

Abstract
In Alzheimer's disease (AD), the mechanisms of neuronal loss remain largely unknown. Although tau pathology is closely correlated with neuronal loss, how its accumulation may lead to activation of neurotoxic pathways is unclear. Here we show that tau increased the levels of ubiquitinated proteins in the brain and triggered activation of the unfolded protein response (UPR). This suggested that tau interferes with protein quality control in the endoplasmic reticulum (ER). Consistent with this, ubiquitin was found to associate with the ER in human AD brains and tau transgenic (rTg4510) mouse brains, but this was not always colocalized with tau. The increased levels of ubiquitinated protein were accompanied by increased levels of phosphorylated protein kinase R-like ER kinase (pPERK), a marker that indicates UPR activation. Depleting soluble tau levels in cells and brain could reverse UPR activation. Tau accumulation facilitated its deleterious interaction with ER membrane and associated proteins that are essential for ER-associated degradation (ERAD), including valosin-containing protein (VCP) and Hrd1. Based on this, the effects of tau accumulation on ERAD efficiency were evaluated using the CD3δ reporter, an ERAD substrate. Indeed, CD3δ accumulated in both in vitro and in vivo models of tau overexpression and AD brains. These data suggest that soluble tau impairs ERAD and the result is activation of the UPR. The reversibility of this process, however, suggests that tau-based therapeutics could significantly delay this type of cell death and therefore disease progression.
Introduction
The number of dead neurons in the Alzheimer's disease (AD) brain exceeds the number of tangles composed of the microtubule associated protein tau (Gómez-Isla et al., 1997). Despite this, tau accumulation tracks best with clinical progression (Braak and Braak, 1991). This suggests that there is likely a secondary route of cell death brought on by the simultaneous accumulation of soluble tau species that cannot be detected histologically. The pathogenic effects of these tau species that can propagate from neuron to neuron remain unclear (Goedert et al., 2010; Liu et al., 2012a; de Calignon et al., 2012). However, recent evidence from studies of other neurodegenerative diseases suggests that endoplasmic reticulum (ER) stress may link proteinopathies together (Hoozemans et al., 2007; Colla et al., 2012).
The most compelling evidence for an association between ER stress and neurodegeneration is that pretangle neurons from tauopathies such as AD, frontotemporal dementia with tau inclusions (FTD-tau), and progressive supranuclear palsy (PSP) have increased levels of protein kinase R-like ER kinase (pPERK; Hoozemans et al., 2009; Nijholt et al., 2012). Although there are no reports showing that tau is processed in the ER, a recent genome-wide association study found a single nucleotide polymorphism (SNP) in the gene coding for PERK with risk for PSP (Hoglinger et al., 2011), providing further evidence for a pathogenic link between the ER system and tau accumulation. One mechanism of ER stress induction is blockage of ER-associated degradation (ERAD), a cellular process necessary for proteostasis. Consequently, PERK becomes activated to attenuate protein translation and decrease nascent protein input to the ER.
PERK is phosphorylated when ERAD is impaired due to activation of the unfolded protein response (UPR) initiated by accretion of unfolded proteins inside the ER (Weihl et al., 2006). The UPR reduces the input of nascent proteins into the ER and facilitates the output of mature proteins from the ER (Schröder and Kaufman, 2005). Two major events in the UPR are activation of PERK and upregulation of the profolding ER chaperone immunoglobulin heavy-chain binding protein (BiP; Bertolotti et al., 2000; Okamura et al., 2000). PERK rests on the cytoplasmic interface of the ER anchored to BiP, which is in the ER lumen (Bertolotti et al., 2000; Okamura et al., 2000). Upon activation of the UPR, PERK and BiP dissociate, allowing PERK to inhibit eukaryotic initiation factor 2; Harding et al., 2000). Through this dissociation, BiP becomes available to triage unfolded proteins (Harding et al., 1999; Harding et al., 2000).
We tested the hypothesis that tau interferes with ERAD, thereby contributing to disease pathogenesis. In tau models and human AD brains, there was significant evidence suggesting that tau accumulation can facilitate an UPR and that ER protein quality control is impaired. We also determined that ERAD was in fact blocked by tau accumulation, a process that led to the UPR, but was reversible when tau levels were reduced. These findings suggest that tau accumulation interferes with ER proteostasis, which activates the UPR.
Materials and Methods
Antibodies.
The following antibodies were purchased from Cell Signaling Technology: ubiquitin (anti-mouse), calnexin, VCP, pPERK, and hrd1. Flotillin-1 antibody was obtained from BD Transduction Laboratories. The ubiquitin antibody used in the experiment shown in Figure 6 was anti-goat (Santa Cruz Biotechnology), which was selected to prevent nonspecific binding of mouse IgG because of inflammatory markers that are induced in the mouse brain after surgical procedures. The CD3δ antibody and the tau-150 antibody (Fig. 2, Fig. 7) were also purchased from Santa Cruz Biotechnology. PHF1 was a generous gift from Dr. Peter Davies and was used for the experiments shown in Figure 1 and Figure 4. Tau5 was a generous gift from Dr. Lester Binder and used in Figure 1 and Figure 6. E1 is an antibody that detects human tau and was generously provided by Dr. Leonard Petrucelli and used in the experiment shown in Figure 5K. The tau antibody used in Figure 5A was purchased from Dako. GAPDH was obtained from Meridian Life Sciences. Actin was purchased from Sigma. Secondary antibodies conjugated to HRP were purchased from SouthernBiotech. Secondary antibodies conjugated to a fluorophore (used at 1:1500) and Neurotrace dye for neuronal-specific staining were purchased from Invitrogen.
Figure 6.

Tau associates with the ERAD heterocomplex members VCP and Hrd1. A, Representative blots of tau and actin from iHEK-Tau cells that were induced to express tau for 96 h and fractionated to separate microsomal and cytoplasmic proteins. B, C, Representative blots from IP:WB experiments in which tau or VCP was immunoprecipitated from mouse hippocampus and samples were immunoblotted for VCP, Hrd1, and tau. The control lane corresponds to Tg lysate that was not incubated tau antibody. Input lanes show bands representative of VCP, Hrd1, tau, and GAPDH. Quantification of the input blots shows a 3-fold increase in VCP levels (n = 4 nontransgenic [Non], n = 7 Tg; *p < 0.05). D–G, Representative immunofluorescent microscopy images and colocalization plots of 9MO Tg mice. D–F, Sections were costained with anti-tau (green) and anti-VCP (red) antibodies and DAPI (blue) was used to identify cell nuclei. F, Colocalized signal appears in yellow of the merged images. Scale bars, 20 μm (low-magnification image) and 10 μm (insets). G, Plot confirming colocalization of signals (Pearson's coefficient > 0.9).
Figure 2.

Increased ubiquitin precedes activation of ER stress. A, Representative Western blot of pPERK, BiP, ubiquitin, tau, and actin from iHEK-Tau cells induced to express tau by adding Tet to the media for 4 d. Samples were collected at the indicated times. B, Linear regression analysis representing the relative quantitation of pPERK, BiP, and ubiquitin levels in A over time; values are relative and normalized to GAPDH and tau. Significance was derived from four experiments; *p < 0.05, **p < 0.01, ***p < 0.001 for C, E, G. Representative immunofluorescent images of Tg hippocampi of the indicated ages and stained for ubiquitin (C; green), pPERK (E; red), and tau (G; red). Scale bar, 100 μm. D, F, H, Quantitative analysis of C, E, and G. Immunofluorescence in 3-month-old (3MO) sections was virtually undetectable above background control slide, which consisted of immunofluorescent staining without primary antibody incubation. Compared with 6MO mice, ubiquitin, pPERK, and tau levels increased by 150% (9MO; †p < 0.0001) and 186% (12MO; †p < 0.0001), 266% and 243% (9MO; †p < 0.0001), and 194% (9MO; ***p < 0.001) and 215% (12MO; ***p < 0.001), respectively. Comparing 9MO with 12MO mice, ubiquitin and tau levels were significantly different (†p < 0.0001 and ***p < 0.001, respectively), but pPERK levels were not (p > 0.05) (n = 3 mice for each condition). Statistical significance was determined using Student's t test.
Figure 7.
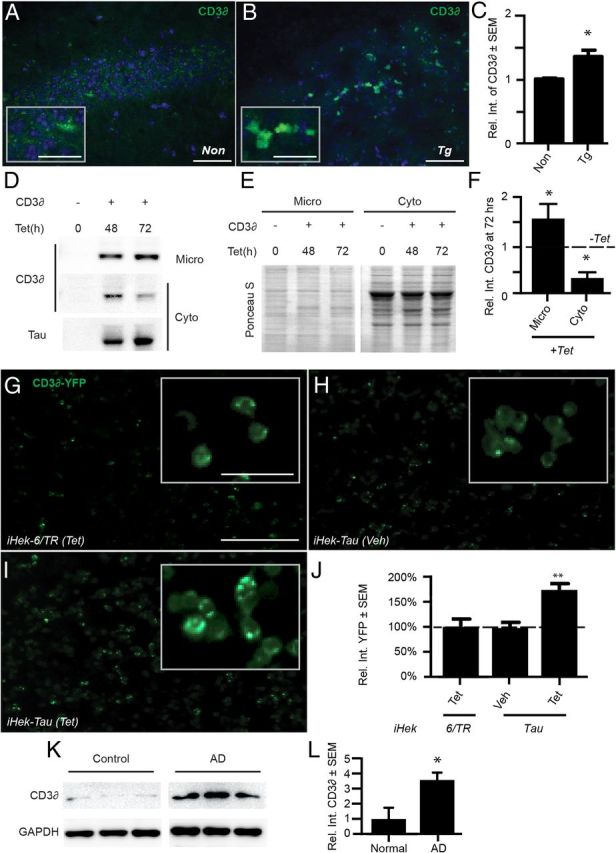
The ERAD substrate CD3δ accumulates in tau-overexpressing models and human AD brains. A, B, Representative immunofluorescent images of CA3 from nontransgenic (Non; A) or Tg (B) mice stained for CD3δ (green). DAPI (blue) was used to identify cell nuclei; scale bars, 50 μm (low-magnification images) and 25 μm (insets). C, Quantification showed a 35% increase of CD3δ-positive signal in the CA3 of Tg brains compared with Non controls (*p < 0.05, n = 3 mice for each condition). D, Representative Western blots of CD3δ and tau in iHEK-Tau cells transfected with CD3δ or control vector plasmids and treated with Tet or vehicle for 48 or 72 h. ER-containing microsomes (Micro) were isolated from cytoplasmic (Cyto) protein fractions and samples were run in separate gels. E, Ponceau S stain of C showing equal loading. F, Relative quantitation of CD3δ signal normalized to the integrated density of Ponceau S for the entire lane. Tau-expressing cells had 50% more CD3δ in the microsomal fraction and 58% less in the cytoplasmic fraction (*p < 0.05). G–I, Live-cell images of iHek-6/TR and iHEK-Tau cells transfected with CD3δ-YFP and treated with Tet or vehicle for 24 h. J, Quantitation of punctate YFP signal using five 10× images from each condition (1.7-fold increase; **p < 0.001; this experiment was done in triplicate). Scale bar, 100 μm (50 μm for insets). K, L, Western blot of CD3δ in AD (stage 5 and 6) and nondemented controls (stage 1 and 2) showing a 3.5-fold increase of CD3δ in AD brains (*p < 0.05). Statistical significance was determined using Student's t test.
Figure 1.

Tau is correlated with accumulation of ubiquitin and activation of PERK. A, Representative Western blots of ubiquitin and pPERK and PERK in the brains of 9-month old Tg and nontransgenic (Non) controls. Tau bands confirm tau overexpression; GAPDH was used as a loading control. B, Quantification of A (n = 6 Non and n = 7 Tg; 4- and 3-fold increases of **p < 0.01 and *p < 0.05, respectively). C–J, Representative immunofluorescent microscopy images of ubiquitin (C,D; green), pPERK (E,F; red), BiP (G,H; red), and tau (I,J; green) in the hippocampus of Non (C,E,G,I) and Tg (D,F,H,J) mice. DAPI (blue) was used to identify cell nuclei. Scale bar, 20 μm. K, Graph showing quantification of immunofluorescent-positive signal in C–J. Ubiquitin, pPERK, and BiP levels in the Tg brains were increased by 3.7-fold (n = 3 Non and n = 6 Tg; ***p < 0.001), 2.4-fold (n = 3 Non and n = 5 Tg; ***p = 0.0001), and 0.3-fold (n = 5 Non and n = 4 Tg; *p < 0.05), respectively; PHF1 tau levels were virtually undetectable above background in I. Statistical significance was determined using Student's t test.
Figure 4.

Ubiquitin colocalizes with tau, the ER, and other proteins in AD brains. A–E, Representative quadruple immunofluorescent stain of tau (A,B,D,E; red), ubiquitin (B–E; green), calnexin (A,C–E; cyan), and neurons (E; blue) in an AD brain (scale bar, 5 μm). Images represent a progression of three z-stacks at 1 μm apart showing partial colocalization of ubiquitin with either tau or calnexin and that tau and calnexin do not colocalize. F, Graphical representation of the amount of colocalization in which 30% and 31% of ubiquitin (top green bar) colocalizes with tau or calnexin, respectively (Pearson > 0.5); however, tau and calnexin do not colocalize (Pearson < 0.5). The remaining ubiquitin signal did not colocalize with either tau or calnexin (∼29%). Red and cyan bars bar show that 39% of tau and 49% of calnexin are ubiquitinated, respectively. Analysis is based on images taken from 3 AD brains in Braak stage 5 or 6; multiple images were taken from each brain and three randomly selected neurons were analyzed to obtain Pearson's, Manders', and Costes' coefficients, as described in the Materials and Methods. G, Representative Western blot of ubiquitin and flotillin-1 in the microsomal fractions of medial temporal gyrus of nondemented controls and AD brains.
Figure 5.
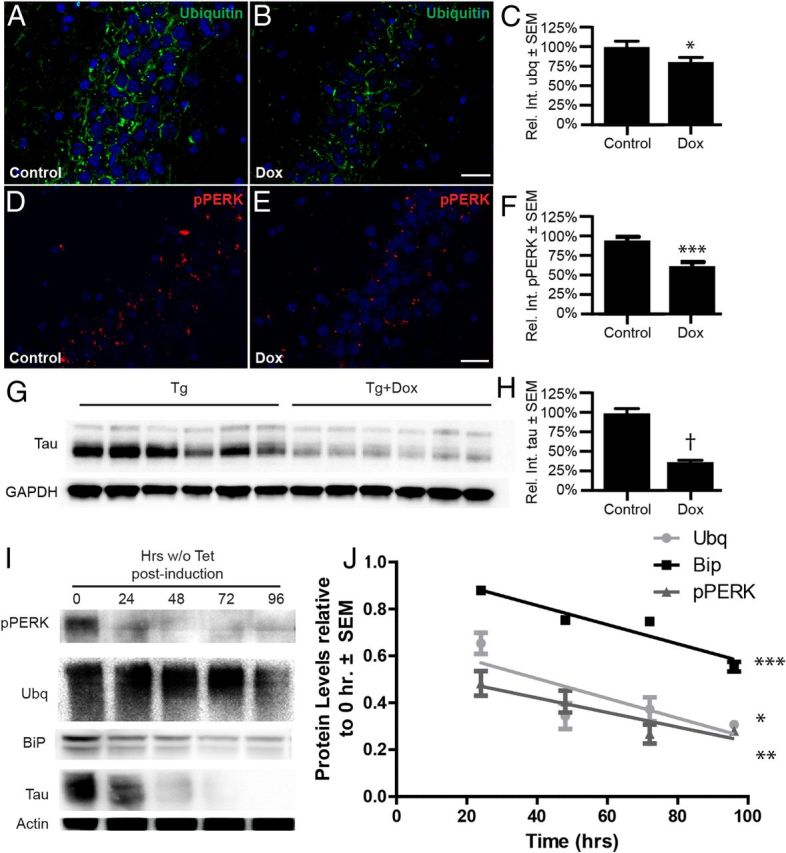
Depletion of soluble tau reverses the accumulation of ubiquitin and pPERK signal in vivo and in vitro. A–H, rTg4510 mice were fed a control or Dox diet for 35 d to suppress tau transgene expression. A, B, D, E, Representative immunofluorescent images of ubiquitin (A,B; green), pPERK (D,E; red), and DAPI (blue) in the CA2 of Tg brains (scale bar, 40 μm). C, F, Graphs showing that ubiquitin and pPERK aggregates were decreased by 20% (*p < 0.05; n = 3 control and n = 4 Dox) and 37% (***p < 0.0001; n = 5 control and n = 6 Dox), respectively. G, H, Representative immunoblot of tau from brains of Dox-fed mice and controls showing that tau levels were decreased by 64% (†p < 0.0001; n = 6 for each condition). I, Representative Western blot of pPERK, BiP, ubiquitin, tau, and actin from iHEK-Tau cells. iHEK-Tau cells were induced to express tau by adding Tet to the media. After 4 d, Tet-containing medium was removed, cells were washed, and Tet-free media was added for 4 d. Cells were harvested at different time points after removing the Tet. J, Linear regression analysis showing reductions in of pPERK, BiP, and ubiquitin levels over time after Tet removal; values are relative and normalized to actin. Significance was derived from four experiments (*p < 0.05, **p < 0.01, ***p < 0.001).
Mice.
All animal studies were approved by the University of South Florida's Institutional Animal Care and Use Committee and abided by that committee's policies on animal care and use in accordance with the Guide for the Care and Use of Laboratory Animals, the Animal Welfare Regulations Title 9 Code of Federal Regulations Subchapter A, “Animal Welfare, ” Parts 1–3, and the Public Health Service Policy on Humane Care and Use of Laboratory Animals. This University of South Florida program and the facilities for animal care and use are fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. The animals were kept in standard housing on a 12 h light dark cycle and received food and water ad libitum. The tau transgenic (rTg4510) and parental mice were maintained and genotyped as described previously (Santacruz et al., 2005). All experiments were done in mice of either sex.
Dox diet.
Six rTg4510 mice (five males and one female) were fed a doxycycline (Dox) diet (TD.00502; Harlan Laboratories) and another six transgenic (Tg) mice (five males and one female) were fed a control diet for 35 d, at which point mice were transcardially perfused, brains removed, and hemispheres separated. One hemisphere was fixed and used for immunohistochemistry and the other half was stripped of cerebellum and midbrain and homogenized.
Human brain tissues.
Frozen medial temporal brain samples were obtained from the Brain Bank and Tissue Repository in Irvine, California. Nondemented and AD cases were catalogued as tangle stages 1 and 2 and 5 and 6, respectively. They ranged in age from 81 to 90 years and both groups had three males and one female. Tissues were processed for immunofluorescence as described above.
Western blot.
Western blot experiments were performed as described previously (Abisambra et al., 2010b). Membranes were incubated with the following primary antibodies: PHF1, ubiquitin, and pPERK were used at 1:500; human tau 150, Tau5, calnexin, VCP, Hrd1, Flotillin-1, and CD3δ, were used at 1:1000; E1 at 1:1500; GAPDH and actin were used at 1:5000.
Immunohistochemistry.
Mouse brains were harvested as described previously (Dickey et al., 2009). Briefly, mice were overdosed with pentobarbital and perfused transcardially with 0.9% normal saline solution. Brains were removed and immersed in 4% paraformaldehyde. Fixed brains were cryoprotected in successive 24 h increments of 10%, 20%, and 30% sucrose gradients as described previously (Gordon et al., 2002). Brains were frozen on a temperature-controlled freezing stage, sagittally sectioned (25–50 μm) on a sliding microtome, and stored in a solution of PBS containing 0.02% NaN3 at 4°C. Immunostaining was performed following protocols described previously with minor modifications (Gordon et al., 1997; Abisambra et al., 2010a). Brain sections were mounted on glass slides with medium (30% ethanol in PBS). Once dry, sections were blocked and permeabilized in blocking buffer (4% donkey serum, 1.83% lysine, and 0.2% Triton X-100 in TBS) for 1 h. Slides were then incubated overnight at 4°C with the following antibodies: CD3δ (mouse 1:50), ubiquitin mouse (1:25), ubiquitin goat (1:50), calnexin (1:50), PHF1 (1:500), VCP (1:50), BiP (1:50), and pPERK (1:25). Slides were washed with TBS and then incubated with Alexa Fluor 488 nm and Alexa Fluor 594 nm secondary antibodies (Invitrogen) at 1:1500 for 2 h at room temperature. Slides were then washed again and incubated with DAPI (1:20,000; Sigma) or Neurotrace as indicated. Nonspecific reaction product formation was negligible as assessed by omitting the primary antibody. Coverslips were placed as described previously (Abisambra et al., 2010b). Human AD tissue staining went through an extra stain with Sudan black to block autofluorescence inherent to the tissue.
Microscopy.
All slides were imaged using the Carl Zeiss AxioImager.Z1 microscope with Apotome, the Olympus Fluoview confocal microscope, or the Olympus FV1000 MPE multiphoton laser scanning microscope as described previously (Abisambra et al., 2010a; Abisambra et al., 2010b). AxioVision 4.8 was used to adjust the brightness and contrast to all images within the same experiment equally and simultaneously; the same procedure was done for the Olympus images but using ImageJ. For this, brightness and contrast settings were obtained by eliminating nonspecific signal of images from the no-primary control tissues; the same settings were then applied to all other images. Images with artifacts were excluded. The human brain tissue analysis was based on sections from three AD and three control brains. For all other immunofluorescent experiments except methylene blue (MB) rescue (which used multiple images from two mice), multiple images were taken from at least three mice for each condition. Data analysis for the Dox rescue experiment was performed as follows: multiple sections from six mice of each condition (Dox treated and controls) were stained for ubiquitin, pPERK, BiP, and tau, and sections were imaged using the Zeiss AxioImager.Z1. Images of DG, CA1, CA2, CA3, and frontal cortex for each section were taken using the same fixed exposure time, which was set above background generated by a negative control slide (no primary incubation). Images were analyzed using ImageJ software as described above by quantitating the positive signal normalized to the amount of cells (DAPI signal) and averaged per condition. All data were plotted as relative to control, which was set to 100%. Data analysis for the MB rescue experiment (Fig. 5) was performed as follows: images were taken from several sections of each mouse brain in the DG of treated and contralateral untreated sides. All images were analyzed in ImageJ to obtain quantitative values for the integrated density and the area. The ratio of integrated density of the ubiquitin signal to that of the DAPI-positive staining was assigned as the amount of ubiquitin signal per image. All values that were two SDs above or below the mean corresponding to each group were discarded. Positive staining was determined using an automatic segmentation based on a set threshold value defined by darker staining (AxioVision 4.8). Colocalization analysis was performed as described previously (Jinwal et al., 2010) and was determined based on Pearson's and Manders' coefficients. To rule out haphazard colocalization, Costes' analysis was also performed. Data analysis for CD3δ-yellow fluorescent protein (YFP) experiments (Fig. 7G–J) was performed as follows: five random images of each condition were captured using the AMG EVOSfl system at 10× magnification using GFP and transmitted light channels. Each image was analyzed as described previously (Abisambra et al., 2010a). The positive signal of all images was processed at a high constant for stringent threshold (level 1) to reveal only punctate staining; this facilitated evident quantifiable peaks of signal. The area of transmitted light signal was used as a correlate of the number of cells in each region of interest (ROI) and this number was used to normalize the data representative of punctate YFP. Moreover, this value was considered to represent the amount of CD3δ in the cells of each condition. Manders' coefficient analysis was performed as follows: between 7 and 12 z-stacked images at different magnifications (1 μm depth between each image) were captured with the Olympus FV1000 MPE multiphoton laser scanning microscope. Files were analyzed using the JACoP plugin in ImageJ. Pearson's coefficients higher than 0.5 but lower than 0.9 were considered to partially colocalize (Bolte and Cordelières, 2006). Manders' coefficients for the green and red channels (M1 or M2) were tabulated and plotted to obtain an average, SD, and minimum/maximum values. ROIs were obtained by magnifying the original images, cropping the cell or cells within the ROI, splitting all channels, and comparing the cyan with green, red with green, and cyan with red channels using the ImageJ toolbox JACoP (Bolte and Cordelières, 2006). This yielded several values for ROIs within the same image. The total number of images for each condition was three or greater, and the amount of ROIs for each image was nine or greater.
Cell culture.
To create tetracycline (Tet)-regulated tau stable cell lines, we inserted a human wild-type tau 4R0N or 6/TR (six repeats of TET repressor sequence) DNA into a pCDNA 4/TO plasmid vector carrying Zeocin selection (Invitrogen). The Tau/TO plasmid construct was then transfected into the Tet-regulated expression (T-REx; Invitrogen) HEK cell lines stably expressing the Tet repressor under the selection of blasticidin. After 24 h of DNA transfection, zeocin (400 μg/ml) and blasticidin (5 μg/ml) were added to the media (DMEM containing 10% FBS, 2 mm l-glutamine, 1% Pen-Strep). Every 3–4 d, the medium was replaced with fresh medium containing zeocin and blasticidin. After 3 weeks, stable cell colonies were transferred to 6-well plates. These colonies were induced by Tet (1 μg/ml, Sigma) and tau expression was assessed by Western blot. Tau- or 6/TR-expressing stable HEK cells were maintained under the selection of zeocin and blasticidin and used for experimentation. Proteins were obtained by harvesting cells in mammalian protein extraction reagent buffer (M-PER; Pierce) containing 1× protease inhibitor mixture (Calbiochem), 100 mm phenylmethylsulfonyl fluoride, and 1× phosphatase inhibitor II and III cocktails (Sigma). This procedure was similar to one described previously (Khlistunova et al., 2006).
Microsome isolation.
For iHek cell culture experiments, cells were plated and allowed to grow to confluency with the indicated treatments (±Tet) in either six-well plates or 10 cm dishes. Cells were scraped at the indicated time points and collected and centrifuged at 1500 rpm for 5 min to remove the medium. Cells were then homogenized in sucrose (0.25 m) containing 1× protease inhibitor mixture (Calbiochem), 100 mm phenylmethylsulfonyl fluoride, and 1× phosphatase inhibitor II and III cocktails (Sigma) by pipetting 30 times. For human brain processing, 200 mg of medial temporal gyrus from 3 male Braak stage 6 brains, 2 male Braak stage 2 brains, and 1 stage 1 brain were dounce homogenized in the same sucrose buffer described above. All types of samples were then centrifuged at 16,000 × g for 15 min at 4°C. The supernatant was transferred and centrifuged at 100,000 × g for 1 h at 4°C. The new supernatant contained the cytoplasmic protein fraction. The pellet corresponded to the microsomal protein fraction and was resuspended in RIPA buffer containing 1× protease inhibitor mixture (Calbiochem), 100 mm phenylmethylsulfonyl fluoride, and 1× phosphatase inhibitor II and III cocktails (Sigma).
Statistical analyses.
Statistical significance for all experiments was determined with Student's t tests (*p < 0.05, **p < 0.01, ***p < 0.001, †p < 0.0001). Linear regression analyses were performed for cell culture time courses. For all data analyses, values above or below two SDs of the mean were considered outliers and discarded from the dataset. This procedure removed ≤two values from each set at the most, which maintained ≥three data points for each condition. Therefore, unless otherwise specified, all experiments were conducted in triplicate and at least three samples were used for each assay. Graphs and statistical analyses were plotted and performed using Prism software (GraphPad).
Results
Studies in cell culture and yeast models have shown that unstructured neurotoxic proteins such as α-synuclein and poly-glutamine lead to accumulation of ubiquitinated proteins and activation of ER stress (Cooper et al., 2006; Bennett et al., 2007; Duennwald and Lindquist, 2008). We investigated whether tau accumulation may also result in similar sequelae. For these studies, we used the rTg4510 mouse model of tauopathy, which have an aggressive phenotype featuring severe tangle pathology and neuronal function deficits at 3 months (Abisambra et al., 2010b) and significant neurodegeneration that is coincident with cognitive decline deficits (Santacruz et al., 2005) at 5.5 months of age. The levels of ubiquitinated proteins, PERK, and pPERK were assessed in the brains of rTg4510 mice at an advanced stage of disease (9 months of age). Although PERK levels did not change, both ubiquitin and pPERK were significantly increased compared with age-matched nontransgenic controls (Fig. 1A,B). Furthermore, the ratio of active (pPERK) to inactive PERK maintained its magnitude difference between control and Tg mice. Ubiquitinated proteins, pPERK, and another marker of ER stress, BiP, were significantly increased by 3.7-fold, 2.4-fold, and 0.3-fold in the hippocampus and cortex of transgenic mice, respectively; these are regions where tau pathology is most robust in these mice (Fig. 1C–K). This was consistent with pathological data from AD brains showing similar increases in pPERK and ubiquitin immunoreactivity (Mori et al., 1987; Perry et al., 1987; Bancher et al., 1989; Kertesz et al., 2000; Hoozemans et al., 2009; Nijholt et al., 2012).
Our results suggested that increased levels of ubiquitinated proteins and increased UPR activation accompanied tau accumulation. However, to determine whether tau could promote these same abnormalities directly and show that this was not just an indirect downstream consequence of tau accretion, we developed a cell model of inducible tau expression. This HEK cell line (iHEK-Tau) is stably transfected with human 4R0N wild-type tau with a Tet-On controllable response element built into the promoter. Therefore, tau expression can be induced in these cells by adding Tet to the medium. We investigated whether acute upregulation of tau could activate the ER stress response using this system. These cells were treated with Tet and harvested at the indicated time points after treatment. Interestingly, we found that the levels of ubiquitinated proteins occurred after just 24 h of tau induction and increased levels of the two ER stress markers followed this at 72 and 96 h (Fig. 2A,B). This result suggested that tau precipitated an initial accumulation of ubiquitinated proteins that then led to UPR activation, which is suggestive of ERAD disruption. We then sought to assess changes in the accumulation of UPR-associated proteins over time in vivo. We stained for tau, pPERK, and ubiquitin in brains of Tg mice at 3, 6, 9, and 12 months. Accumulation of pPERK and ubiquitin was concomitant with the increases in tau aggregates (Fig. 2C–H), suggesting that aged neurons are more susceptible to UPR stress than young neurons.
We then investigated the localization of ubiquitin and tau in brain tissue from 9-month old tau transgenic mice using co-immunofluorescently labeled ubiquitin (green) and tau (red) (Fig. 3A–C). We performed a rigorous and unbiased colocalization routine using Costes' and Manders' analyses. Costes' analysis uses an automatic threshold to generate Pearson coefficients of colocalization and Manders' coefficients (M1 and M2) determine the amount of each signal that colocalizes with the other (Bolte and Cordelières, 2006). We found that ubiquitin and tau indeed colocalized but, surprisingly, this interaction was only partial. In fact, only 24% of the ubiquitin signal associated with tau. The reciprocal analysis showed that 60% of total tau colocalized with the ubiquitin signal. These data suggest that there is a large amount of ubiquitin (∼76%) that accumulated in the cell yet was independent of tau. We then investigated where this other pool of ubiquitinated proteins was accumulating within neurons. Because impairment of ERAD can cause the accumulation of ubiquitinated proteins on the ER membrane (Weihl et al., 2006), we performed a quadruple co-immunofluorescent stain for ubiquitin (green), tau (red), the ER-resident protein calnexin (cyan; Fig. 3D), and a neuron-specific dye (blue) in rTg4510 brains. Pathologically visible tau did not colocalize with the ER marker (Fig. 3E). However, ubiquitin-positive signal significantly colocalized with >42% of the ER membrane protein, accounting for 17% of the total ubiquitin-positive signal present in neurons (Fig. 3F). Therefore, a large area of the ER was covered by ubiquitin-positive immunofluorescence. The remaining 59% of ubiquitin-positive signal did not colocalize with either tau or calnexin, suggesting that tau might disrupt other processes involving the clearance of ubiquitinated proteins.
Figure 3.

Ubiquitin colocalizes with tau, the ER, or other proteins in Tg brains. A–G, Quadruple immunofluorescent stain of tau (A,C,E,G; red), ubiquitin (B,C,F,G; green), calnexin (D–G; cyan), and neurons (G; blue) in the hippocampus of 9-month-old Tg mice (low-magnification scale bar, 100 μm). Insets show higher-magnification images (inset scale bar, 10 μm). G, Inset of the merged image shows tau-ubiquitin colocalization (white arrowhead), non-colocalizing ubiquitin (yellow arrowhead), and calnexin-ubiquitin colocalization (orange arrowhead). H, Graphical representation of the amount of colocalization, where 24% and 17% of ubiquitin (top green bar) colocalizes with tau or calnexin, respectively (Pearson > 0.5); however, tau and calnexin do not colocalize (Pearson < 0.5). Approximately 59% of the ubiquitin signal did not colocalize with either tau or calnexin. Red and cyan bars show that 60% of tau and 42% of calnexin are ubiquitinated, respectively; the remainder of the signal did not colocalize with ubiquitin.
To confirm that these ubiquitin/ER stress abnormalities were not simply artifacts of tau overexpression as in the rTg4510 mice and iHEK cell model, we performed the same quadruple labeling experiment using fixed human cortical sections of advanced stage AD (Braak stage 5–6), which have robust accumulation of aberrant tau that is not driven by overexpression (Fig. 4). Similar to mice, we found that tau-positive fluorescence did not associate with the ER signal (Fig. 4A). Moreover, ubiquitin-positive signal partially colocalized with immunofluorescent signals for tau (30% of total ubiquitin) and calnexin (31% of total ubiquitin; Fig. 4B,C). Therefore, sporadic cases of AD showed even stronger colocalization of ubiquitin-positive signal with the ER marker calnexin. Further analysis showed that, similar to the mice, almost half (49%) of the ER was decorated by ubiquitin-positive signal, whereas 39% of tau was ubiquitin positive (Fig. 4F). We then assessed ubiquitin levels in AD and nondiseased medial temporal gyrus after subcellular fractionation. The ubiquitin signal was markedly accumulated in the AD microsomes, whereas the cytoplasmic fraction showed no noticeable difference in the levels of ubiquitinated proteins (Fig. 4G).
We then investigated whether removing tau could reverse these ER and UPR defects that were caused by tau accumulation. Recent findings suggest that some species of soluble tau are toxic in tauopathies, whereas insoluble tau accumulation may be protective to some extent (Oddo et al., 2006; Goedert et al., 2010; Liu et al., 2012a; de Calignon et al., 2012). Based on this information and our findings in the iHEK cell model shown in Figure 2, in which acute upregulation of wild-type tau could drive accumulation of ubiquitinated proteins and UPR activation, we abrogated tau levels in the Tg mice using Dox. The rTg4510 mouse has a TET-off system that allows for suppression of tau transgene expression upon administration of Dox (Santacruz et al., 2005). We fed mice a Dox-enriched diet for 35 d and measured changes in tau, ubiquitin, and pPERK levels, particularly in areas where tau deposition is prevalent, such as the hippocampus and the frontal cortex (Fig. 5A–F). Indeed, ubiquitin, pPERK, and tau levels were significantly reduced with Dox treatment by 20% (*p < 0.05; n = 3 control and n = 4 Dox), 37% (*p < 0.05; n = 5 control and n = 6 Dox), and 64% (†p < 0.0001; n = 6 control and n = 6 Dox), respectively (Fig. 5C,F,H). To further confirm these results, we investigated whether reducing tau levels acutely could also reduce ER stress in the iHEK cell model. Cells were induced to express tau for 4 d to achieve maximal induction and the Tet was then washed out to suppress tau expression. Cell lysates were collected at the indicated time points after Tet wash out and analyzed by Western blot (Fig. 5I,J). As expected, tau levels were reduced over time. Similarly, we observed concomitant decreases of pPERK, BiP, and ubiquitin, thus confirming the in vivo results: tau-induced ER stress is reversible.
Our data thus far suggested that tau impedes the clearance of ubiquitinated proteins from the ER and causes an ER stress response. However, we sought to identify a mechanism by which tau could be causing this defect. Based on previous work with α-synuclein and poly-glutamine expansion proteins, we speculated that tau accumulation might be able to interfere with ERAD through a nonspecific interaction with proteins associated with the ER membrane. Indeed, separation of cellular lysates into membrane and cytosolic containing fractions showed that a substantial amount of tau associated with membranes, supporting this hypothesis (Fig. 6A). Based on this result, we explored whether tau could interact with one of the major contributors to the ERAD pathway, the VCP/p97 complex. This is a large heteromeric protein complex consisting of VCP, ubiquitin ligases, adaptor proteins, translocation machinery, and other trafficking proteins involved in exporting misfolded ER proteins to the proteasome (Bays et al., 2001; Ye et al., 2001; Braun et al., 2002; Jarosch et al., 2002; Rabinovich et al., 2002; Carvalho et al., 2006). Indeed, coimmunoprecipitation experiments with brain lysates from rTg4510 mice and wild-type littermates revealed that tau was associated with the VCP complex, as evidenced by its association with VCP itself and with Hrd1, a ubiquitin ligase component of the VCP complex (Carvalho et al., 2006; Fig. 6B,C). Increased levels of VCP (3-fold, *p < 0.05) in rTg4510 mice were also observed (Fig. 6C). Immunofluorescent colocalization analyses also revealed an association of tau with VCP in the brains of rTg4510 mice (Fig. 6D–G). In this way, tau accumulation drives its aberrant interaction with membrane-associated proteins. In fact, its interaction with the VCP complex would be one mechanism by which this aberrant membrane association triggers ER stress.
To determine directly whether tau could impair ERAD, we assessed the levels of CD3δ, a subunit of the T-cell antigen receptor that is a well characterized substrate of ERAD, in the rTg4510 brain (Yang and Parkhouse, 1998; Fig. 7A–C). CD3δ was increased by ∼35% in the CA3 region of the hippocampus of rTg4510 mice compared with wild-type controls (*p < 0.05, n = 3 for each condition). To better evaluate whether tau could impair ERAD directly, we transfected iHEK-Tau cells with CD3δ-YFP, a reporter that is used to assess ERAD function, or vector-only plasmids and induced tau expression for 48 and 72 h. Cells were fractionated to separate ER-containing microsomes from cytoplasmic proteins. Increased tau expression caused ∼50% of the CD3δ signal to shift from the cytoplasmic to the microsomal fraction (Fig. 7D–F; *p < 0.05, n = 3 independent experiments). Next, iHEK-Tau cells and the control iHEK-6/TR line were transfected with CD3δ-YFP and treated with Tet. Live-cell images were captured after 24 h of tau expression (Fig. 7G–J). Compared with Tet-treated iHEK-6/TR cells (Fig. 7G) and vehicle-treated iHEK-Tau cells (Fig. 7H), Tet-treated iHEK-Tau cells showed a 74% increase in punctate YFP-positive staining (Fig. 7I,J; **p < 0.01). Therefore, tau was able to impair ERAD directly, leading to UPR stress and the associated cell death that occurs when this pathway is activated.
Discussion
Brains from AD patients and tau transgenic mice have common pathological features: abnormal accumulation of the ERAD substrate CD3δ in the ER, accretion of ubiquitin on the ER membrane, and activation of the ER stress sensor PERK. One possible mechanism by which tau impairs ERAD is through interference with the ERAD complex, which contains proteins such as VCP and Hrd1. Therefore, we propose a pathological cascade model in tauopathies and AD that begins with tau accumulation impairing ERAD and results in activation of the UPR. Interestingly, this cascade could be stopped if soluble tau was depleted, suggesting that strategies aimed at reducing soluble tau could be beneficial for tauopathies including AD.
Our observation that PERK activation was increased in the AD and tau transgenic brain supports recent evidence suggesting that this UPR kinase plays an important role in neurodegenerative disorders (Hoglinger et al., 2011). For example, the largest genome-wide association study for PSP ever performed revealed that a SNP on the gene coding for PERK (EIF2AK3) was associated with this tauopathy. Interestingly, the SNP is present in the 5′UTR of EIF2AK3, suggesting that altered PERK expression may participate in the disease process. In addition, pathological studies have shown increased levels of pPERK in brain tissues of individuals suffering from tauopathies such as AD, FTD, and PSP (Hoozemans et al., 2007; Nijholt et al., 2012). However, signs of ER stress in neurodegenerative diseases are not exclusive to tauopathies: pathological studies from diseases such as Parkinson's disease, pol-glutamine diseases, and even glaucoma have shown that PERK and the ER stress system play a role in their pathogenesis as well (Ryu et al., 2002; Bennett et al., 2007; Doh et al., 2010).
The mechanisms contributing to ER stress and PERK activation in neurodegenerative diseases are not entirely clear, but several recent studies suggest that it is soluble, oligomeric species of disease-associated proteins such as Aβ42 and α-synuclein that are most likely responsible (Casas-Tinto et al., 2011; Castillo-Carranza et al., 2012; Colla et al., 2012). In one study, the investigators found that toxic prefibrillar α-synuclein oligomers form from within the ER lumen before the onset of disease signs and symptoms. Therefore, ER-associated soluble α-synuclein oligomers were the early driving force behind activation of ER stress. Here, we show that it is likely that soluble tau conformers also lead to tau-induced ER stress. This is important because recent evidence suggests that soluble oligomeric tau, but not insoluble tau, is most likely responsible for tau-induced neurotoxicity (O'Leary et al., 2010; de Calignon et al., 2012). Therefore, the formation of soluble tau intermediates may activate a cell damage pathway that is only visible pathologically using ER stress markers. This could help to explain why the number of neurons lost in the AD brain far exceeds the number of tau tangles. It is likely that multiple mechanisms of cell death are activated when tau begins to accumulate, and ER stress appears to be one of the major routes.
ER stress is triggered when there is an increase in nascent protein input and a diminution of output, and the latter effect is due to alterations in either protein secretion or ERAD. Because there is no evident change in protein secretion in AD or the tauopathic brain, we focused on ERAD damage as a possible mechanism for the ER stress. Indeed, ER stress can arise due to impairment of ERAD in in vitro models of synucleinopathy and a polyglutamine expansion disease, as shown by Cooper et al. (2006) and Duennwald and Lindquist (2008). In the latter study, polyglutamine caused toxicity via ER stress in cell culture models by trapping essential components of the ERAD machinery comprised largely of the VCP heterocomplex (Duennwald and Lindquist, 2008). With this in mind, we speculated that tau could similarly interfere with ERAD machinery. Indeed, we found that tau could associate with members of the VCP complex (Fig. 6), suggesting a possible mechanism for the ER stress occurring in the AD brain and tau transgenic mouse brain.
VCP is part of a dynamic heteroprotein complex that participates in varied cellular functions, including transport of ERAD proteins from the ER membrane to the proteasome, endolysosomal transport, and mitochondrial DNA repair. There are thus far 30 proteins that have been identified as binding the VCP complex (Liu et al., 2012b). Two such proteins, hrd1 and doa10, are associated with the ER membrane (Carvalho et al., 2006). They serve as adaptor proteins that anchor the VCP complex for it to “pick up” ubiquitinated proteins or “clients” and they do so independently of each other. Both proteins recruit specific client types for degradation. Interestingly, autosomal-dominant, gain-of-function mutations on the VCP gene are associated with the disease known as inclusion body myopathy with early-onset Paget disease and frontotemporal dementia (IBMPFD; for review, see Ju and Weihl, 2010; Hsueh, 2012). Individuals with these mutations suffer from three major pathologies that effectively compromise the musculoskeletal and nervous systems. IBMPFD patients present with signs of FTD, linking VCP malfunction to neurological and cognitive damage, but these patients lack any tau pathology. In light of our findings, the IBMPFD neurological phenotype may indicate a major route for tau toxicity: as tau accumulates, soluble toxic conformers are produced that interfere with normal VCP function, resulting in neurotoxicity and neurologic impairment. The mutations in VCP are sufficient on their own to cause toxicity without the need for tau accumulation. This is similar to how tau mutations are sufficient to cause neurotoxicity and dementia without the need for amyloid β accumulation.
The association of tau with hrd1, a VCP-associated E3 ubiquitin ligase, suggests yet another layer to the dysregulation of ERAD by tau. There are three types of ERAD, and each one requires the use of distinct protein assemblies. Hrd1 participates in ERAD-L and ERAD-M, degradation pathways that target proteins with lesions in their luminal or transmembrane domains, respectively (Hampton et al., 1996; Knop et al., 1996; Vashist and Ng, 2004; Carvalho et al., 2006; Denic et al., 2006). However, hrd1 is not required for ERAD-C, which disposes of membrane-bound proteins with lesions in their cytoplasmic tails. Therefore, the hrd1–tau association (Fig. 6) may implicate tau in specific impairment of ERAD-L and ERAD-M, but not ERAD-C. This would cause accumulation of proteins that have the same type of defects, potentiating disequilibrium of proteostasis. Alternatively, tau may exert a general blockage of ERAD, accounting for overall accumulation of ERAD-bound proteins in the ER. Identification of the type of ERAD affected will provide insight into valuable therapeutic approaches.
This study may help to explain the inconsistencies between tangle number and neuronal loss that exist in tauopathic brains (Gómez-Isla et al., 1997; Kril et al., 2002). We propose that the tangle may serve as a seed for additional toxic tau conformers to spawn. These conformers are then able to assault the neuron from within on multiple fronts, one being ERAD impairment and ER stress-mediated cell death. In addition, these tau species can spread to neighboring neurons, as suggested in recent studies (Clavaguera et al., 2009; Liu et al., 2012a; de Calignon et al., 2012). The effects of these spreading tau species once they are taken up by neurons are unknown; however, we speculate that they may have an even greater impact on ER stress given their likely involvement with the endocytic pathway that can converge on the ER machinery. Ultimately, the involvement of tau with the ER could lead to new insights about the mechanisms of tau dysfunction in tauopathies.
Footnotes
J.F.A. was supported by the Alzheimer's Association (Grant NIRGD-12-242642) and the Foundation for PSP/CBD and Related Brain Diseases. C.A.D. was supported by the National Institute of Neurological Disorders and Stroke–National Institutes of Health (Grant R01NS073899) and National Institute on Aging–National Institutes of Health (Grant R00AG031291). We thank Drs. Peter Davies, Lester Binder, and Leonard Petrucelli for the PHF1, Tau5, and E1 antibodies, respectively, and Dr. David Cribbs for facilitating the transfer of materials from the Brain Bank and Tissue Repository of the ADRC at University of California, Irvine.
The authors declare no competing financial interests.
References
- Abisambra JF, Fiorelli T, Padmanabhan J, Neame P, Wefes I, Potter H. LDLR expression and localization are altered in mouse and human cell culture models of Alzheimer's disease. PLoS One. 2010a;5:e8556. doi: 10.1371/journal.pone.0008556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abisambra JF, Blair LJ, Hill SE, Jones JR, Kraft C, Rogers J, Koren J, 3rd, Jinwal UK, Lawson L, Johnson AG, Wilcock D, O'Leary JC, Jansen-West K, Muschol M, Golde TE, Weeber EJ, Banko J, Dickey CA. Phosphorylation dynamics regulate Hsp27-mediated rescue of neuronal plasticity deficits in tau transgenic mice. J Neurosci. 2010b;30:15374–15382. doi: 10.1523/JNEUROSCI.3155-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bancher C, Brunner C, Lassmann H, Budka H, Jellinger K, Seitelberger F, Grundke-Iqbal I, Iqbal K, Wisniewski HM. Tau and ubiquitin immunoreactivity at different stages of formation of Alzheimer neurofibrillary tangles. Prog Clin Biol Res. 1989;317:837–848. [PubMed] [Google Scholar]
- Bays NW, Wilhovsky SK, Goradia A, Hodgkiss-Harlow K, Hampton RY. HRD4/NPL4 is required for the proteasomal processing of ubiquitinated ER proteins. Mol Biol Cell. 2001;12:4114–4128. doi: 10.1091/mbc.12.12.4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett EJ, Shaler TA, Woodman B, Ryu KY, Zaitseva TS, Becker CH, Bates GP, Schulman H, Kopito RR. Global changes to the ubiquitin system in Huntington's disease. Nature. 2007;448:704–708. doi: 10.1038/nature06022. [DOI] [PubMed] [Google Scholar]
- Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- Bolte S, Cordelières FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Braun S, Matuschewski K, Rape M, Thoms S, Jentsch S. Role of the ubiquitin-selective CDC48(UFD1/NPL4)chaperone (segregase) in ERAD of OLE1 and other substrates. EMBO J. 2002;21:615–621. doi: 10.1093/emboj/21.4.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho P, Goder V, Rapoport TA. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell. 2006;126:361–373. doi: 10.1016/j.cell.2006.05.043. [DOI] [PubMed] [Google Scholar]
- Casas-Tinto S, Zhang Y, Sanchez-Garcia J, Gómez-Velazquez M, Rincon-Limas DE, Fernandez-Funez P. The ER stress factor XBP1s prevents amyloid-beta neurotoxicity. Hum Mol Genet. 2011;20:2144–2160. doi: 10.1093/hmg/ddr100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo-Carranza DL, Zhang Y, Guerrero-Muñoz MJ, Kayed R, Rincon-Limas DE, Fernandez-Funez P. Differential activation of the ER stress factor XBP1 by oligomeric assemblies. Neurochem Res. 2012;37:1707–1717. doi: 10.1007/s11064-012-0780-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M, Jucker M, Goedert M, Tolnay M. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909–913. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colla E, Coune P, Liu Y, Pletnikova O, Troncoso JC, Iwatsubo T, Schneider BL, Lee MK. Endoplasmic reticulum stress is important for the manifestations of alpha-synucleinopathy in vivo. J Neurosci. 2012;32:3306–3320. doi: 10.1523/JNEUROSCI.5367-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, Cao S, Caldwell KA, Caldwell GA, Marsischky G, Kolodner RD, Labaer J, Rochet JC, Bonini NM, Lindquist S. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science. 2006;313:324–328. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Calignon A, Polydoro M, Suárez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, Spires-Jones TL, Hyman BT. Propagation of tau pathology in a model of early Alzheimer's disease. Neuron. 2012;73:685–697. doi: 10.1016/j.neuron.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denic V, Quan EM, Weissman JS. A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell. 2006;126:349–359. doi: 10.1016/j.cell.2006.05.045. [DOI] [PubMed] [Google Scholar]
- Dickey C, Kraft C, Jinwal U, Koren J, Johnson A, Anderson L, Lebson L, Lee D, Dickson D, de Silva R, Binder LI, Morgan D, Lewis J. Aging analysis reveals slowed tau turnover and enhanced stress response in a mouse model of tauopathy. Am J Pathol. 2009;174:228–238. doi: 10.2353/ajpath.2009.080764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doh SH, Kim JH, Lee KM, Park HY, Park CK. Retinal ganglion cell death induced by endoplasmic reticulum stress in a chronic glaucoma model. Brain Res. 2010;1308:158–166. doi: 10.1016/j.brainres.2009.10.025. [DOI] [PubMed] [Google Scholar]
- Duennwald ML, Lindquist S. Impaired ERAD and ER stress are early and specific events in polyglutamine toxicity. Genes Dev. 2008;22:3308–3319. doi: 10.1101/gad.1673408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Clavaguera F, Tolnay M. The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosci. 2010;33:317–325. doi: 10.1016/j.tins.2010.04.003. [DOI] [PubMed] [Google Scholar]
- Gómez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- Gordon MN, Schreier WA, Ou X, Holcomb LA, Morgan DG. Exaggerated astrocyte reactivity after nigrostriatal deafferentation in the aged rat. J Comp Neurol. 1997;388:106–119. doi: 10.1002/(SICI)1096-9861(19971110)388:1<106::AID-CNE7>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Gordon MN, Holcomb LA, Jantzen PT, DiCarlo G, Wilcock D, Boyett KW, Connor K, Melachrino J, O'Callaghan JP, Morgan D. Time course of the development of Alzheimer-like pathology in the doubly transgenic PS1+APP mouse. Exp Neurol. 2002;173:183–195. doi: 10.1006/exnr.2001.7754. [DOI] [PubMed] [Google Scholar]
- Hampton RY, Gardner RG, Rine J. Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol Biol Cell. 1996;7:2029–2044. doi: 10.1091/mbc.7.12.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5:897–904. doi: 10.1016/S1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- Höglinger GU, Melhem NM, Dickson DW, Sleiman PM, Wang LS, Klei L, Rademakers R, de Silva R, Litvan I, Riley DE, van Swieten JC, Heutink P, Wszolek ZK, Uitti RJ, Vandrovcova J, Hurtig HI, Gross RG, Maetzler W, Goldwurm S, Tolosa E, et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet. 2011;43:699–705. doi: 10.1038/ng.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoozemans JJ, van Haastert ES, Eikelenboom P, de Vos RA, Rozemuller JM, Scheper W. Activation of the unfolded protein response in Parkinson's disease. Biochem Biophys Res Commun. 2007;354:707–711. doi: 10.1016/j.bbrc.2007.01.043. [DOI] [PubMed] [Google Scholar]
- Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, Eikelenboom P, Scheper W. The unfolded protein response is activated in pretangle neurons in Alzheimer's disease hippocampus. J Pathol. 2009;174:1241–1251. doi: 10.2353/ajpath.2009.080814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsueh YP. From neurodevelopment to neurodegeneration: the interaction of neurofibromin and valosin-containing protein/p97 in regulation of dendritic spine formation. J Biomed Sci. 2012;19:33. doi: 10.1186/1423-0127-19-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarosch E, Taxis C, Volkwein C, Bordallo J, Finley D, Wolf DH, Sommer T. Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat Cell Biol. 2002;4:134–139. doi: 10.1038/ncb746. [DOI] [PubMed] [Google Scholar]
- Jinwal UK, Koren J, 3rd, Borysov SI, Schmid AB, Abisambra JF, Blair LJ, Johnson AG, Jones JR, Shults CL, O'Leary JC, 3rd, Jin Y, Buchner J, Cox MB, Dickey CA. The Hsp90 cochaperone, FKBP51, increases Tau stability and polymerizes microtubules. J Neurosci. 2010;30:591–599. doi: 10.1523/JNEUROSCI.4815-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju JS, Weihl CC. Inclusion body myopathy, Paget's disease of the bone and fronto-temporal dementia: a disorder of autophagy. Hum Mol Genet. 2010;19:R38–R45. doi: 10.1093/hmg/ddq157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kertesz A, Kawarai T, Rogaeva E, St George-Hyslop P, Poorkaj P, Bird TD, Muñoz DG. Familial frontotemporal dementia with ubiquitin-positive, tau-negative inclusions. Neurology. 2000;54:818–827. doi: 10.1212/WNL.54.4.818. [DOI] [PubMed] [Google Scholar]
- Khlistunova I, Biernat J, Wang Y, Pickhardt M, von Bergen M, Gazova Z, Mandelkow E, Mandelkow EM. Inducible expression of Tau repeat domain in cell models of tauopathy: aggregation is toxic to cells but can be reversed by inhibitor drugs. J Biol Chem. 2006;281:1205–1214. doi: 10.1074/jbc.M507753200. [DOI] [PubMed] [Google Scholar]
- Knop M, Finger A, Braun T, Hellmuth K, Wolf DH. Der1, a novel protein specifically required for endoplasmic reticulum degradation in yeast. EMBO J. 1996;15:753–763. [PMC free article] [PubMed] [Google Scholar]
- Kril JJ, Patel S, Harding AJ, Halliday GM. Neuron loss from the hippocampus of Alzheimer's disease exceeds extracellular neurofibrillary tangle formation. Acta Neuropathol. 2002;103:370–376. doi: 10.1007/s00401-001-0477-5. [DOI] [PubMed] [Google Scholar]
- Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, Duff K. Trans-synaptic spread of tau pathology in vivo. PLoS One. 2012a;7:e31302. doi: 10.1371/journal.pone.0031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Hei Y, Shu Q, Dong J, Gao Y, Fu H, Zheng X, Yang G. VCP/p97, down-regulated by microRNA-129–5p, could regulate the progression of hepatocellular carcinoma. PLoS One. 2012b;7:e35800. doi: 10.1371/journal.pone.0035800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori H, Kondo J, Ihara Y. Ubiquitin is a component of paired helical filaments in Alzheimer's disease. Science. 1987;235:1641–1644. doi: 10.1126/science.3029875. [DOI] [PubMed] [Google Scholar]
- Nijholt DA, van Haastert ES, Rozemuller AJ, Scheper W, Hoozemans JJ. The unfolded protein response is associated with early tau pathology in the hippocampus of tauopathies. J Pathol. 2012;226:693–702. doi: 10.1002/path.3969. [DOI] [PubMed] [Google Scholar]
- Oddo S, Vasilevko V, Caccamo A, Kitazawa M, Cribbs DH, LaFerla FM. Reduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem. 2006;281:39413–39423. doi: 10.1074/jbc.M608485200. [DOI] [PubMed] [Google Scholar]
- Okamura K, Kimata Y, Higashio H, Tsuru A, Kohno K. Dissociation of Kar2p/BiP from an ER sensory molecule, Ire1p, triggers the unfolded protein response in yeast. Biochem Biophys Res Commun. 2000;279:445–450. doi: 10.1006/bbrc.2000.3987. [DOI] [PubMed] [Google Scholar]
- O'Leary JC, 3rd, Li Q, Marinec P, Blair LJ, Congdon EE, Johnson AG, Jinwal UK, Koren J, 3rd, Jones JR, Kraft C, Peters M, Abisambra JF, Duff KE, Weeber EJ, Gestwicki JE, Dickey CA. Phenothiazine-mediated rescue of cognition in tau transgenic mice requires neuroprotection and reduced soluble tau burden. Mol Neurodegener. 2010;5:45. doi: 10.1186/1750-1326-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry G, Friedman R, Shaw G, Chau V. Ubiquitin is detected in neurofibrillary tangles and senile plaque neurites of Alzheimer disease brains. Proc Natl Acad Sci U S A. 1987;84:3033–3036. doi: 10.1073/pnas.84.9.3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich E, Kerem A, Fröhlich KU, Diamant N, Bar-Nun S. AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol Cell Biol. 2002;22:626–634. doi: 10.1128/MCB.22.2.626-634.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu EJ, Harding HP, Angelastro JM, Vitolo OV, Ron D, Greene LA. Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson's disease. J Neurosci. 2002;22:10690–10698. doi: 10.1523/JNEUROSCI.22-24-10690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat Res. 2005;569:29–63. doi: 10.1016/j.mrfmmm.2004.06.056. [DOI] [PubMed] [Google Scholar]
- Vashist S, Ng DT. Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J Cell Biol. 2004;165:41–52. doi: 10.1083/jcb.200309132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihl CC, Dalal S, Pestronk A, Hanson PI. Inclusion body myopathy-associated mutations in p97/VCP impair endoplasmic reticulum-associated degradation. Hum Mol Genet. 2006;15:189–199. doi: 10.1093/hmg/ddi426. [DOI] [PubMed] [Google Scholar]
- Yang H, Parkhouse RM. Differential activation requirements associated with stimulation of T cells via different epitopes of CD3. Immunology. 1998;93:26–32. doi: 10.1046/j.1365-2567.1998.00396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, Rapoport TA. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 2001;414:652–656. doi: 10.1038/414652a. [DOI] [PubMed] [Google Scholar]