

Abstract
Elastomeric polypeptides are very interesting biopolymers and are characterized by rubber-like elasticity, large extensibility before rupture, reversible deformation without loss of energy, and high resilience upon stretching. Their useful properties have motivated their use in a wide variety of materials and biological applications. This chapter focuses on elastin and resilin – two elastomeric biopolymers – and the recombinant polypeptides derived from them (elastin-like polypeptides and resilin-like polypeptides). This chapter also discusses the applications of these recombinant polypeptides in the fields of purification, drug delivery, and tissue engineering.
Keywords: Elastin, Elastin-like polypeptides, Elastomeric polypeptides, Resilin, Resilin-like polypeptides
1 Introduction
Elastomeric polypeptides are a class of very interesting biopolymers and are characterized by rubber-like elasticity, large extensibility before rupture, reversible deformation without loss of energy, and high resilience upon stretching. Their useful properties have motivated their use in a wide variety of materials and biological applications. Here, we focus on two elastomeric proteins and the recombinant polypeptides derived thereof.
The first elastomeric protein is elastin, this structural protein is one of the main components of the extracellular matrix, which provides structural integrity to the tissues and organs of the body. This highly crosslinked and therefore insoluble protein is the essential element of elastic fibers, which induce elasticity to tissue of lung, skin, and arteries. In these fibers, elastin forms the internal core, which is interspersed with microfibrils [1, 2]. Not only this biopolymer but also its precursor material, tropoelastin, have inspired materials scientists for many years. The most interesting characteristic of the precursor is its ability to self-assemble under physiological conditions, thereby demonstrating a lower critical solution temperature (LCST) behavior. This specific property has led to the development of a new class of synthetic polypeptides that mimic elastin in its composition and are therefore also known as elastin-like polypeptides (ELPs).
Resilin is the second elastomeric polypeptide that is discussed here. This protein, found in specialized regions of the insect cuticle, similarly demonstrates extraordinary extensibility and elasticity; its role as an elastic spring in insect organs provides clear illustration of its characteristic fatigue resistance and resilience. In fact, its mechanical properties are in near-agreement with classic rubber-elasticity theory, a remarkable feature for a hydrophilic biopolymer network. Apart from the outstanding mechanical properties, resilin provides an interesting comparison to other protein elastomers due to its hydrophilicity, but also due to the fact that resilin shares certain compositional characteristics. In particular, the high number of proline and glycine residues found in resilin as well as other protein elastomers could indicate some sort of structural importance. While silks and elastin have received the most attention from the scientific community over the past few decades, the development of recombinant resilins, facilitated by advances made in recombinant DNA technologies and protein engineering, has provided invaluable opportunities for the application of resilin as a biomaterial. Recombinant resilin could potentially see application in a variety of fields, including tissue engineering, drug delivery, biosensors, and nanobiotechnology.
This chapter will discuss the basic aspects of elastin and resilin and will address their biological role, biochemical processing, and properties. The materials inspired by elastin and resilin, such as elastin-like polypeptides and resilin-like polypeptides, and applications thereof, will also be covered.
2 Elastin and Elastin-Like Polypeptides
2.1 Elastin Biosynthesis
Elastin is a heavily crosslinked biopolymer that is formed in a process named elastogenesis. In this section, the role of elastin and the different steps of elastin production will be described, starting with transcription of the genetic code and processing of the primary transcript, followed by translation into the elastin precursor protein and its transport to the extracellular matrix. Finally, the crosslinking and fiber formation, which result in the transition from tropoelastin to elastin, are described.
Tropoelastin is the soluble precursor of elastin and consists of alternating hydrophobic and hydrophilic peptide domains. The most common amino acids in the hydrophobic domains are Gly, Val, Ala, and Pro, which are often present in repeats of tetra-, penta-, and hexapeptides, such as Gly-Gly-Val-Pro, Gly-Val-Gly-Val-Pro, Gly-Val-Pro-Gly-Val, and Gly-Val-Gly-Val-Ala-Pro, respectively [3, 4]. The hydrophilic domains are mainly composed of lysines interspersed by alanines.
Tropoelastin is encoded by a single copy gene and the alternating hydrophobic and hydrophilic domains are generally encoded by different expressed regions, or exons (Fig. 1). Those exons are alternated by introns (intragenic regions), which are not present in the final transcript. Even though a single gene is coding for tropoelastin, various types (isoforms) of tropoelastin exist due to extensive alternative splicing [5, 6]. Six exons were shown to be subject to this process in a cassette-like fashion in which an exon is either included or excluded, and this results in at least 11 human isoforms (Fig. 2, arrows) [7]. The elastin genes of human, bovine, chick, and rat have been sequenced and the amino acid sequences were determined [8]. Even though a strong similarity was found between tropoelastins from different sources, some variation does exist. Some exons are specific for a certain species, for example exon 26A is only present in human elastin. This exon is rich in charged and polar amino acids and is thought to influence the structural and biological functions of elastin [6].
Fig. 1.

Primary structure of human tropoelastin isoform 3 (EBI accession no. P15502). The highlighted regions correspond to the signal peptide and hydrophobic and hydrophilic domains. Based on [2]
Fig. 2.

cDNA structure of human tropoelastin. The alternating hydrophobic and hydrophilic domains are generally encoded by different exons, shown in white and black, respectively. The arrows indicate the six exons that are subject to alternative splicing in a cassette-like fashion. Reproduced from [8] with permission from John Wiley and Sons, copyright 1998
After mRNA splicing, the tropoelastin mRNA is translated at the surface of the rough endoplasmic reticulum (RER) in a variety of cells: smooth muscle cells, endothelial and microvascular cells, chondrocytes and fibroblasts. The approximately 70 kDa precursor protein (depending on isoform) is synthesized with an N-terminal 26-amino-acid signal peptide. This nascent polypeptide chain is transported into the lumen of the RER, where the signal peptide is removed cotranslationally [9].
Very few post-translational modifications have been found on tropoelastin. However, hydroxylation of 25% of the proline residues is observed [10]. The enzymatic modification of proline to hydroxyproline (Hyp) is performed by prolyl hydroxylase [11]. The purpose of this hydroxylation remains unclear and it is even proposed that Hyps in tropoelastin are a by-product of collagen hydroxylation as this occurs in the same cellular compartment [8].
Once the tropoelastin is synthesized, it is bound by the elastin-binding protein (EBP). This binding of EBP to tropoelastin is believed to prevent intracellular aggregation and to protect it from proteolytic degradation [12]. EBP is a 67 kDa multifunctional peripheral membrane protein and is part of the elastin receptor [13]. The two other subunits of the receptor complex are transmembrane proteins of 61 kDa and 55 kDa [14]. EBP is also equipped with a binding site for β-galactosaccharides, and binding of these sugars allosterically leads to reduced affinity for tropoelastin and results in dissociation of EBP from the 61 kDa and 55 kDa membrane proteins. While complexed with EBP, elastin is transported from the RER to the cell surface via the Golgi apparatus. Upon reaching the cell surface, it is secreted from the cell. Galactosugar moieties on microfibrils at the cell surface are then bound by EBP, which leads to release of tropoelastin (Fig. 3).
Fig. 3.

Binding and release of tropoelastin. The elastin receptor consists of a 67 kDa peripheral subunit (EBP) with two transmembrane proteins of 61 and 55 kDa. The 67 kDa protein binds tropoelastin and galactosugars through two separate sites. (a) Tropoelastin binds to the intact EBP complex. (b) Upon binding of a galactosugar, the EBP loses its affinity for both tropoelastin and the membrane-bound protein, which leads to the release of tropoelastin. Reproduced from [8] with permission from John Wiley and Sons, copyright 1998
The microfibrils act as a scaffold onto which the tropoelastin molecules are deposited. In order for tropoelastin to be incorporated into the elastic fibers it needs to be crosslinked. To facilitate the crosslinking, these elastin precursors must associate and align. This alignment is thought to proceed via the process of coacervation. This process occurs via an inverse temperature transition, which induces the aggregation of tropoelastin. At low temperatures tropoelastin is soluble in aqueous solutions; upon raising the temperature the solution becomes cloudy because the tropoelastin molecules aggregate and become ordered by the interactions between their hydrophobic domains. This phenomenon is completely reversible (by cooling) and is thermodynamically controlled. This process is also known as lower critical solution temperature (LCST) behavior and can be influenced by protein concentration, salt concentration, and pH.
After the self-assembly process, the tropoelastin molecules are enzymatically crosslinked via the lysine residues in the hydrophilic domains. In this last step of the elastogenesis, the amines of the lysine residues are enzymatically converted to aldehydes (allysine) by lysyl oxidase via oxidative deamination (Fig. 4). Lysyl oxidase is a copper-dependent amine oxidase. In the following spontaneous condensations, allysine can react with another allysine or with lysine to form di-, tri- and tetrafunctional crosslinks, to eventually yield desmosine and isodesmosine linkages [15]. The interchain crosslinking and the high number of hydrophobic residues immediately lead to insolubility of the elastin.
Fig. 4.

Structures and formation routes of crosslinks in elastin. In the first step, lysine is catalytically converted to allysine by lysyl oxidase; all subsequent condensation steps are spontaneous
2.2 General Properties of Elastin and Tropoelastin
Research into elastin, its properties, and the fiber formation was for a considerable period of time hindered due to its insolubility. However, discovery of the soluble tropoelastin precursor made new investigations possible. The tropoelastin protein can be isolated from copper-deficient animals. However, this is a very animal-unfriendly and low yielding process [2]. Therefore, it is preferred to obtain tropoelastin from overexpression in microbial hosts such as Escherichia coli (E. coli). Most studies are thus based on tropoelastin obtained via bacterial production.
The main biological function of elastin is to provide elasticity to organs and tissues. However, it has been shown that elastin and peptides derived from elastin also have additional biological properties. Elastin can be degraded by several protein-degrading enzymes, such as elastases, matrix metalloproteinases, and cathepsins. The resulting elastin-derived peptides (EDP) can interact with other extracellular matrix proteins to induce a broad range of biological activities. EDPs are chemotactic for various cells, such as fibroblasts and monocytes, by binding to the EBP [16, 17]. The consensus sequence GXXPG (X being any natural amino acid) of EDPs was proposed to be crucial for the interaction with the EBP.
The coacervation of tropoelastin plays a crucial role in the assembly into elastic fibers. This coacervation is based on the LCST behavior of tropoelastin, which causes tropoelastins structure to become ordered upon raising the temperature. The loss of entropy of the biopolymer is compensated by the release of water from its chain [2, 18, 19]. This release of water results in dehydration of the hydrophobic side chains, and this is the onset of the self-assembly leading to the alignment of tropoelastin molecules.
Weiss and coworkers intensively investigated the coacervation behavior of a single isoform of tropoelastin. They produced tropoelastin by overexpression in E. coli, and the coacervation was then assayed by monitoring turbidity while varying protein concentration, NaCl concentration, and pH. All these factors influence the temperature at which the coacervation is initiated. A typical tropoelastin coacervation curve is shown in Fig. 5. Increasing tropoelastin concentration resulted in a shift towards lower temperatures and a reduction of the temperature range over which coacervation took place. A lowering of coacervation temperature was also found upon increasing NaCl concentration. It is suggested that this is caused by NaCl decreasing the effective concentration of water by binding it in hydration shells, which therefore leads to an increase in protein concentration. Impurities that are able to bind the hydrophobic domains of the tropoelastin also had a major influence on the coacervation [20].
Fig. 5.
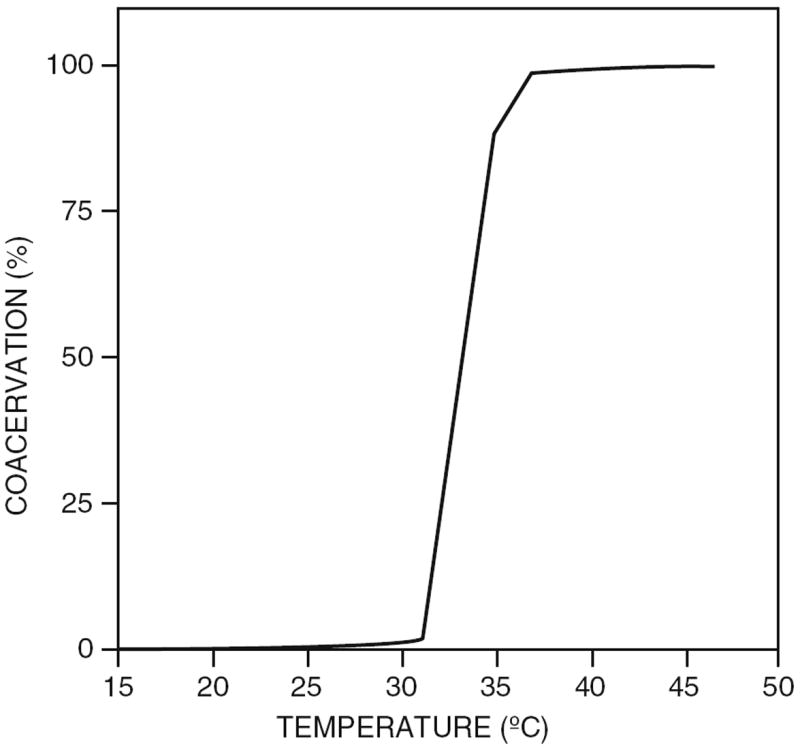
Coacervation curve of 40 mg/mL tropoelastin in 10 mM sodium phosphate pH 7.4, containing 150 mM NaCl. Maximal coacervation was obtained at 37 °C. Reproduced from [20] with permission from John Wiley and Sons, copyright 1997
This coacervation process forms the basis for the self-assembly, which takes place prior to the crosslinking. The assembly of tropoelastin is based on an ordering process, in which the polypeptides are converted from a state with little order to a more structured conformation [8]. The insoluble elastic fiber is formed via the enzymatic crosslinking of tropoelastin (described in Sect. 2.1). Various models have been proposed to explain the mechanism of elasticity of the elastin fibers. The main models are described in a review by Vrhovski and Weiss [8]. For ideal elastomers in the extended mode, all the energy resides on the backbone and can therefore be recovered upon relaxation [18]. Generally, it is believed that the mechanism of elasticity is entropy-driven, thus the stretching decreases the entropy of the system and the recoil is then induced by a spontaneous return to the maximal level of entropy [8].
2.3 Development and Properties of Recombinant Elastins
In order to obtain a better understanding of the origin of the interesting characteristics of elastin and its precursor, several groups have investigated model polymers with elastin-based sequences. Urry and coworkers performed pioneering work in this field. They were the first to synthesize the repetitive peptides, their oligomers, and the polymers that were found in the hydrophobic domains of elastin. Via this approach they determined the conformation and interactions of these domains. Of particular interest proved to be the repeat sequence of Val-Pro-Gly-Xaa-Gly (VPGXG), where the fourth residue can be any natural amino acid except proline. They observed that polymers of this pentapeptide repeat coacervate in a similar way to tropoelastin. Polypeptides containing this type of sequence were coined elastin-like polypeptides [21]. In some articles and reviews the terms elastin-mimetic polypeptides or elastin-like recombinamers are used instead of elastin-like polypeptides, however in this chapter we will refer to these polymers as elastin-like polypeptides.
Poly(VPGVG) (Fig. 6) has been studied most thoroughly and it was shown that it exhibits an inverse phase transition. The biopolymer undergoes phase separation from solution upon increasing temperature, resulting in a β-spiral structure and simultaneous release of water molecules associated with the polymer chain (Fig. 7).
Fig. 6.
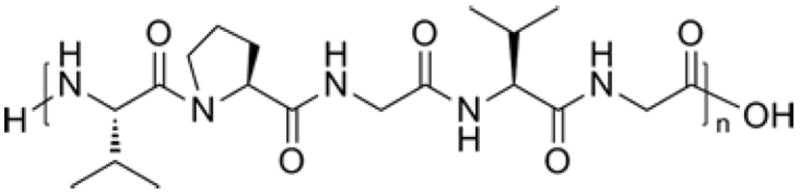
Chemical structure of poly(Val-Pro-Gly-Val-Gly)
Fig. 7.

(a) Recurring β-turn found in poly(VPGVG). (b) β-spiral structure adopted by the poly(VPGVG) upon raising the temperature above the inverse transition temperature. Reprinted from [22] with permission from Elsevier, copyright 1992
The effects of varying the fourth amino acid and the biopolymer length have been extensively investigated and models have been proposed [23-26]. ELP constructs are usually described using the notation ELP[XiYjZk-n], where the capital letters between the brackets indicate the single letter amino acid code for the Xaa-replacing residue in the pentapeptide Val-Pro-Gly-Xaa-Gly. The subscript stands for the ratio of the guest residues and the n represents the total number of pentapeptide repeats.
Figure 8a shows the turbidity measurements for different guest residues in ELP [V8X2]. Lower transition temperatures (Tt) correlate with increased hydrophobicity of the guest residue [24, 25]. This data was extrapolated to other ratios of Val:Xaa (Fig. 8b). The transition temperature could also be influenced by the molecular weight of the ELP. The Tt was shown to increase with decreasing polymer length (Fig. 8c) [23, 26].
Fig. 8.

(a) Effects of varying the amino acid at fourth position in ELP[V8X2]. (b) Extrapolation of transition temperature for other ratios of Val:Xaa. (a) and (b) Reprinted from [24] with permission from American Chemical Society, copyright 1993 (c) Molecular weight dependence of transition temperature. Reprinted from [23] with permission from Elsevier, copyright 2002
ELPs can be produced via chemical synthesis and biosynthetically. For chemical synthesis via solid phase peptide synthesis, the attainable polymer length is limited, and if long polymers with a defined length are required then the biosynthetic approach is more appropriate. An advantage of chemical synthesis is, however, that it enables the facile introduction of functional residues in the polypeptide [27]. In the biosynthetic approach, protein expression in E. coli [27], yeast [28, 29], and plants [30, 31] have been employed. This approach requires the construction of genes encoding for these repetitive polypeptides. Different methods for gene construction have been published: multimerization [32], recursive directional ligation [33], and recursive directional ligation via plasmid reconstruction [23, 34].
Several studies were performed on the optimization of expression levels of ELP proteins in E. coli. In a recent example, the expression protocol was optimized for an ELP fusion with green fluorescent protein (GFP). This fusion protein was expressed and purified in a yield of 1.6 g/L of bacterial culture, which finally yielded ~400 mg GFP/L bacterial culture. This extremely high yield was found after uninduced expression in nutrient-rich medium supplemented with phosphate, glycerol and certain amino acids, such as proline and alanine [234]. The influence of fusion order was also examined and it was found that positioning the ELP at the C-terminus of target protein resulted in significantly higher expression levels [35].
2.4 Applications of Elastin-Like Polypeptides
2.4.1 Temperature-Triggered Purification
After expression of poly(VPGXG) genes, the biopolymer can easily be purified from a cellular lysate via a simple centrifugation procedure, because of the inverse temperature transition behavior. This causes the ELPs to undergo a reversible phase transition from being soluble to insoluble upon raising the temperature above the Tt and then back to soluble by lowering the temperature below Tt (Fig. 9). The insoluble form can be induced via addition of salt [27]. The inverse transition can be assayed by monitoring the solution turbidity as function of temperature. The complete purification process is called inverse transition cycling (ITC), which is achieved by repeated centrifugation while alternating the temperature above and below the Tt[33].
Fig. 9.

Purification of ELPs by ITC is based on the reversible inverse phase transition. Left: Protein purification via direct ELP fusions. A soluble ELP fused to a target protein becomes reversibly insoluble upon increasing temperature above Tt. Center: Protein purification via ELP coaggregation. An excess of free ELPs enhances the aggregation of trace quantities of ELP-fusions. Right: Purification via ELP-mediated affinity capture (EMAC). ELPs are fused to capture proteins, which bind specifically and reversibly to a target protein. This target protein can then be aggregated at temperatures above the Tt. Adapted from [38] with permission from Elsevier, copyright 2010
The inverse transition temperature of the elastomeric polypeptide motivated Meyer and Chilkoti to design ELP fusion proteins. They hypothesized that the temperature-dependent inverse transition of the free biopolymer could be transferred to the fusion protein and would allow nonchromatographic separation of recombinant fusion proteins via the ITC procedure [33]. This hypothesis was furthermore supported by the results of previous studies on protein conjugates of poly(N-isopropylacrylamide), a synthetic polymer that undergoes a similar thermally driven phase transition, which showed that the transition behavior of the free polymer is retained in the conjugate [36, 37]. This founded the use of ELPs for purification of primarily recombinant proteins but also other molecules. The ELP-mediated purification procedures can be sorted into three categories: protein purification via direct ELP fusions (Fig. 9, left), protein purification via ELP coaggregation (Fig. 9, center), and purification via ELP-mediated affinity capture (EMAC) (Fig. 9, right).
Protein Purification via Direct ELP Fusion
As an example of purification via the ELP fusion approach Meyer and Chilkoti (Fig. 9, left), purified the proteins thioredoxin and tendamistat. For this purpose these target proteins and ELP were genetically fused via a short peptide sequence that included a thrombin cleavage site, which allows the removal of the ELP tag after the purification is completed. The general outline of the purification procedure is depicted in Fig. 10. They showed that ELPs could be used for purification of target proteins with technical simplicity, low cost, ease of scale-up, and capacity for multiplexing [33].
Fig. 10.

Inverse transition cycling (ITC) purification. The target proteins are genetically fused to an ELP via a short peptide linker that includes a protease cleavage site. After expression, the fusion protein is separated from the other proteins in the cellular lysate by inducing the ELP inverse temperature phase transition. The solution is then centrifuged at elevated temperature to pellet the aggregated fusion protein. The pellet is resolubilized by cooling below the Tt and the solution is centrifuged at low temperature to remove the remaining insoluble matter. This cycle of heated and cooled centrifugation is repeated several times to obtain higher purity. The target protein can then be liberated from the ELP via proteolytic cleavage and the cleaved ELP is then removed with another round of ITC. Reproduced from [39] with permission from John Wiley and Sons, copyright 2001
In a subsequent study, the effect of reducing the ELP molecular weight on the expression and purification of a fusion protein was investigated. Two ELPs, ELP [V-20] and ELP[V5A2G3-90], both with a transition temperature at ~40°C in phosphate-buffered saline (PBS) containing 1 M NaCl, were applied for the purification of thioredoxin. Similar yields were observed for both fusion proteins, resulting in a higher thioredoxin yield for the ELP[V-20] fusion, since the ELP fraction was smaller. However, a more complex phase transition behavior was observed for this ELP and therefore a selection of an appropriate combination of salt concentration and solution temperature was required [39].
The ELP expression system was compared to the conventional oligohistidine fusion, which is traditionally applied for purification by immobilized metal affinity chromatography (IMAC). Both techniques were shown to have a similar yield of the recombinant protein. The temperature-triggered approach offers a fast and inexpensive nonchromatographic separation with the possibility for larger scale purification. Although the ELP expression system may not be applicable to all types of recombinant proteins, numerous examples have already been shown [40].
In the previous examples, protease-dependent cleavage was used to remove the ELP tag and to obtain the purified target protein. However, several problems are associated with the use of a protease-dependent cleavage: additional cost of the purification process, an additional purification step to separate the protease from the target protein and possible cleavage sites in the target protein. Therefore, the methodology was optimized by introducing self-cleaving inteins for protease-independent cleavage of the target protein. Both Wood and coworkers [41] and the research groups of Chilkoti and Filipe [42] simultaneously reported on this self-cleaving ELP tag. The latter demonstrated this approach by the use of the mini-intein from Mycobacterium xenopi GyrA gene (Mxe) to purify thioredoxin [42]. Wood and coworkers published several extensive protocols describing every aspect of how to use cleavable elastin-like polypeptide tags [43, 44]. Some other proteins were also expressed as a direct ELP fusion protein and were then purified by ITC [45, 46].
Protein Purification via ELP Coaggregation
The previous ELP fusions all are examples of protein purification in which the ELP is covalently connected to the protein of choice. This approach is suitable for the purification of recombinant proteins that are expressed to high levels, but at very low concentrations of ELP the recovery becomes limited. Therefore this approach is not applicable for proteins expressed at micrograms per liter of bacterial culture, such as toxic proteins and complex multidomain proteins. An adjusted variant of ITC was designed to solve this problem. This variant makes use of coaggregation of free ELPs with ELP fusion proteins. In this coaggregation process, an excess of free ELP is added to a cell lysate to induce the phase transition at low concentrations of ELP fusion protein (Fig. 9, center). It was shown that picomolar levels of ELP fusion proteins could be purified via ELP coaggregation [47, 48]. The value of this technique was shown by the purification of low levels of ELP fused to an anti-atrazine antibody [49].
Purification via ELP-Mediated Affinity Capture
The third purification procedure is based on the combination of temperature-triggered aggregation and affinity capture and has been used to not only purify proteins [50-54] but also other molecules [55, 56], and for the removal of pollutants from a solution [57-59]. For this procedure, ELP is conjugated to a capture reagent, which can be done either genetically or chemically. This procedure eliminates the need for cleavage of ELP after purification and introduces the potential to recycle the ELP.
Various purification systems of this type were developed by Chen and coworkers, such as a new plasmid purification method. This system is based on the interaction between a DNA-binding protein and its cognate DNA sequence harbored on the target plasmid. Thus, an ELP is genetically fused to a DNA-binding protein. After binding of the fusion protein to the plasmid via the recognition sequence, the complex can be collected by triggering the inverse phase transition of ELP. Subsequently, the plasmid can be eluted from the DNA-binding protein by heating to ~60°C, after which the ELP-DNA binding protein is removed and recycled via centrifugation to obtain the purified plasmid in solution. A schematic overview of the plasmid purification via temperature-triggered affinity capture is depicted in Fig. 11 [55, 56].
Fig. 11.
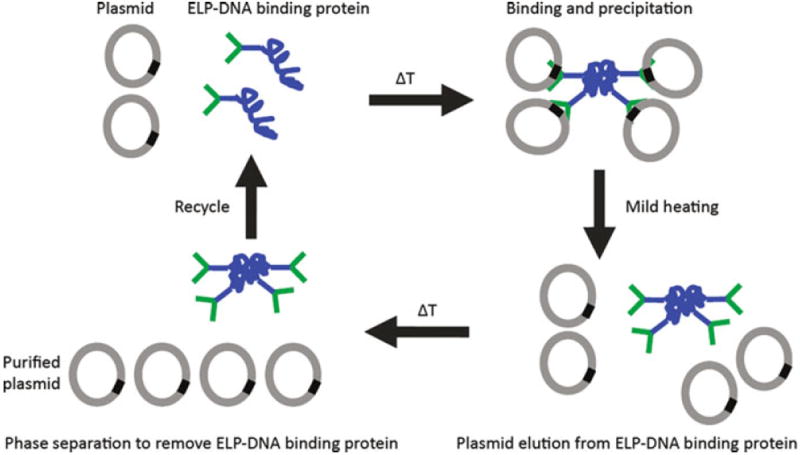
Principle of temperature-triggered affinity purification of plasmid DNA. An ELP fused to a DNA binding protein (ELP-DNA binding protein) was used to bind a plasmid. The ELP-DNA binding protein–plasmid complex is aggregated by increasing the temperature and is then collected by centrifugation. Subsequently, the plasmid is eluted from the DNA binding protein by mild heating, after which the ELP-DNA binding protein is removed and recycled via centrifugation to obtain the purified plasmid in solution. Adapted from [56] by permission from Macmillan Publishers Ltd: Nature Protocols, copyright 2007
Proteins have been purified in a similar way, for example antibodies. For this purpose, ELPs were combined with antibody-binding domains, such as Protein G, Protein L, or Protein A [50-52, 54]. The antibodies can be bound via the ELP-antibody binding domain fusion and then those complexes can be purified via temperature-triggered precipitation. Also histidine-tagged proteins were purified by temperature-triggered purification. An ELP with repeating sequences of [(VPGVG)2(VPGKG)(VPGVG)2]21 was chemically modified by reacting the amino groups of the lysines with imidazole-2-carboxaldehyde to introduce metal-binding ligands. Subsequently, the biopolymer was charged with Ni2+ to enable binding of histidine-tagged proteins. This combined the advantages of histidine-tag purification with inverse transition cycling [53].
The last example of EMAC also makes use of metal-binding groups, although here they are used to remove heavy metal contaminants. The first report on temperature-triggered removal of cadmium made use of histidine-tagged ELPs, because they strongly bind Cd2+[57]. In a later report, ELP was fused to a metal-binding domain, which was shown to bind Cd2+ more effectively and selectively than the histidine-tagged ELPs [59]. A similar approach was followed with the construction of ELP-MerR, where an ELP was fused to the metalloregulatory protein MerR, which is responsible for regulating expression of the mercury detoxification pathway. This fusion protein was used for specific removal and recycling of mercury [58].
2.4.2 Drug Delivery
Numerous experimental therapeutics have shown potency in vitro; however, when they are tested in vivo, they often lack significant efficacy. This is often attributed to unfavorable pharmacokinetic properties and systemic toxicity, which limit the maximum tolerated dose. These limitations can be overcome by use of drug carriers. Two general types of carrier systems have been designed: drug conjugation to macromolecular carriers, such as polymers and proteins; and drug encapsulation in nanocarriers, such as liposomes, polymersomes and micelles.
Over the past decades, ELPs have also been applied in drug delivery as both macromolecular carriers and nanocarriers. The ELP-based carrier systems combine the advantages of drug carriers with the properties of ELPs. In addition to these carrier systems, ELP-based drug depots for controlled release have been designed, such as drug releasing gels and drug-eluting films.
ELPs with different transition temperatures have been employed for drug delivery. These can typically be divided into three categories:
Soluble ELPs with a Tt above the body temperature
Stimulus-responsive ELPs with a Tt in the region where phase transition can be triggered in vivo, thus slightly above body temperature
Insoluble ELPs with a Tt below body temperature
This final class of ELPs is used in nanocarriers and in drug depots. In this section, the emphasis will be on systems that have already been tested in vivo, and on some recent promising developments in the area of ELP-assisted drug delivery.
Macromolecular Carriers
In macromolecular drug delivery systems, drugs are attached to polymeric compounds, such as synthetic polymers [60], dendrimers [61], and antibodies [62], in order to enhance the delivery of the active substance to the diseased tissue and to reduce the toxicity to healthy tissue. The use of macromolecular delivery systems provides several advantages: extension of the half-life of the drug, the ability to introduce targeting moieties into the carrier, the possibility of triggered drug release, and the aforementioned reduced cytotoxicity.
In cancer treatment, passive targeting of macromolecular carriers to tumors is a commonly used approach. This passive targeting is based on the enhanced permeability and retention (EPR) effect, which leads to an accumulation of the high molecular weight carrier in the tumor tissue. The EPR effect arises from the different physiology of tumor vasculature, where the vessel walls are highly porous and lack the tight junctions that are present in healthy tissue. As a result, macromolecular carriers extravasate and accumulate preferentially in tumor tissue relative to normal tissues [63, 64].
All the aforementioned advantages and several additional features apply to ELP-based macromolecular carriers. First, ELP-based carriers are thermally responsive. Second, ELP is a biopolymer and therefore is nontoxic and biodegradable. Third, the gene-based synthesis of ELP allows the creation of genetic fusions with functional peptides and proteins, such as targeting sequences.
Recently, Chilkoti and coworkers showed the effective use of self-assembled ELP-drug conjugates in the treatment of solid tumors in mice. For this purpose, the ELP[VA8G7-160] with a transition temperature of ≫37°C was equipped with a conjugation site for hydrophobic molecules, for example therapeutics. This conjugation segment consisted of eight cysteine residues, interspersed with diglycine spacers. Doxorubicin (Dox), a hydrophobic and clinically effective drug, was modified with a terminal maleimide via an acid-labile hydrazone linker and then coupled to the thiols of the Cys residues via the maleimide–thiol reaction. This macromolecular carrier spontaneously assembled into micellar nanoparticles of ~40 nm diameter (Fig. 12). The half-life, the maximum tolerated dose, and the anti-tumor activity of these nanoparticles in mice were evaluated by administering the drug conjugates systemically. It was found that the ELP-Dox formulation allowed a higher dose than the free drug, which is probably caused by the shielding of ELP to limit the toxicity of Dox to healthy tissue. With a single injection, at the maximum tolerated dose, ELP-Dox outperformed the free drug in reducing tumor volume and this led to a substantial increase in animal survival [65].
Fig. 12.

Left: Structure of ELP[VA8G7-160] conjugated to doxorubicin via the cysteine residues. Right: Self-assembly of ELP-Dox to form nanoparticles. Reprinted from [65] by permission from Macmillan Publishers Ltd: Nature Materials, copyright 2009
The previous example of application of ELPs in drug delivery does not exploit their thermal responsiveness. In most other examples, the ELP phase transition is used to target ELP-drug conjugates towards tumors by applying local heating of tissue (local hyperthermia). Chilkoti and coworkers were the first to employ this principle. They developed a method for targeting of intravenously injected ELPs towards solid tumors by local hyperthermia. In addition, mild hyperthermia also enhanced the delivery of drugs to solid tumors, due to the increase of vascular permeability at temperatures between 40°C and 45°C [66]. Two ELPs were synthesized: ELP[V5A2G3-150] with a Tt of 41°C, which is between the body temperature and the mild hyperthermic temperature; and the control ELP[VA8G7-160] with a Tt well above 55°C, which is unresponsive at the mild hyperthermic temperature. These biopolymers were labeled with either rhodamine or with radioactive iodine for in vivo fluorescence video-microscopy and radiolabel distribution studies in nude mice with implanted human tumors. Thermal targeting of ELP [V5A2G3-150] led to an increase in tumor localization compared to the control ELP with hyperthermia and to the same polymer without hyperthermia [67, 68]. In these labeling studies, the amines in the ELPs were labeled, but in another study 14C radiolabels were incorporated into the backbone of both ELPs during protein expression. This was used to minimize the influence of the labeling while studying the biodistribution and pharmacokinetics and to prevent release of the radiolabel [69, 70]. Next, Dox was conjugated to ELP[V5A2G3-150] via an acid-labile hydrazone linker, but this lowered the Tt to below the body temperature, thus this ELP could not be employed for in vivo thermally triggered targeting [71]. In a subsequent study to minimize perturbation of the linker on the phase transition behavior and to provide an efficient and quantitative release of Dox, the chain length and the hydrophobicity of the hydrazone linker were optimized [72]. Also, the targeting of ELP towards the tumor via local hyperthermia was further optimized by thermal cycling of the tumor. This method relies on heating and cooling cycles of the tumor tissue, whereby the hyperthermia causes an increased vascular concentration of the ELP[V5A2G3-150] aggregates adhering to the vessel walls in the tumor, which upon return to normal temperature leads to an increase in transvascular transport [73]. A recent review by Chilkoti and coworkers gives a comprehensive overview on their efforts in the field of ELP-assisted drug delivery [74].
Generally, the plasma membrane of eukaryotic cells is impermeable to macro-molecular carriers. Therefore, Raucher and coworkers improved the ELP-assisted drug delivery by fusing a cell-penetrating peptide (CPP) to the ELP carrier to facilitate its cellular uptake [75, 76]. CCPs are short peptides that are capable of efficiently crossing the cellular plasma membrane and entering the cell’s cytoplasm. CPPs have been shown to deliver a variety of cargos across the cell membrane [77]. Various CPPs, such as penetratin (Pen) derived from Drosophila transcription factor Antennapaedia, the Tat peptide derived from the HIV-1 Tat protein, the hydrophobic membrane translocating sequence (MTS) from the Kaposi fibroblast growth factor, and the cationic Bac peptide derived from the bactenecin antimicrobial peptide, were fused to the N-terminus of the ELP (for sequences see Fig. 13) [78, 79]. In addition to the use of this system for delivery of Dox, therapeutic peptides were fused to the C-terminus of the CPP-ELP (Fig. 13). This design was examined for a therapeutic peptide that inhibits the oncogenic transcription factor c-Myc [78, 80] and for cell cycle inhibitory peptides [79, 81, 82]. These types of drug delivery systems were shown to be effective in different types of cancer cells, but no in vivo studies have yet been reported. However, in a recent review on CPP-ELPs for therapeutic peptide delivery it is mentioned that a Bac-ELP with a therapeutic peptide for inhibition of c-Myc is currently under investigation in mouse breast cancer models and in a rat glioma model [83].
Fig. 13.

Left: ELP carrier N-terminally fused to a cell-penetrating peptide and C-terminally fused to a therapeutic peptide. Right: Amino acid sequences for several cell-penetrating peptides (see text for details). Reprinted from [83] with permission from Elsevier, copyright 2010
Another approach that also employs the thermal responsiveness of ELPs for drug delivery is the hyperthermia-triggered multivalency. To this end, block copolymers of two ELP regions were synthesized (ELP[VA8G7-n]-ELP[V5-n]), where one of the blocks had a transition temperature at the hyperthermic temperature of the tumor and the other block was insensitive in this temperature range. The end of the insensitive block was genetically modified with a peptidic ligand that targets a specific cellular receptor or a membrane protein that is upregulated by tumor cells. Normally, the effectiveness of this type of peptide is limited due to its systemic toxicity; however, in this approach the block copolymer was triggered by the hyperthermia in the tumor to assemble into a drug delivery vehicle displaying multiple ligands. This triggered multivalency is supposed to increase the binding affinity of the ligand-ELP in tumor tissue without increasing the accumulation in healthy tissue. A schematic overview of the design and rationale is given in Fig. 14[84, 85].
Fig. 14.
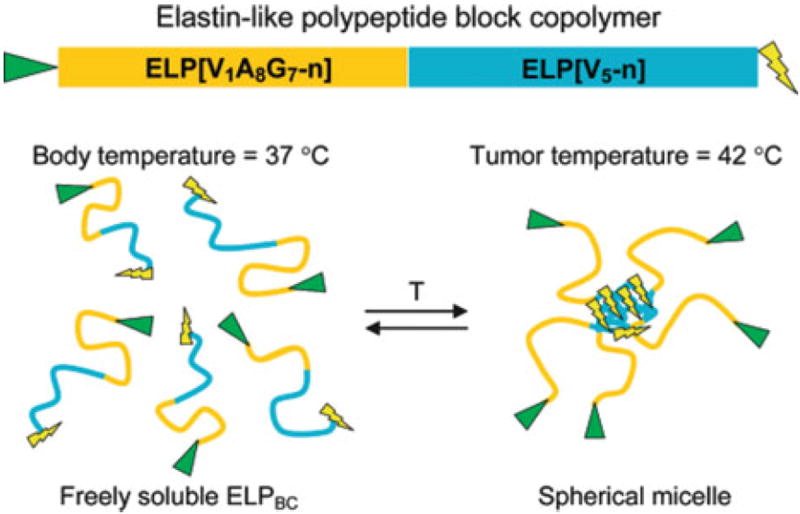
Hyperthermia-triggered multivalency. Block copolymers consisting of two ELP blocks, a hydrophilic ELP[VA8G7-n] block and a hydrophobic ELP[V5-n] block were designed. The N-terminal hydrophilic block was modified with a peptidic ligand (triangle) by gene fusion and the design also allows introduction of a drug or imaging agent at the C-terminus (lightning bolt). T temperature. Reprinted in part from [84] with permission from American Chemical Society, copyright 2008
Nanocarriers
Micellar nanocarriers have already been applied successfully for delivery of hydrophobic drugs [86]. These carriers are usually the product of self-assembled block copolymers, consisting of a hydrophilic block and a hydrophobic block. Generally, an ELP with a transition temperature below body temperature is used as hydrophobic block and the hydrophilic block can be an ELP with a transition temperature above body temperature or another peptide or protein. The EPR effect also directs these types of carriers towards tumor tissue.
In 2000, the first example of ELP diblock copolymers for reversible stimulus-responsive self-assembly of nanoparticles was reported and their potential use in controlled delivery and release was suggested [87]. Later, these type of diblock copolypeptides were also covalently crosslinked through disulfide bond formation after self-assembly into micellar nanoparticles. In addition, the encapsulation of 1-anilinonaphthalene-8-sulfonic acid, a hydrophobic fluorescent dye that fluoresces in hydrophobic environment, was used to investigate the capacity of the micelle for hydrophobic drugs [88]. Fujita et al. replaced the hydrophilic ELP block by a polyaspartic acid chain (Dm). They created a set of block copolymers with varying block lengths, ELP[V-n]-Dm where n = 40, 80, 120, or 160 and m = 22, 44, 88, or 176. They investigated the self-assembled structures and they observed the formation of particles with a homogeneous size of 50–250 nm upon triggering the transition [89].
ELP-based triblock copolypeptides have also been used to produce stimulus-responsive micelles, and Chaikof and coworkers envisioned the possible application of these micelles as controlled drug delivery vehicles. These amphiphilic triblock copolymers were constructed from two identical hydrophobic ELP endblocks and a hydrophilic ELP midblock. Below the transition temperature, loose and monodispersed micelles were formed that reversibly contracted upon heating, leading to more compact micelles with a reduced size [90].
Rodríguez-Cabello and coworkers reported a modified elastin-like polypeptide, in which the glycine at the third position of the repetitive sequence was replaced by an alanine residue. These biopolymers consisting of VPAVG repeats also showed a reversible phase transition; however, strong undercooling was needed to resolubilize this ELP [91]. This biopolymer has already been used to encapsulate the model drug dexamethasone phosphate [92]. A recent study uses this type of ELP, (VPAVG)220, to encapsulate bone morphogenetic proteins (BMPs) in ELP-based nanoparticles for drug delivery. BMPs are signaling proteins that are capable of promoting new bone formation, and the developed delivery system was shown to be effective in an in vitro cell assay [93].
Drug Depots
ELP-assisted local delivery of drugs is realized via several strategies. In the first strategy, soluble ELPs are injected and their coacervation is triggered by the body temperature. Here, the drug can be covalently attached to the ELP or it can just be mixed with the ELP. In another approach, crosslinked ELP depots containing a drug can be produced and then implanted to generate a stable release of the drug.
This first strategy was used by Setton and coworkers to trigger the in situ formation of an ELP-based drug depot for sustained release. Biodistribution studies of radiolabeled ELPs with a Tt below body temperature were performed after intra-articular injection in rats [94]. Later, drug depots of ELPs with covalently attached immunomodulator therapeutics [95] and anti-TNFα therapeutics [96] were created. Respectively, ELP[V-120] and ELP[V-60] biopolymers with a transition below body temperature were used in these studies.
The Chilkoti group applied the local injection approach for intratumoral drug delivery. ELP[V-120], with a transition at 27°C, was designed and labeled with 14C, 125I or 131I for radiotherapy. The first two labels were used to monitor tumor retention of the ELP and the last label was addressed to equip the ELP with anti-tumor activity. It was found that mice treated with 131I-labeled ELP[V-120] experienced a significantly prolonged survival over those treated with saline [97].
For the second type of drug release from depots, ELPs were crosslinked prior to implantation. In an investigation on the controlled release of the antibiotics cefazolin and vancomycin, ELP[KV16-102] was used. This ELP was crosslinked via the amines on the lysine residues using β-[tris(hydroxymethyl)phosphino]-propionic acid (THPP) to enable a high degree of antibiotic entrapment. The functionality of this approach was shown in vitro [98]. Another very recent example of this methodology is the production of drug-loaded ELP microspheres, which are crosslinked with glutaraldehyde. The ELP temperature-responsiveness was then used to open and close the pores of these microspheres [99].
In clinical trials using adenoviral vectors for gene delivery, it was shown that selective delivery of sufficient number of therapeutic gene copies remains difficult and that the lack of this selectivity caused acute toxicity and immunogenicity for the healthy tissue. Cappello and Ghandehari applied silk-elastin-like polypeptide (SELPs) [32] hydrogels to overcome the challenges of cancer gene therapy in head and neck cancer [100]. They found that intratumoral injection of adenoviruses with SELPs enhanced gene expression in tumors up to tenfold compared to viral injection without the biopolymer [101-103]. In mouse models, the SELPs efficiently controlled the duration and extent of transfection in tumors for up to 5 weeks with no detectable spread to the liver. This resulted in a fivefold greater reduction in tumor volume compared to intra-tumoral injection of adenoviruses without the biopolymer [104, 105].
2.4.3 Tissue Engineering
In the field of tissue engineering, the principles of engineering and life sciences are applied for the development of functional substitutes for damaged tissue. To this end, biomaterials have been used to replace, restore, or enhance organ function. Therefore the material needs to be able to match the characteristics of the tissue it is replacing, such as shape, physical properties, and support in cellular processes [106].
The application of ELPs in tissue engineering is attractive for several reasons: first, ELPs are based on elastin, which is a main component of the extracellular matrix. As a result they are non-immunogenic, biocompatible, and biodegradable [107]. ELPs can furthermore be produced in relatively high yields in host cells, such as bacteria, and this type of polymer synthesis provides control over the exact amino acid sequence and polymer length. ELPs can also easily be purified by exploiting their inverse temperature transition. For these reasons, ELPs were used and modified to form hydrogels, films, and fibers for various tissue engineering applications, such as cartilaginous, vascular, ocular, and liver tissue regeneration.
Setton and Chilkoti applied ELPs as a three-dimensional matrix to entrap chondrocytes. In their study, ELP[V5G3A2-90] with a transition temperature of 35°C at 50 mg/mL in PBS was used. This biopolymer can be used to generate a suspension with cells, which upon injection into a defect site will form a scaffold. They showed that in vitro the resulting ELP gel supported the viability of chondrocytes and the synthesis and accumulation of cartilage-specific extracellular matrix material. This suggested that ELPs indeed could be used for in situ formation of a scaffold for cartilaginous tissue repair [108]. Later, they also demonstrated that ELP gels could induce and support chondrocytic differentiation of human adipose-derived adult stem cells in vitro, without the addition of chondrocyte-specific growth factors [109]. Haider et al. [110] also demonstrated the potential of SELPs [32] as scaffold for the encapsulation and chondrogenesis of human mesenchymal stem cells. These SELPs irreversibly form hydrogels upon triggering the transition [110]. One significant problem is that the shear modulus of uncrosslinked ELPs is several orders lower than of articular cartilage. In order to increase the load-bearing capabilities of ELP gels, glutamine- and lysine-containing ELPs that could be enzymatically crosslinked via transglutaminase were developed [111]. Even though the shear modulus was increased and the ability to promote the synthesis and retention of cartilage tissue was retained, the crosslinking reaction proceeded over a long time, which prevented its clinical use. Therefore, a chemical crosslinking method that would proceed within a shorter time was developed. For this method, several lysine-containing ELPs were crosslinked with β-[tris (hydroxymethyl)phosphino]propionic acid (THPP) [112-114]. ELP[V6K1-224] was then used as an injectable scaffold for cartilage repair in a goat model of an osteochondral defect. It was shown that the ELP crosslinker solution was easily delivered and resulted in a stable, well-integrated gel that supported cell infiltration and matrix synthesis, but unfortunately this gel was prone to rapid degradation [115]. In a recent study, many lysine-containing ELPs were evaluated for their mechanical properties in an effort to optimize the ELP formulation for in vivo application [116].
Tirrell and coworkers designed artificial extracellular matrix (aECM) proteins for small-diameter vascular grafts. For vascular grafts, both mechanical integrity and biological interaction with host tissue are important properties. Therefore, the aECM proteins contained alternating CS5 binding domains and elastin-like poly-peptide domains (Fig. 15, aECM 3). The fibronectin CS5 domains provide cell adhesion signals whereas the ELP domains give these materials elasticity and mechanical integrity. The aECMs also contained lysine residues, either incorporated into the ELP domains or near the C- and N-termini, to allow crosslinking without interrupting the CS5 binding domains [117-119]. It was shown that these crosslinked films of aECMs had properties resembling those of the natural elastin [120]. In another study, aECMs with RGD binding domains were produced (Fig. 15, aECM 1) and compared to the CS5 domains. Here, it was found that the cellular response to aECM proteins can be modulated through choice of binding domain and that those with the RGD sequence showed rapid cell spreading and matrix adhesion [121]. Subsequently, variation in ligand density was accomplished by mixing two aECMS (Fig. 15, aECMs 1 and 2) in different ratios prior to crosslinking. Using this method, cell adhesion and spreading could be modulated [122].
Fig. 15.

Amino acid sequences of artificial extracellular matrix (aECM) proteins. Each protein contains a T7 tag, a histidine tag, a cleavage site, and elastin-like domains with lysine residues for crosslinking. The RGD cell-binding domain is found in aECM 1, whereas aECM 3 contains the CS5 cell-binding domain. aECM 2 and aECM 4 are the negative controls with scrambled binding domains for aECM 1 and aECM 3, respectively. Reprinted from [121] with permission from American Chemical Society, copyright 2004
2.4.4 Hybrid Materials
In the previous sections it was demonstrated that the stimulus-responsive behavior of ELP was transferred to ELP fusion proteins and even to non-covalently bound moieties, such as proteins, plasmids, and heavy metals, mostly for biomedical and biotechnological applications. The ability of ELPs to reversibly switch their polarity is also of great interest for the development of stimulus-responsive materials. Many approaches have therefore recently been undertaken to integrate ELPs with, for example, polymers, particles, and surfaces.
In 2003, the van Hest group produced elastin-based side-chain polymers [123]. This research was motivated by the demonstration of the occurrence of an inverse temperature transition in a single repeat of VPGVG [124]. A methacrylate-functionalized VPGVG was synthesized and used as a monomer to perform atom transfer radical polymerization (ATRP) to produce homopolymers (Fig. 16b) or ABA block copolymers (Fig. 16a), in which the A blocks are based on the methacrylate-functionalized VPGVG and the B block is a bifunctional poly(ethylene glycol) ATRP macroinitiator. It was shown that these polymers exhibited behavior similar to that of linear poly(VPGVG) [123, 125]. Later, Cameron and coworkers used another type of controlled radical polymerization (reversible addition-fragmentation chain transfer polymerization, RAFT) to produce homopolymer elastin-based side-chain polymers (Fig. 16c) [126]. They found that binary mixtures of well-defined elastin-based side-chain polymers showed a single transition temperature, which depended on the blend composition [127]. Additionally, they immobilized these elastin-based side-chain polymers on polymerized high internal phase emulsions (polyHIPE), to demonstrate the potential of this methodology to reversibly immobilize biomolecules such as enzymes [128]. Also ring-opening metathesis polymerization (ROMP) was used to prepare elastin-based oligopeptides of low molecular weight and low polydispersity [129]. More recently, random copolymers of elastin-based monomers and oligo(ethylene glycol)-based monomers were generated using ROMP [130].
Fig. 16.

Various type of elastin-based side-chain polymers (a) ABA block copolymer produced via ATRP, (b) homopolymer produced via ATRP, (c) homopolymer produced via RAFT polymerization
Inspired by the elastin-based side-chain polymers, Lemieux et al. prepared elastin-based stimulus-responsive gold nanoparticles. To this end, they capped gold particles with a layer of a single repeat of thiol-functionalized VPGVG peptides (Fig. 17a). These nanoparticles showed LCST behavior, which was modulated by varying the pH of the solution [131].
Fig. 17.

(a) Elastin-based stimulus-responsive gold nanoparticles. Reproduced from [131] by permission of The Royal Society of Chemistry (b) Functionalization of a glass surface with ELP. In the first step, the glass surface is aminosilylated with N-2-(aminoethyl)-3-aminopropyl-trimethoxysilane, then modified with glutaraldehyde. Subsequently, the stimulus-responsive bio-polymer is covalently immobilized using reductive amination. Reproduced from [132] by permission of The Royal Society of Chemistry
Besides short ELPS, longer ELPs have also been conjugated to synthetic polymers. In one approach, Cu(I)-catalyzed azide-alkyne cycloaddition click chemistry was applied. For this purpose, ELPs were functionalized with azides or alkynes via incorporation of azidohomoalanine and homopropargyl glycine, respectively, using residue-specific replacement of methionine in ELP via bacterial expression [133]. More recently, an alternative way to site-selectively introduce azides into ELPs was developed. Here, an aqueous diazotransfer reaction was performed directly onto ELP[V5L2G3-90] using imidazole-1-sulfonyl azide [134].
In another investigation, ELP[V5L2G3-90] with three lysines in the N-terminal region was immobilized on a glass surface in a microreactor to enable temperature-controlled positioning of ELP fusion proteins. For this purpose, the glass surface was first functionalized with N-2-(aminoethyl)-3-aminopropyltrimethoxysilane, followed by glutaraldehyde treatment and reductive amination to immobilize the biopolymer on the surface (Fig. 17b) [132].
Hybrid materials of poly(VPGVG)s and gold were also conveniently produced. ELP[V5A2G3-180] was adsorbed onto colloidal gold particles via electrostatic interactions [135]. Subsequently, these ELPs were covalently immobilized on the gold surface, by reacting the amines in the ELP with N-hydroxysuccinimide-activated carboxyl groups attached to gold. These stimulus-responsive particles were subsequently used to capture and release biomolecules [136].
Huang et al. were able to synthesize optically responsive hybrid materials. They used the block copolymer ELP[V-40]-ELP[V4C1-10], which was assembled onto gold nanorods via the thiol-moieties of the cysteine residues. Exposure of the ELP-functionalized gold nanorods to near-infrared light led to heating of the gold due to surface plasmon resonance (SPR). This resulted in phase transition and aggregation of the ELP-functionalized gold nanorods [137]. Also, the group of Rodriguez-Cabello reported similar hybrid materials based on ELP-functionalized gold nanoparticles [138, 139].
3 Resilin and Resilin-Like Polypeptides
3.1 Resilin Occurrence and Biosynthesis
3.1.1 Occurrence
Resilin was first discovered and brought to the attention of the scientific community by Weis-Fogh in the late 1950s [140, 141]. Through studies into arthropod locomotion, certain proteinaceous patches of the insect cuticle (i.e., exoskeleton) [142] were shown to exhibit perfect long-range elasticity and superior extensibility, supporting rapid deformation in insect organs without hysteresis. The first written descriptions of resilin in 1960 [from tendons of dragonflies (Odonata) and wing hinges of locusts (Orthoptera)] described it as a colorless, swollen isotropic rubber with remarkable elastic recovery and a unique amino acid composition [141]. Resilin did not dissolve in any solvent that did not break peptide bonds and the crosslinks were described as being “extremely stable,” though the chemistry of the crosslinks was unknown at the time. Following the initial discovery and description of resilin in locusts and dragonflies, others quickly began finding the protein throughout the Arthropoda phylum [140].
The solid cuticle of the arthropod is a complex extracellular product made of chitin (a polysaccharide) and tanned or fibrous proteins, and is often hard and colored [142]. However, in 1947, La Greca identified and described a colorless, transparent cuticle found in the wing hinges of locusts that sharply contrasted with the solid cuticle associated with insects. Later to be named “rubber-like cuticle,” these elastic hyaline structures largely went unnoticed until Weis-Fogh’s investigation of locust flight during the 1950 s [141, 143]. Detailed analysis of several elastic elements of the locust flight system revealed small cuticular patches that possessed long-range elasticity; these patches, which consisted of protein and chitin, could undergo extreme deformation before quickly returning to their original shape [144]. Through careful analysis of similar rubber-like cuticle regions prepared from different insect species, it was determined that the protein was responsible for the elastic properties of the cuticle. Further, it was determined that the protein exhibited a unique amino acid composition that differed from other insect cuticle proteins as well as other structural proteins such as collagen and elastin [141, 145]. The protein was named resilin, derived from the latin resilire, which means “to spring back” [144, 145].
Resilin is often found in the insect cuticle as part of a relatively simple composite material containing only the protein and chitin [141, 144]. Chitin is found in nearly all types of arthropod cuticle in the form of stiff filaments that run parallel to the surface of the cuticle, but that gradually change direction to form a helicoidal pattern. The resilin-containing cuticle in locust wing-hinge ligament contains the helicoidal chitin structures and makes the ligament difficult to stretch, but capable of bending and twisting deformations [144]. It was determined that the elasticity of the rubber-like cuticle was derived from the resilin protein as the treatment of the cuticle with proteases, strong acids, or bases left only the chitinous component, which was delicate and inextensible [141].
Following the discovery of resilin in the locust and dragonfly [141, 146], resilin was more widely found in other arthropods. Resilin was discovered in the salivary pump of assassin bugs and in the feeding pump of Rhodnius prolixus where, in both cases, it acted as an elastic antagonist to muscle action [140, 147]. Other instances of resilin serving the function of an elastic spring in feeding/pumping mechanisms include the tsetse fly [148], reduviid bugs [149], and honey bees [150]. These elastic resilin springs act as an energy-saving mechanism for the insects by providing a passive means of emptying the pump cavities [148, 149]. In the honey bee, resilin was found to provide resistance to the power stroke of the venom-dispensing pump and also served to facilitate the stinging action by providing cushioning for the stinger apparatus [150].
Resilin has also been found to serve an important role in the sound production of arthropods, notably in the Cicadae family, but also in Pyralidae family (moths) and in scorpions [151-157]. Relatively thick patches of resilin (100–150 μm) are found in the tymbals, which are the sound production organs of the cicada. As seen in the feeding/pumping mechanisms, these elastic patches provide spring-like elasticity that returns the tymbal to its rest position following release of tension in the tymbal muscle. The resilin contained in these structures must operate under very rapid stress-release cycles, in the 1–10 kHz range for some cicadas, but despite these demanding mechanical requirements, it has been found that energy losses can be less than 20% [140, 151, 154, 156, 157].
The elasticity, rapid response, and low energy dissipation serve a particularly important role in the mechanically active tissues responsible for the locomotion of insects. In addition to the description of resilin in the flight systems of the locust and the dragonfly [146], the protein was found to contribute to insect flight in damselflies [158], beetles [159], and other species of the Dermapteran order [160]. Resilin was found in the components of the wing in these insects where it served to provide spring-like recovery of different wing elements following deformation, as well as to dampen the aerodynamic forces felt by the wing [158-160]. Apart from flight, resilin has been found in the ambulatory systems of cockroaches [161, 162] and house flies [163]; resilin provides elasticity to the joints and thereby facilitates rapid movement in both species [161, 162]. Resilin in insect tarsus has been indicated in additional taxa [164], including ants and bees [165]. Perhaps the most notable application of resilin-based rubber-like elasticity in the locomotion of insects is seen in the jumping mechanism of fleas [166-171] and other jumping arthropods [172-176]. Initial investigations of these jumping mechanisms suggested a spring-like model, with resilin serving as the major elastic element responsible for storing the energy required for a jump [169, 171, 174, 176]; however, competing evidence has suggested that the chitinous components are actually responsible for most of the energy storage [168, 173].
The rubber-like elasticity of resilin is only one of its outstanding mechanical properties; it also demonstrates a remarkable capacity for stretching. The extensibility of resilin can be seen in the ability of some insects to swell their abdomens to many times the original size. For example, resilin has been found in the cuticle of the physogastric queen termite, which has been shown to increase the size of its abdomen up to 50 times following fertilization [177]. Honey ants, which swell their abdomens when gorging on food, have also been shown to contain resilin [178]. The one unifying characteristic with respect to the occurrence of resilin in arthropods is that it can almost always be found when rubber-like elasticity is a prerequisite for function. However, resilin also appears in unexpected places such as the lens cuticle of the firefly, Photinus pyralis Linnaeus [179], and even outside of the insect community as in the case of the maxilliped flagella of crabs and crayfish [180].
3.1.2 Biosynthesis
Interest in the composition of resilin, specifically in its primary structure, grew in the early 1990s when Lombardi and Kaplan published the amino acid sequences of tryptic digests of cockroach resilin [181]. These sequences were found to resemble the tryptic digest sequences of locust resilin investigated by Ardell and Andersen, who subsequently used both sets of sequences to search for genes that may code for resilin in Drosophila melanogaster. This effort led to the discovery of two genes that produce peptides homologous to the tryptic digest sequences from both the cockroach and locust [144, 182]. One of the genes, CG15920, was suggested to encode the precursor protein for resilin. The gene product had an amino acid composition and an isoelectric point that closely resembled resilin [182]. It contained a 17-residue signal peptide sequence at the N-terminus, indicating that it may be secreted into the extracellular space [182, 183], and it contained a sequence that resembled the R&R-2 variant of the Rebers–Riddiford consensus sequence that has been identified in the binding of proteins to chitin in the insect cuticle [182, 184]. Many cuticle proteins from evolutionary disparate species of insect were found to contain some variant of this sequence, G-X7-[DEN]-G-X6-[FY]-X-A-[DGN]-X2,3-G-[FY]-X-[AP] (X is any amino acid and the numerical subscript is the number of residues) [185], leading to the belief that the highly conserved sequence served an important biological function such as the binding to chitin in the cuticle [186]. Subsequent expression and characterization of this domain has provided convincing evidence for its role in binding to chitin [185, 187].
However, the most important indication that the CG15920 gene encoded a resilin precursor was the discovery of highly conserved repeat sequences. These resilin “motif” sequences are repeated in domains on either side of the chitin binding domain (see Fig. 18). The first motif sequence, GGRPSDSYGAPGGGN, is located in the N-terminal region (exon 1) with respect to the chitin binding domain (exon 2), while the second motif sequence, GYSGGRPGGQDLG, dominates the C-terminal region (exon 3) of the CG15920 gene product [182]. As discussed in Sect. 3.3, these sequences have provided the basis for the development of recombinant resilin-like polypeptides that attempt to recreate the long-range elasticity of natural resilin. Furthermore, the modules of the CG15920 gene appear to mirror the protein-chitin-protein morphology of the insect cuticle. Additionally, it has been shown that there are two splices of the CG15920 gene: one that contains the chitin binding domain and one where it is absent [182, 187, 188]. It is possible that the synthesis of the two variants depends on whether chitin is being synthesized in the rubber-like cuticle. For instance, the synthesis of chitin-binding resilin may be unregulated during the formation of chitin lamellae while the alternative variant is synthesized primarily to form the embedding proteinaceous matrix. Recent research into putative resilin genes has revealed a high level of conservation for the resilin genes amongst Drosophila species, but these putative resilin genes can vary once outside a particular genus of insect [188].
Fig. 18.
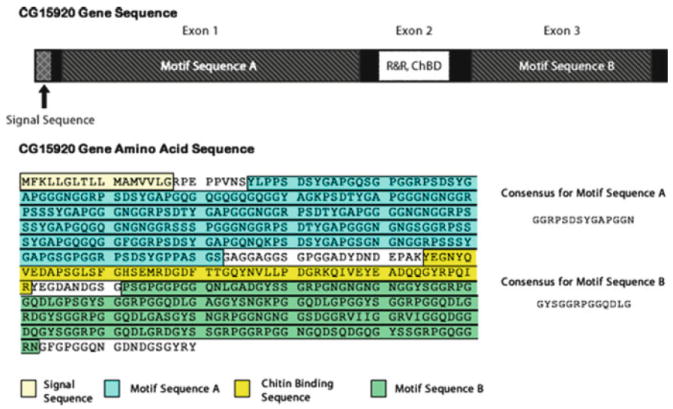
CG15920 gene sequence and primary structure. The consensus repeat sequences are also represented. The highlighted regions correspond to the signal sequence, R&R chitin-binding domain, and the elastomeric domains containing repeat motifs A and B. Reproduced from [182, 188] with permission from Elsevier, copyright Elsevier 2001, 2010
The amino acid composition of resilin was elucidated shortly after its first description and was shown to be unique among other structural proteins found in Nature; a summary of the amino acid sequence is given in Table 1.
Table 1.
Amino acid composition of resilin from wing-hinge and prealar arm of locust
| Amino acid | Resilin
|
Comparison
|
|||
|---|---|---|---|---|---|
| Residues/105 g protein
|
Residues/105 g protein
|
||||
| Average wing-hinge | Prealararm | Collagen (oxhide) | Elastin (oxligamentum nuchae) | Silk fibroin (Bombyxmori) | |
| Asp | 113 | 122 | 52 | 4.5 | 16 |
| Thr | 35 | 34 | 19 | 8 | 12 |
| Ser | 91 | 91 | 41 | 8 | 160 |
| Glu | 53 | 54 | 76 | 14 | 14 |
| Pro | 87 | 89 | 125 | 156 | 5 |
| Gly | 448 | 414 | 354 | 398 | 590 |
| Ala | 129 | 120 | 116 | 212 | 389 |
| Val | 30 | 36 | 21 | 148 | 30 |
| Met | Nil | Nil | 6.5 | (<1) | Nil |
| iLeu | 20 | 19 | 14 | 31 | 9 |
| Leu | 26 | 27 | 28 | 66 | 7 |
| Tyr | 29 | 35 | 5 | 9 | 69 |
| Phe | 30 | 27 | 14 | 30 | 8 |
| Amide N | (102) | – | (46) | (3) | – |
| Lys | 6 | – | 27 | 3 | 4 |
| His | 8.5 | – | 4.5 | 0.5 | 2 |
| Arg | 40 | – | 47 | 5 | 6 |
| Total | 1,145 | 1,128 | 1,063a | 1,093 | 1,321 |
Results of amino acid analysis performed on resilin from the locust Schistorcerca gregaria, compared with values for collagen, elastin, and silk fibroin. Reproduced from [145] with permission from Elsevier, copyright Elsevier 1961
Includes 106 hydroxyproline and 7 hydroxylysine
Resilin has a greater percentage of acidic residues than collagen, elastin, and silk fibroin and contains fewer non-polar residues (i.e., Gly, Ala, Val, Ile, Leu, Pro, Met, and Phe) than silk fibroin and elastin. The significant content of acidic residues might account for resilin’s hydrophilicity as well as its low isoelectric point, as indicated through swelling experiments [141, 145]. Resilin contains little to no methionine, cysteine, hydroxyproline, and tryptophan. Except for silk fibroin, resilin contains more tyrosine residues than the other structural proteins compared in the Table 1. The levels of tyrosine in the insect cuticle can vary from 4% to 13% (by weight) and in resilin, tyrosine accounts for approximately 5% of the total weight [145, 189, 190].
Andersen demonstrated that two novel fluorescent compounds served as the chemical crosslinks in resilin [189-192]. These aromatic α-amino acids contained a phenolic group and were shown to be a diaminodicarboxylic acid and a triamino-tricarboxylic acid: dityrosine and trityrosine (see Fig. 19) [192]. Physical measurements determined the average molecular weight between junction points to be ~5,000 Da in the case of dragonfly resilin [193], which corresponds closely to the value of ~3,000 Da derived from the amino acid analysis, assuming that the fluorescent amino acids constitute the crosslinks in resilin. Other various forms of crosslinking, such as disulphide bridges, ester groups, amide bonds, etc., had been ruled out, leading to the conclusion that these novel fluorescent compounds were indeed the only crosslinks for the network protein [189-192, 194, 195]. The formation of di- and trityrosine or homolog crosslinks was later discovered in other elastic tissues, including those originating from higher organisms. Dityrosine has been isolated from elastin [196], collagen [197], fibroin, keratin [198], and other structural proteins and tissues [199-201]. The crosslinking of the tyrosine residues can result from the phenolic coupling of two phenoxy radicals of the residues [202]; this is initiated by a radical species (i.e., •OH) and was shown to be mediated, ex vivo, by the reaction of a peroxidase and hydrogen peroxide [203].
Fig. 19.
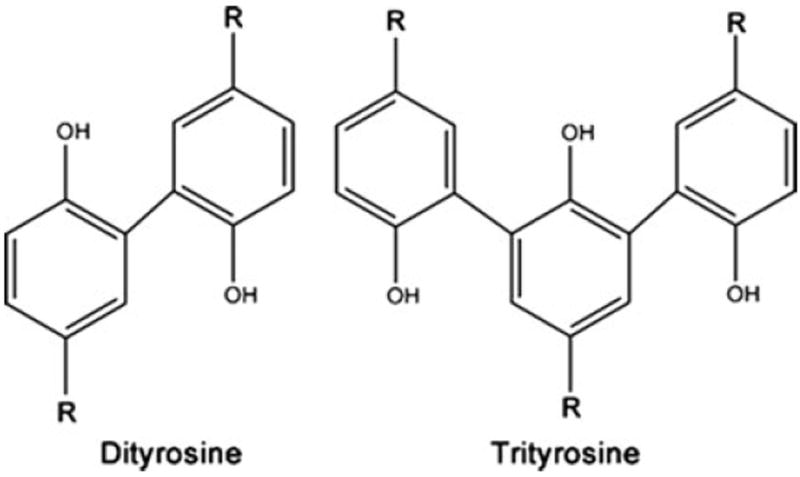
Dityrosine and trityrosine crosslinks found in resilin. The R groups connect to the backbone of the polypeptide chain
Additionally, the crosslinking and immobilization of resilin in the insect cuticle appears to occur very quickly, perhaps as soon as it is deposited. Temperature also plays a role; it has been found that the rate and the extent to which resilin is crosslinked increases with increasing temperatures [204-206]. The exact mechanism by which resilin is actually crosslinked is still largely unknown and although peroxidase has been demonstrated to form di- and trityrosines there is no substantiated proof of this mechanism for the formation of the resilin network [144, 207]. The discovery of the resilin gene and interest in the development of recombinant resilins (discussed in Sect. 3.3) may reinvigorate the study of the crosslinking of resilin [144].
3.2 General Properties of Resilin
Crosslinked resilin is a highly stable, swollen isotropic rubber that is resistant to both high temperatures and protein coagulants. Resilin in neutral water is unaffected by heating up to 125°C and does not begin to degrade until 140–150°C. Additionally, a myriad of fixatives, and tanning and coagulating agents are virtually unsuccessful in bringing about permanent change in resilin from the rubbery state to the solid state, even with prolonged treatment [141]. Only under the most extreme conditions, outside physiological ranges, is the protein irreparably altered; for example, the protein degrades under high or low pH conditions or when incubated with proteases. In fact, resilin is quite easily digested by proteolytic enzymes including papain, various trypsins, chymotrypsin, pepsin, elastase from porcine sources, and the mixture of enzymes found in the saliva of Reduviid bugs [141]. Resilin is transparent and absent of any color; however, the refractive index of resilin cuticle exceeds 1.4 and the water content at neutral pH is 50–60%, which indicates a very concentrated material. Resilin swells in alkaline buffers and shrinks in acidic buffer, but the material remains rubbery under all conditions unless it is dried out or dehydrated in pure alcohol [141]. Dried resilin behaves like a glassy polymer, but when reintroduced to water it regains its rubbery nature and demonstrates all the properties of an amorphous polymer network above its glass transition. Nonpolar solvents have proven completely incapable of penetrating the resilin cuticle; very few polar solvents will plasticize the protein matrix, which is insoluble in all solvents. Resilin will shrink from dehydration in alcohols and also become glassy (as when dried), but the protein will swell and become rubbery in ethylene glycol, formic acid and formamide. Resilin is also capable of isotropic, reversible swelling; resilin can be swollen in alkaline buffer, shrunk in acidic buffer and then swollen once again when returned to an alkaline buffer [141]. In addition to the absolute insolubility of resilin in nonpolar solvents, the protein also proved to be resistant to chaotropic solutes such as urea, guanidine hydrochloride, and alkaline cupric ethylenediamine, the latter of which demonstrated the ability to disrupt hydrogen bonding in silk fibroin without destroying peptide bonds. Additionally, resilin was unaffected by agents that disrupt the disulphide bridges of cystine. The insolubility of resilin under all of these conditions and its remarkable resistance to heat supports the suggestion that the protein exists as a covalently crosslinked network with very chemically stable crosslinks [141, 145].
Resilin and elastin, unlike other structural proteins, fulfill both definitions of an “elastic” material. Colloquially speaking, resilin and elastin are stretchy or flexible. They also fulfill the strict definition of an elastic material, i.e., the ability to deform in proportion to the magnitude of an applied stress without a loss of energy, and the recovery of the material to its original state when that stress is removed. Resilin and elastin are alone in the category of structural proteins (e.g., collagen, silk, etc.) in that they have the correct blend of physical properties that allow the proteins to fulfill both definitions of elasticity. Both proteins have high extensibility and combine that property with remarkable resilience [208].
Resilin from dragonfly tendon is capable of stretching 3–4 times its resting length before breaking and will completely recover even after weeks of an applied tensile stress. The tensile strength was measured to be at least 30 kg cm−2 (2.94 MPa) and the elastic modulus has been reported to be approximately 6 kg cm.−2 (0.588 MPa) [141, 144, 193], but the initial elastic modulus has been reported to be as high as 20 kg cm.−2 (1.96 MPa) [146, 208]. The toughness and the resilience of dragonfly tendon has been reported to be 4 MJ m−3 and 92%, respectively [208]. Possibly due to the addition of stiffer elements (i.e., the chitin lamellae), the prealar arm of the wing-hinge of the locust was found to have a slightly higher elastic modulus at 9 kg cm−2 (0.882 MPa). The mechanical properties of resilin are dependent on pH and on the swelling of the material; resilin swelled in alkaline buffers becomes increasingly stiff. Dynamic testing of the prealar arm revealed that the loss factor, defined as the ratio of energy irreversibly lost as heat to the total change in stored elastic energy, was exceedingly low at about 3.5% per half cycle when strained at biologically relevant frequencies (15–20 cycles/s). Even at high frequencies, such as 200 cycles/s, the loss factor remained less than 10% and, as mentioned previously, the loss factor is low even in the extreme case of cicada sound production [140, 146, 151, 156, 157].
Resilin and elastin have relatively high extensibility and resilience, but as compared to the collagen and the silks, the proteins sacrifice stiffness (elastic modulus) and strength (see Table 2). Collagen and dragline silk are much stiffer materials, but lack the extensibility that is characteristic of the rubber-like proteins. On the other hand, the mussel byssus fibers and the viscid silk have the extensibility of resilin and elastin, but lack the resilience [208].
Table 2.
Material properties of structural proteins and synthetics
| Material | Modulus, Einit (GPa) | Strength, σmax (GPa) | Extensibility, εmax | Toughness (MJ m−3) | Resilience (%) |
|---|---|---|---|---|---|
| Elastin (bovine ligament)a | 0.0011 | 0.002 | 1.5 | 1.6 | 90 |
| Resilin (dragonfly tendon)a | 0.002 | 0.004 | 1.9 | 4 | 92 |
| Collagen (mammalian tendon)a | 1.2 | 0.12 | 0.13 | 6 | 90 |
| Mussel byssus, distal (M. californianus)a | 0.87 | 0.075 | 1.09 | 45 | 28 |
| Mussel byssus, proximal (M. californianus)a | 0.016 | 0.035 | 2.0 | 35 | 53 |
| Dragline silk (A. diadematus)a | 10 | 1.1 | 0.3 | 160 | 35 |
| Viscid silk (A. diadematus)a | 0.003 | 0.5 | 2.7 | 150 | 35 |
| Kevlara | 130 | 3.6 | 0.027 | 50 | – |
| Carbon fibrea | 300 | 4 | 0.013 | 25 | – |
| High-tensile steela | 200 | 1.5 | 0.008 | 6 | – |
| Poly(glycerol-sebacate)b | 0.00028 | – | 2.67 | – | – |
| Poly(PEG-citrate)b | 0.00191 | 0.00151 | 15.05 | – | – |
| Natural rubberc | 0.002 | 0.030 | 9 | – | – |
| Chloroprene rubberc | 0.003 | 0.022 | 10 | – | – |
Table summarizes the material properties of structural proteins, high performance synthetic materials and synthetic elastomers
Table and material data redrawn from [208] with permission from The Royal Society, copyright 2002
Maximum values presented. Data collected from [209]
Unfilled, vulcanized rubber. Maximum values presented. Data collected from [210]
The most notable physical property of natural resilin is the near-ideal rubber elasticity it exhibits when swollen in water or other polar solvents [141]. Its long-range elasticity, insolubility in various solvents, and chemical stability of the di-and trityrosine crosslinks provided the first indications that resilin might be an ideal network [141, 193, 211]. As mentioned above for elastin, in rubber elasticity theory, the force applied to deform a network is made up of an energetic contribution and an entropic contribution; the entropic contribution accounts for much of the applied force in rubbers and therefore, drives recovery following deformation [211, 212]. In a perfect rubber, energy used to deform the network decreases the entropy of the system; little to no energy is lost in other interactions [144]. Resilin demonstrates remarkable agreement with rubber elasticity theory and, in fact, the agreement between theory and experiment was even closer than that for natural and synthetic rubbers studied at the time of the resilin experiments [193], despite the fact that resilin can undergo a variety of hydrophobic and hydrogen bonding interactions [144, 211]. This agreement suggests that the disappearance and creation of these interactions must be balanced during deformation [144].
Electron microscopy and X-ray diffraction experiments conducted on resilin-containing insect cuticle provided further support for resilin existing in the rubbery state as a crosslinked random network of protein chains. No fine structure was revealed by the electron microscopy experiments and zero crystallinity could be detected from the X-ray diffraction experiments. Furthermore, the diffraction measurements taken for strained resilin, which might have some microcrystalline structure due to chain alignment, were little changed from the relaxed resilin measurements. The complete lack of any indication of structure from either the microscopy or diffraction experiments supports the conclusion that natural resilin lacks any regular structural pattern [213].
These remarkable mechanical properties thus probably arise owing to the flexibility of the chain, imparted by conserved amino acid sequences. A survey of the amino acid sequences of other elastomeric proteins reveals one often-conserved motif, the proline-glycine dyad. The proline-glycine motif has been found in numerous proteins possessing long range elasticity, including vertebrae elastins, molluscan byssus fibers, high molecular weight glutenin, spider flagelliform silk, and spider dragline silk (see Table 3) [188, 214, 215]. It has been suggested that the conformational features of the proline-glycine dyad, particularly the propensity of the motif to form β-turns, confers elastomeric behavior to these structural proteins [214, 215]. To this end, specific mechanisms have been proposed, though not without controversy, as to how the β-turn structure might bestow long-range elasticity. Studies on the repeat motifs found in elastin indicated that the β-turn structure was the major conformational feature and that elastin might be made up of tandem β-turns, which form a β-spiral structure. The dampening of internal chain dynamics upon the extension of elastin was subsequently proposed for the mechanism of elasticity [216]. The non-crystalline regions of spider silks have also been predicted to contain repetitive proline-glycine β-turn structures and are believed to be responsible for the elasticity of the materials [217, 218]. The repetitive domains of glutenin have demonstrated a propensity to form β-turns and it has been proposed that it forms a β-spiral type similar to elastin. However, it has not been unequivocally established that this is the contributing factor to the material’s elasticity [215].
Table 3.
Sequences of repeat motifs of elastomeric domains
| Protein | Repeat motif |
|---|---|
| Abductin | GGFGGMGGGX |
| Elastin | VPGG |
| VPGVG | |
| APGVGV | |
| Byssus | GPGGG |
| Flagelliform silk | GPGGX |
| Dragline silk | GPGQQ |
| GPGGY | |
| GGYGPGS | |
| HMW subunits of wheat gluten | PGQGQQ |
| GYYPTSPQQ | |
| GQQ | |
| Resilin | GGRPSDSYGAPGGGN |
| GYSGGRPGGQDLG |
Partially reproduced from [215] with permission from The Royal Society, copyright 2002
The conservation of proline-glycine repeats in proresilin suggests some form of structural importance, as in other elastomeric proteins [182], but subsequent investigations on resilin-like motifs have provided little evidence for the β-spiral model [219]. More rigorous analysis on an array of resilin-like polypeptides pointed to an equilibrium between folded β-turns and extended structures [220]. This would suggest, in accordance with the historic evidence, that resilin behaves as an entropic spring whereby stretching the material causes the chains to shift to extended conformations and recover via entropically driven recoil. In this light, the role of proline and glycine might not be to impose a particular conformation, and therefore a repeated structure, but as recently discussed by Rausher et al. [221], the true role of these residues in elastomeric proteins might be to disrupt the occurrence of regular structures. Prolines have the tendency to disrupt secondary structure because of their rigid conformation, whereas glycines preclude the formation of secondary structure because they are simply too flexible [220, 221]. However, the one missing element is corroboration from mechanical studies. Structural studies and molecular dynamic investigations, although informative, cannot conclude a direct relationship between structure and function. Additionally, resilin exists as a swollen network and the crosslinking of the resilin sequences into a network may alter the favored conformational states of the peptide chain. An approach that combines structural studies and mechanical testing on an array of slightly different resilin-like motifs might provide more conclusive evidence for or against a proline-glycine structure–function relationship.
3.3 Development and Properties of Recombinant Resilins
As highlighted throughout this volume and in the sections on elastin in this review, progress in recombinant DNA technologies over the past three decades has made the development of biosynthetic protein polymers increasingly simple and promising. These methodologies permit the straightforward design of de novo sequences that endow protein polymers with tailored properties and with applications ranging from medicine to the development of high performance materials [222]. The development of recombinant resilins has likewise benefited from the increasingly facile nature of protein engineering and biosynthetic protein polymer production [182, 187, 219, 223-230]. Following the first description of the Drosophila CG15920 gene, Elvin et al. cloned, expressed, and purified the first exon of the gene as a soluble protein in E. coli. The authors demonstrated that this soluble protein, rec1-resilin, could be cast into a highly concentrated network by reacting tyrosines using a peroxidase from Arthomyces ramosus or through Ru(II)-mediated photo-crosslinking. The initial yields from isopropyl β-d-thiogalacto-pyranoside (IPTG)-induced expression of the rec1-resilin gene were relatively low at about 15 mg/L culture volume, too low to be useful for many applications especially as gels could only be formed at concentrations greater than 20 wt% [226]. However, improvements in expression have permitted production of relatively pure rec1-resilin at levels as high as 300 mg/L culture volume. Expression was conducted under high cell density fermentation conditions and with a two-step induction method inspired by Studier auto-induction methods [227, 231]. Affinity chromatography was replaced by a facile non-chromatographic “salting-out and heating” method that precipitated rec1-resilin through initial ammonium sulfate precipitation, followed by heating the resolubilized precipitate. The extraordinary heat stability and hydrophilic properties of resilin allowed the protein to remain in solution while other proteins denatured, aggregated, and precipitated [227]. This non-chromatographic purification provides a possible cost-effective pathway to the scaling up of the production of recombinant resilin.
Two new resilin genes based upon the consensus repeat motifs of the Drosophila CG15920 gene and from an Anopheles gambiae (African malaria mosquito) homolog have also been constructed. The consensus sequences from Anopheles (AQTPSSQYGAP) contain a slightly different combination of amino acids to the Drosophila sequence (GGRPSDSYGAPGGGN), with conservation of the YGAP motif. To ensure the fidelity of the final genes, short doubled-stranded oligonucleotides were designed to contain the consensus repeat motifs and were recursively ligated to develop genes containing 2, 4, 8, 16, 32, and 64 copies of the repeat in tandem. The genes containing 16 repeats (Dros16, from the Drosophila gene and An16, from the Anopheles gene) were expressed and purified using the non-chromatographic method [228].
Recombinant resilins appear to closely match native resilin in both physical and mechanical properties. Following the crosslinking of the first recombinant resilin, rec1-resilin, Elvin et al. were able to demonstrate the presence of dityrosine as well as the characteristic blue fluorescence [226]; the crosslinking and fluorescence of An16 and Dros16 were confirmed in a following study [229]. As with native resilin, the rec1-resilin, An16, and Dros16 recombinant proteins all demonstrated remarkable resistance to heat [227, 228]. Investigations into the structure of the An16 resilin through Raman spectroscopy, 2D-NMR and small angle X-ray scattering indicated that the protein did not form a consistent secondary structure either in concentrated crosslinked materials or as soluble protein. The results suggest that the An16 resilin is a dynamic and heterogeneous protein with possibly a small degree of order in the YGAP region of the sequence [219]. Later circular dichroism (CD) studies on all three recombinant resilins revealed that An16 and the rec1-resilin were populated with a number of conformations, including β-sheets, turns, and poly (l-proline II) (PPII) structures, but that approximately 60% of the protein was disordered (see Table 4). However, solutions of the An16 and rec1-resilin protein in urea, which tends to stabilize PPII structure, failed to provide any conclusive results as to whether it was a dominant structure in the recombinant resilins. It is possible that the two proteins exchange between PPII and another conformation or that the contribution of the PPII conformation is overestimated by the traditional secondary structure prediction programs employed to deconvolute the CD spectra [232]. Interestingly, the CD of Dros16 demonstrated a more ordered protein with more β-structure than the other two resilin-like polypeptides [229].
Table 4.
Secondary structure analysis: a comparison of resilin-like polypeptides
| Resilin-like polypeptide and sequence motif | Technique | Helices (%) | Strands (%) | Turns (%) | Unordered (%) (or random coil) | Beta sheet (%) | PPII (%) |
|---|---|---|---|---|---|---|---|
| An 16a (AQTPSSQYGAP) | CD | 5 | – | 11 | 64 | 10 | 10 |
| Dros 16b (GGRPSDSYGAPGGGN) | CD | 5.3 | – | 11.8 | 45.7 | 27.3 | 9.9 |
| Rec1 resilina (GGRPSDSYGAPGGGN, but irregular)c | CD | 2 | – | 11 | 58 | 18 | 11 |
| Full-length Drosophila Resilin (two motifs)d | FTIR | 11.9 | 15.3 | 29.1 | 43.8 | – | – |
| CD | 9.8 | 16.1 | 17.5 | 41.6 | – | – | |
| Crosslinked Drosophila resilin (two motifs)d | FTIR | 14.2 | 19.9 | 24.4 | 41.4 | – | – |
| CD | 9.3 | 15.7 | 17.9 | 42.7 | – | – |
Data reproduced from [229] with permission from The American Chemical Society, copyright 2009
Reported in supplementary material from Lyons et al. [229]
Sequence derived from first exon of CG15920 gene; therefore, it contains irregularities, but consensus repeat GGRPSDSYGAPGGGN
Sequence derived from complete CG15920 gene; contains two consensus repeat motifs: GGRPSDSYGAPGGGN and GYSGGRPGGQDLG [187] with permission from The American Chemical Society, copyright 2009
Perhaps the most important indicator of a successful recombinant resilin is whether its mechanical properties match those of native resilin. In most of the studies, tensile testing and either scanning probe microscopy (SPM) or AFM were utilized to investigate the mechanical properties of crosslinked gels of recombinant resilin. The SPM studies of rec1-resilin, An16, and Dros16 demonstrated that the crosslinked materials had negligible hysteresis and resilience of 97±3%, 98±4% and 91±5%, respectively. Tensile testing confirmed the resilience as well as provided elastic moduli and strain-to-break values for rec1-resilin and An16. In both cases, the recombinant polypeptides showed good extension-to-break values: up to 250±43% for rec1-resilin and 347±61% for An16. The rec1-resilin exhibited the greater modulus of the two, with a modulus at 26±9 kPa as compared to the 5.7±2 kPa for An16. The degree of crosslinking was only slightly lower in the An16 than in the rec1-resilin; however, An16 has 14.3% dityrosine content and rec1-resilin has 18.8–21% distyrosine content [226, 229].
3.4 Applications of Resilin-Like Polypeptides
Multiple applications for resilin-like polypeptides have garnered renewed research interest since the report of the first recombinant resilin in 2005. The excellent mechanical properties of the resilin-like polypeptides has directed investigation toward their use as high-performance materials and in tissue engineering applications. It is widely acknowledged that cells interact and take cues from their microenvironment and, therefore, the development of polymeric scaffolds to mimic the extracellular matrix and drive desired cell or tissue responses has been of wide interest. To this end, our laboratories have developed a modular resilin-like polypeptide (RLP12) (see Fig. 20) that contains not only twelve repeats of the putative resilin-like motif from the first exon of the Drosophila CG15920 gene, but also sequences for cell binding, proteolytic degradation, and heparin immobilization. Additionally, the tyrosine residues in the resilin motif were replaced with phenylalanine to facilitate future studies involving photochemical crosslinking using the non-natural amino acids. The well-characterized cell-binding domain from the tenth type-III domain of fibronectin, RGDSP, was included in several locations along the polypeptide chain. A matrix metalloproteinase sequence, GPQG↓IWGQ, and heparin-interacting domain, KAAKRPKAAKDKQTK, were also included to confer specific degradation and heparin immobilization properties. The heparin immobilization could potentially facilitate the sequestration of growth factors such as vascular endothelial growth factor (VEGF) or basic fibroblast growth facor (bFGF). This construct was expressed using a BL21Star(DE3)/pET28a expression system and Studier auto-induction; the protein was purified using Ni-nitrilotriacetic acid (NTA) chromatography. Structural investigations of the RLP12 using CD and Fourier transform infrared (FTIR) spectroscopy demonstrated that the recombinant protein was largely unordered, but there was evidence for a small population of β-turns [223]. The RLP12 construct has subsequently been recursively ligated to create higher molecular weight polypeptides that might, as they include multiple heparin-binding domains, facilitate the formation of noncovalent networks with heparin. Unlike the other recombinant resilins, the RLP12 is crosslinked through the Mannich-type reaction of lysine side chains with the hydroxyl residues of the THPP (β-[Tris(hydroxymethyl) phosphino] propionic acid) crosslinker. The mechanical properties of 25 wt% gels were analyzed via oscillatory rheology and tensile testing. The oscillatory rheology indicated the storage modulus was approximately 10 kPa, while the tensile testing exhibited a Young’s modulus in the 30–60 kPa range and a strain-to-break ratio of 180% [223]. Recently, more thorough analysis of the properties of the RLP12 gels revealed the expected strong dependence of the mechanical properties on the extent of crosslinking and has resulted in materials with storage moduli relevant to soft tissue engineering, but with significantly increased resilience [233].
Fig. 20.

Modular resilin-like polypeptide containing domains conferring elastomeric properties, heparin molecule interaction, cell adhesion, and matrix metalloproteinase (MMP) proteolysis. Lysine residues are encoded periodically to permit crosslinking
The modular design of the RLP, containing domains conferring both desired mechanical and biological properties, offers the opportunity to target specific tissues as well as providing a more complex material that better captures salient features of native extracellular matrix. Initial studies using mouse NIH 3T3 fibroblasts provided initial evidence for the adhesion and proliferation of cells on these novel matrices, but more rigorous cell culture studies are necessary [223]. In particular, the culturing of other cell lines such as those found in vocal fold or cardiac tissue; varying gel stiffness; and possibly the culturing of cells with mechanical stimulation would all be important studies to elucidate whether these gels can serve as tissue engineering scaffolds in mechanically demanding applications.
Recently, Qin et al. cloned and expressed the full native resilin protein of Drosophila CG15920 gene from cDNA created from RNA extracted from Drosophila melanogaster pupa [187]. In addition, the authors cloned two genes encoding only the first exon alone (resilin) and the first two exons (resilin and the chitin-binding domain) into plasmids containing histidine fusion tags for purification purposes. The purified proteins derived from these two genes were used to demonstrate the efficacy of the R&R-2 chitin-binding domain. The results demonstrated that the resilin protein containing the domain bound strongly to chitin-coated beads while the resilin lacking the sequence showed no affinity (see Fig. 21). All of the genes were expressed using IPTG induction; the full length resilin was purified via heating and salt precipitation and the His-tagged gene products were purified using Ni-NTA purification. Full-length recombinant resilin, analyzed through FTIR and CD, exhibited the greatest degree of structure, both as soluble protein and in crosslinked gels. Approximately 60% of the protein was reported to adopt either helical, strand, or turn structure [187]. The full-length resilin was also crosslinked into a gel using horseradish peroxidase, and the mechanical properties of the polypeptide were probed via AFM. These constructs demonstrated similarly high resilience to recombinant resilin and other resilin-like polypeptides [187]. Despite modest differences between the recombinant resilins, it appears that they all exist largely in a disordered state, which would be expected from an ideal isotropic rubber.
Fig. 21.

Chitin binding of 6×His-tagged resilin with chitin-binding domain (6 H-resChBD) as compared to 6×His-tagged resilin without chitin-binding domain (6 H-res). T total protein after affinity chromatography purification, B bound protein eluted from chitin beads, UB unbound protein. Reproduced from [187] with permission from The American Chemical Society, copyright 2009
Another group has looked into incorporating resilin-like sequences into a protein polymer that could be used as a biomaterial or for tissue engineering purposes. Lv et al. designed a co-block polypeptide combining resilin-like sequences with folded immunoglobulin-like domains in order to mimic the passive mechanical properties of muscle (see Fig. 22). The giant muscle protein, titin, is a complex molecular spring consisting of a series of immunoglobulin-like domains separated by largely unstructured sequences. Under high stretching forces, the immunoglobulin-like domains can unfold, dissipate energy, and prevent damage due to overstretching. The recombinant proteins produced by the authors mimicked the alternating structure of titin and, through the use of AFM techniques, they demonstrated the unfolding of immunoglobulin-like domains. Furthermore, the titin-mimetic polypeptide could be crosslinked into a hydrated biomaterial through a photochemical [Ru(bpy)3]2+ strategy. The resilin-like motifs provided the unstructured region and potential crosslinking sites, but whether the resilin served a mechanically important role is not clear [235].
Fig. 22.

Photo-crosslinked titin-mimetic hydrogel, with the circles indicating the folded immuno-globulin-like domains and the wavy lines indicating the resilin-like domains. Reproduced from [235] with permission from Nature Publishing Group, copyright 2010
The rec1-resilin protein has also seen application in other areas. The ability to control the organization of proteins to various surfaces is crucial in many biological applications and in particular, clinical diagnostics [225]. Dutta et al. demonstrated that the assembly, structure, and morphology of adsorbed rec1-resilin could be tuned by altering the physical properties of the substrate surface, specifically by altering the pH and substrate hydrophilicity [225]. Truong et al. investigated the pH-dependent adsorption of rec1-resilin onto gold substrates via SPR and a quartz crystal microbalance with dissipation monitoring (QCM-D) [230]. It was shown that the recombinant resilin adsorbs to the gold substrate with different orientations and conformations, based upon varying electrostatic interactions that are influenced by the pH. The resilin could be induced to form compact or brush-like conformations; furthermore, these changes were completely reversible and fast. The authors attributed these properties to the dual phase transition behavior of rec1-resilin. These resilin-modified surfaces, and their demonstrated pH-responsiveness, could have potential applications in nanobiotechnology as a means to control cell adhesion and migration, or for creating a functional surface for biosensors [230].
4 Conclusions
Elastin is a structural protein with outstanding properties, and therefore it has inspired many investigations, with special interest in its elastomeric properties. This behavior can be mimicked with ELPs and these have found wide application in bioengineering, biomedicine, and nanobiotechnology. It has been shown that the inverse phase transition of ELPs could be transferred to almost any type of material, such as biopolymers, small molecules, synthetic polymers, metals, and inorganic particles. A wide variety of applications for elastin-based polypeptides have been explored.
Similarly, resilin is a remarkable protein with characteristic resilience, high extensibility, and near-perfect rubber elasticity; it has demonstrated ability as an energy-storing material in numerous insect organs. Analysis of resilin gene products across an array of sources has demonstrated that although the resilin sequences can differ widely amongst insects, they maintain their useful mechanical behavior [188]. A small number of resilin-like polypeptides have been reported, with applications directed toward tissue engineering, composites, and surface modification. The development of new sequences will undoubtedly expand the toolbox and applications for biosynthetic resilins and resilin-like polypeptides.
Contributor Information
Mark B. van Eldijk, Institute for Molecules and Materials, Radboud University Nijmegen, 6525 AJ Nijmegen, The Netherlands
Christopher L. McGann, Department of Materials Science and Engineering, University of Delaware, Newark, DE 19716, USA
Kristi L. Kiick, Email: kiick@udel.edu, Department of Materials Science and Engineering, University of Delaware, Newark, DE 19716, USA.
Jan C.M. van Hest, Email: j.vanhest@science.ru.nl, Institute for Molecules and Materials, Radboud University Nijmegen, 6525 AJ Nijmegen, The Netherlands.
References
- 1.Mithieux SM, Weiss AS. Elastin. In: Parry DAD, Squire JM, editors. Fibrous proteins: coiled-coils, collagen and elastomers Advances in protein chemistry. Vol. 70. Elsevier Academic; San Diego: 2005. pp. 437–461. [Google Scholar]
- 2.Daamen WF, Veerkamp JH, van Hest JCM, van Kuppevelt TH. Biomaterials. 2007;28:4378–4398. doi: 10.1016/j.biomaterials.2007.06.025. [DOI] [PubMed] [Google Scholar]
- 3.Foster JA, Bruenger E, Gray WR, Sandberg LB. J Biol Chem. 1973;248:2876–2879. [PubMed] [Google Scholar]
- 4.Gray WR, Sandberg LB, Foster JA. Nature. 1973;246:461–466. doi: 10.1038/246461a0. [DOI] [PubMed] [Google Scholar]
- 5.Indik Z, Yeh H, Ornsteingoldstein N, Sheppard P, Anderson N, Rosenbloom JC, Peltonen L, Rosenbloom J. Proc Natl Acad Sci U S A. 1987;84:5680–5684. doi: 10.1073/pnas.84.16.5680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Z, Shin MH, Moon YJ, Lee SR, Kim YK, Seo JE, Kim JE, Kim KH, Chung JH. Exp Dermatol. 2009;18:378–386. doi: 10.1111/j.1600-0625.2008.00799.x. [DOI] [PubMed] [Google Scholar]
- 7.Sato F, Wachi H, Starcher BC, Murata H, Amano S, Tajima S, Seyama Y. Clin Biochem. 2006;39:746–753. doi: 10.1016/j.clinbiochem.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 8.Vrhovski B, Weiss AS. Eur J Biochem. 1998;258:1–18. doi: 10.1046/j.1432-1327.1998.2580001.x. [DOI] [PubMed] [Google Scholar]
- 9.Saunders NA, Grant ME. Biochem J. 1984;221:393–400. doi: 10.1042/bj2210393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Getie M, Schmelzer CEH, Neubert RHH. Proteins: Structure, Function and Bioinformatics. 2005;61:649–657. doi: 10.1002/prot.20643. [DOI] [PubMed] [Google Scholar]
- 11.Uitto J, Hoffmann HP, Prockop DJ. Arch Biochem Biophys. 1976;173:187–200. doi: 10.1016/0003-9861(76)90249-6. [DOI] [PubMed] [Google Scholar]
- 12.Hinek A, Rabinovitch M. J Cell Biol. 1994;126:563–574. doi: 10.1083/jcb.126.2.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mecham RP. Ann N Y Acad Sci. 1991;624:137–146. doi: 10.1111/j.1749-6632.1991.tb17013.x. [DOI] [PubMed] [Google Scholar]
- 14.Rodgers UR, Weiss AS. Pathologie Biologie. 2005;53:390–398. doi: 10.1016/j.patbio.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 15.Reiser K, McCormick RJ, Rucker RB. FASEB J. 1992;6:2439–2449. doi: 10.1096/fasebj.6.7.1348714. [DOI] [PubMed] [Google Scholar]
- 16.Moroy G, Ostuni A, Pepe A, Tamburro AM, Alix AJP, Hery-Huynh S. Proteins-Structure Function and Bioinformatics. 2009;76:461–476. doi: 10.1002/prot.22361. [DOI] [PubMed] [Google Scholar]
- 17.Senior RM, Griffin GL, Mecham RP, Wrenn DS, Prasad KU, Urry DW. J Cell Biol. 1984;99:870–874. doi: 10.1083/jcb.99.3.870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Urry DW, Parker TM. J Muscle Res Cell Motil. 2002;23:543–559. [PubMed] [Google Scholar]
- 19.van Hest JCM, Tirrell DA. Chem Commun. 2001;19:1897–1904. doi: 10.1039/b105185g. [DOI] [PubMed] [Google Scholar]
- 20.Vrhovski B, Jensen S, Weiss AS. Eur J Biochem. 1997;250:92–98. doi: 10.1111/j.1432-1033.1997.00092.x. [DOI] [PubMed] [Google Scholar]
- 21.Urry DW, Long MM, Cox BA, Ohnishi T, Mitchell LW, Jacobs M. Biochim Biophys Acta. 1974;371:597–602. doi: 10.1016/0005-2795(74)90057-9. [DOI] [PubMed] [Google Scholar]
- 22.Urry DW. Prog Biophys Mol Biol. 1992;57:23–57. doi: 10.1016/0079-6107(92)90003-o. [DOI] [PubMed] [Google Scholar]
- 23.Chilkoti A, Dreher MR, Meyer DE. Adv Drug Deliv Rev. 2002;54:1093–1111. doi: 10.1016/s0169-409x(02)00060-1. [DOI] [PubMed] [Google Scholar]
- 24.Urry DW. J Phys Chem B. 1997;101:11007–11028. [Google Scholar]
- 25.Urry DW, Gowda DC, Parker TM, Luan CH, Reid MC, Harris CM, Pattanaik A, Harris RD. Biopolymers. 1992;32:1243–1250. doi: 10.1002/bip.360320913. [DOI] [PubMed] [Google Scholar]
- 26.Meyer DE, Chilkoti A. Biomacromolecules. 2002;3:357–367. doi: 10.1021/bm015630n. [DOI] [PubMed] [Google Scholar]
- 27.McPherson DT, Xu J, Urry DW. Protein Expr Purif. 1996;7:51–57. doi: 10.1006/prep.1996.0008. [DOI] [PubMed] [Google Scholar]
- 28.Schipperus R, Teeuwen RLM, Werten MWT, Eggink G, de Wolf FA. Appl Microbiol Biotechnol. 2009;85:293–301. doi: 10.1007/s00253-009-2082-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sallach RE, Conticello VP, Chaikof EL. Biotechnol Prog. 2009;25:1810–1818. doi: 10.1002/btpr.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Floss DM, Mockey M, Zanello G, Brosson D, Diogon M, Frutos R, Bruel T, Rodrigues V, Garzon E, Chevaleyre C, Berri M, Salmon H, Conrad U, Dedieu L. J Biomed Biotechnol. 2010;2010:274346. doi: 10.1155/2010/274346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Floss DM, Sack M, Arcalis E, Stadlmann J, Quendler H, Rademacher T, Stoger E, Scheller J, Fischer R, Conrad U. Plant Biotechnol J. 2009;7:899–913. doi: 10.1111/j.1467-7652.2009.00452.x. [DOI] [PubMed] [Google Scholar]
- 32.Cappello J, Crissman J, Dorman M, Mikolajczak M, Textor G, Marquet M, Ferrari F. Biotechnol Prog. 1990;6:198–202. doi: 10.1021/bp00003a006. [DOI] [PubMed] [Google Scholar]
- 33.Meyer DE, Chilkoti A. Nat Biotechnol. 1999;17:1112–1115. doi: 10.1038/15100. [DOI] [PubMed] [Google Scholar]
- 34.McDaniel JR, MacKay JA, Quiroz FG, Chilkoti A. Biomacromolecules. 2010;11:944–952. doi: 10.1021/bm901387t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Christensen T, Amiram M, Dagher S, Trabbic-Carlson K, Shamji MF, Setton LA, Chilkoti A. Protein Sci. 2009;18:1377–1387. doi: 10.1002/pro.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen JP, Hoffman AS. Biomaterials. 1990;11:631–634. doi: 10.1016/0142-9612(90)90020-q. [DOI] [PubMed] [Google Scholar]
- 37.Chen JP, Yang HJ, Hoffman AS. Biomaterials. 1990;11:625–630. doi: 10.1016/0142-9612(90)90019-m. [DOI] [PubMed] [Google Scholar]
- 38.Floss DM, Schallau K, Rose-John S, Conrad U, Scheller J. Trends Biotechnol. 2010;28:37–45. doi: 10.1016/j.tibtech.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 39.Meyer DE, Trabbic-Carlson K, Chilkoti A. Biotechnol Prog. 2001;17:720–728. doi: 10.1021/bp010049o. [DOI] [PubMed] [Google Scholar]
- 40.Trabbic-Carlson K, Liu L, Kim B, Chilkoti A. Protein Sci. 2004;13:3274–3284. doi: 10.1110/ps.04931604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Banki MR, Feng L, Wood DW. Nat Methods. 2005;2:659–661. doi: 10.1038/nmeth787. [DOI] [PubMed] [Google Scholar]
- 42.Ge X, Yang DSC, Trabbic-Carlson K, Kim B, Chilkoti A, Filipe CDM. J Am Chem Soc. 2005;127:11228–11229. doi: 10.1021/ja0531125. [DOI] [PubMed] [Google Scholar]
- 43.Wu WY, Fong BA, Gilles AG, Wood DW. Recombinant protein purification by self-cleaving elastin-like polypeptide fusion tag. Curr Protoc Prot Sci. 2009;26.4:21–18. doi: 10.1002/0471140864.ps2604s58. [DOI] [PubMed] [Google Scholar]
- 44.Wu WY, Mee C, Califano F, Banki R, Wood DW. Nat Protoc. 2006;1:2257–2262. doi: 10.1038/nprot.2006.314. [DOI] [PubMed] [Google Scholar]
- 45.Hu F, Ke T, Li X, Mao PH, Jin X, Hui FL, Ma XD, Ma LX. Appl Biochem Biotechnol. 2010;160:2377–2387. doi: 10.1007/s12010-009-8850-2. [DOI] [PubMed] [Google Scholar]
- 46.Shimazu M, Mulchandani A, Chen W. Biotechnol Bioeng. 2003;81:74–79. doi: 10.1002/bit.10446. [DOI] [PubMed] [Google Scholar]
- 47.Ge X, Filipe CDM. Biomacromolecules. 2006;7:2475–2478. doi: 10.1021/bm060507n. [DOI] [PubMed] [Google Scholar]
- 48.Christensen T, Trabbic-Carlson K, Liu W, Chilkoti A. Anal Biochem. 2007;360:166–168. doi: 10.1016/j.ab.2006.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim JY, Mulchandani A, Chen W. Anal Biochem. 2003;322:251–256. doi: 10.1016/j.ab.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 50.Gao D, McBean N, Schultz JS, Yan YS, Mulchandani A, Chen WF. J Am Chem Soc. 2006;128:676–677. doi: 10.1021/ja056364e. [DOI] [PubMed] [Google Scholar]
- 51.Kim JY, Mulchandani A, Chen W. Biotechnol Bioeng. 2005;90:373–379. doi: 10.1002/bit.20451. [DOI] [PubMed] [Google Scholar]
- 52.Kim JY, O’Malley S, Mulchandani A, Chen W. Anal Chem. 2005;77:2318–2322. doi: 10.1021/ac0484326. [DOI] [PubMed] [Google Scholar]
- 53.Stiborova H, Kostal J, Mulchandani A, Chen W. Biotechnol Bioeng. 2003;82:605–611. doi: 10.1002/bit.10609. [DOI] [PubMed] [Google Scholar]
- 54.Lao UL, Mulchandani A, Chen W. J Am Chem Soc. 2006;128:14756–14757. doi: 10.1021/ja065343x. [DOI] [PubMed] [Google Scholar]
- 55.Kostal J, Mulchandani A, Chen W. Biotechnol Bioeng. 2004;85:293–297. doi: 10.1002/bit.10890. [DOI] [PubMed] [Google Scholar]
- 56.Lao UL, Kostal J, Mulchandani A, Chen W. Nat Protoc. 2007;2:1263–1268. doi: 10.1038/nprot.2007.171. [DOI] [PubMed] [Google Scholar]
- 57.Kostal J, Mulchandani A, Chen W. Macromolecules. 2001;34:2257–2261. [Google Scholar]
- 58.Kostal J, Mulchandani A, Gro KE, Chen W. Environ Sci Technol. 2003;37:4457–4462. doi: 10.1021/es034210y. [DOI] [PubMed] [Google Scholar]
- 59.Lao UL, Chen A, Matsumoto MR, Mulchandani A, Chen W. Biotechnol Bioeng. 2007;98:349–355. doi: 10.1002/bit.21478. [DOI] [PubMed] [Google Scholar]
- 60.Pillai O, Panchagnula R. Curr Opin Chem Biol. 2001;5:447–451. doi: 10.1016/s1367-5931(00)00227-1. [DOI] [PubMed] [Google Scholar]
- 61.Patri AK, Majoros IJ, Baker JR. Curr Opin Chem Biol. 2002;6:466–471. doi: 10.1016/s1367-5931(02)00347-2. [DOI] [PubMed] [Google Scholar]
- 62.Teicher BA. Curr Cancer Drug Targets. 2009;9:982–1004. doi: 10.2174/156800909790192365. [DOI] [PubMed] [Google Scholar]
- 63.Matsumura Y, Maeda H. Cancer Res. 1986;46:6387–6392. [PubMed] [Google Scholar]
- 64.Maeda H, Bharate GY, Daruwalla J. Eur J Pharm Biopharm. 2009;71:409–419. doi: 10.1016/j.ejpb.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 65.MacKay JA, Chen MN, McDaniel JR, Liu WG, Simnick AJ, Chilkoti A. Nat Mater. 2009;8:993–999. doi: 10.1038/nmat2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hauck ML, Dewhirst MW, Bigner DD, Zalutsky MR. Clin Cancer Res. 1997;3:63–70. [PubMed] [Google Scholar]
- 67.Meyer DE, Kong GA, Dewhirst MW, Zalutsky MR, Chilkoti A. Cancer Res. 2001;61:1548–1554. [PubMed] [Google Scholar]
- 68.Meyer DE, Shin BC, Kong GA, Dewhirst MW, Chilkoti A. J Control Release. 2001;74:213–224. doi: 10.1016/s0168-3659(01)00319-4. [DOI] [PubMed] [Google Scholar]
- 69.Liu W, Dreher MR, Chow DC, Zalutsky MR, Chilkoti A. J Control Release. 2006;114:184–192. doi: 10.1016/j.jconrel.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 70.Liu WE, Dreher MR, Furgeson DY, Peixoto KV, Yuan H, Zalutsky MR, Chilkoti A. J Control Release. 2006;116:170–178. doi: 10.1016/j.jconrel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 71.Dreher MR, Raucher D, Balu N, Colvin OM, Ludeman SM, Chilkoti A. J Control Release. 2003;91:31–43. doi: 10.1016/s0168-3659(03)00216-5. [DOI] [PubMed] [Google Scholar]
- 72.Furgeson DY, Dreher MR, Chilkoti A. J Control Release. 2006;110:362–369. doi: 10.1016/j.jconrel.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 73.Dreher MR, Liu WG, Michelich CR, Dewhirst MW, Chilkoti A. Cancer Res. 2007;67:4418–4424. doi: 10.1158/0008-5472.CAN-06-4444. [DOI] [PubMed] [Google Scholar]
- 74.McDaniel JR, Callahan DJ, Chilkoti A. Adv Drug Deliv Rev. 2010;62:1456–1467. doi: 10.1016/j.addr.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bidwell GL, Davis AN, Fokt I, Priebe W, Raucher D. Invest New Drugs. 2007;25:313–326. doi: 10.1007/s10637-007-9053-8. [DOI] [PubMed] [Google Scholar]
- 76.Bidwell GL, Fokt I, Priebe W, Raucher D. Biochem Pharmacol. 2007;73:620–631. doi: 10.1016/j.bcp.2006.10.028. [DOI] [PubMed] [Google Scholar]
- 77.Temsamani J, Vidal P. Drug Discov Today. 2004;9:1012–1019. doi: 10.1016/S1359-6446(04)03279-9. [DOI] [PubMed] [Google Scholar]
- 78.Bidwell GL, Davis AN, Raucher D. J Control Release. 2009;135:2–10. doi: 10.1016/j.jconrel.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 79.Massodi I, Bidwell GL, Raucher D. J Control Release. 2005;108:396–408. doi: 10.1016/j.jconrel.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 80.Bidwell GL, Raucher D. Mol Cancer Ther. 2005;4:1076–1085. doi: 10.1158/1535-7163.MCT-04-0253. [DOI] [PubMed] [Google Scholar]
- 81.Massodi I, Moktan S, Rawat A, Bidwell GL, Raucher D. Int J Cancer. 2010;126:533–544. doi: 10.1002/ijc.24725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bidwell GL, Whittom AA, Thomas E, Lyons D, Hebert MD, Raucher D. Peptides. 2010;31:834–841. doi: 10.1016/j.peptides.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bidwell GL, Raucher D. Adv Drug Deliv Rev. 2010;62:1486–1496. doi: 10.1016/j.addr.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dreher MR, Simnick AJ, Fischer K, Smith RJ, Patel A, Schmidt M, Chilkoti A. J Am Chem Soc. 2008;130:687–694. doi: 10.1021/ja0764862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Simnick AJ, Valencia CA, Liu RH, Chilkoti A. ACS Nano. 2010;4:2217–2227. doi: 10.1021/nn901732h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Torchilin VP. Pharm Res. 2007;24:1–16. doi: 10.1007/s11095-006-9132-0. [DOI] [PubMed] [Google Scholar]
- 87.Lee TAT, Cooper A, Apkarian RP, Conticello VP. Adv Mater. 2000;12:1105. [Google Scholar]
- 88.Kim W, Thevenot J, Ibarboure E, Lecommandoux S, Chaikof EL. Angew Chem Int Ed. 2010;49:4257–4260. doi: 10.1002/anie.201001356. [DOI] [PubMed] [Google Scholar]
- 89.Fujita Y, Mie M, Kobatake E. Biomaterials. 2009;30:3450–3457. doi: 10.1016/j.biomaterials.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 90.Sallach RE, Wei M, Biswas N, Conticello VP, Lecommandoux S, Dluhy RA, Chaikof EL. J Am Chem Soc. 2006;128:12014–12019. doi: 10.1021/ja0638509. [DOI] [PubMed] [Google Scholar]
- 91.Reguera J, Lagaron JM, Alonso M, Reboto V, Calvo B, Rodriguez-Cabello JC. Macromolecules. 2003;36:8470–8476. [Google Scholar]
- 92.Herrero-Vanrell R, Rincon AC, Alonso M, Reboto V, Molina-Martinez IT, Rodriguez-Cabello JC. J Control Release. 2005;102:113–122. doi: 10.1016/j.jconrel.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 93.Bessa PC, Machado R, Nurnberger S, Dopler D, Banerjee A, Cunha AM, Rodriguez-Cabello JC, Redl H, van Griensven M, Reis RL, Casal M. J Control Release. 2010;142:312–318. doi: 10.1016/j.jconrel.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 94.Betre H, Liu W, Zalutsky MR, Chilkoti A, Kraus VB, Setton LA. J Control Release. 2006;115:175–182. doi: 10.1016/j.jconrel.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 95.Shamji MF, Whitlatch L, Friedman AH, Richardson WJ, Chilkoti A, Setton LA. Spine. 2008;33:748–754. doi: 10.1097/BRS.0b013e3181695773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shamji MF, Chen J, Friedman AH, Richardson WJ, Chilkoti A, Setton LA. J Control Release. 2008;129:179–186. doi: 10.1016/j.jconrel.2008.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu WG, MacKay JA, Dreher MR, Chen MN, McDaniel JR, Simnick AJ, Callahan DJ, Zalutsky MR, Chilkoti A. J Control Release. 2010;144:2–9. doi: 10.1016/j.jconrel.2010.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Adams SB, Shamji MF, Nettles DL, Hwang P, Setton LA. J Biomed Mater Res B Appl Biomater. 2009;90B:67–74. doi: 10.1002/jbm.b.31254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Na K, Jung J, Lee J, Hyun J. Langmuir. 2010;26:11165–11169. doi: 10.1021/la1013285. [DOI] [PubMed] [Google Scholar]
- 100.Megeed Z, Haider M, Li DQ, O’Malley BW, Cappello J, Ghandehari H. J Control Release. 2004;94:433–445. doi: 10.1016/j.jconrel.2003.10.027. [DOI] [PubMed] [Google Scholar]
- 101.Hatefi A, Cappello J, Ghandehari H. Pharm Res. 2007;24:773–779. doi: 10.1007/s11095-006-9200-5. [DOI] [PubMed] [Google Scholar]
- 102.Greish K, Araki K, Li DQ, O’Malley BW, Dandu R, Frandsen J, Cappello J, Ghandehari H. Biomacromolecules. 2009;10:2183–2188. doi: 10.1021/bm900356j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gustafson J, Greish K, Frandsen J, Cappello J, Ghandehari H. J Control Release. 2009;140:256–261. doi: 10.1016/j.jconrel.2009.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Greish K, Frandsen J, Scharff S, Gustafson J, Cappello J, Li DQ, O’Malley BW, Ghandehari H. J Gene Med. 2010;12:572–579. doi: 10.1002/jgm.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gustafson JA, Price RA, Greish K, Cappello J, Ghandehari H. Mol Pharm. 2010;7:1050–1056. doi: 10.1021/mp100161u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Langer R, Vacanti JP. Science. 1993;260:920–926. doi: 10.1126/science.8493529. [DOI] [PubMed] [Google Scholar]
- 107.Nettles DL, Chilkoti A, Setton LA. Adv Drug Deliv Rev. 2010;62:1479–1485. doi: 10.1016/j.addr.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Betre H, Setton LA, Meyer DE, Chilkoti A. Biomacromolecules. 2002;3:910–916. doi: 10.1021/bm0255037. [DOI] [PubMed] [Google Scholar]
- 109.Betre H, Ong SR, Guilak F, Chilkoti A, Fermor B, Setton LA. Biomaterials. 2006;27:91–99. doi: 10.1016/j.biomaterials.2005.05.071. [DOI] [PubMed] [Google Scholar]
- 110.Haider M, Cappello J, Ghandehari H, Leong KW. Pharm Res. 2008;25:692–699. doi: 10.1007/s11095-007-9282-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.McHale MK, Setton LA, Chilkoti A. Tissue Eng. 2005;11:1768–1779. doi: 10.1089/ten.2005.11.1768. [DOI] [PubMed] [Google Scholar]
- 112.Lim DW, Nettles DL, Setton LA, Chilkoti A. Biomacromolecules. 2007;8:1463–1470. doi: 10.1021/bm061059m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lim DW, Nettles DL, Setton LA, Chilkoti A. Biomacromolecules. 2008;9:222–230. doi: 10.1021/bm7007982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Trabbic-Carlson K, Setton LA, Chilkoti A. Biomacromolecules. 2003;4:572–580. doi: 10.1021/bm025671z. [DOI] [PubMed] [Google Scholar]
- 115.Nettles DL, Kitaoka K, Hanson NA, Flahiff CM, Mata BA, Hsu EW, Chilkoti A, Setton LA. Tissue Eng Part A. 2008;14:1133–1140. doi: 10.1089/ten.tea.2007.0245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nettles DL, Haider MA, Chilkoti A, Setton LA. Tissue Eng Part A. 2010;16:11–20. doi: 10.1089/ten.tea.2009.0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Welsh ER, Tirrell DA. Biomacromolecules. 2000;1:23–30. doi: 10.1021/bm0002914. [DOI] [PubMed] [Google Scholar]
- 118.Heilshorn SC, DiZio KA, Welsh ER, Tirrell DA. Biomaterials. 2003;24:4245–4252. doi: 10.1016/s0142-9612(03)00294-1. [DOI] [PubMed] [Google Scholar]
- 119.Heilshorn SC, Liu JC, Tirrell DA. Biomacromolecules. 2005;6:318–323. doi: 10.1021/bm049627q. [DOI] [PubMed] [Google Scholar]
- 120.Di Zio K, Tirrell DA. Macromolecules. 2003;36:1553–1558. [Google Scholar]
- 121.Liu JC, Heilshorn SC, Tirrell DA. Biomacromolecules. 2004;5:497–504. doi: 10.1021/bm034340z. [DOI] [PubMed] [Google Scholar]
- 122.Liu JC, Tirrell DA. Biomacromolecules. 2008;9:2984–2988. doi: 10.1021/bm800469j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ayres L, Vos MRJ, Adams PHHM, Shklyarevskiy IO, van Hest JCM. Macromolecules. 2003;36:5967–5973. [Google Scholar]
- 124.Reiersen H, Clarke AR, Rees AR. J Mol Biol. 1998;283:255–264. doi: 10.1006/jmbi.1998.2067. [DOI] [PubMed] [Google Scholar]
- 125.Ayres L, Koch K, Adams PHHM, van Hest JCM. Macromolecules. 2005;38:1699–1704. [Google Scholar]
- 126.Fernandez-Trillo F, Dureault A, Bayley JPM, van Hest JCM, Thies JC, Michon T, Weberskirch R, Cameron NR. Macromolecules. 2007;40:6094–6099. [Google Scholar]
- 127.Fernandez-Trillo F, van Hest JCM, Thies JC, Michon T, Weberskirch R, Cameron NR. Chem Commun. 2008;2008:2230–2232. doi: 10.1039/b800266e. [DOI] [PubMed] [Google Scholar]
- 128.Fernandez-Trillo F, van Hest JCM, Thies JC, Michon T, Weberskirch R, Cameron NR. Adv Mater. 2009;21:55–59. [Google Scholar]
- 129.Roberts SK, Chilkoti A, Setton LA. Biomacromolecules. 2007;8:2618–2621. doi: 10.1021/bm0702713. [DOI] [PubMed] [Google Scholar]
- 130.Conrad RM, Grubbs RH. Angew Chem Int Ed. 2009;48:8328–8330. doi: 10.1002/anie.200903888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lemieux V, Adams PH, van Hest JCM. Chem Commun (Camb) 2010;46:3071–3073. doi: 10.1039/b923280j. [DOI] [PubMed] [Google Scholar]
- 132.Teeuwen RLM, Zuilhof H, de Wolf FA, van Hest JCM. Soft Matter. 2009;5:2261–2268. [Google Scholar]
- 133.Teeuwen RLM, van Berkel SS, van Dulmen THH, Schoffelen S, Meeuwissen SA, Zuilhof H, de Wolf FA, van Hest JCM. Chem Commun. 2009;2009:4022–4024. doi: 10.1039/b903903a. [DOI] [PubMed] [Google Scholar]
- 134.Schoffelen S, van Eldijk MB, Rooijakkers B, Raijmakers R, Heck AJR, van Hest JCM. Chem Sci. 2011;2:701–705. [Google Scholar]
- 135.Nath N, Chilkoti A. J Am Chem Soc. 2001;123:8197–8202. doi: 10.1021/ja015585r. [DOI] [PubMed] [Google Scholar]
- 136.Hyun J, Lee WK, Nath N, Chilkoti A, Zauscher S. J Am Chem Soc. 2004;126:7330–7335. doi: 10.1021/ja049721e. [DOI] [PubMed] [Google Scholar]
- 137.Huang HC, Koria P, Parker SM, Selby L, Megeed Z, Rege K. Langmuir. 2008;24:14139–14144. doi: 10.1021/la802842k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Alvarez-Rodriguez R, Alonso M, Girotti A, Reboto V, Rodriguez-Cabello JC. Eur Polym J. 2010;46:643–650. [Google Scholar]
- 139.Alvarez-Rodriguez R, Arias FJ, Santos M, Testera AM, Rodriguez-Cabello JC. Macromol Rapid Commun. 2010;31:568–573. doi: 10.1002/marc.200900727. [DOI] [PubMed] [Google Scholar]
- 140.Bennet-Clark HC. J Exp Biol. 2007;210:3879–3881. doi: 10.1242/jeb.001339. [DOI] [PubMed] [Google Scholar]
- 141.Weis-Fogh T. J Exp Biol. 1960;37:889–907. [Google Scholar]
- 142.Moore J, Overhill R. An introduction to the invertebrates. Cambridge University Press; Cambridge: 2001. [Google Scholar]
- 143.Anderesen SO, Weis-Fogh T. In: Advances in insect physiology. Beament J, Treherne JE, Wigglesworth VB, editors. Vol. 2. Academic; London: 1964. pp. 1–65. [Google Scholar]
- 144.Andersen SO. In: Elastomeric proteins: structures, biomechanical properties, and biological roles. Shewry PR, Tatham AS, Bailey AJ, editors. Cambridge University Press; Cambridge: 2003. pp. 259–278. [Google Scholar]
- 145.Bailey K, Weis-Fogh T. Biochim Biophys Acta. 1961;48:452. doi: 10.1016/0006-3002(61)90043-9. [DOI] [PubMed] [Google Scholar]
- 146.Jensen M, Weis-Fogh T. Philos Trans R Soc Lond B Biol Sci. 1962;245:137. [Google Scholar]
- 147.Bennet-Clark HC. J Exp Biol. 1963;40:223–229. [Google Scholar]
- 148.Rice MJ. Nature. 1970;228:1337–1338. doi: 10.1038/2281337a0. [DOI] [PubMed] [Google Scholar]
- 149.Edwards HA. J Exp Biol. 1983;105:407. [Google Scholar]
- 150.Hermann HR, Willer DE. Int J Insect Morphol Embryol. 1986;15:107–114. [Google Scholar]
- 151.Young D, Bennet-Clark HC. J Exp Biol. 1995;198:1001–1020. doi: 10.1242/jeb.198.4.1001. [DOI] [PubMed] [Google Scholar]
- 152.Skals N, Surlykke A. J Exp Biol. 1999;202:2937–2949. doi: 10.1242/jeb.202.21.2937. [DOI] [PubMed] [Google Scholar]
- 153.Scott JA. Pan-Pac Entomol. 1970;46:225. [Google Scholar]
- 154.Nahirney PC, Forbes JG, Morris HD, Chock SC, Wang K. FASEB J. 2006;20:2017–2026. doi: 10.1096/fj.06-5991com. [DOI] [PubMed] [Google Scholar]
- 155.Govindarajan S, Rajulu GS. Experientia. 1974;30:908–909. [Google Scholar]
- 156.Fonseca PJ, Bennet-Clark HC. J Exp Biol. 1998;201:717–730. [Google Scholar]
- 157.Bennet-Clark HC. J Exp Biol. 1997;200:1681–1694. doi: 10.1242/jeb.200.11.1681. [DOI] [PubMed] [Google Scholar]
- 158.Gorb SN. Naturwissenschaften. 1999;86:552–555. doi: 10.1007/s001140050674. [DOI] [PubMed] [Google Scholar]
- 159.Haas F, Gorb S, Blickhan R. Proc R Soc Lond B Biol Sci. 2000;267:1375–1381. doi: 10.1098/rspb.2000.1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Haas F, Gorb S, Wootton RJ. Arthropod Struct Dev. 2000;29:137–146. doi: 10.1016/s1467-8039(00)00025-6. [DOI] [PubMed] [Google Scholar]
- 161.Frazier SF, Larsen GS, Neff D, Quimby L, Carney M, DiCaprio RA, Zill SN. J Comp Physiol A. 1999;185:157–172. [Google Scholar]
- 162.Neff D, Frazier SF, Quimby L, Wang RT, Zill S. Arthropod Struct Dev. 2000;29:75–83. doi: 10.1016/s1467-8039(00)00014-1. [DOI] [PubMed] [Google Scholar]
- 163.Niederegger S, Gorb S. J Insect Physiol. 2003;49:611–620. doi: 10.1016/s0022-1910(03)00048-9. [DOI] [PubMed] [Google Scholar]
- 164.Gorb SN. J Morphol. 1996;230:219–230. doi: 10.1002/(SICI)1097-4687(199611)230:2<219::AID-JMOR8>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 165.Federle W, Brainerd EL, McMahon TA, Holldobler B. Proc Natl Acad Sci U S A. 2001;98:6215–6220. doi: 10.1073/pnas.111139298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Rothschild M, Schlein Y, Parker K, Neville C, Sternber S. Sci Am. 1973;229:92. [Google Scholar]
- 167.Rothschild M, Schlein Y, Sternber S, Parker K. Nature. 1972;239:45. [Google Scholar]
- 168.Burrows M. J Exp Biol. 2009;212:2881–2883. doi: 10.1242/jeb.022855. [DOI] [PubMed] [Google Scholar]
- 169.Bennet-Clark HC, Lucey ECA. J Exp Biol. 1967;47:59–76. doi: 10.1242/jeb.47.1.59. [DOI] [PubMed] [Google Scholar]
- 170.Rothschild M, Schlein J. Philos Trans R Soc Lond B Biol Sci. 1975;271:457. doi: 10.1098/rstb.1975.0062. [DOI] [PubMed] [Google Scholar]
- 171.Rothschild M, Schlein J, Parker K, Neville C, Sternberg S. Philos Trans R Soc Lond B Biol Sci. 1975;271:499. doi: 10.1098/rstb.1975.0064. [DOI] [PubMed] [Google Scholar]
- 172.Bennet-Clark HC. J Exp Biol. 1975;63:53–83. doi: 10.1242/jeb.63.1.53. [DOI] [PubMed] [Google Scholar]
- 173.Burrows M, Shaw SR, Sutton GP. BMC Biol. 2008;6:16. doi: 10.1186/1741-7007-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Gorb SN. Arthropod Struct Dev. 2004;33:201–220. doi: 10.1016/j.asd.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 175.Burrows M. J Exp Biol. 2010;213:469–478. doi: 10.1242/jeb.037861. [DOI] [PubMed] [Google Scholar]
- 176.Alexander RM, Bennet-Clark HC. Nature. 1977;265:114–117. doi: 10.1038/265114a0. [DOI] [PubMed] [Google Scholar]
- 177.Varman AR. Experientia. 1980;36:564. [Google Scholar]
- 178.Varman AR. Journal of the Georgia Entomological Society. 1981;16:11–13. [Google Scholar]
- 179.Sannasi A. Experientia. 1970;26:154. doi: 10.1007/BF01895549. [DOI] [PubMed] [Google Scholar]
- 180.Burrows M. BMC Biol. 2009;7:17. doi: 10.1186/1741-7007-7-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lombardi EC, Kaplan DL. Mater Res Soc Symp Proc. 1993;292:3–7. [Google Scholar]
- 182.Ardell DH, Andersen SO. Insect Biochem Mol Biol. 2001;31:965–970. doi: 10.1016/s0965-1748(01)00044-3. [DOI] [PubMed] [Google Scholar]
- 183.Nielsen H, Engelbrecht J, Brunak S, von Heijne G. Protein Eng. 1997;10:1–6. doi: 10.1093/protein/10.1.1. [DOI] [PubMed] [Google Scholar]
- 184.Rebers JE, Riddiford LM. J Mol Biol. 1988;203:411–423. doi: 10.1016/0022-2836(88)90009-5. [DOI] [PubMed] [Google Scholar]
- 185.Rebers JE, Willis JH. Insect Biochem Mol Biol. 2001;31:1083–1093. doi: 10.1016/s0965-1748(01)00056-x. [DOI] [PubMed] [Google Scholar]
- 186.Andersen SO, Hojrup P, Roepstorff P. Insect Biochem Mol Biol. 1995;25:153–176. doi: 10.1016/0965-1748(94)00052-j. [DOI] [PubMed] [Google Scholar]
- 187.Qin GK, Lapidot S, Numata K, Hu X, Meirovitch S, Dekel M, Podoler I, Shoseyov O, Kaplan DL. Biomacromolecules. 2009;10:3227–3234. doi: 10.1021/bm900735g. [DOI] [PubMed] [Google Scholar]
- 188.Andersen SO. Insect Biochem Mol Biol. 2010;40:541–551. doi: 10.1016/j.ibmb.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 189.Andersen SO. Biochim Biophys Acta. 1963;69:249. doi: 10.1016/0006-3002(63)91258-7. [DOI] [PubMed] [Google Scholar]
- 190.Andersen SO. Acta Chem Scand. 1963;17:869. [Google Scholar]
- 191.Andersen SO, Kristens B. Acta Physiol Scand. 1963;59(suppl 213):15. [Google Scholar]
- 192.Andersen SO. Biochim Biophys Acta. 1964;93:213–215. doi: 10.1016/0304-4165(64)90289-2. [DOI] [PubMed] [Google Scholar]
- 193.Weis-Fogh T. J Mol Biol. 1961;3:648. [Google Scholar]
- 194.Andersen SO. Acta Physiol Scand. 1966;66(suppl 263):1–81. [PubMed] [Google Scholar]
- 195.Andersen SO. Federation Proc. 1966;25:715. [Google Scholar]
- 196.LaBella F, Keeley F, Vivian S, Thornhil D. Biochem Biophys Res Commun. 1967;26:748. doi: 10.1016/s0006-291x(67)80137-2. [DOI] [PubMed] [Google Scholar]
- 197.LaBella F, Waykole P, Queen G. Biochem Biophys Res Commun. 1968;30:333–338. doi: 10.1016/0006-291x(68)90746-8. [DOI] [PubMed] [Google Scholar]
- 198.Raven DJ, Earland C, Little M. Biochim Biophys Acta. 1971;251:96–99. doi: 10.1016/0005-2795(71)90065-1. [DOI] [PubMed] [Google Scholar]
- 199.Devore DP, Gruebel RJ. Biochem Biophys Res Commun. 1978;80:993–999. doi: 10.1016/0006-291x(78)91343-8. [DOI] [PubMed] [Google Scholar]
- 200.Garciacastineiras S, Dillon J, Spector A. Science. 1978;199:897–899. doi: 10.1126/science.622574. [DOI] [PubMed] [Google Scholar]
- 201.Nomura K, Suzuki N, Matsumoto S. Biochemistry. 1990;29:4525–4534. doi: 10.1021/bi00471a004. [DOI] [PubMed] [Google Scholar]
- 202.Gross AJ, Sizer IW. J Biol Chem. 1959;234:1611–1614. [PubMed] [Google Scholar]
- 203.Aeschbach R, Amadoò R, Neukom H. Biochim Biophys Acta Protein Structure. 1976;439:292–301. doi: 10.1016/0005-2795(76)90064-7. [DOI] [PubMed] [Google Scholar]
- 204.Kristensen BI. J Insect Physiol. 1966;12:173–176. [Google Scholar]
- 205.Kristensen BI. J Insect Physiol. 1968;14:1135–1138. [Google Scholar]
- 206.Neville AC. J Insect Physiol. 1963;9:265–272. [Google Scholar]
- 207.Coles GC. J Insect Physiol. 1966;12:679. doi: 10.1016/0022-1910(66)90114-4. [DOI] [PubMed] [Google Scholar]
- 208.Gosline J, Lillie M, Carrington E, Guerette P, Ortle C, Savage K. Philos Trans R Soc Lond B Biol Sci. 2002;357:121–132. doi: 10.1098/rstb.2001.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Amsden B. Soft Matter. 2007;3:1335–1348. doi: 10.1039/b707472g. [DOI] [PubMed] [Google Scholar]
- 210.Brostow W. In: Physical properties of polymers handbook. Mark JE, editor. Springer; New York: 2007. pp. 443–444. [Google Scholar]
- 211.Weis-Fogh T. J Mol Biol. 1961;3:520. [Google Scholar]
- 212.Rubinstein M, Colby RH. Polymer physics. Oxford University Press; Oxford: 2006. [Google Scholar]
- 213.Elliott GF, Huxley AF, Weis-Fogh T. J Mol Biol. 1965;13:791. [Google Scholar]
- 214.Kim W, Conticello VP. Polym Rev. 2007;47:93–119. [Google Scholar]
- 215.Tatham AS, Shewry PR. Philos Trans R Soc Lond B Biol Sci. 2002;357:229–234. doi: 10.1098/rstb.2001.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Urry DW, Hugel T, Seitz M, Gaub HE, Sheiba L, Dea J, Xu J, Parker T. Philos Trans R Soc Lond B Biol Sci. 2002;357:169–184. doi: 10.1098/rstb.2001.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Hayashi CY, Lewis RV. J Mol Biol. 1998;275:773–784. doi: 10.1006/jmbi.1997.1478. [DOI] [PubMed] [Google Scholar]
- 218.Hayashi CY, Lewis RV. Bioessays. 2001;23:750–756. doi: 10.1002/bies.1105. [DOI] [PubMed] [Google Scholar]
- 219.Nairn KM, Lyons RE, Mulder RJ, Mudie ST, Cookson DJ, Lesieur E, Kim M, Lau D, Scholes FH, Elvin CM. Biophys J. 2008;95:3358–3365. doi: 10.1529/biophysj.107.119107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Tamburro AM, Panariello S, Santopietro V, Bracalello A, Bochicchio B, Pepe A. Chembiochem. 2010;11:83–93. doi: 10.1002/cbic.200900460. [DOI] [PubMed] [Google Scholar]
- 221.Rauscher S, Baud S, Miao M, Keeley FW, Pomes R. Structure. 2006;14:1667–1676. doi: 10.1016/j.str.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 222.Kiick KL. Polym Rev. 2007;47:1–7. [Google Scholar]
- 223.Charati MB, Ifkovits JL, Burdick JA, Linhardt JG, Kiick KL. Soft Matter. 2009;5:3412–3416. doi: 10.1039/b910980c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Dudek DM, Gosline JM, Michal CA, Depew TA, Elvin C, Kim M, Lyons R, Dumsday G. Integr Comp Biol. 2009;49:E50. [Google Scholar]
- 225.Dutta NK, Choudhury NR, Truong MY, Kim M, Elvin CM, Hill AJ. Biomaterials. 2009;30:4868–4876. doi: 10.1016/j.biomaterials.2009.06.019. [DOI] [PubMed] [Google Scholar]
- 226.Elvin CM, Carr AG, Huson MG, Maxwell JM, Pearson RD, Vuocolo T, Liyou NE, Wong DCC, Merritt DJ, Dixon NE. Nature. 2005;437:999–1002. doi: 10.1038/nature04085. [DOI] [PubMed] [Google Scholar]
- 227.Kim M, Elvin C, Brownlee A, Lyons R. Protein Expr Purif. 2007;52:230–236. doi: 10.1016/j.pep.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 228.Lyons RE, Lesieur E, Kim M, Wong DCC, Huson MG, Nairn KM, Brownlee AG, Pearson RD, Elvin CM. Protein Eng Des Sel. 2007;20:25–32. doi: 10.1093/protein/gzl050. [DOI] [PubMed] [Google Scholar]
- 229.Lyons RE, Nairn KM, Huson MG, Kim M, Dumsday G, Elvin CM. Biomacromolecules. 2009;10:3009–3014. doi: 10.1021/bm900601h. [DOI] [PubMed] [Google Scholar]
- 230.Truong MY, Dutta NK, Choudhury NR, Kim M, Elvin CM, Hill AJ, Thierry B, Vasilev K. Biomaterials. 2010;31:4434–4446. doi: 10.1016/j.biomaterials.2010.02.019. [DOI] [PubMed] [Google Scholar]
- 231.Studier FW. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 232.Tamburro AM, Bochicchio B, Pepe A. Biochemistry. 2003;42:13347–13362. doi: 10.1021/bi034837t. [DOI] [PubMed] [Google Scholar]
- 233.Li L, Teller S, Clifton RJ, Jia X, Kiick KL. Biomacromolecules. 2011;12:2302–2310. doi: 10.1021/bm200373p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Chow DC, Dreher K, Trabbic-Carlson, Chilkoti A. Biotechnol Prog. 2006;22:638–646. doi: 10.1021/bp0503742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Lv S, Dudek DM, Cao Y, Balamurali MM, Gosline J, Li HB. Nature. 2010;465:69–73. doi: 10.1038/nature09024. [DOI] [PubMed] [Google Scholar]