

Abstract
Human immunodeficiency virus type 1 reverse transcriptase (HIV-1 RT) is a major target for currently approved anti-HIV drugs. These drugs are divided into two classes: nucleoside and non-nucleoside reverse transcriptase inhibitors (NRTIs and NNRTIs). This study illustrates the synthesis and biochemical evaluation of a novel bifunctional RT inhibitor utilizing d4T (NRTI) and a TMC-derivative (a diarylpyrimidine NNRTI) linked via a poly(ethylene glycol) (PEG) linker. HIV-1 RT successfully incorporates the triphosphate of d4T-4PEG-TMC bifunctional inhibitor in a base-specific manner. Moreover, this inhibitor demonstrates low nanomolar potency that has 4.3-fold and 4300-fold enhancement of polymerization inhibition in vitro relative to the parent TMC-derivative and d4T, respectively. This study serves as a proof-of-concept for the development and optimization of bifunctional RT inhibitors as potent inhibitors of HIV-1 viral replication.

INTRODUCTION
The human immunodeficiency virus (HIV) epidemic still remains a major public health concern since more than 30 million people around the world continue to live with HIV and 2.7 million people become newly infected with HIV each year.1 There have been fundamental improvements in the treatment of HIV infection through highly active antiretroviral therapy (HAART), leading to significant reductions in HIV-1 related morbidity and mortality.2 HAART is a combination of several antiretroviral drugs from different drug classes, yet eradication of HIV has not been possible due to the rapid emergence of drug-resistant HIV variants.3 The majority of Food and Drug Administration (FDA) approved antiretroviral drugs target HIV-1 reverse transcriptase (RT), the viral polymerase essential for HIV-1 replication, and these drugs are classified into two distinct classes: nucleoside reverse transcriptase inhibitors (NRTIs) and non-nucleoside reverse transcriptase inhibitors (NNRTIs). HIV-1 RT possesses a high mutation rate due to its low fidelity caused by the lack of an intrinsic exonucleolytic proofreading activity, therefore influencing the development of rapid drug resistance against these inhibitors.4 In addition to high mutation rate, multiple other factors may also give rise to the high level of drug resistance, including the enormous replication and recombination rates of HIV-1.5–9 As a result, there is a continued need for novel anti-HIV drugs.
NRTIs are nucleoside analogue prodrugs that are converted intracellularly into their respective triphosphate forms and incorporated into the growing viral DNA chain by HIV-1 RT. Consequently, these nucleotide analogues cause chain termination due to the lack of a 3′-hydroxyl (3′-OH) group in the sugar moiety that is necessary for incorporation of the next incoming nucleotide by RT.10 During this process, NRTIs compete with their natural counterparts, deoxyribonucleoside triphosphates (dNTPs), for binding to the polymerase active site of RT in order to be incorporated into viral DNA. Unfortunately, these competitive inhibitors could also act as substrates for host polymerases like human mitochondrial DNA polymerase γ (pol γ), which is responsible for replication and repair of mitochondrial DNA,11,12 as well as DNA polymerase β (pol β) that is responsible for nuclear DNA repair in vivo.13 Previous studies illustrate NRTI-induced mitochondrial toxicity that causes serious adverse effects ranging from lactic acidosis to hepatic steatosis during long-term HAART treatment.14,15 On the other hand, NNRTIs bind to an allosteric hydrophobic site, approximately 10 Å away from the polymerase active site, which is unique to HIV-1 RT and absent in host cell polymerases.16 Therefore, unlike NRTIs, NNRTIs specifically target RT while excluding any binding interaction with other polymerases. In this paper, we utilized the high specificity of NNRTIs as an anchor to design bifunctional inhibitors consisting of an NRTI unit that is joined to an NNRTI moiety via a noncleavable linker with the goal of achieving complementary action at each distinct target site.
The concept of joining the two drugs via a linker was originally proposed by Smerdon et al.,17 based on observation of the close proximity of the respective NRTI and NNRTI binding sites. Further rationale includes mechanistic studies establishing that the two sites could be simultaneously occupied18,19 and studies on the mechanism of synergistic inhibition of RT by NRTIs and NNRTIs.20–22 Earlier work on developing chimeric NRTI/NNRTI inhibitors with a non-cleavable linker showed limited success where there has been no evidence for a pronounced synergistic antiviral effect in cell-based assays.23–25 For interpretation of these results, one important factor to be considered is the prerequisite phosphorylation of the nucleoside portion of the bifunctional inhibitor into the pharmacologically active triphosphate form. Therefore, it is unclear whether the observed lack of activity enhancement compared to the NNRTI alone in previous reports was due to lack of phosphorylation by cellular kinases or lack of binding affinity to RT. In the present study, we test this issue directly by preparing the triphosphate form of bifunctional chimeric NRTI/NNRTI and examining the interaction with HIV-1 RT at the biochemical level.
Among the key issues in the design of a chimeric NRTI/ NNRTI joined by a flexible tether are (1) selection of optimal NRTI and NNRTIs, (2) linkage points for the NRTI and NNRTI, and (3) linker length and composition. We have designed chimeric NRTI-linker-NNRTI bifunctional inhibitors aided by computer modeling and the available structural information.26,27
In the design of the bifunctional NRTI-linker-NNRTI, 2′,3′-didehydrothymidine (d4T, Figure 1) was chosen as the NRTI, on the basis of our mechanistic studies showing that d4T triphosphate (d4TTP) binds to HIV-1 RT with comparable binding affinity to the natural substrate.28 More importantly, d4T continues to play a major role especially in resource-limited settings, although d4T-induced mitochondrial toxicity may cause serious health risks for HIV-positive patients after chronic administration.29 In selecting a point of attachment for the linker on the d4T, substitution at the C5 position of the thymine ring was chosen due to an anticipated low interference with base pairing, in addition to the established evidence showing that addition of various linker entities at this position does not interfere with antiviral activity.24,30–32 In previous work, we described our initial efforts toward the design and synthesis of novel bifunctional RT inhibitors utilizing tetrahydroimidazobenzodiazepinone (TIBO), an early-generation NNRTI with nanomolar antiviral potency, as the NNRTI portion attached to d4T via a PEG linker.33 Although we demonstrated the incorporation of the triphosphate form of an N3-substituted d4T-6PEG-TIBO bifunctional compound, the use of 8-Cl-TIBO as a pharmacophore for our bifunctional inhibitor was less than ideal since the addition of a 6PEG alcohol at the 8-position substantially diminished the antiviral activity.33
Figure 1.
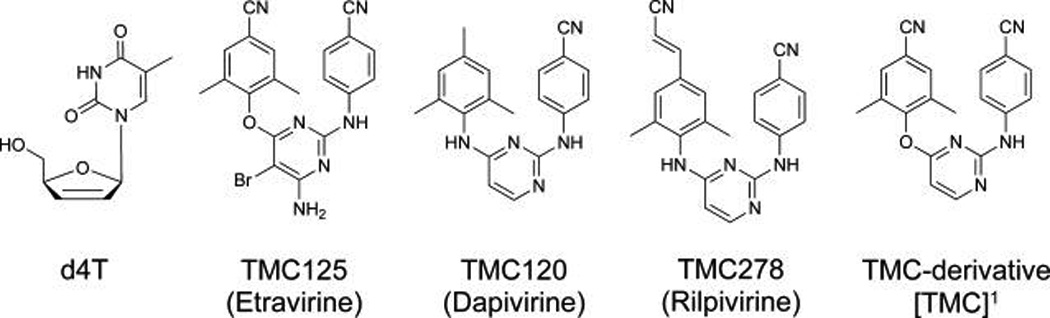
Structures of d4T and various TMC-derivatives.1 For simplicity, we refer to [TMC-derivative] as TMC in text.
Our results from functionalizing 8-Cl-TIBO lead us toward utilizing new flexible NNRTIs for the bifunctional inhibitor, concomitant with the paradigm shift in NNRTI design. Not only is the concept of flexibility important for overcoming antiviral resistance mutations, but also one would anticipate that this would allow the inhibitor to better accommodate the addition of a linker moiety. Therefore, we selected the diarylpyrimidine (DAPY) series because of their conformational flexibility and high potency against wild type (WT) and various NNRTI-resistant HIV-1 viruses that resulted in two FDA-approved drugs (Figure 1); TMC125 and TMC278.34 DAPY compounds present a common binding mode termed as “butterfly-like” conformation, with the structural components defined as wing I, wing II, and a body/linker.27 To ease synthetic complexity for our bifunctional inhibitor, the bromine and amino functionalities on the pyrimidine ring of TMC125 were omitted, rendering a TMC120-like compound that incorporates several aspects of TMC125. Our TMC-derivative includes an ether linkage from the central pyrimidine ring to wing II and a cyano group at the para position as shown in Figure 1. With the aid of computer modeling, we have used the available three-dimensional structures of the catalytic RT/ DNA/dNTP complex along with the RT/TMC inhibitor complexes to guide our selection of the most feasible NRTI and NNRTI linker positions as well as to estimate the optimal linker length between the two binding sites (Figure 2).27 Selection of the NNRTI attachment point to the linker and the route from the NNRTI-binding pocket (NBP) to the polymerization site poses a challenge, since the opening of the NBP points away from the nucleotide-binding site.
Figure 2.
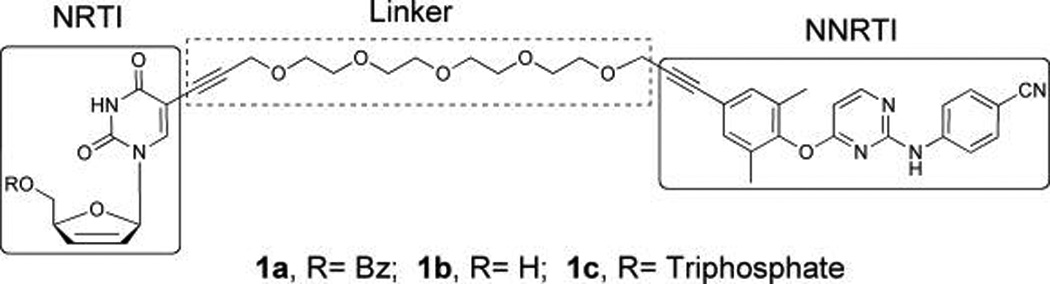
Structures of [d4T-4PEG-TMC] bifunctional inhibitor derivatives.
Modeling and structural information of RT/TMC inhibitor complexes have demonstrated the importance of the two aryl substituents on the pyrimidine ring, often referred to as wings I and II, and their contact with key hydrophobic residues in the NBP.26,27,35 Especially, the cyanodimethylphenoxy ring (wing I) of TMC125 interacts with Y181, Y188, and the conserved W229 residues. The recently approved TMC278 demonstrates that the long cyanovinyl functionality at the para position of wing I maintains low nanomolar potency and even improves the antiviral activity against mutant strains.36 Therefore, a plausible possibility for NNRTI linker attachment would involve a linker attached to the para position of the dimethylphenoxy ring of TMC125 leading out of the pocket via alkynyl attachment in the region of the W229 and F227 interactions. The flexibility of the residues lining the NBP is an important feature in regard to linker attachment. Upon NNRTI binding, the side chains of Y181 and Y188 flip from being pointed toward the entrance of the putative binding pocket nearly 180° from their initial position, to point toward the polymerization active site, forming the NBP.37 The pocket itself is formed upon NNRTI binding, and the pocket is not seen in structures of RT without NNRTI bound. We predicted, on the basis of our molecular model and aforementioned literature, that this flexibility of Y181, Y188, and W229 could accommodate a long linker and allow an NRTI-linker-NNRTI bifunctional inhibitor to span the NBP and the active site (Figure 3A,B). The feasibility of this position on the TMC scaffold was further validated by the crystal structure of the HIV-1 RT/TMC278 complex, which demonstrates that the pcyanovinyl arm of wing I of TMC278 protrudes into a hydrophobic tunnel in the pocket that opens to the active site (Figure 3C).35 This study focuses on the development of novel bifunctional compounds utilizing a TMC scaffold as the NNRTI portion, which is coupled to a d4T moiety with a PEG linker (Figure 2). We have utilized an integrated approach combining organic synthesis, molecular modeling, and in vitro enzyme inhibition assays in order to realize our goal of identifying a bifunctional inhibitor that demonstrates binding at both sites simultaneously with a concomitant increase in potency.
Figure 3.
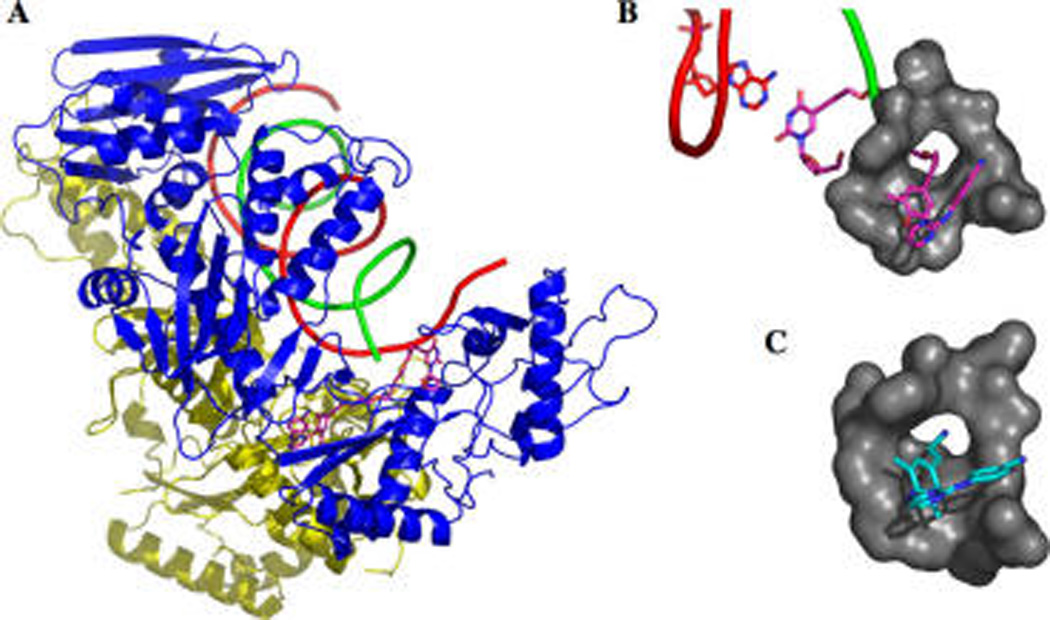
Molecular modeling predicts binding of d4T-4PEG-TMC. (A) Molecular modeling suggests a 4PEG linker would be long enough to span the NNRTI binding pocket and the active site. (B) The PEG linker is predicted to extend from the NNRTI binding pocket toward the active site through a hydrophobic tunnel, similar to the tunnel identified in the TMC278 bound structure (pdb ID 2ZD1).35 This would allow proper base-pairing of the NRTI portion with the templating A. (C) TMC278 bound structure.
RESULTS AND DISCUSSION
A PEG linker was chosen for the bifunctional inhibitor design, in view of optimal water solubility while retaining significant organic lipophilicity and flexibility. Molecular modeling using the X-ray structures of RT/TMC12027 and the ternary structure of RT38 together suggested a four-PEG spacer was optimal to bridge both binding sites and allow proper base pairing at the active site (Figure 3B). In addition, we recognize the importance of linker addition and the attachment point to the NNRTI entity from our previous study with the TIBO bifunctional series with decreased antiviral activities compare to 8-Cl-TIBO.33 Alignment of the RT/8-Cl-TIBO structure39 with the RT/TMC278 structure35 shows that the chlorine atom on the benzene ring of 8-Cl-TIBO projects into the base of the hydrophobic tunnel to some extent, where the cyanovinyl substituent of wing I of TMC278 binds in a subpocket generated by residues F227 and W229. Although the functionalization at the 8-Cl position could point toward the active site of the enzyme, our results demonstrated that placing a PEG tether at this position severely impairs potency by 2000-fold.33 This result implies that either the 8-Cl-TIBO lacks flexibility or the neighboring residues in this region are perhaps less flexible than the residues lining the hydrophobic tunnel accepting the cyanovinyl substituent of TMC278. Therefore, the present study focused on utilizing d4T-4PEG-TMC as our bifunctional prototype. Previous studies on C-5-substituted d4T derivatives yielded mixed results but mostly resulted in inactive analogues.24,25,40–43 Moreover, earlier attempts to show efficient HIV replication inhibition of bifunctional compounds all failed.44,45 We hypothesized that one of the possible reasons for these unsuccessful attempts could be impaired phosphorylation of the nucleoside end of bifunctional analogues catalyzed by cellular kinases. Consequently, the synthesis of a metabolically active triphosphate form of our d4T-4PEG-TMC bifunctional nucleoside was conducted to directly address the phosphorylation requirement for in vitro incorporation assays catalyzed by HIV-1 RT.
The synthesis of 5′-benzoyl-d4T-4PEG-TMC (1a, Figure 2) began with targeting quantities of the known pyrimidine derivative 2,46 which underwent nucleophilic substitution in high yield to 3 with 4-iodo-2,6-dimethylphenol by use of cesium carbonate as base (Scheme 1). Subsequent Sonogashira coupling with the monopropargyl ether of 4-PEG-diol47 gave tethered TMC-derivative 4, followed by hydroxyl group propargylation to give 5, which was subjected to a further Sonogashira coupling with 5′-benzoyl-5-iodo-d4U48 to afford bifunctional benzoate (1a) in around 60% isolated yield. Deprotection of 1a with sodium methoxide, quenching with acetic acid, and direct silica-gel flash chromatography without a workup afforded bifunctional nucleoside, d4T-4PEG-TMC (1b). Transformation to its triphosphate derivative (1c) proved to be problematic in that direct phosphorylation by Yoshikawa’s procedure49 with POCl3 and trimethyl phosphate failed to give the desired product. Convergent cross-coupling of the 5’-triphosphate of 5-iodo-d4U as a tetra(triethylammonium) salt with alkyne 5 via a homogeneous50 Sonogashira reaction in N,N-dimethylformamids (DMF) appeared to produce the product by thin-layer chromatography (TLC), but separation from the copper reagent proved to be problematic. Finally, the bifunctional nucleoside (1b) was successfully converted into d4TTP-4PEG-TMC (1c) via the procedure of Ludwig and Eckstein.51 The product was purified by Sephadex (diethylaminoethyl, DEAE) ion-exchange chromatography with aqueous triethylammonium bicarbonate (TEAB, 1 M)/H2O as the mobile phase to afford 1c as a film. All final compounds for in vitro testing—TMC-derivative (Figure 1), the propargyl-4PEG-TMC-derivative (5), and d4TTP-4PEG-TMC (1c)— were obtained in moderate yields, and conformation of structure and purity were provided by NMR spectroscopy (1H, 13C, and 31P) as well as HPLC and high-resolution mass spectrometry (HRMS), in preparation for the inhibition assays.
Scheme 1. Synthesis of d4TTP-4PEG-TMC Bifunctional Derivativea.
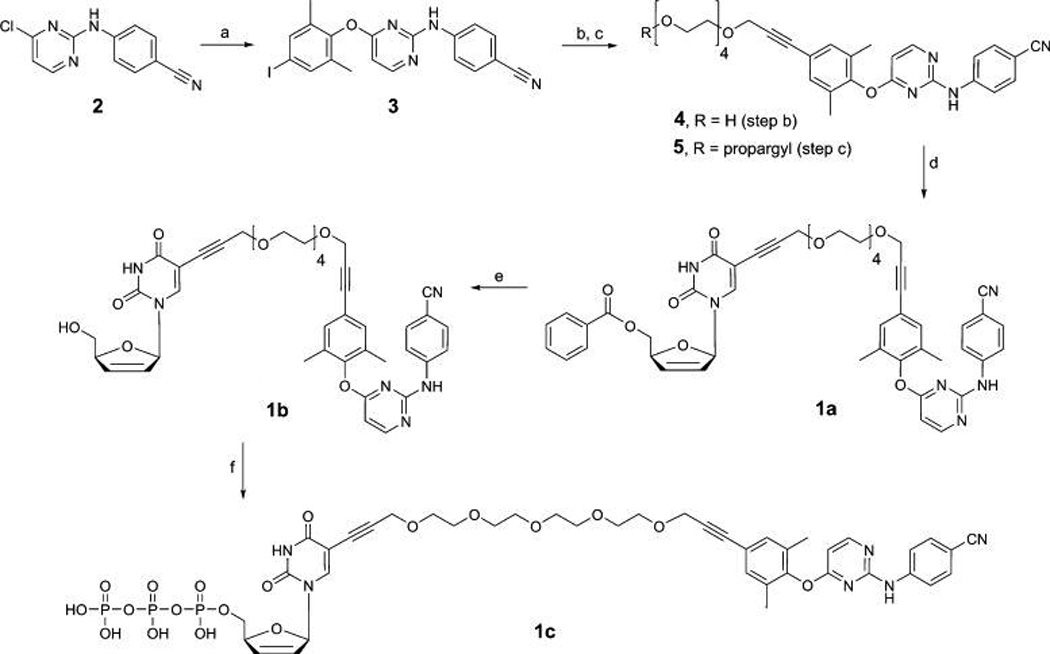
aReagents and conditions: (a) 4-Iodo-2,6-dimethylphenol, Cs2CO3, DMF, Δ, 85%. (b) Monopropargyl ether of 4-PEG diol (or n-PEG diol), Pd(PPh3)4 (10 mol %), CuI (50 mol %), THF, DMF, 85%. (c) (i) TsCl, NEt3, CH2Cl2; (ii) propargyl alcohol, NaH, THF, Δ, 92%. (d) 5′-Benzoyl-5-iodo-d4U, Pd(PPh3)4 (10 mol %), CuI (50 mol %), THF, DMF, 59%. (e) NaOMe, MeOH, 94%. (f) (i) 2-Chloro-1,3,2-benzodioxaphosphorin-4-one, pyr, DMF, 0 °C to rt; (ii) bis(tributylammonium) pyrophosphate; (iii) I2; (iv) 1 M Et3NH+ HCO2−, 37%.
Initially, we examined the ability of d4TTP-4PEG-TMC to serve as a substrate for incorporation by HIV-1 RT. Incorporation assays under single-turnover conditions demonstrated 100% conversion of the D23 substrate to an elongated primer chain terminated with the bifunctional monophosphate, indicated as a D23-d4TMP-4PEG-TMC product band that migrates significantly higher than the D23 primer substrate or a D24-mer elongated by one dNTP in Figure 4A. Control experiments lacking magnesium chloride were also performed, demonstrating that formation of the elongated product required divalent metal ions. We next sought to address whether the linker location at the C-5 position on d4TTP would affect base paring. In other words, was incorporation of d4TTP-4PEG-TMC base-specific? For this purpose, another primer/template (P/T) for G incorporation (D24/D36) was utilized, which failed to produce an elongated primer opposite a template dCMP as shown in lane 3 (Figure 4B). The negative control experiment lacking magnesium chloride was repeated for D24/D36 P/T (lane 2), and no elongation was observed as well. These findings illustrate the requirement of Mg2+ as a divalent metal ion and, more importantly, the base-specific incorporation of the bifunctional molecule possessing a thymine nucleobase when utilizing D23/D36 but not D24/ D36 primer/template containing a guanine nucleobase. Moreover, as mentioned above, the altered mobility of the D23-d4TMP-4PEG-TMC band that is higher in distance compared to a standard mobility distance of a single nucleotide elongated product band strongly supports the incorporation of the large bifunctional compound.
Figure 4.
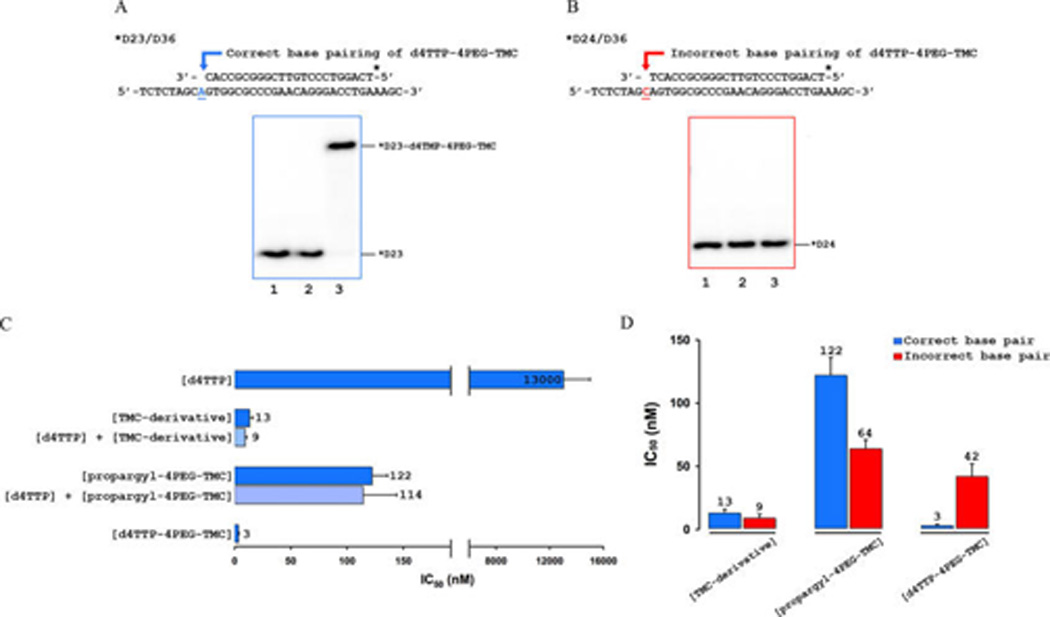
Incorporation and potent HIV-1 RT inhibition of d4TTP-4PEG-TMC is base-specific. (A) Sequence of D23/D36 P/T for correct base pairing used in biochemical assays. d4TTP-4PEG-TMC serves as a substrate for HIV-1 RT-catalyzed polymerization when it is used as a primer for dTTP incorporation. Lane 1 represents the 0 time point, and lane 2 serves as a negative control lacking Mg2+. Lane 3 represents the incorporated bifunctional triphosphate (indicated as *D23-d4TMP-4PEG-TMC) product band by HIV-1 RT. (B) Sequence of D24/D36 P/T for incorrect base pairing used in biochemical assays. d4TTP-4PEG-TMC does not serve as a substrate for HIV-1 RT when it is used as a primer for dGTP incorporation. Lane 1 represents the 0 time point, and lane 2 serves as a negative control lacking Mg2+. Lane 3 shows the lack of an incorporated bifunctional triphosphate product band. (C) Inhibition of dTTP incorporation is shown in bar graphs for d4TTP (13 000 nM), TMC-derivative (13 nM), propargyl-4PEG-TMC (122 nM), and d4TTP-4PEG-TMC (3 nM). Additionally, the calculated IC50 values for 1:1 mixtures of d4TTP/TMC-derivative and d4TTP/propargyl-4PEG-TMC are 9 nM and 114 nM, respectively. IC50 values were determined in triplicate and averaged. (D) Inhibition of dTTP incorporation (correct base pairing) is shown in blue; inhibition of dGTP incorporation (incorrect base pairing) is shown in red. Whereas d4TTP-4PEG-TMC is very potent with proper base pairing (3 nM), its ability to inhibit dGTP incorporation is only slightly more potent (42 nM) than the propargyl-4PEG-TMC (64 nM). Results for TMC-derivative and propargyl-4PEG-TMC are similar for inhibition of dTTP incorporation or dGTP incorporation. Potent RT inhibition of d4TTP-4PEG-TMC is therefore base-specific.
After illustrating that d4TTP-4PEG-TMC bound to the active site and served as a substrate for incorporation by HIV-1 RT, we evaluated the inhibition activity of the bifunctional inhibitor at the biochemical level via a steady-state competition assay to determine its potency (Figure 4C). The IC50 values were determined in triplicate and averaged for in vitro inhibition of RT for TMC-derivative, TMC-4PEG-propargyl, and triphosphate of the bifunctional nucleoside. The parent TMC-derivative had an IC50 value of 13 ± 3 nM. Elaboration of the TMC-derivative with a 4-PEG linker, to give the NNRTI tether propargyl-4PEG-TMC, results in a 10-fold decrease in potency (122 ± 14 nM). Analysis of the metabolically active d4TTP-4PEG-TMC exhibited a low nanomolar inhibition (3 ± 1 nM). This enhancement is approximately 4.3-fold improvement relative to the TMC-derivative, a 40-fold improvement over propargyl-4PEG-TMC, and a 4300-fold improvement relative to d4TTP (13 000 ± 2000 nM). True binding affinity may in fact be higher, as this value may represent stoichiometric binding to the enzyme (as our experimental setup utilized 6 nM active sites of HIV-1 RT). Additional experiments demonstrated that 1:1 mixtures of d4TTP/TMC-derivative (9 ± 1.5 nM) and d4TTP/propargyl-4PEG-TMC (114 ± 30 nM) showed no significant change in inhibition compared to the NNRTI or NNRTI tether component alone (Figure 4C), implying that having both components physically attached as one molecule is essential for the increase in potency.
After demonstrating that proper base pairing is essential for incorporation of the NRTI component of our bifunctional triphosphate, we hypothesized that the increased potency of d4TTP-4PEG-TMC was linked to proper base pairing at the active site. Therefore, we performed a similar set of biochemical experiments to explore this by simply changing the nucleotide and primer/template used in the assay. We would predict that since the bifunctional nucleoside triphosphate was not incorporated opposite a templating C, it would be less potent in inhibiting dGTP incorporation due to the diminished binding interactions at the active site. Under these conditions, the IC50 values of the TMC-derivative (9 ± 3 nM) and propargyl-4PEG-TMC (64 ± 7 nM) were similar to results for inhibition of dTTP incorporation, as expected since these inhibitors are not predicted to interact with the active site (Figure 4D). However, when the bifunctional triphosphate would not be properly base-paired for incorporation at the active site, the polymerase inhibition potency of d4TTP-4PEG-TMC was significantly reduced (42 ± 10 nM), indicating that the potent inhibition seen during dTTP incorporation is linked to the proper base pairing of the NRTI moiety. Our findings suggest that simultaneous occupation of the NNRTI binding pocket and the active site is occurring to yield the high level of potency with d4TTP-4PEG-TMC bifunctional inhibitor.
CONCLUSIONS AND FUTURE DIRECTIONS
Although we have shown that the nucleoside triphosphate moiety of the bifunctional can bind at the active site and be incorporated, further work is required to prove simultaneous binding at both sites. One potential avenue to establish the binding of the inhibitor will be through obtaining an X-ray crystal structure of the bifunctional triphosphate bound to HIV-1 RT. Crystallization trials are currently underway in an attempt to determine structures of d4TTP-4PEG-TMC bound to HIV-1 RT in the presence and absence of primer/template. Further evidence suggesting interaction at both the active site and the NNRTI binding pocket could be obtained by determining the effect of NRTI resistance mutations (Q151M) or NNRTI resistance mutations (Y181C) on the activity of d4TTP-4PEG-TMC in a steady-state inhibition assay.
We believe this study offers a major advance toward establishing proof-of-concept and the feasibility of designing bifunctional RT inhibitors with a binding mode that confirms that the NRTI (d4T) moiety is binding in the dNTP pocket in a base-specific manner and with improved potency relative to the parent NRTI and NNRTI moieties. Building on these results, further optimization studies are in progress to more fully exploit the potential additive binding energies of the NRTI and NNRTI. Additional work is in progress to prepare various lengths of PEG linker for the bifunctional compounds in nucleoside form, as well as their prodrug versions to obtain optimal potency at the cellular level utilizing HIV-1 infected human T-cells. The triphosphates of these derivatives will also be synthesized and evaluated via similar biochemical experiments. Since intracellular phosphorylation may be a potential barrier for these bifunctional RT inhibitors, we are currently exploring an alternative approach for delivery involving the encapsulation of the bifunctional inhibitor triphosphates in biodegradable nanogels. These nanogel formulations have been successfully used in cancer chemotherapy as drug delivery systems for cytotoxic nucleoside analogues.52,53 Another avenue that we are exploring is using nucleotide-competing RT inhibitors (NcRTIs)54 instead of NRTIs in our bifunctional inhibitor designs, since NcRTIs are distinct from the chain-terminator NRTIs and do not require any phosphorylation by cellular kinases.
EXPERIMENTAL SECTION
Chemistry
Unless stated otherwise, reactions were performed in oven-dried glassware under a nitrogen atmosphere (house nitrogen, dried with Drierite and KOH) with dry, deoxygenated solvents (distilled or passed over a column of activated alumina). 2-Thiouracil and 4-aminobenzonitrile were purchased from Sigma-Aldrich Chemical Co. and used as received. The key starting material, 4-(4-chloropyrimidin-2-ylamino)benzonitrile (2), and the TMC-derivative, 4-[4-(4-iodo-2,6-dimethylphenoxy)pyrimidin-2-ylamino]benzonitrile, that we used in our bifunctional compounds were synthesized according to literature procedures.46,55,56 Sephadex ion-exchange (DEAE) adsorbent was purchased from Sigma, while 4-iodo-2,6-dimethylphenol,57 2-(2-{2-[2-(prop-2-ynyloxy)ethoxy]ethoxy}-ethoxy)ethanol,47 and 5-iodo-5′-benzoyl-d4U48,58 were also prepared according to literature procedures. All reactions were magnetically stirred and monitored by thin-layer chromatography. TLC was performed on E. Merck silica-gel 60 F254 precoated plates (0.25 mm) or RediSep amine-functionalized silica gel containing a 254 nm fluorescent indicator for triphosphate synthesis and visualized by UV fluorescence quenching or Hanessian staining. Except for triphosphate synthesis, traditional flash chromatography and a CombiFlash Companion system were used for all of the separations with normal-phase silica gel utilizing repackable columns with 35–60 µm average particle size (230–400 mesh). The triphosphate synthesis purification was carried out on DEAE-Sephadex with increasing percentages of aqueous TEAB (1 M, triethylammonium bicarbonate)/H2O mixtures as the mobile phase. TLC for the latter utilized TEAB/MeOH = 6:4 on RediSep amine-functionalized plates. All synthesized final compounds were determined to be ≥95% pure by LC-MS. Each compound identity was verified by 1H NMR, 13C NMR, and HRMS, and additionally by 31P NMR for 1c. 1H and 13C NMR spectra were recorded on a Bruker Avance DPX (at 500 and 400 MHz, respectively) in solvents CDCl3 and MeOH-d4, with the resonances for CDCl3 taken as δ 7.24 ppm for 1H and δ 77.0 ppm for 13C, and for MeOH-d4 as δ 3.30 ppm for 1H and δ 49.0 ppm for 13C to serve as internal references (Cambridge Isotope Laboratories, Inc.). The data for 1H NMR spectra are reported as follows: chemical shift in parts per million, ppm (multiplicity, coupling constant in hertz, integration). Those for 13C NMR spectra are reported in terms of chemical shifts to one significant figure relative to the solvent references as described above. High-resolution mass spectra (ESI/MS) were obtained from the Keck Biotechnology Resource Laboratory, Yale University.
4-[4-(4-Iodo-2,6-dimethylphenoxy(pyrimidin-2-ylamino]-benzonitrile (3)
A mixture of 4-(4-chloropyrimidin-2-ylamino)-benzonitrile46 (2) (600.0 mg, 2.60 mmol), 4-iodo-2,6-dimethylphenol57 (838.0 mg, 3.38 mmol), and cesium carbonate (2.20 g, 6.76 mmol) in dry DMF (12 mL) was heated to 90 °C.59 After 3 h, TLC (EtOAc/hexane = 50:50) revealed a more polar product and consumption of compound 2. The mixture was poured into water (100 mL) and the product was extracted into EtOAc (3 × 50 mL). The combined extracts were washed with brine (50 mL), followed by water (2 × 50 mL), and then dried over Na2SO4. The solvent was evaporated in vacuo to furnish a residue that was subjected to silica-gel column chromatography with EtOAc/hexane (50:50) as eluent to produce 3 as a white solid (982.0 mg, 2.22 mmol, 85%). 1H NMR (CDCl3, 500 MHz) δ (ppm) 8.31 (d, J = 6.0 Hz, 1H), 7.48 (s, 2H), 7.42 (d, J = 8.5 Hz, 1H), 7.36 (d, J = 8.5 Hz, 1H), 7.31 (s, 1H, NH), 6.47 (d, J = 6.0 Hz, 1H), 2.06 (s, 6H). 13C NMR (CDCl3, 500 MHz) δ (ppm) 168.9, 159.7, 159.1, 149.9, 143.3, 137.5, 133.4, 133.1, 119.3, 118.1, 104.7, 99.5, 89.9, 16.0. HRMS (EI+) m/z calcd for C19H15IN4O [M + H]+ 443.0363, found 443.0357.
4-(4-{4-[3-(2-{2-[2-(2-Hydroxyethoxy(ethoxy]ethoxy}ethoxy(prop-1-ynyl]-2,6-dimethylphenoxy}pyrimidin-2-ylamino(benzonitrile (4)
To a deoxygenated solution of iodopyrimidine (3) (494 mg, 1.12 mmol) and 2-(2-{2-[2-(prop-2-ynyloxy)ethoxy]ethoxy}ethoxy)-ethanol47 (780 mg, 3.36 mmol) in dry DMF (1.5 mL) and dry THF (3.0 mL) were added successively dry triethylamine (562.0 mg, 5.55 mmol, 0.80 mL), copper(I) iodide (105.7 mg, 0.56 mmol), and tetrakis(triphenylphosphine)palladium(0) (128.3 mg, 0.11 mmol). The reaction mixture was stirred at 25 °C under a nitrogen atmosphere for 3 h, after which TLC (DCM/MeOH = 3:97) revealed a more polar product and consumption of starting material 3. The mixture was poured into ethylenediaminetetraacetic acid (EDTA) solution (20 mL, 5%) and the organic product was extracted into EtOAc (3 × 50 mL). The combined organic extracts were washed with brine (20 mL) and dried over Na2SO4. The solvent was evaporated in vacuo to furnish a residue that was subjected to silica-gel column chromatography with DCM/MeOH (95:5) as eluent to produce 4 as a light-brown oil (522.0 mg, 0.956 mmol, 85%). 1H NMR (CD3OD, 400 MHz) δ (ppm) 8.37 (d, J = 5.6 Hz, 1H), 7.44 (d, J = 9.2 Hz, 2H), 7.35 (d, J = 9.2 Hz, 2H), 7.27 (s, 2H), 6.60 (d, J = 5.6 Hz, 1H), 4.46 (s, 2H), 3.71-3.79 (m, 4H), 3.53–3.66 (m, 12H), 2.08 (s, 6H). 13C NMR (CDCl3, 500 MHz) δ (ppm) 169.1, 159.6, 159.3, 150.0, 143.4, 133.0, 132.1, 131.2, 120.3, 119.3, 118.1, 104.6, 99.5, 85.6, 85.1, 72.5, 70.7, 70.6, 70.6, 70.5, 70.4, 69.2, 61.8, 59.2, 16.3. HRMS (EI+) m/z calcd for C30H34N4O6 [M + H]+ 547.2551, found 547.2528.
4-[4-(4-{3-[2-(2-{2-[2-(Prop-2-ynyloxy)ethoxy]ethoxy}ethoxy)-ethoxy]prop-1-ynyl}-2,6-dimethylphenoxy)pyrimidin-2-ylamino]-benzonitrile (5)
Dry triethylamine (278 mg, 2.75 mmol, 0.4 mL) followed by p-toluenesulfonyl chloride (349.0 mg, 1.83 mmol) and 4-dimethylaminopyridine (11.2 mg, 0.009 mmol) were added to alcohol 4 (500.0 mg, 0.92 mmol) dissolved in dry dichloromethane (5 mL). The reaction was left stirring for 3 h at room temperature, after which time TLC (DCM/MeOH = 5:95) revealed a less polar product and consumption of starting material 4. The mixture was poured into saturated aqueous sodium bicarbonate (20 mL) and extracted with dichloromethane (3 × 50 mL). The combined organic extracts were washed with brine (20 mL) and then dried (Na2SO4). Evaporation of the solvent in vacuo gave a light brown oil (658.0 mg), which was taken to the next step directly by adding it to a solution of propargyl alcohol (526 mg, 9.39 mmol, 0.54 mL) dissolved in dry THF (5 mL) to which sodium hydride (375 mg, 60%, 9.39 mmol) had been added. The reaction mixture was heated to 60 °C for 3 h, after which time TLC (DCM/MeOH = 5:95) indicated the formation of a more polar product and consumption of the intermediate. The mixture was poured into saturated aqueous sodium bicarbonate (20 mL) and extracted with EtOAc (3 × 50 mL), and the combined organic extracts were washed with brine (20 mL) and then dried (Na2SO4). Evaporation of the solvent in vacuo gave a residue that was subjected to column chromatography on silica gel with DCM/MeOH mixtures to furnish 5 as a light-yellow oil (493.0 mg, 0.84 mmol, 92% for two steps). 1H NMR (CDCl3, 500 MHz) δ (ppm) 8.28 (d, J = 6.0 Hz, 1H), 8.05 (s, 1H, NH), 7.35 (m, 4H), 7.18 (s, 2H), 6.44 (d, J = 6.0 Hz, 1H), 4.38 (s, 2H), 4.11 (d, J = 2.5 Hz, 2H), 3.72-3.67 (m, 4H), 3.66-3.58 (m, 12H), 2.37 (t, J = 2.5 Hz, 1H), 2.03 (s, 6H). 13C NMR (CDCl3, 500 MHz) δ (ppm), 168.9, 159.4, 159.1, 149.9, 143.5, 132.8, 131.9, 131.1, 120.0, 119.2, 118.1, 104.2, 99.2, 85.5, 84.9, 79.5, 74.4, 70.5, 70.4, 70.4, 70.4, 70.3, 70.2, 69.1, 68.9, 59.0, 58.2, 16.1. HRMS (EI+) m/z calcd for C33H36N4O6Na [M + Na]+ 607.2527, found 607.2500.
Benzoyl-d4U-propynyl-4-PEG-propynyl-TMC (1a)
(For simplicity, we refer to this structure as 5′-benzoyl-d4T-4PEG-TMC in text, while formally it might best be considered as a uridine rather than thymidine derivative.) A mixture of (tetrakis)triphenylphosphinepalladium(0) (30.0 mg, 0.026 mmol) and copper(I) iodide (30.0 mg, 0.16 mmol) was added rapidly to a solution of 5-iodo-5′-benzoyl-d4U58 (110 mg, 0.25 mmol) and alkyne 5 (99.0 mg, 0.17 mmol) in a degassed mixture of THF and DMF (6 mL, 2:1) at room temperature under nitrogen. TLC indicated reaction to a more polar product to be complete after 3 h with complete consumption of alkyne. Workup involved adding EDTA solution (20 mL, 5%) and extracting the product into EtOAc (3 × 50 mL). Drying (Na2SO4) and evaporation of solvent, followed by column chromatography of the residue on silica gel with EtOAc/ hexane mixtures as eluent, furnished the bifunctional compound 1a (91.0 mg, 59%). 1H NMR (CDCl3, 500 MHz) δ (ppm) 11.82 (1H, s), 8.95 (1H, s), 8.42 (1H, d, J = 5.7 Hz), 7.99 (2H, m), 7.67 (1H, s), 7.56 (1H, m), 7.43 (m, 2H), 7.32 (m, 4H), 7.22 (s, 2H), 6.94 (m, 1H), 6.47 (d, J = 5.7 Hz, 1H), 6.39 (m, 1H), 5.96 (m, 1H), 5.19 (m, 1H), 4.67 (dd, J = 4.5, 10.0 Hz, 1H), 4.49 (dd, J = 3.0, 10.0 Hz, 1H), 4.43 (s, 2H), 4.20 (s, 2H), 3.58-3.80 (m, 16H), 2.06 (s, 6H). 13C NMR (CDCl3, 500 MHz) δ (ppm) 169.2, 166.2, 162.2, 159.0, 158.9, 150.3, 150.1, 143.6, 142.5, 133.7, 133.4, 132.8, 132.1, 131.2, 129.7, 129.1, 128.7, 126.8, 120.2, 119.5, 118.2, 104.2, 100.2, 99.0, 90.6, 90.1, 85.6, 85.2, 85.1, 77.0, 70.6, 70.5, 70.5, 70.5, 70.4, 70.3, 69.2, 69.2, 65.0, 59.2, 59.0, 16.2. HRMS (EI+) m/z calcd for C49H48N6O11 [M + H]+ 897.3453, found 897.3464.
d4U-Propynyl-4-PEG-propynyl-TMC (1b)
(For simplicity, we refer to this structure as d4T-4PEG-TMC in text; see note above.) To a solution of benzoate 1a (38.0 mg, 0.042 mmol) dissolved in methanol (3 mL) at 0 °C was added a solution of sodium methoxide in methanol (0.5 M, 0.1 mL). After 2 h, the solution was allowed to warm to room temperature. Once TLC indicated the complete consumption of starting material, acetic acid was added (3.0 mg, 0.05 mmol) and methanol was removed on the rotoevaporator. The residue was immediately chromatographed on silica gel with EtOAc/MeOH mixtures to obtain nucleoside 1b (31.5 mg, 94%) as a colorless oil. 1H NMR (CDCl3, 500 MHz), δ (ppm) 11.54 (br s, 1H), 8.88 (br s, 1H), 8.38 (d, J = 5.2 Hz, 1H), 8.34 (s, 1H), 7.33 (m, 4H), 7.22 (s, 2H), 6.98 (m, 1H), 6.47 (d, J = 5.2 Hz, 1H), 6.34 (d, J = 5.8 Hz, 1H), 5.82 (d, J = 5.8 Hz, 1H), 4.94 (m, 1H), 4.44 (s, 2H), 4.32 (s, 2H), 3.94 (dd, J = 3.0, 11.2 Hz, 1H), 3.84 (dd, J = 2.0, 11.2 Hz, 1H), 3.60–3.80 (m, 16H), 3.48 (br s, 1H, OH), 2.06 (s, 6H). 13C NMR (CDCl3, 500 MHz) δ (ppm) 169.2, 162.4, 159.1, 158.9, 150.5, 150.2, 145.2, 143.6, 135.0, 132.8, 132.1, 131.3, 126.0, 120.1, 119.5, 118.3, 104.2, 99.4, 99.0, 90.1, 89.1, 87.7, 85.7, 85.0, 77.9, 70.5, 70.5, 70.5, 70.4, 70.4, 70.3, 69.2, 69.0, 62.7, 59.2, 59.2, 16.3. HRMS (EI+) m/z calcd for C42H44N6O10 [M + H]+ 793.3191, found 793.3204.
d4U-Propynyl-4-PEG-propynyl-TMC-triphosphate (1c)
(For simplicity, we refer to this structure as d4TTP-4PEG-TMC in text.) The bifunctional nucleoside 1b (95.0 mg, 0.120 mmol) was dissolved in a 1:1 mixture of DMF/pyridine (1.0 mL) and the flask was cooled to −20 °C. 2-Chloro-1,3,2-benzodioxaphosphorin-4-one51 (30.0 mg, 0.148 mmol) dissolved in THF (0.5 mL) was added slowly, and the reaction was left to warm to room temperature. After 60 min, bis(tributylammonium) pyrophosphate (92.0 mg, 0.168 mmol) in DMF (0.5 mL) was added, followed by triethylamine (0.5 mL), and the solution was left stirring for 1 h. I2 (43.0 mg, 0.170 mmol) dissolved in pyridine/water (2 mL, 98:2) was then added, and the solution was left stirring for 15 min before being quenched by aqueous Na2S2O3 (0.5 M, 0.5 mL). TEAB (1 M, 3.0 mL) was immediately added and the solution was left stirring for 2 h at room temperature. The solution was then evaporated to dryness before being chromatographed on a Sephadex ion-exchange (DEAE) column (3.0 g) with aqueous TEAB as the mobile phase. The bifunctional triphosphate eluted at around 0.6 M. Fractions identified from TLC (on amine-impregnated silica gel plates with TEAB/MeOH = 3.5/6.5) were combined and the solvent was evaporated. Following several additions of methanol with pumping, tris(tiethylammonium) triphosphate 1c (60.0 mg, 0.045 mmol, 37%) was obtained as a colorless oil. 1H NMR (CD3OD, 500 MHz) δ (ppm) 8.32 (m, 1H), 7.78 (s, 1H), 7.43 (m, 2H), 7.33 (m, 2H), 7.23 (s, 2H), 6.85 (m, 1H), 6.55 (m, 2H), 5.88 (m, 1H), 5.02 (m, 1H), 4.42 (s, 2H), 4.32 (s, 2H), 4.12 (m, 2H), 3.54– 3.74 (m, 16H), 3.13 (br s, 18H), 2.04 (s, 6H), 1.25 (br s, 27H). 13C NMR (CD3OD, 75 MHz) δ (ppm) 170. 4, 164.1, 161.2, 160.8, 151.8, 151.6, 145.9, 145.4, 136.6, 133.7, 133.1, 132.8, 126.5, 121.5, 120.4, 119.6, 104.6, 100.5, 100.1, 91.8, 90.9, 87.5, 86.6, 86.0, 78.4, 71.5, 71.5, 71.5, 71.5, 71.5, 71.4, 70.2, 70.2, 68.4, 59.8, 59.7, 47.2, 16.4, 9.1. 31P NMR (CD3OD, 202.4 MHz) δ (ppm) −8.9 (d, J = 20.2 Hz), −9.8 (d, J = 20.2 Hz), −22.1 (t, J = 20.2 Hz). HRMS (EI+) m/z calcd for tetraphosphoric acid C42H46N6O19P3 [M − H]+ 1031.2036, found 1031.2035.
High-Pressure Liquid Chromatography of d4TTP-4PEG-TMC (1c)
d4TTP-4PEG-TMC was further analyzed by HPLC on a DNA Pac PA-100 analytical column (Dionex, Sunnyvale, CA) under the following conditions: mobile phase A, 0.05 M triethylammonium bicarbonate (TEAB), pH 8.0; mobile phase B, 0.5 M TEAB, pH 8.0. Mobile phase flow rate was 1 mL/min with a gradient of 100% A to 50% A/50% B for 10 min and then to 100% B for 5 min, followed by 100% B for 10 min. One Gaussian peak was seen with a retention time of 20 min (Figure S1, Supporting Information). Absorbance was measured at 260 nm.
Biology
Expression and Purification of HIV-1 RT
C-Terminal histidine-tagged heterodimeric p66/p51 wild-type HIV-1 RT was expressed and purified as described previously60 from a clone generously provided by Stephen Hughes, Paul Boyer, and Andrea Ferris (Frederick Cancer Research and Development Center, Frederick, MD).
Nucleoside Triphosphates
dTTP and dGTP were purchased from GE Biosciences. d4TTP was purchased from Moravek Biochemicals (Brea, CA). Concentrated stocks were diluted and concentrations were verified spectrophotometrically by determining absorbance at 260 nM by use of known extinction coefficients for each nucleotide.
Labeling and Annealing of Oligonucleotides
Primers and templates used for incorporation and removal studies were synthesized at the Keck Facility at Yale University and purified by 20% polyacrylamide denaturing gel electrophoresis. The sequences of primers and templates used in this study are as follows: D23 (5′-TCA GGT CCC TGT TCG GGC GCC AC-3′), D24 (5′-TCA GGT CCC TGT TCG GGC GCC ACT-3′), and D36 (5′-TCT CTA GCA GTG GCG CCC GAA CAG GGA CCT GAA AGC-3′). The primers D23 and D24 were 5′-32P-labeled with T4 polynucleotide kinase (New England Biolabs) as previously described.61 [γ-32P]ATP was purchased from GE Healthcare. Bio-Spin columns for the removal of excess [γ-32P]ATP were purchased from Bio-Rad. Annealing of the primers, D23 and D24, and template D36 were carried out by mixing a 1:1.4 molar ratio of purified primer/template at 90 °C for 5 min, 50 °C for 10 min, and 0 °C for 10 min. The annealed primer and template were then analyzed by 15% nondenaturing polyacrylamide gel electrophoresis to ensure complete annealing. Concentrations of the oligonucleotides were determined by UV absorbance at 260 nm by use of calculated extinction coefficients.
Incorporation Assays
Incorporation experiments were performed under single-turnover conditions: 50 µM d4TTP-4PEG-TMC and 10 mM MgCl2 were mixed with 250 nM RT (active sites) and 50 nM 5′-labeled primer/template to initiate the reaction. Negative controls were performed under identical conditions without MgCl2. The reaction was allowed to proceed for 30 min at 37 °C, after which the reaction was quenched with 0.3 M EDTA. Reaction products were subjected to 20% denaturing polyacrylamide gel electrophoresis and analyzed on a Bio-Rad Molecular Imager FX.
IC50 Determination
RT (6 nM active sites, based on pre-steady-state active-site determination) was preincubated for at least 15 min with 1 µM 5′-labeled primer/template prior to mixing with appropriate concentrations of inhibitor and allowed to incubate for a minimum of 15 additional minutes on ice. DMSO concentrations were kept constant at less than 2%. DMSO alone was added as a no-inhibitor control for each set of experiments. Reactions were initiated with the addition of 5 µM dNTP and 10 mM MgCl2 and were quenched after 15 min at 37 °C with 0.3 M EDTA. All concentrations represent final concentrations after mixing. Reaction products were subjected to 20% denaturing polyacrylamide gel electrophoresis and quantitated on a Bio-Rad Molecular Imager FX. Product formation was plotted as a function of inhibitor concentration and fitted to a hyperbola to generate IC50 curves. IC50 values are defined as the concentration of inhibitor that inhibits steady-state single nucleotide incorporation by 50%.
Molecular Modeling of [d4T-4PEG-TMC] Bound to HIV-1 RT/Template/Primer
Creation of the molecular model of the bifunctional nucleoside bound to the binary complex of RT/template/primer was based on a composite of structural data. Since there is no crystal structure of RT complexed with both a NRTI and a NNRTI, a model was created by starting from the crystal structures of the RT template/ primer complex from Huang et al. (PDB entry 1rtd) and the Das et al. complex of TMC120-R147681 NNRTI (PDB entry 1s6q). First, both of these files were read into the UCSF Chimera program62 and superimposed by use of the Matchmaker utility. A composite protein was then created by combining residues 1a−92a, 108a−178a, 241a− 554a, and 3b−249b, template, primer, and four Mg2+ ions from 1rtd and residues 93a−107a and 179a−240a and the ligand from 1s6q.
The initial 3D structure of the bifunctional ligand bf4 was created by drawing it in ChemDraw (version 6.0, CambridgeSoft Corp., Cambridge, MA, 2000), followed by energy minimization using the MM2 force field in Chem3D (version 5.0, CambridgeSoft Corp., Cambridge, MA, 2000) and writing as a PDB file. After reading this structure in Chimera, the nucleotide end of this molecule was superimposed on the dNTP of the composite protein created above, and a few of the dihedrals on the poly(ethylene glycol) linker were manually changed to g+ or g− to superimpose as best as possible the NNRTI end on the TMC120 crystallographic ligand. The side-chain torsions χ1 and χ2 of Y188a were adjusted by −30° and +30°, respectively, to avoid a severe steric clash. Finally, the crystallographic dNTP and TMC120 were then removed and the resulting complex was exported as a PDB file.
The creation of the model was completed by reading the PDB files created by Chimera in Schrödinger’s Maestro 7.5 (version 7.5, Schrödinger, LLC, New York, NY, 2006) and adding the hydrogen atoms needed at protonation states appropriate to pH = 7. The following set of energy minimizations were sequentially run with the Impact program (version 4.0, Schrödinger LLC, New York, 2006) using the OPLS_2001 force field with a distance-dependent dielectric ε = 4r, a 12 Å cutoff for nonbonded interactions, and the steepest-descent algorithm. First, the newly created ligand and the segments of the protein chains originally from the 1s6q structure (93a−107a and 179a−240a) as well as all the residues within 4 Å of those were allowed to move, while the rest were kept fixed at their original positions during a 100-step optimization during which the total energy of the system decreased from 6.1 × 1014 to 927.0 kcal/mol. The resulting structure was checked for consistency, and a second run of 300 steps was restarted form this point using the same degrees of freedom, during which the energy decreased further to −187.2 kcal/ mol. The entire system was then allowed to relax for successive optimizations of 100 and 300 steps, which reduced the total energy from 8490.5 to −3398.0 kcal/mol. The Cα rms deviation of the final structure to the initial 1rtd is only 0.27 Å, which shows that there was relatively little distortion created by the process.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH Grant GM49551 to K.S.A., NIH Grants AI44616 and GM32136 to W.L.J., and a Fulbright Fellowship to R.H..
ABBREVIATIONS USED
- HIV-1
human immunodeficiency virus type 1
- RT
reverse transcriptase
- NRTIs
nucleoside reverse transcriptase inhibitors
- NNRTIs
non-nucleoside reverse transcriptase inhibitors
- HAART
highly active antiretroviral therapy
- dNTP
deoxynucleoside triphosphate
- d4T
β-d-(+)−2′,3′-didehydro-3′-deox-ythymidine
- 8-Cl-TIBO
8-chlorotetrahydroimidazobenzodiaze-pinone
- WT
wild type
- TEAB
triethylammonium bicarbonate
- DMSO
dimethyl sulfoxide
- PEG
poly(ethylene glycol)
- TBDMS
tert-butyldimethylsilyl
- MW
molecular weight
- Tris
tris(hydroxymethyl)aminomethane
Footnotes
ASSOCIATED CONTENT
Supporting Information
One figure showing the HPLC trace of d4TTP-4PEG-TMC (1c). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- 1.World Health Organization. [accessed January 14, 2013];Progress Report 2011: Global HIV/ AIDS response. http://www.who.int/hiv/pub/progress_report2011/en/index.html.
- 2.World Health Organization. [accessed January 14, 2013];Antiretroviral therapy for HIV infection in adults and adolescents: Recommendations for a public health approach, 2010 revision. http://www.who.int/hiv/pub/arv/adult2010/en/index.html. [PubMed]
- 3.Menendez-Arias L. Molecular basis of human immunodeficiency virus drug resistance: an update. Antiviral Res. 2010;85:210–231. doi: 10.1016/j.antiviral.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 4.Martinez-Cajas JL, Petrella M, Wainberg MA. Antimicrobial Resistance and Implications for the Twenty-First Century. New York: Springer; 2008. Clinical significance and biological basis of HIV drug resistance; pp. 231–261. [Google Scholar]
- 5.Mansky LM. Retrovirus mutation rates and their role in genetic variation. J. Gen. Virol. 1998;79(Pt. 6):1337–1345. doi: 10.1099/0022-1317-79-6-1337. [DOI] [PubMed] [Google Scholar]
- 6.Coffin JM. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science. 1995;267:483–489. doi: 10.1126/science.7824947. [DOI] [PubMed] [Google Scholar]
- 7.Perelson AS, Neumann AU, Markowitz M, Leonard JM, Ho DD. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science. 1996;271:1582–1586. doi: 10.1126/science.271.5255.1582. [DOI] [PubMed] [Google Scholar]
- 8.Rhodes T, Wargo H, Hu WS. High rates of human immunodeficiency virus type 1 recombination: near-random segregation of markers one kilobase apart in one round of viral replication. J. Virol. 2003;77:11193–11200. doi: 10.1128/JVI.77.20.11193-11200.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levy DN, Aldrovandi GM, Kutsch O, Shaw GM. Dynamics of HIV-1 recombination in its natural target cells. Proc. Natl. Acad. Sci. U.S.A. 2004;101:4204–4209. doi: 10.1073/pnas.0306764101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goody RS, Muller B, Restle T. Factors contributing to the inhibition of HIV reverse transcriptase by chain-terminating nucleotides in vitro and in vivo. FEBS Lett. 1991;291:1–5. doi: 10.1016/0014-5793(91)81089-q. [DOI] [PubMed] [Google Scholar]
- 11.Bienstock RJ, Copeland WC. Molecular insights into NRTI inhibition and mitochondrial toxicity revealed from a structural model of the human mitochondrial DNA polymerase. Mitochondrion. 2004;4:203–213. doi: 10.1016/j.mito.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 12.Bailey CM, Anderson KS. A mechanistic view of human mitochondrial DNA polymerase gamma: providing insight into drug toxicity and mitochondrial disease. Biochim. Biophys. Acta. 2010;1804:1213–1222. doi: 10.1016/j.bbapap.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown JA, Pack LR, Fowler JD, Suo Z. Pre-steady-state kinetic analysis of the incorporation of anti-HIV nucleotide analogs catalyzed by human X- and Y-family DNA polymerases. Antimicrob. Agents Chemother. 2011;55:276–283. doi: 10.1128/AAC.01229-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koczor CA, Lewis W. Nucleoside reverse transcriptase inhibitor toxicity and mitochondrial DNA. Expert Opin. Drug Metab. Toxicol. 2010;6:1493–1504. doi: 10.1517/17425255.2010.526602. [DOI] [PubMed] [Google Scholar]
- 15.Desai VG, Lee T, Delongchamp RR, Leakey JE, Lewis SM, Lee F, Moland CL, Branham WS, Fuscoe JC. Nucleoside reverse transcriptase inhibitors (NRTIs)-induced expression profile of mitochondria-related genes in the mouse liver. Mitochondrion. 2008;8:181–195. doi: 10.1016/j.mito.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 16.Sarafianos SG, Marchand B, Das K, Himmel DM, Parniak MA, Hughes SH, Arnold E. Structure and function of HIV-1 reverse transcriptase: molecular mechanisms of polymerization and inhibition. J. Mol. Biol. 2009;385:693–713. doi: 10.1016/j.jmb.2008.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smerdon SJ, Jager J, Wang J, Kohlstaedt LA, Chirino AJ, Friedman JM, Rice PA, Steitz TA. Structure of the binding site for nonnucleoside inhibitors of the reverse transcriptase of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. U.S.A. 1994;91:3911–3915. doi: 10.1073/pnas.91.9.3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spence RA, Kati WM, Anderson KS, Johnson KA. Mechanism of inhibition of HIV-1 reverse transcriptase by nonnucleoside inhibitors. Science. 1995;267:988–993. doi: 10.1126/science.7532321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rittinger K, Divita G, Goody RS. Human immunodeficiency virus reverse transcriptase substrate-induced conformational changes and the mechanism of inhibition by nonnucleoside inhibitors. Proc. Natl. Acad. Sci. U.S.A. 1995;92:8046–8049. doi: 10.1073/pnas.92.17.8046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iyidogan P, Anderson KS. Understanding the molecular mechanism of sequence dependent tenofovir removal by HIV-1 reverse transcriptase: differences in primer binding site versus polypurine tract. Antiviral Res. 2012;95:93–103. doi: 10.1016/j.antiviral.2012.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Basavapathruni A, Bailey CM, Anderson KS. Defining a molecular mechanism of synergy between nucleoside and nonnucleoside AIDS drugs. J. Biol. Chem. 2004;279:6221–6224. doi: 10.1074/jbc.C300523200. [DOI] [PubMed] [Google Scholar]
- 22.Basavapathruni A, Vingerhoets J, de Bethune MP, Chung R, Bailey CM, Kim J, Anderson KS. Modulation of human immunodeficiency virus type 1 synergistic inhibition by reverse transcriptase mutations. Biochemistry. 2006;45:7334–7340. doi: 10.1021/bi052362v. [DOI] [PubMed] [Google Scholar]
- 23.Velazquez S, Alvarez R, San-Felix A, Jimeno ML, De Clercq E, Balzarini J, Camarasa MJ. Synthesis and anti-HIV activity of [AZT]-[TSAO-T] and [AZT]-[HEPT] dimers as potential multifunctional inhibitors of HIV-1 reverse transcriptase. J. Med. Chem. 1995;38:1641–1649. doi: 10.1021/jm00010a008. [DOI] [PubMed] [Google Scholar]
- 24.Renoud-Grappin M, Fossey C, Fontaine G, Laduree D, Aubertin AM, Kirn A. Imidazo[1,5-b]pyridazine-d4T conjugates: synthesis and anti-human immunodeficiency virus evaluation. Antiviral Chem. Chemother. 1998;9:205–223. doi: 10.1177/095632029800900302. [DOI] [PubMed] [Google Scholar]
- 25.Pontikis R, Dolle V, Guillaumel J, Dechaux E, Note R, Nguyen CH, Legraverend M, Bisagni E, Aubertin AM, Grierson DS, Monneret C. Synthesis and evaluation of “AZT-HEPT”, “AZT-pyridinone”, and “ddC-HEPT” conjugates as inhibitors of HIV reverse transcriptase. J. Med. Chem. 2000;43:1927–1939. doi: 10.1021/jm991125l. [DOI] [PubMed] [Google Scholar]
- 26.Udier-Blagovic M, Tirado-Rives J, Jorgensen WL. Validation of a model for the complex of HIV-1 reverse transcriptase with nonnucleoside inhibitor TMC125. J. Am. Chem. Soc. 2003;125:6016–6017. doi: 10.1021/ja034308c. [DOI] [PubMed] [Google Scholar]
- 27.Das K, Clark AD, Jr., Lewi PJ, Heeres J, De Jonge MR, Koymans LM, Vinkers HM, Daeyaert F, Ludovici DW, Kukla MJ, De Corte B, Kavash RW, Ho CY, Ye H, Lichtenstein MA, Andries K, Pauwels R, De Bethune MP, Boyer PL, Clark P, Hughes SH, Janssen PA, Arnold E. Roles of conformational and positional adaptability in structure-based design of TMC125-R165335 (etravirine) and related non-nucleoside reverse transcriptase inhibitors that are highly potent and effective against wild-type and drug-resistant HIV-1 variants. J. Med. Chem. 2004;47:2550–2560. doi: 10.1021/jm030558s. [DOI] [PubMed] [Google Scholar]
- 28.Vaccaro JA, Parnell KM, Terezakis SA, Anderson KS. Mechanism of inhibition of the human immunodeficiency virus type 1 reverse transcriptase by d4TTP: an equivalent incorporation efficiency relative to the natural substrate dTTP. Antimicrob. Agents Chemother. 2000;44:217–221. doi: 10.1128/aac.44.1.217-221.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.World Health Organization. [accessed January 14, 2013];Antiretroviral therapy for HIV infection in adults and adolescents, 2010. http://www.who.int/hiv/pub/arv/adult2010/en/index.html. [PubMed]
- 30.Ruth JL, Cheng Y. Selective antiviral agents. The metabolism of 5-propyl-2′-deoxyuridine and effects on DNA synthesis in herpes simplex virus type 1 infections. J. Biol. Chem. 1982;257:10261–10266. [PubMed] [Google Scholar]
- 31.Rong F-G, Soloway AH. Synthesis of 5-tethered carborane-containing pyrimidine nucleosides as potential agents for DNA incorporation. Nucleosides Nucleotides. 1994;13:2021–2034. [Google Scholar]
- 32.Rong F-G, Soloway AH, Ikeda S, Ives DH. Synthesis and biochemical activity of 5-tethered carborane-containing pyrimidine nucleosides as potential agents for DNA incorporation. Nucleosides Nucleotides. 1995;14:1873–1887. [Google Scholar]
- 33.Piao D, Basavapathruni A, Iyidogan P, Dai G, Hinz W, Ray AS, Murakami E, Feng JY, You F, Dutschman GE, Austin DJ, Parker KA, Anderson KS. Bifunctional inhibition of HIV-1 reverse transcriptase: a first step in designing a bifunctional triphosphate. Bioorg. Med. Chem. Lett. 2013;23:1511–1518. doi: 10.1016/j.bmcl.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li D, Zhan P, De Clercq E, Liu X. Strategies for the design of HIV-1 non-nucleoside reverse transcriptase inhibitors: Lessons from the development of seven representative paradigms. J. Med. Chem. 2012;55:3595–3613. doi: 10.1021/jm200990c. [DOI] [PubMed] [Google Scholar]
- 35.Das K, Bauman JD, Clark AD, Jr., Frenkel YV, Lewi PJ, Shatkin AJ, Hughes SH, Arnold E. High-resolution structures of HIV-1 reverse transcriptase/TMC278 complexes: strategic flexibility explains potency against resistance mutations. Proc. Natl. Acad. Sci. U.S.A. 2008;105:1466–1471. doi: 10.1073/pnas.0711209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Janssen PA, Lewi PJ, Arnold E, Daeyaert F, de Jonge M, Heeres J, Koymans L, Vinkers M, Guillemont J, Pasquier E, Kukla M, Ludovici D, Andries K, de Bethune MP, Pauwels R, Das K, Clark AD, Jr., Frenkel YV, Hughes SH, Medaer B, De Knaep F, Bohets H, De Clerck F, Lampo A, Williams P, Stoffels P. In search of a novel anti-HIV drug: multidisciplinary coordination in the discovery of 4-[[4-[[4-[(1E)-2-cyanoethenyl]-2,6-dimethylphenyl]amino]-2- pyrimidinyl]amino]benzonitrile (R278474, rilpivirine) J. Med. Chem. 2005;48:1901–1909. doi: 10.1021/jm040840e. [DOI] [PubMed] [Google Scholar]
- 37.Hsiou Y, Ding J, Das K, Clark AD, Jr., Hughes SH, Arnold E. Structure of unliganded HIV-1 reverse transcriptase at 2.7 A resolution: implications of conformational changes for polymerization and inhibition mechanisms. Structure. 1996;4:853–860. doi: 10.1016/s0969-2126(96)00091-3. [DOI] [PubMed] [Google Scholar]
- 38.Huang H, Chopra R, Verdine GL, Harrison SC. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science. 1998;282:1669–1675. doi: 10.1126/science.282.5394.1669. [DOI] [PubMed] [Google Scholar]
- 39.Ding J, Das K, Moereels H, Koymans L, Andries K, Janssen PA, Hughes SH, Arnold E. Structure of HIV-1 RT/TIBO R 86183 complex reveals similarity in the binding of diverse nonnucleoside inhibitors. Nat. Struct. Biol. 1995;2:407–415. doi: 10.1038/nsb0595-407. [DOI] [PubMed] [Google Scholar]
- 40.Younis Y, Hunter R, Muhanji CI, Hale I, Singh R, Bailey CM, Sullivan TJ, Anderson KS. [d4U]-Spacer-[HI-236] doubledrug inhibitors of HIV-1 reverse-transcriptase. Bioorg. Med. Chem. 2010;18:4661–4673. doi: 10.1016/j.bmc.2010.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Delbederi Z, Fossey C, Fontaine G, Benzaria S, Gavriliu D, Ciurea A, Lelong B, Laduree D, Aubertin AM, Kirn A. Synthesis and antiviral activity of C-5 substituted beta-D- and beta-L-D4T analogues. Nucleosides Nucleotides Nucleic Acids. 2000;19:1441–1461. doi: 10.1080/15257770008033853. [DOI] [PubMed] [Google Scholar]
- 42.Ciurea A, Fossey C, Benzaria S, Gavriliu D, Delbederi Z, Lelong B, Laduree D, Aubertin AM, Kirn A. Synthesis of 5-alkenylated D4T analogues via the Pd-catalyzed cross-coupling reaction. Nucleosides Nucleotides Nucleic Acids. 2001;20:1655–1670. doi: 10.1081/NCN-100105902. [DOI] [PubMed] [Google Scholar]
- 43.Laduree D, Fossey C, Renoud-Grappin M, Fontaine G, Camara F, Gavriliu D, Ciurea A, Aubertin AM, Kirn A. Synthesis of novel C-5 substituted d4T analogues bearing linker arms as potential anti-HIV agents. Nucleosides Nucleotides. 1999;18:883–884. doi: 10.1080/15257779908041592. [DOI] [PubMed] [Google Scholar]
- 44.Velazquez S, Tunon V, Jimeno ML, Chamorro C, De Clercq E, Balzarini J, Camarasa MJ. Potential multifunctional inhibitors of HIV-1 reverse transcriptase. Novel [AZT]-[TSAO-T] and [d4T]-[TSAO-T] heterodimers modified in the linker and in the dideoxynucleoside region. J. Med. Chem. 1999;42:5188–5196. doi: 10.1021/jm991092+. [DOI] [PubMed] [Google Scholar]
- 45.Gavriliu D, Fossey C, Ciurea A, Delbederi Z, Sugeac E, Laduree D, Schmidt S, Laumond G, Aubertin AM. Synthesis and anti-HIV activity of [d4U]-[Trovirdine analogue] and [d4T]-[Trovirdine analogue] heterodimers as inhibitors of HIV-1 reverse transcriptase. Nucleosides Nucleotides Nucleic Acids. 2002;21:505–533. doi: 10.1081/NCN-120015066. [DOI] [PubMed] [Google Scholar]
- 46.Ludovici DW, De Corte BL, Kukla MJ, Ye H, Ho CY, Lichtenstein MA, Kavash RW, Andries K, de Bethune MP, Azijn H, Pauwels R, Lewi PJ, Heeres J, Koymans LM, de Jonge MR, Van Aken KJ, Daeyaert FF, Das K, Arnold E, Janssen PA. Evolution of anti-HIV drug candidates. Part 3: Diarylpyrimidine (DAPY) analogues. Bioorg. Med. Chem. Lett. 2001;11:2235–2239. doi: 10.1016/s0960-894x(01)00412-7. [DOI] [PubMed] [Google Scholar]
- 47.Hernandez E, Galan A, Rovira C, Veciana J. Synthesis of a series of symmetrically disubstituted diacetylenes with polychlor-ophenyl rings as side groups and linear polyether chains as spacers. Synthesis. 1992;1992:1164–1169. [Google Scholar]
- 48.Hunter R, Muhanji CI, Hale I, Bailey CM, Basavapathruni A, Anderson KS. [d4U]-Butyne-[HI-236] as a non-cleavable, bifunctional NRTI/NNRTI HIV-1 reverse-transcriptase inhibitor. Bioorg. Med. Chem. Lett. 2007;17:2614–2617. doi: 10.1016/j.bmcl.2007.01.107. [DOI] [PubMed] [Google Scholar]
- 49.Burgess K, Cook D. Syntheses of nucleoside triphosphates. Chem. Rev. 2000;100:2047–2060. doi: 10.1021/cr990045m. [DOI] [PubMed] [Google Scholar]
- 50.Thoresen LH, Jiao GS, Haaland WC, Metzker ML, Burgess K. Rigid, conjugated, fluoresceinated thymidine triphos-phates: syntheses and polymerase mediated incorporation into DNA analogues. Chemistry. 2003;9:4603–4610. doi: 10.1002/chem.200304944. [DOI] [PubMed] [Google Scholar]
- 51.Ludwig J, Eckstein F. Rapid and efficient synthesis of nucleoside 5′-O-(1-thiotriphosphates), 5′-triphosphates and 2′,3′-cyclophosphorothioates using 2-chloro-4H-1,3,2-benzodioxaphosphor-in-4-one. J. Org. Chem. 1989;54:631–635. [Google Scholar]
- 52.Vinogradov SV, Zeman AD, Batrakova EV, Kabanov AV. Polyplex nanogel formulations for drug delivery of cytotoxic nucleoside analogs. J. Controlled Release. 2005;107:143–157. doi: 10.1016/j.jconrel.2005.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Galmarini CM, Warren G, Kohli E, Zeman A, Mitin A, Vinogradov SV. Polymeric nanogels containing the triphosphate form of cytotoxic nucleoside analogues show antitumor activity against breast and colorectal cancer cell lines. Mol Cancer Ther. 2008;7:3373–3380. doi: 10.1158/1535-7163.MCT-08-0616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maga G, Radi M, Gerard MA, Botta M, Ennifar E. HIV-1 RT inhibitors with a novel mechanism of action: NNRTIs that compete with the nucleotide substrate. Viruses. 2010;2:880–899. doi: 10.3390/v2040880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Spychala J. A Facile preparation of N2-arylisocytosines. Synth. Commun. 1997;27:1943–1949. [Google Scholar]
- 56.Okada M, Yoden T, Kawaminami E, Shimada Y, Kudoh M, Isomura Y. Studies on aromatase inhibitors. III. Synthesis and biological evaluation of [(4-bromobenzyl)(4-cyanophenyl)amino]-azoles and their azine analogs. Chem. Pharm. Bull. 1997;45:482–486. doi: 10.1248/cpb.45.482. [DOI] [PubMed] [Google Scholar]
- 57.Chabrier P, Seyden-Penne J, Fouage AM. C. R. Hebd. Seances Acad. Sci. 1957;245:174–175. [Google Scholar]
- 58.Mansuri MM, Starrett JE, Wos JA, Tortolani DR, Brodfuehrer PR, Howell HG, Martin JC. Preparation of 1-(2,3-dideoxy-β-D-glyceropent-2-enofuranosyl)thymine (d4T) and 2′,3′-dideoxyadenosine (ddA): General methods for the synthesis of 2′,3′-olefinic and 2′,3′-dideoxynucleoside analogs active against HIV. J. Org. Chem. 1989;54:4780–4785. [Google Scholar]
- 59.Zhang YM, Razler T, Jackson PF. Synthesis of pyrimido[4,5-b]indoles and benzo[4,5]furo[2,3-d]pyrimidines via palladium-catalyzed intramolecular arylation. Tetrahedron Lett. 2002;43:8235–8239. [Google Scholar]
- 60.Kerr SG, Anderson KS. Pre-steady-state kinetic characterization of wild type and 3′-azido-3′-deoxythymidine (AZT) resistant human immunodeficiency virus type 1 reverse transcriptase: implication of RNA directed DNA polymerization in the mechanism of AZT resistance. Biochemistry. 1997;36:14064–14070. doi: 10.1021/bi9713862. [DOI] [PubMed] [Google Scholar]
- 61.Murakami E, Feng JY, Lee H, Hanes J, Johnson KA, Anderson KS. Characterization of novel reverse transcriptase and other RNA-associated catalytic activities by human DNA polymerase gamma: importance in mitochondrial DNA replication. J. Biol. Chem. 2003;278:36403–36409. doi: 10.1074/jbc.M306236200. [DOI] [PubMed] [Google Scholar]
- 62.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.