cell transfer, recipients were either maintained on gfd (gliadin/gfd), or challenged with gluten (gliadin/gluten, ovalbumin/gluten). b) Changes in body weight in relation to individual starting weights of Rag1−/− mice (n=8–18 per group) during 8.5 weeks after transfer. Significantly lower weights in the gliadin/gluten vs. gliadin/gfd group (***p=0.001) or vs. ovalbumin/gfd (***p=0.001); while differences between gliadin/gfd vs. ovalbumin/gluten, although significant (+p<0.05), were small and transient.
Exacerbation of duodenitis in Rag1−/− mice challenged with oral gluten after adoptive transfer of gliadin-presensitized T cells
At necropsy, mice from all 3 groups displayed duodenal inflammation, but mice in the gliadin/gluten group were most severely affected. Also, there was a trend towards increased full-length small bowel weight in the gliadin/gluten group (supplementary Fig.S1c). Microscopically, moderate to severe duodenitis was found in the majority of mice in the gliadin/gluten group, whereas in both control groups duodenitis was mild to moderate, and rarely severe (Fig. 2a–c). Duodenal pathology was characterized by infiltration of the basal lamina propria with neutrophils, leading to cryptitis and crypt abscesses (Fig. 2d), lymphocytic infiltration of the villus and basal lamina propria (Fig. 2e,f), and crypt hyperplasia and villus atrophy (Fig. 3a–d). While erosions or ulcerations were not seen, multinucleated giant cells were noted in the lamina propria in moderate to severe duodenitis (Fig. 2d). With increasing duodenitis severity, similar histologic changes were more frequently present in the proximal jejunum, but rarely the ileum. To assess the severity of duodenitis, three parameters (crypt/villus (C/V) ratio, villus lamina propria mononuclear cell infiltration and basal infiltration with neutrophils) were integrated into a histological duodenitis score. Confirming the clinical and macroscopic findings, duodenitis scores in the gliadin/gluten group were significantly increased vs. the gliadin/gfd or ovalbumin/gluten control groups (Fig. 4). Low-level duodenitis in controls was comparable to baseline duodenal inflammation seen in a separate experiment after transfer of unprimed CD4+CD45RBlowCD25− T cells (data not shown). Histological analysis of additional organs revealed no alternative explanation other than duodenitis for the observed differences in weight gain. Few mice only showed signs of mild colitis, leading to very low colitis activity indices or histological colitis scores (means <1 on scales from 0 to 8 for all groups, n.s.).
Fig. 2. Duodenitis in Rag1−/− recipients of CD4+CD45RBlowCD25− T cells.
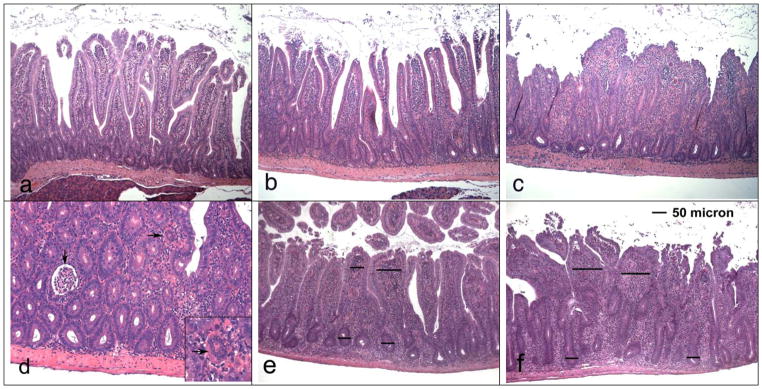
Duodenal samples were stained with H/E. a) Mild duodenitis (section taken from mouse in the ovalbumin/gluten group). Mild infiltration of villus lamina propria with mononuclear cells and presence of cryptitis, but normal crypt/villus ratio (magn. 100x). b) Moderate duodenitis (from gliadin/gfd group). Increased infiltration of the villus and basal lamina propria with mononuclear cells, presence of cryptitis (but no crypt abscesses), increase of crypt/villus ratio. c) Severe duodenitis (from gliadin/gluten group). Loss of villus structure due to severe infiltration of the lamina propria with mononuclear cells, pronounced villus shortening and elongation of crypts, cryptitis (crypt abscesses present at other locations of the same section). d) Severe duodenitis (from gliadin/gluten group; magn. 200x) with cryptitis, crypt abscess (↓) and multinucleated giant cell in lamina propria (→; see insert magn. 400x). e, f) Scoring of villus mononuclear cell infiltration (magn. 100 x), relating villus lamina propria diameters to crypt diameters. Examples for e) villus diameter 1–2x crypt diameter (score 2) and f) >2x crypt diameter (score 3).
Fig. 3. Changes of crypt/villus ratios in Rag1−/− recipients of T cells.

Duodenal sections were stained for Ki-67 to delineate the lengths of labeled proliferative (crypt) and unlabeled non-proliferative (villus) zones using image analysis software. Shown are examples for different crypt/villus (C/V) ratio scores (higher scores reflecting increased severity) derived from villus/crypt (V/C) ratios (100x). a) C/V ratio score 0 (V/C ratio >3), b) C/V ratio score 1 (V/C ratio 2.0–2.49), c) C/V ratio score 2 (V/C ratio 1.0–1.49), d) C/V ratio score 3 (V/C ratio <0.5).
Fig. 4. Exacerbation of histological duodenitis in gluten challenged Rag1−/− recipients of gliadin-presensitized CD4+CD45RBlowCD25− T cells.

Composite duodenitis scores integrating C/V ratio, mononuclear cell and neutrophil infiltration scores (max. score 3+3+3=9). Significant differences between the gliadin/gluten vs. the gliadin/gfd group (*p=0.02), and the ovalbumin/gluten group (*p=0.02; n=8–17).
Th1 polarization of gluten-reactive T cells in Rag1−/− mice challenged with oral gluten after adoptive transfer of gliadin-presensitized T cells
We now determined the T cell cytokine secretion patterns in response to gliadin restimulation in vitro. Splenocytes from individual mice in the gliadin/gluten group produced high levels of interferon γ (IFNγ), accompanied by smaller increases for interleukin (IL-) 2, IL-4 and IL-10 (Fig. 5a–d). Importantly, levels of IFNγ were markedly increased in supernatants from the gliadin/gluten group compared to control groups (Fig. 5a), whereas production of the regulatory cytokine IL-10 was decreased compared to gliadin/gfd, and only non-significantly increased compared to ovalbumin/gluten (Fig. 5d). Thus, the gliadin-specific T cell cytokine secretion pattern in the gliadin/gluten group was characterized by increased Th1 polarization.
Fig. 5. Th1/Th17 polarization of T cells in Rag1−/− recipients of gliadin-presensitized CD4+CD45RBlowCD25− T cells orally challenged with gluten.



a–d) Splenocytes from individual mice (n=5–12 per group) were restimulated with gliadin or ovalbumin (negative control). Cytokine concentrations in supernatants (ELISA) were refered to internal standard, yielding two ratios for gliadin- and ovalbumin-stimulation. Elevated levels in response to gliadin of a) IFNγ, b) IL-2 and c) IL-4 in the gliadin/gluten vs. the gliadin/gfd and the ovalbumin/gluten groups, and of d) IL-10 in the gliadin/gfd vs. the ovalbumin/gluten and the gliadin/gluten groups (*p<0.05, **p<0.01, ***p<0.001). e) Increased transcript levels of IFNγ (p=0.06, n.s), IL-17A (p=0.15, n.s.), IL-17F (p=0.33, n.s.) in the duodenum of the gliadin/gluten vs. the gliadin/gfd group, but unchanged levels of TGFβ1 and IL-10 mRNA (RT-PCR). Significant increases for the ratios of IFNγ/TGFβ1 mRNA (**p=0.008) and IFNγ/IL-10 mRNA (*p=0.02). IL-15 mRNA levels were decreased (*p=0.03, n=11–14).
These findings were confirmed in vivo by quantification of cytokine transcript levels in duodenal samples of the gliadin/gluten and gliadin/gfd groups. While transcript levels of IL-4 were at the limit of detection, expression of the following cytokines was correlated with duodenal histological damage, as assessed by duodenitis scores: IFNγ (r=0.84, ***p<0.001), IL-17A (r=0.74, ***p<0.001), IL-17F (r=0.60, ***p<0.001), TNFα (r=0.59, ***p<0.001), TGFβ1 (r=0.67, ***p<0.001) and IL-10 (r=0.53, *p=0.01). Similarly, significant correlations were found for the functionally relevant ratios of IFNγ over TGFβ1 mRNA (r=0.90, ***p<0.001, (supplementary Fig.S2)) and IFNγ over IL-10 mRNA (r=0.66, **p<0.01), but not of IL-17A or IL-17F over TGFβ1 or IL-10 (not shown). Expression of IL-2 was not significantly correlated, while IL-15 (r=−0.65, ***p<0.001) was negatively correlated with histological damage. When the gliadin/gluten was compared with the gliadin/gfd group, significant increases were found for the transcript ratios of IFNγ over both TGFβ1 and IL-10 (Fig. 5e). Similar to human cd19, these findings strongly suggested that overproduction of IFNγ (and possibly IL-17) drives small intestinal inflammation in response to luminal gluten in our model.
Introduction of a gfd leads to the reversal of gluten-induced inflammatory changes in Rag1−/− recipients of gliadin-presensitized T cells
To prove that the observed differences in severity of duodenal inflammation were caused by immune recognition of oral gluten and therefore would reverse after elimination of gluten from the diet, 3 groups of Rag1−/− mice were injected with gliadin-presensitized CD4+CD45RBlowCD25− T cells and maintained on a gluten-free (gliadin/gfd group) or a gluten-containing diet. At week 10.5, when the weight difference between the gliadin/gfd and the first of 2 groups on gluten-containing diet became statistically significant (*p<0.05), this group was changed to a gfd (diet switch-over group). Subsequently, the diet switch-over group steadily gained weight, approaching values of the gliadin/gfd group and reaching a statistical weight gain (***p<0.001) over the remaining gliadin/gluten group until week 21.5 (Fig. 6a). Duodenitis scores in the diet switch-over group did not differ significantly from the gliadin/gfd group (p=0.11), and were reduced in comparison to the gliadin/gluten group (p=0.35) that suffered from significantly increased duodenitis (*p<0.05) vs. the gliadin/gfd group (Fig. 6b). Furthermore, duodenal IFNγ and IL-17A transcript levels decreased significantly in the diet switch-over vs. the gliadin/gluten group (Fig. 6c), confirming gluten-specificity of our model.
Fig. 6. Switch-over to a gfd leads to reversal of weight loss and improvement of enteropathy.



a) Changes in body weight of Rag1−/− mice during 21.5 weeks after transfer of gliadin-presensitized CD4+CD45RBlowCD25− T cells. Immediately after introduction of a gfd at week 10.5 (↑), the diet switch-over group is gaining weight compared to the gliadin/gluten group (***p<0.001). Significantly lower weights in the gliadin/gluten and the diet-switch-over vs. the gliadin/gfd group (+++p<0.001, °°°p<0.001, n=8–9). b) Exacerbation of histological duodenitis in the gliadin/gluten vs. the gliadin/gfd group at 21.5 weeks (*p<0.05), but non-significant difference for the diet switch-over vs. the gliadin/gfd group (p=0.11, n=8–9). c) Decreased duodenal transcript levels for IFNγ and IL-17A (both *p<0.05), and non-significant decreases for IL-17F (p=0.13) and IL-2 (p=0.15) in the diet switch-over vs. gliadin/gluten group (n=8).
Adoptive transfer of naïve T cells into Rag1−/− mice does not lead to a breach of tolerance to oral gluten
Next, we asked whether oral gluten challenge exacerbates small intestinal pathology in recipients of naïve instead of gliadin-presensitized effector/memory T cells. 2 groups of Rag1−/− mice (n=6) were adoptively transferred with CD4+CD45RBhigh (naïve) T cells from non-sensitized donors, followed by a single injection with gliadin/CFA on day 1 and oral gluten challenge (or gfd). After 8.5 weeks, recipient mice in both groups suffered from colitis and mild to moderate duodenitis, as described for this model previously9,20. Importantly, both groups of recipients did not differ in any of the following parameters: Body weight, colitis activity index, small and large bowel histology and cytokine secretion by splenocytes restimulated in vitro (data not shown). In conclusion, gliadin priming of donor mice and selection of CD4+CD45RBlowCD25− memory T cells for adoptive transfer is crucial for the induction of gluten-sensitive enteropathy.
Nude mice challenged with oral gluten after adoptive transfer of gliadin-presensitized T cells produce serum gliadin antibodies
While our model showed that gluten-reactive memory T cells lead to increased Th1/Th17 deviation and small intestinal pathology in Rag1−/− mice fed gluten, abrogation of B cell tolerance to gluten, as occurs in human cd, remained to be demonstrated. Therefore, we adoptively transferred CD4+CD45RBlowCD25− T cells from gliadin-sensitized donors into B cell-competent nude mice, kept either on gfd or a gluten-containing diet. At 9 weeks post-transfer, duodenitis scores ranged only from 1–4 in individual mice, without differences between the gliadin/gluten and gliadin/gfd groups (mean 2.25+/−0.31 vs. 2.25+/−0.16, n.s.), suggesting significant protection of nude mice from baseline duodenitis and, consequently, lack of gluten-induced enteropathy. Few mice showed signs of colitis without differences between groups (not shown), while histology of other organs was normal. Interestingly, however, nude mice in the gliadin/gluten group produced very high titers of serum anti-gliadin IgA, indicative of intestinal production, as well as serum anti-gliadin IgG, namely IgG1 and IgG2c (Fig. 7). Serum titers in the gliadin/gfd group were negative. No serum IgA or IgG autoantibodies against mouse tTG were detected (not shown). Thus, nude recipients of gliadin-presensitized T cells produce serum anti-gliadin IgA and IgG (including Th1-associated IgG2c subclass) antibodies only when challenged with oral gluten, confirming breach of both T and B cell tolerance to gluten in our model, and its similarity with human cd.
Fig. 7. Serum anti-gliadin antibodies in nude mice after adoptive transfer of gliadin-presensitized CD4+CD45RBlowCD25− T cells.

Serum levels of anti-gliadin IgA and IgG were determined in 2 groups of nude mouse recipients (gliadin/gluten, gliadin/gfd), as well as in C57BL/6 control mice raised on a gluten-free diet (C57BL/6-gfd) or normal (gluten-containing) chow (C57BL/6-gluten; each n=8). Differences for anti-gliadin IgG and IgA between gliadin/gluten and every other group were highly significant (***p<0.001). Levels of anti-gliadin IgG1 and IgG2c were determined in sera positive for anti-gliadin IgG (i.e. in the gliadin/gluten group only).
Discussion
The development of models in which non-dietary therapies can be tested has become a priority for cd research. We transferred gliadin-presensitized CD4+CD45RBlowCD25− T cells into Rag1−/− mice, which caused exacerbation of baseline duodenitis9,20 only in recipients challenged with oral gluten, but not in mice kept on a gfd. Gluten-specificity was further demonstrated by lack of effect in recipients of naive T cells or memory T cells presensitized with irrelevant food antigen, and importantly, by reversal of weight loss, improvement of enteropathy and normalization of immunological changes after switch-over to a gfd. Gluten-induced inflammatory changes in small bowel histology were associated with increased Th1 (and Th17) polarization in the duodenum, closely mimicking cytokine patterns in the mucosa of patients with active cd4,19. The production of high serum anti-gliadin IgA and IgG titers (including the Th1-associated IgG2c subclass) in nude recipients further demonstrated a breach of intestinal tolerance to gluten in this model.
Disturbance of oral tolerance to harmless antigens, usually maintained by intestinal regulatory T cells, causes autoimmune inflammatory disease in the intestine8–10. Accordingly, regulatory CD4+CD25+ T cells (Treg) were depleted in our model by FACS selection of antigen-experienced CD4+CD45RBlowCD25− T cells from gliadin-immunized donors. Previous reports had shown that CD4+CD45RBlowCD25− T cells (but not CD4+CD45RBlowCD25+ T cells) from naïve donors can induce wasting disease in Rag2−/− mice21, and do not suppress colitis in the CD4+CD45RBhigh T cell transfer model22. Our results using CD4+CD45RBlowCD25− T cells from unprimed or control-immunized donors confirmed the intrinsic pathogenicity of this T cell fraction, leading to low-level baseline duodenitis in Rag1−/− mice. Gluten-induced specific changes therefore had to be measured against an inflammatory background.
Proximal small intestinal pathology in our model resembled duodenitis observed after adoptive transfer of CD4+CD45RBhigh T cells into lymphopenic mice9,20, generally regarded a model of Crohn’s disease. Pathology was characterized by mucosal infiltration with lymphocytes, crypt hyperplasia and villus atrophy, changes that are considered typical for cd, but also by infiltration with neutrophils, leading to cryptitis and crypt abscesses. While crypt abscesses are untypical of cd, neutrophil infiltration is a characteristic feature of celiac small intestinal lesions24,25. Recently, gene expression profiling in biopsy samples from celiac patients revealed chronic recruitment of activated neutrophils both in active disease or remission26, indicating that neutrophils may provide (early) innate immune signals for cd development2,27. Interestingly, nude mice were partially protected from duodenitis when compared to Rag1−/− recipients, similar to partial protection from colitis observed in B cell-competent vs. -incompetent TCRα −/− mice23, an effect likely ascribable to B cell regulatory function.
As regards adaptive immunity, cd is characterized by dysregulation of gluten-specific T cell responses. The celiac autoantigen tTG can deamidate certain glutamine residues of ingested gluten peptides (mainly gliadins)17, and these modified gliadin peptides bind with increased affinity only to HLA-DQ2 and -DQ8 molecules on antigen-presenting cells1,3. The result is an activation of gliadin-specific CD4+ Th1 cells, seemingly unchecked by regulatory T cells. Leading to a similar outcome, our model is based on adoptive transfer of CD4+CD45RBlowCD25− T cells from mice immunized with gliadin in CFA, expressing the murine MHC H2b haplotype. Therefore, it lacks the components of HLA-DQ2- or -DQ8-restricted (deamidated) gluten peptide presentation and autoimmunity to tTG. Nonetheless, the model can be further refined by introduction of cd-associated HLA class II transgenes.
Feeding soluble antigen to naive mice leads to the induction of antigen-specific, tolerogenic Treg28,29 that suppress inflammatory responses in the gastrointestinal tract29. To date only one single study reported the induction of T cell mediated enteropathy by feeding soluble antigen30, resulting from blockade of immunomodulatory arachidonic acid metabolites, e.g. prostaglandin E2. Accordingly, both the depletion of tolerogenic Treg and the provision by the innate immune system of costimulatory inflammatory signals are likely necessary for activation of gliadin-specific memory T cells and subsequent gluten-dependent exacerbation of duodenitis in our model. The former is further suggested by the observation that unfractionated T cells do not lead to intestinal inflammation similar to the CD4+CD45RBhigh transfer model9,10.
The feeding dose of ~12.5mg gluten/mouse/day corresponded to 2.7 times the average body weight-adjusted gluten intake in humans, calculated at ~13g gluten/person/day for Western Europe31. In naive animals, ingestion of such high amounts of food protein would lead to oral tolerance, as shown in healthy or colitic mice (CD4+CD45RBhigh model)29,32 that were fed specific food protein (ovalbumin), eliciting the production of regulatory cytokines TGFβ1 and IL-10 and suppression of Th1 cells. In clear contrast, our strategy of selective transfer of gliadin-primed T cells resulted in abrogation of tolerance to dietary gluten and increased duodenal pathology that was characterized by overexpression of IFNγ and IL-17. The production of both IFNγ and IL-17 by CD4+ T cells was recently reported in murine gvhd33. Relative increases of TGFβ1 and IL-10 vs. IFNγ transcript levels in Rag1−/− recipients at 21.5 vs. 8.5 weeks, and pronounced increases of IL-17 and IL-2 respectively, may indicate increased regulation of effector T cell responses, and possibly a shift from Th1 towards Th17 cytokine production during chronification of duodenitis in our model.
In conclusion, we present a small animal model of gluten-induced enteropathy, resembling human cd. We suggest that this model can be used to study mechanisms of oral tolerance and cd pathogenesis, but also for the testing of non-dietary therapies of cd that aim at the degradation, neutralization or mucosal exclusion of gluten after ingestion. This is important, since 1) the standard therapy, a strict gluten-free diet, is difficult to maintain, and 2) hidden traces of gluten in food pose continuous dietary risks for celiac patients. Therapeutic approaches such as oral enzyme therapies34,35 or inhibition of intestinal gluten uptake36 have recently gained attention. Testing their applicability and efficacy should be feasible in this novel animal model.
Supplementary Material
Acknowledgments
We thank John Daley and Suzan Lazo-Kallanian (Dana Farber Cancer Institute, Boston, MA) for technical support with cell sorting, Jessica Zaks and Anisha Y. Sharma for performing RT-PCR and Suzanne White, Wendy Dasgupta and Justine Cohen (all Beth Israel Deaconess Medical Center, Boston, MA) for help with histology and immunohistochemistry. We are grateful to Dr. Daniele Sblattero (University of Eastern Piedmont, Novara, Italy) for providing recombinant mouse tTG.
Funding:
This work was supported by NIH grant # 1 R21 DKO73254-01 to Detlef Schuppan.
Non-standard abbreviations
- cd
Celiac disease
- gfd
gluten-free diet
- tTG
tissue transglutaminase
Footnotes
Competing interests:
None to declare.
References
- 1.Schuppan D. Current concepts of celiac disease pathogenesis. Gastroenterology. 2000;119:234–42. doi: 10.1053/gast.2000.8521. [DOI] [PubMed] [Google Scholar]
- 2.Green PH, Cellier C. Celiac disease. N Engl J Med. 2007;357:1731–43. doi: 10.1056/NEJMra071600. [DOI] [PubMed] [Google Scholar]
- 3.Sollid LM. Molecular basis of celiac disease. Annu Rev Immunol. 2000;18:53–81. doi: 10.1146/annurev.immunol.18.1.53. [DOI] [PubMed] [Google Scholar]
- 4.Nilsen EM, Jahnsen FL, Lundin KE, Johansen FE, Fausa O, Sollid LM, Jahnsen J, Scott H, Brandtzaeg P. Gluten induces an intestinal cytokine response strongly dominated by interferon gamma in patients with celiac disease. Gastroenterology. 1998;115:551–63. doi: 10.1016/s0016-5085(98)70134-9. [DOI] [PubMed] [Google Scholar]
- 5.Chen Z, Dudek N, Wijburg O, Strugnell R, Brown L, Deliyannis G, Jackson D, Koentgen F, Gordon T, McCluskey J. A 320-kilobase artificial chromosome encoding the human HLA DR3-DQ2 MHC haplotype confers HLA restriction in transgenic mice. J Immunol. 2002;168:3050–6. doi: 10.4049/jimmunol.168.6.3050. [DOI] [PubMed] [Google Scholar]
- 6.Black KE, Murray JA, David CS. HLA-DQ determines the response to exogenous wheat proteins: a model of gluten sensitivity in transgenic knockout mice. J Immunol. 2002;169:5595–600. doi: 10.4049/jimmunol.169.10.5595. [DOI] [PubMed] [Google Scholar]
- 7.Mowat AM. Anatomical basis of tolerance and immunity to intestinal antigens. Nat Rev Immunol. 2003;3:331–41. doi: 10.1038/nri1057. [DOI] [PubMed] [Google Scholar]
- 8.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–64. [PubMed] [Google Scholar]
- 9.Morrissey PJ, Charrier K, Braddy S, Liggitt D, Watson JD. CD4+ T cells that express high levels of CD45RB induce wasting disease when transferred into congenic severe combined immunodeficient mice. Disease development is prevented by cotransfer of purified CD4+ T cells. J Exp Med. 1993;178:237–44. doi: 10.1084/jem.178.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Powrie F, Leach MW, Mauze S, Menon S, Caddle LB, Coffman RL. Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity. 1994;1:553–62. doi: 10.1016/1074-7613(94)90045-0. [DOI] [PubMed] [Google Scholar]
- 11.Matsuda JL, Gapin L, Sydora BC, Byrne F, Binder S, Kronenberg M, Aranda R. Systemic activation and antigen-driven oligoclonal expansion of T cells in a mouse model of colitis. J Immunol. 2000;164:2797–806. doi: 10.4049/jimmunol.164.5.2797. [DOI] [PubMed] [Google Scholar]
- 12.Dummer W, Niethammer AG, Baccala R, Lawson BR, Wagner N, Reisfeld RA, Theofilopoulos AN. T cell homeostatic proliferation elicits effective antitumor autoimmunity. J Clin Invest. 2002;110:185–92. doi: 10.1172/JCI15175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.King C, Ilic A, Koelsch K, Sarvetnick N. Homeostatic expansion of T cells during immune insufficiency generates autoimmunity. Cell. 2004;117:265–77. doi: 10.1016/s0092-8674(04)00335-6. [DOI] [PubMed] [Google Scholar]
- 14.Burns RC, Rivera-Nieves J, Moskaluk CA, Matsumoto S, Cominelli F, Ley K. Antibody blockade of ICAM-1 and VCAM-1 ameliorates inflammation in the SAMP-1/Yit adoptive transfer model of Crohn’s disease in mice. Gastroenterology. 2001;121:1428–36. doi: 10.1053/gast.2001.29568. [DOI] [PubMed] [Google Scholar]
- 15.Hollander GA, Simpson SJ, Mizoguchi E, Nichogiannopoulou A, She J, Gutierrez-Ramos JC, Bhan AK, Burakoff SJ, Wang B, Terhorst C. Severe colitis in mice with aberrant thymic selection. Immunity. 1995;3:27–38. doi: 10.1016/1074-7613(95)90156-6. [DOI] [PubMed] [Google Scholar]
- 16.Popov Y, Patsenker E, Fickert P, Trauner M, Schuppan D. Mdr2 (Abcb4)−/− mice spontaneously develop severe biliary fibrosis via massive dysregulation of pro- and antifibrogenic genes. J Hepatol. 2005;43:1045–54. doi: 10.1016/j.jhep.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 17.Dieterich W, Ehnis T, Bauer M, Donner P, Volta U, Riecken EO, Schuppan D. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat Med. 1997;3:797–801. doi: 10.1038/nm0797-797. [DOI] [PubMed] [Google Scholar]
- 18.Bachmanov AA, Reed DR, Beauchamp GK, Tordoff MG. Food intake, water intake, and drinking spout side preference of 28 mouse strains. Behav Genet. 2002;32:435–43. doi: 10.1023/a:1020884312053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tiittanen M, Westerholm-Ormio M, Verkasalo M, Savilahti E, Vaarala O. Infiltration of forkhead box P3-expressing cells in small intestinal mucosa in celiac disease but not in type 1 diabetes. Clin Exp Immunol. 2008;152:498–507. doi: 10.1111/j.1365-2249.2008.03662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ostanin DV, Pavlick KP, Bharwani S, D’Souza D, Furr KL, Brown CM, Grisham MB. T cell-induced inflammation of the small and large intestine in immunodeficient mice. Am J Physiol Gastrointest Liver Physiol. 2006;290:G109–19. doi: 10.1152/ajpgi.00214.2005. [DOI] [PubMed] [Google Scholar]
- 21.Annacker O, Pimenta-Araujo R, Burlen-Defranoux O, Barbosa TC, Cumano A, Bandeira A. CD25+ CD4+ T cells regulate the expansion of peripheral CD4 T cells through the production of IL-10. J Immunol. 2001;166:3008–18. doi: 10.4049/jimmunol.166.5.3008. [DOI] [PubMed] [Google Scholar]
- 22.Mottet C, Uhlig HH, Powrie F. Cutting edge: cure of colitis by CD4+CD25+ regulatory T cells. J Immunol. 2003;170:3939–43. doi: 10.4049/jimmunol.170.8.3939. [DOI] [PubMed] [Google Scholar]
- 23.Mizoguchi A, Mizoguchi E, Smith RN, Preffer FI, Bhan AK. Suppressive role of B cells in chronic colitis of T cell receptor alpha mutant mice. J Exp Med. 1997;186:1749–56. doi: 10.1084/jem.186.10.1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dhesi I, Marsh MN, Kelly C, Crowe P. Morphometric analysis of small intestinal mucosa. II. Determination of lamina propria volumes; plasma cell and neutrophil populations within control and celiac disease mucosae. Virchows Arch A Pathol Anat Histopathol. 1984;403:173–80. doi: 10.1007/BF00695233. [DOI] [PubMed] [Google Scholar]
- 25.Loft DE, Marsh MN, Sandle GI, Crowe PT, Garner V, Gordon D, Baker R. Studies of intestinal lymphoid tissue. XII. Epithelial lymphocyte and mucosal responses to rectal gluten challenge in celiac sprue. Gastroenterology. 1989;97:29–37. doi: 10.1016/0016-5085(89)91411-x. [DOI] [PubMed] [Google Scholar]
- 26.Diosdado B, van Bakel H, Strengman E, Franke L, van Oort E, Mulder CJ, Wijmenga C, Wapenaar MC. Neutrophil recruitment and barrier impairment in celiac disease: a genomic study. Clin Gastroenterol Hepatol. 2007;5:574–81. doi: 10.1016/j.cgh.2006.11.014. [DOI] [PubMed] [Google Scholar]
- 27.Jabri B, Sollid LM. Mechanisms of disease: immunopathogenesis of celiac disease. Nat Clin Pract Gastroenterol Hepatol. 2006;3:516–25. doi: 10.1038/ncpgasthep0582. [DOI] [PubMed] [Google Scholar]
- 28.Zhang X, Izikson L, Liu L, Weiner HL. Activation of CD25(+)CD4(+) regulatory T cells by oral antigen administration. J Immunol. 2001;167:4245–53. doi: 10.4049/jimmunol.167.8.4245. [DOI] [PubMed] [Google Scholar]
- 29.Zhou P, Borojevic R, Streutker C, Snider D, Liang H, Croitoru K. Expression of dual TCR on DO11.10 T cells allows for ovalbumin-induced oral tolerance to prevent T cell-mediated colitis directed against unrelated enteric bacterial antigens. J Immunol. 2004;172:1515–23. doi: 10.4049/jimmunol.172.3.1515. [DOI] [PubMed] [Google Scholar]
- 30.Newberry RD, Stenson WF, Lorenz RG. Cyclooxygenase-2-dependent arachidonic acid metabolites are essential modulators of the intestinal immune response to dietary antigen. Nat Med. 1999;5:900–6. doi: 10.1038/11341. [DOI] [PubMed] [Google Scholar]
- 31.van Overbeek FM, I, Uil-Dieterman G, Mol IW, Kohler-Brands L, Heymans HS, Mulder CJ. The daily gluten intake in relatives of patients with coeliac disease compared with that of the general Dutch population. Eur J Gastroenterol Hepatol. 1997;9:1097–9. doi: 10.1097/00042737-199711000-00013. [DOI] [PubMed] [Google Scholar]
- 32.Marth T, Ring S, Schulte D, Klensch N, Strober W, Kelsall BL, Stallmach A, Zeitz M. Antigen-induced mucosal T cell activation is followed by Th1 T cell suppression in continuously fed ovalbumin TCR-transgenic mice. Eur J Immunol. 2000;30:3478–86. doi: 10.1002/1521-4141(2000012)30:12<3478::AID-IMMU3478>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 33.Chen X, Vodanovic-Jankovic S, Johnson B, Keller M, Komorowski R, Drobyski WR. Absence of regulatory T cell control of TH1 and TH17 cells is responsible for the autoimmune-mediated pathology in chronic graft-versus-host disease. Blood. 2007;110:3804–13. doi: 10.1182/blood-2007-05-091074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pyle GG, Paaso B, Anderson BE, Allen DD, Marti T, Li Q, Siegel M, Khosla C, Gray GM. Effect of pretreatment of food gluten with prolyl endopeptidase on gluten-induced malabsorption in celiac sprue. Clin Gastroenterol Hepatol. 2005;3:687–94. doi: 10.1016/s1542-3565(05)00366-6. [DOI] [PubMed] [Google Scholar]
- 35.Stepniak D, Spaenij-Dekking L, Mitea C, Moester M, de Ru A, Baak-Pablo R, van Veelen P, Edens L, Koning F. Highly efficient gluten degradation with a newly identified prolyl endoprotease: implications for celiac disease. Am J Physiol Gastrointest Liver Physiol. 2006;291:G621–9. doi: 10.1152/ajpgi.00034.2006. [DOI] [PubMed] [Google Scholar]
- 36.Paterson BM, Lammers KM, Arrieta MC, Fasano A, Meddings JB. The safety, tolerance, pharmacokinetic and pharmacodynamic effects of single doses of AT-1001 in coeliac disease subjects: a proof of concept study. Aliment Pharmacol Ther. 2007;26:757–66. doi: 10.1111/j.1365-2036.2007.03413.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.