


Abstract
DNA ligase I (Lig I) has key roles in chromosomal DNA replication and repair in the eukaryotic cell nucleus. In the budding yeast Saccharomyces cerevisiae the Lig I enzyme Cdc9p is also required for mitochondrial DNA replication and repair. In this report, dual nuclear–mitochondrial localization is demonstrated to be a property of the essential Lig I enzyme Cdc17 from the distantly related fission yeast Schizosaccharomyces pombe. Expression of nuclear and mitochondrial forms of Cdc17 from separate genes shows that, whereas expression of either protein alone is insufficient to restore viability to cells lacking endogenous Cdc17, co-expression restores full viability. In the nucleus, Lig I interacts with the sliding clamp proliferating cell nuclear antigen (PCNA) via a conserved PCNA interacting sequence motif known as a PIP box. Deletion of the PIP motif from the N-terminus of the nuclear form of Cdc17 fails to abolish Cdc17 function, indicating that PCNA binding by Cdc17 is not an absolute requirement for completion of S-phase.
INTRODUCTION
Successful chromosomal DNA replication in eukaryotic cells requires the complex interplay of a variety of distinct essential factors in a spatially and temporally coordinated manner. One such factor is DNA ligase I (Lig I), the ATP-dependent enzyme responsible for the ligation of the Okazaki fragments generated during discontinuous synthesis of the lagging strand (1,2). In yeasts, Lig I is essential for the completion of chromosomal DNA replication: budding or fission yeast cells carrying temperature-sensitive mutant forms of Lig I undergo S-phase arrest when shifted to the restrictive temperature (3–5), accumulating unligated Okazaki fragments (6). Yeast Lig I also has a key role in a number of DNA repair processes, including nucleotide excision repair and base excision repair. As a consequence, yeast Lig I mutants are hypersensitive to a range of DNA damaging agents, such as UV, bleomycin and MMS (7,8). In higher eukaryotes, Lig I is believed to play similar roles to the yeast enzyme, in both DNA replication and repair (9,10). However, unlike the situation in yeast, the mammalian Lig I protein is not essential for viability of cultured cells, although it is required for embryonic development (11,12).
On the basis of comparative sequence analysis, the Lig I proteins can be considered to be composed of three distinct functional domains (1) (Fig. 1A): a non-catalytic N-terminal domain (NTD) that is poorly conserved from species to species but which contains a nuclear localization signal (NLS) as well as a binding site for the essential replication factor proliferating cell nuclear antigen (PCNA) (see below), a non-catalytic central domain (NCD) that is reasonably well conserved between other ATP-dependent DNA ligase proteins but whose function is unknown, and a catalytic domain related in sequence, and presumably structure, to that of other ATP-dependent ligases (13,14). While the importance of the catalytic domain for Lig I function in vivo is not in doubt, the importance of the non-conserved N-terminal domain and the conserved NCD is less clear. It has been reported that a truncated form of the Cdc9 protein lacking the N-terminal 99 amino acids is able to support growth of cdc9Δ cells, albeit when expressed at levels considerably higher than the endogenous Cdc9 protein (15).
Figure 1.

(A) Domain structure of fission yeast Cdc17 protein. (Top) Schematic of Cdc17 protein structure showing the extent of the NTD, the conserved NCD and the catalytic domain (CD). The position of the key catalytic lysine residue (K416) is indicated. (Bottom) Expanded view of NTD, showing the position of the predicted MTS, the PCNA binding motif (PIP box) and part of the NLS. Note that the numbering of amino acid residues begins with the first methionine encoded by the ORF, i.e. the predicted initiating methionine for the mitochondrial Cdc17 pre-protein. (B) Helical wheel representation of first 18 amino acids of Cdc17, showing the amphipathic nature of the predicted MTS. Amino acids with hydrophobic side chains are shaded; those with polar or charged side chains are not, although charged amino acids are indicated by the + signs. (C) Sequence alignment of PIP box of Lig I proteins from S.pombe (Sp, Swiss-Prot accession no. P12000), S.cerevisiae (Sc, P04819), human (Hs, P18858), mouse (Mm, P37913) and Xenopus (Xl, P51892). The regions shown correspond to amino acids 20–35 (Sp), 34–49 (Sc) and 1–13 (Hs, Mm and Xl). (D) In vitro PCNA binding assay. Retention of recombinant Pcn1 by GST or GST-Cdc17(1–175) proteins determined by immunoblotting using anti-MRGS antibodies (see Materials and Methods). Lanes 1 and 2, GST alone incubated with (lane 2) or without Pcn1 (lane 1); lanes 3 and 4, GST-Cdc17(1–175) incubated with (lane 4) or without Pcn1 (lane 3). See Materials and Methods for details.
Higher eukaryotic Lig I has been shown to bind to the toroidal sliding clamp protein PCNA via a p21Cip1-like PCNA binding motif located at its extreme N-terminus (16). This eight-residue protein sequence motif, sometimes called a PIP (PCNA interacting protein) box (17), is required for localization of Lig I to sites of ongoing DNA replication (so-called replication factories) in the mammalian cell nucleus (18–20). Experiments with mammalian cell extracts point to important roles for the interaction between Lig I and PCNA in both DNA replication and repair (21).
In addition to playing vital roles in nuclear DNA replication and repair, the Saccharomyces cerevisiae Lig I protein Cdc9p also plays an important role in mitochondria (22,23). Saccharomyces cerevisiae cells express distinct nuclear and mitochondrial forms of Cdc9p from sequential in-frame start codons in the CDC9 ORF (22). The mitochondrial form of the protein is expressed from the first AUG codon in the ORF and is later processed to remove its N-terminal mitochondrial targeting sequence (MTS), giving rise to the mature mitochondrial Cdc9 protein. The nuclear form is expressed from a second AUG codon in the ORF and is unprocessed (22). Campbell and co-workers have characterized the in vivo role of mitochondrial Cdc9, elegantly demonstrating vital roles for the enzyme in mtDNA maintenance (23). In the absence of mitochondrial Cdc9 function, budding yeast cells cannot grow on medium containing glycerol as the sole carbon source, indicative of impairment of mitochondrial function (23). Moreover, inactivation of mitochondrial Cdc9p function during growth on glucose, or in stationary phase, results in a sharp decrease in mitochondrial DNA levels. Specific evidence for Cdc9 playing a key role in the repair of broken mtDNA was provided by targeting the restriction endonuclease EcoRI to mitochondria (23).
In this paper, the results of analysis of the structure and function of Cdc17, the essential Lig I protein in the fission yeast Schizosaccharomyces pombe (3,5,24,25) are presented. It is shown that, as in budding yeast (22), the NTD of Cdc17 is sufficient to target GFP to both the nucleus and mitochondria, consistent with a model in which the use of alternate in-frame start codons results in the production of distinct nuclear and mitochondrial forms of the Cdc17 protein. Mitochondrial function is known to be essential for fission yeast viability (26). Here it is shown that co-expression of nuclear and mitochondrial forms in vivo restores full viability to cdc17Δ cells, whereas expression of either protein alone does not. In addition, it is shown that the PCNA-binding PIP box at the N-terminus of the Cdc17 protein is not essential for nuclear Cdc17 function: cells co-expressing mitochondrial Cdc17 with a nuclear Cdc17 protein lacking the PIP box are viable. In contrast, Cdc17 proteins lacking the NCD are inactive in vivo, indicating that this domain plays a vital role in Cdc17 function.
MATERIALS AND METHODS
Fission yeast strains and media
The fission yeast strains used in this study were: cdc17-K42, cdc17-L16 and cdc17-M75 (27), leu1-32/leu1-32 ura4-D18/ura4-D18 his7-366/his7-366 ade6-M210/ade6-M216 h–/h+, and leu1-32 h–. Schizosaccharomyces pombe media and general techniques were essentially as described (28).
Construction of Cdc17-GFP expression plasmids
Plasmid pBluescript-GFP containing an Aequorea victoria GFP-S65T cDNA (a gift of Dr K. Hardwick, WTCCB, University of Edinburgh) was used as the template in a PCR with oligonucleotides GFP-E5 (5′-GTGTGTGAATTC ATGAGTAAAGGAGAAGAACT-3′) and GFP-B3 (5′-GTGTGTGGATCCTTATTTGTATAGTTCATCCA-3′). The resulting PCR product was restricted with EcoRI and BamHI, both sites being located in the primers (underlined), cloned into plasmid pGEX4T (Pharmacia) to make pGEX4T-GFP, and sequenced to confirm the absence of errors. Portions of the cloned cdc17 gene (as indicated in Fig. 4) were then amplified using oligonucleotide primer pairs with built-in XhoI (5′ primer) and EcoRI (3′ primer) sites. Full oligonucleotide sequences can be obtained from the authors on request. The XhoI–EcoRI fragments were then cloned into pGEX4T-GFP and sequenced, before the entire XhoI–BamHI Cdc17-GFP region was transferred into fission yeast expression plasmid pREP3X (29–32). All Cdc17-GFP expressing plasmids were then transformed into S.pombe leu1-32 h– cells and plated onto selective medium containing 5 µM thiamine to repress the nmt1 promoter. Individual colonies were then used to inoculate liquid cultures. Expression of the Cdc17-GFP proteins was induced by transferring cells into thiamine-free minimal medium and growing for a further 12–16 h, to an OD600nm of 0.1–0.2.
Figure 4.

Summary of subcellular localization studies. A schematic representation of the N-terminal part of the Cdc17 protein showing, in cartoon form, the structures of the mutant proteins whose localization was examined by GFP fusion. In two cases, although nuclear fluorescence was detectable, this was only at a very low level; these are indicated as (+). PIP box, PCNA binding motif. NLS, nuclear localization sequence. MTS, mitochondrial targeting sequence.
Deletion of cdc17+ from the chromosome
To delete the cdc17+ gene, the PCR-based gene targeting method of Bähler et al. was used (33). The kanMX6 cassette was PCR amplified using DeepVent polymerase (New England Biolabs) from plasmid pFA6a-kanMX6 using the following oligonucleotides: IM3 (5′-CGTACACGTGTTA CTTGTTCTTCAGGTGTACCCTCACTATTGTCAAATCT TATGAAATTCTTTACTCGTTGACTTGATTCGGATCC CCGGGTTAATTAA-3′) and IM4 (5′-ATTCATATTAGC CTCTTTGGGCAAATTTTAGCCGAACATTACCCTTTA TTATGACAGTAGCTCTCAGCAGTAACTCTCAGAAT TCGAGCTCGTTTAAAC-3′). A total of 15 µg of purified PCR product was transformed into S.pombe leu1-32/leu1-32 ura4-D18/ura4-D18 his7-366/his7-366 ade6-M210/ade6-M216 h–/h+ and transformants selected on YE agar plates containing G418 (GibcoBRL, used at 0.1 mg/ml in YE medium) according to the published method (33). Four stable integrants were recovered and screened by PCR using oligos IM5 (5′-GAGACAAGGAACACTCATACTATAT CAATG-3′), IM6 (5′-GCTAGGATACAGTTCTCACAT CACATCCG-3′), IM7 (5′-AATAGCCATCAGCTAGA GCA-3′) and IM8 (5′-GATCGCAGTGGTGAGTAACCA TGCATCATC-3′).
Cloning of cdc17+ by complementation
Plasmid pUR19-Cdc17 was isolated by screening an S.pombe genomic DNA library (34) for plasmids able to rescue the temperature sensitivity of a cdc17-K42 strain. cdc17-K42 leu1-32 h+ cells transformed with 1 µg of library DNA (a gift of Dr A.M.Carr, MRC Genome Stability Unit, University of Sussex, UK) were plated onto selective medium and incubated overnight at the permissive temperature of 28°C, then shifted to the restrictive temperature of 36.5°C. After 5 days, four colonies able to grow at 36.5°C were identified from ∼20 000 transformants. Plasmids were recovered from these and the vector-insert boundaries sequenced (data not shown). All four colonies were found to contain the same cdc17+ containing plasmid, designated pUR19-Cdc17.
Construction of Cdc17 expression plasmids
The cdc17+ ORF was PCR amplified from pUR19-Cdc17 using primers IM1 (5′-GATCGGATCCTTGTTATGCGAAC AGTATTTTCG-3′) and IM2 (5′-GATCGGATCCCAAACT TAATAATCTTCGGCAGCTGGGGAC-3′) with built-in BamHI restriction sites (underlined), subcloned into pTZ19R (Fermentas) and sequenced, to generate pTZ19R-Cdc17. Mutant cdc17 alleles expressing truncated Cdc17 proteins (see Fig. 5B) were generated by introducing a BamHI restriction site at the appropriate position in the gene by oligonucleotide-directed mutagenesis, using the MutaGene Phagemid kit, version 2 (BioRad), and sequenced. Full oligonucleotide sequences can be obtained from the authors on request. Point mutants of Cdc17 were also generated by oligonucleotide-directed mutagenesis and processed as above, as was the Cdc17-NΔ31R allele, in which sequences encoding the first 30 amino acids of Cdc17 were replaced by those encoding the first 10 amino acids of fission yeast Rfc1, including the Rfc1 PIP box (Rfc1 protein sequence: MSNSDIRSFF, with conserved residues underlined). The only exception to this was the gene encoding Cdc17-M20A/ΔNLS, which was produced by PCR overlap extension mutagenesis. As with the earlier alleles, the final product was sequenced to confirm the absence of errors introduced during the PCR process. Finally, the BamHI fragments were subcloned into the BamHI site of various pREP plasmids (29–32), as indicated in the text.
Figure 5.
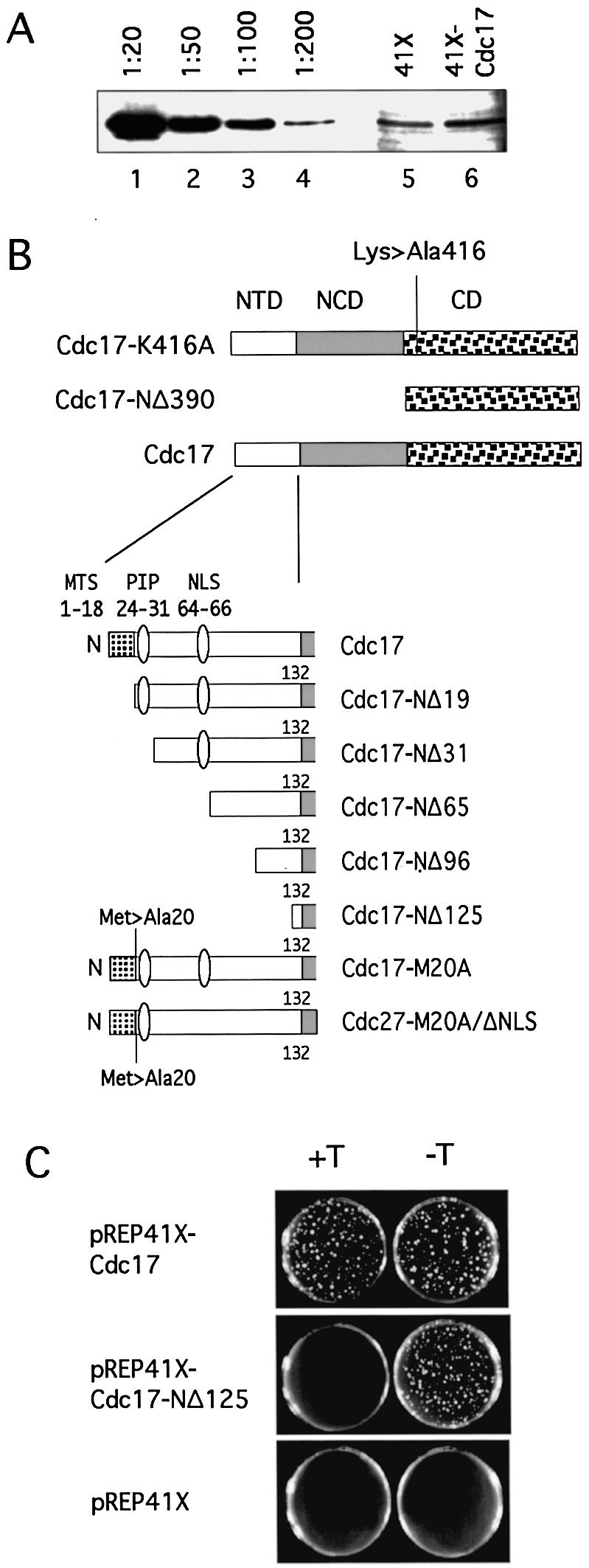
(A) Immunoblotting using affinity-purified anti-Cdc17 C-terminal peptide antibody of protein extracts prepared from wild-type fission yeast cells growing in the presence of thiamine and carrying either pREP41X (lane 5) or pREP41X-Cdc17 (lane 6). Expression of Cdc17 from the repressed nmt41 promoter leads to a small (<2-fold) increase in the amount of Cdc17 in cells. Lanes 1–4 show immunoblotting of various dilutions of an extract prepared from wild-type cells carrying pREP41X-Cdc17 but grown in the absence of thiamine. Consistent with previous estimates, derepression of the nmt41 promoter in the absence of thiamine leads to a 50–100-fold increase in the amount of expressed protein. (B) Schematic representation of the structures of truncated and mutant Cdc17 proteins. (C) Rescue of haploid cdc17-kanMX6 cells by pREP41X-Cdc17, pREP41X-Cdc17-NΔ125 and pREP41X in the presence or absence of thiamine (+T, –T, respectively). In each case, 1000 cells were plated per plate. Phenotypes were scored after 5 days incubation at 32°C. See text for details.
Plasmid shuffling protocol
First, the heterozygous cdc17+/cdc17::kanMX6 leu1-32/leu1-32 ura4-D18/ura4-D18 his7-366/his7-366 ade6-M210/ade6-M216 h–/h+ diploid was transformed with pUR19-Cdc17. Transformants were sporulated and the spores subjected to random spore analysis to identify G418-resistant ura+ ade– haploids of genotype cdc17::kanMX6 leu1-32 ade6– his7-366 ura4-D18 (pUR19-Cdc17). This strain was then transformed with various pREP41X-Cdc17 constructs. Transformant cells carrying both plasmids were then replica plated twice on supplemented EMM containing uracil before being grown up in supplemented liquid EMM medium to mid-log phase. Cell number was determined using a Coulter counter and cells were plated (∼1000 cells per plate) onto EMM containing uracil and 5′-fluoroorotic acid (5-FOA) with or without thiamine. 5-FOA plates were prepared by adding the solid (to a final concentration of 1 mg/ml) to supplemented EMM held at 55°C and keeping it at 55°C until dissolved. After plating, plates were incubated at 32°C for the indicated times and the results were judged according to colony number, size and microscopic examination.
Fluorescence microscopy
To visualize GFP fluorescence, cells were grown to an OD of 0.1–0.2 in EMM medium; 1 ml of culture was then centrifuged briefly and ∼2 µl of the loose cell pellet pipetted onto a glass slide, dried and covered with a cover slip. The cells were then observed using a Zeiss Axioplan 2 fluorescence microscope. For MitoTracker staining, 1 ml of cell culture was spun down before the cells were resuspended in 1 ml of EMM containing 200 nM MitoTracker Red CM-H2Xros (Molecular Probes), incubated for 30 min in the dark, washed once with EMM, then visualized as above.
In vitro PCNA binding assays
Assays were performed as described previously (35) using recombinant MRGS(His6)-tagged S.pombe Pcn1 expressed from pQE32-Pcn1 and GST-Cdc17(1–175) expressed from pGEX4T-3-Cdc17(1–175), except that bound PCNA was visualized by immunoblotting using anti-MRGS antibodies (Qiagen). pGEX4T-3-Cdc17(1–175) was constructed by amplifying sequences encoding the first 175 amino acids of Cdc17 from an S.pombe cDNA library using oligonucleotdies with in-built BamHI (5′ oligo) and EcoRI (3′ oligo) sites, as above, and cloning into pGEX4T-3 (Amersham Pharmacia).
Anti-Cdc17 antibodies
For production of anti-Cdc17 antibodies, the catalytic domain of Cdc17 was expressed as a GST fusion protein as follows. First, the BamHI fragment of pTZ19R-Cdc17-NΔ390 was cloned into plasmid pGEX6P-1B, a modified form of pGEX6P-1 (Amersham Biosciences) in which the reading frame of the polylinker BamHI site is altered and several additional polylinker restriction sites are absent (S.A.MacNeill, unpublished results). The GST-Cdc17-NΔ390 fusion protein was then expressed in Escherichia coli BL21 (DE3) pLysS at 37°C for 3 h. The recombinant protein produced in this way formed insoluble inclusion bodies that were purified by the following method (Y. Adachi, personal communication). Harvested cell pellets were frozen at –80°C, thawed and resuspended in TBS containing 1 mM β-mercaptoethanol and 0.2 mM PMSF. Following sonication for 20 s at high power using an ultrasonic processor, the suspension was spun down, resuspended in the same buffer supplemented with 2% Triton X-100, and sonicated briefly (5 s) at low power. The cells were then pelleted, washed in the same buffer, and resuspended in TBS containing 1 mM β-mercaptoethanol. This crude extract was then mixed with 2× SDS–PAGE sample buffer, heated to 95°C for 5 min, and run on a small preparative SDS–PAGE gel. The gel was stained with Coomassie blue, and the GST-Cdc17-NΔ390 band excised, crushed and incubated overnight in elution buffer (15 mM NH4HCO3, 0.025% SDS, 1 mM DTT, 0.1 mM PMSF) at room temperature on a rotating wheel. The crushed gel was then washed once more in elution buffer, before the supernatants were pooled and lyophilized. Two rabbits were each immunized with 400 µg of GST-Cdc17-NΔ390 by Diagnostics Scotland (Edinburgh, UK) according to standard procedures.
Anti-peptide antisera were raised in two rabbits against a synthetic peptide corresponding to the C-terminal 20 amino acids of Cdc17 by Sigma-Genosys (Cambridge, UK). Approximately 2–3 mg of peptide coupled to keyhole limpet haemocyanin (KLH) was injected into two rabbits according to standard protocols. Both antisera were then affinity purified against recombinant GST-Cdc17-NΔ390 according to standard procedures.
Immunofluorescence
Cells expressing various forms of Cdc17 were grown in minimal medium in the absence of thiamine to mid-log phase at 32°C, fixed with formaldehyde/glutaraldehyde and processed according to previously published procedures (36). Affinity-purified rabbit anti-Cdc17 C-terminal peptide antisera were used (see above), followed by anti-rabbit Cy3 (Jackson Immunoresearch) or Alexa488 (Santa Cruz, a gift of Dr H.Ohkura, WTCCB) secondary antibodies. Cells were viewed using a Zeiss Axioplan microscope. Images were captured using a CCD camera and analysed using SmartCapture VP software (Vysis Ltd, UK).
Cloning and sequencing of temperature-sensitive cdc17 alleles
Genomic DNA was prepared from the temperature-sensitive cdc17-K42, cdc17-M75 and cdc17-L16 strains and amplified using DeepVent polymerase (New England Biolabs) and primers IM1 and IM2 described above. The resulting PCR products were cloned into pTZ19R (BamHI site) and multiple isolated of each sequenced. A single deviation from the wild-type cdc17+ sequence was observed in each case (see Results).
RESULTS
Domain structure of Cdc17
The fission yeast cdc17+ gene has the capacity to encode a protein of 768 amino acids in length that can be considered as having three distinct domains (1,24,25): an NTD of ∼132 amino acids, a central domain (the NCD) spanning amino acids 133–390, and a C-terminal catalytic domain, from amino acid 391 to 768 (Fig. 1A and data not shown). The catalytic domain contains a highly conserved lysine residue, lysine 416 (K416), to which AMP is covalently bound in the first (adenylation) step of the ligation reaction (37).
Mitochondrial matrix targeting of budding yeast Lig I protein Cdc9p requires an N-terminal MTS spanning amino acids 1–23 (22). This sequence has the potential to form an amphipathic α-helix of the type commonly found in mitochondrial preproteins (38). As noted previously (22), a similar sequence is found at the N-terminus of fission yeast Cdc17 (illustrated in Fig. 1B), suggesting that Cdc17 may also be localized to mitochondria. Further evidence in support of this comes from computational analysis of the Cdc17 protein sequence: both the iPSORT (http://hypothesiscreator.net/iPSORT/) and Mitoprot II (http://ihg.gsf.de/ihg/mitoprot.html) applications predict Cdc17 to be localized to mitochondria (39,40).
In common with other eukaryotic Lig I proteins, the N-terminus of Cdc17 contains a PIP box (17), a canonical PCNA binding motif of the type first identified in the mammalian DNA replication inhibitor p21Cip1 (41). Figure 1C shows the sequence of the PCNA binding motif of Cdc17 aligned with similar sequences from the Lig I proteins from human, mouse, Xenopus and budding yeast. In vitro binding assays using recombinant proteins confirm that Cdc17 and Pcn1, the fission yeast PCNA protein, do indeed interact via the NTD (Fig. 1D).
Nuclear and mitochondrial targeting of Cdc17
To examine the in vivo role of the NTD in localizing Cdc17, a series of Cdc17-GFP fusion proteins were expressed in wild-type S.pombe cells. In these experiments expression of the fusion protein genes was under the control of the thiamine-repressible nmt1 promoter (29–31) in plasmid pREP3X (see Materials and Methods). Transformed cells were routinely cultured in the presence of 5 µM thiamine (no fluorescence was detectable under these growth conditions), before being transferred to thiamine-free medium at 30°C for 12–16 h to induce expression of the Cdc17-GFP fusion protein. Live cells were then viewed under the fluorescence microscope to determine GFP localization.
Initially, amino acids 1–175 of Cdc17 were fused to GFP (S65T) to generate a Cdc17(1–175)-GFP fusion protein. Amino acids 1–175 correspond to the entire NTD of Cdc17 and a small part of the conserved NCD. Figure 2A shows cells expressing the Cdc17(1–175)-GFP fusion protein: bright nuclear fluorescence is seen, with weaker signals in ribbon-like patterns in the cytoplasm. GFP alone localizes throughout the cell (data not shown). The cytoplasmic fluorescence pattern seen with Cdc17(1–175)-GFP resembles previously published images of S.pombe mitochondria (42–44), suggesting that Cdc17(1–175)-GFP is targeted to both nucleus and mitochondria, as predicted by Stirling and co-workers (22). To confirm this, cells expressing Cdc17(1–175)-GFP were stained with the mitochondrion-specific dye MitoTracker Red (see Materials and Methods). The cytoplasmic Cdc17-GFP and mitochondrial signals were seen to colocalize (Fig. 2B and G). Thus, the N-terminal 175 amino acids of Cdc17 are sufficient to target GFP to both the nucleus and mitochondria of fission yeast cells.
Figure 2.

Subcellular localization of Cdc17-GFP fusion proteins. (A and B) Wild-type cells expressing Cdc17(1–175)-GFP viewed under fluorescence. In (B), a cell is shown under fluoresence (left) and stained with MitoTracker (centre), with the merged image shown at the right. (C–F) Wild-type cells expressing Cdc17(1–175)-GFP [shown in (C)], Cdc17(31–175)-GFP (D), Cdc17(66–175)-GFP (E) and Cdc17(1–175/ΔNLS)-GFP (F). (G–J) Cells expressing Cdc17(1–175)-GFP [shown in part (G)], Cdc17(20–175)-GFP (H), Cdc17(1–175/M20A)-GFP (I) and Cdc17(1–175/M20A/ΔNLS)-GFP (J), with GFP fluorescence on the left, Mitotracker Red staining in the center and the merged image on the right.
Mitochondrial targeting sequence
To examine the role of amino acids 1–20 for mitochondrial targeting, the localization of a Cdc17-GFP fusion protein in which these amino acids were deleted was examined. Consistent with expectations, the Cdc17(20–175)-GFP fusion protein was exclusively nuclear in location (compare Fig. 2G with H), indicating that amino acids 1–20, encompassing the putative MTS, are required for mitochondrial targeting.
Support for this result was obtained by indirect immunofluorescence microscopy (Fig. 3). Wild-type cells expressing either full-length Cdc17 or Cdc17-NΔ31 from the intermediate strength nmt41 promoter in plasmid pREP41X (see below) were probed with affinity purified anti-Cdc17 C-terminal peptide antisera (see Materials and Methods). Note that in this case the proteins are not present as GFP fusions. Bound antibody was detected using fluorescent secondary antibodies conjugated with Cy3 (Fig. 3A and B). In cells expressing full-length Cdc17, both nuclear and cytoplasmic (mitochondrial) fluorescence was detected, whereas in cells expressing Cdc17-NΔ31, the cytoplasmic signal is absent suggesting that the latter protein is incapable of localizing to mitochondria.
Figure 3.
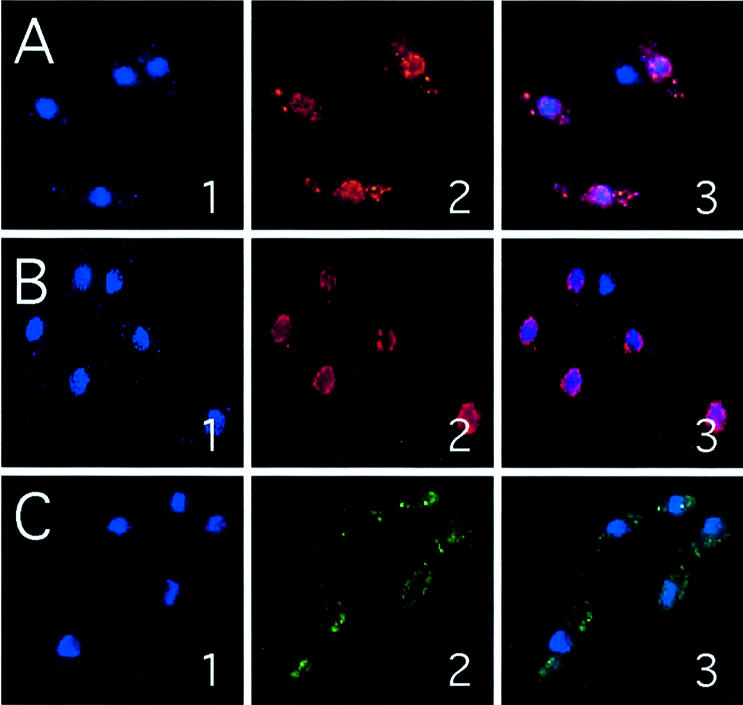
Indirect immunofluorescence images of cells expressing Cdc17, Cdc17-NΔ31 or Cdc17-M20A. Cells expressing full-length Cdc17 (A), Cdc17-NΔ31 (B) or Cdc17-M20A (C) from plasmid pREP41X were fixed and stained with the DNA binding dye DAPI (panel 1), or anti-Cdc17 C-terminal peptide antisera followed by fluorescent secondary antibody (panel 2). Merged images are shown in the panel 3. For technical reasons, different secondary antibodies were used for Cdc17 and Cdc17-NΔ31 (Cy3) versus Cdc17-M20A (Alexa 388). See Materials and Methods for details.
Nuclear localization signal
In order to map sequences responsible for nuclear localization of Cdc17, additional N-terminal truncated GFP fusion proteins were tested (Fig. 2, results summarized in Fig. 4). These results implicated sequences between amino acids 33 and 65 as being responsible for nuclear localization of Cdc17: while Cdc17(32–175)-GFP was nuclear localized (Fig. 2D), Cdc17(66–175)-GFP was not (Fig. 2E).
Inspection of the Cdc17 sequence shows that this region does not contain a classical NLS but does include a short stretch of basic amino acids (K62R63K64) that could form part of a bipartite or non-canonical NLS. To test this, the localization of a fusion protein in which these three basic amino acids were replaced with alanine was examined: Cdc17(1–175/ΔNLS)-GFP. Expression of this protein resulted in significantly reduced nuclear fluorescence (compare Fig. 2F with C). Amino acid residues 62–64 are therefore necessary for effective nuclear targeting of Cdc17. Whether amino acids 32–65 were sufficient for nuclear targeting was also examined, but they were not: the localization of a Cdc17(32–65)-GFP fusion protein was indistinguishable from that of GFP alone (data not shown). Thus amino acids 62–64, while being necessary for nuclear localization of Cdc17, are not sufficient, indicating that sequences beyond amino acid 65 play a vital role in nuclear targeting (see Discussion).
Requirement for in-frame AUG codon for nuclear Cdc17 localization
In budding yeast, translation of nuclear Cdc9p is initiated from an in-frame AUG codon (22). To determine if this is also likely to be the case in fission yeast, the effect of mutating the second in-frame AUG codon in the cdc17+ ORF on the localization of the Cdc17(1–175)-GFP fusion protein was examined. Replacing the AUG with GCG (resulting in a methionine to alanine change at amino acid 20 in the encoded protein) was found to cause a significant decrease in the amount of the Cdc17-GFP fusion detectable in the nucleus without affecting mitochondrial GFP levels. Cells expressing Cdc17(1–175/M20A) are shown in Figure 2I. Similar results (normal mitochondrial localization, greatly reduced nuclear fluorescence) were also seen with Cdc17-M20A expressed in wild-type cells by indirect immunofluorescence (Fig. 3C). Interestingly, simultaneous mutation of the second in-frame AUG and deletion of the putative NLS [the Cdc17(1–175/M20A/ΔNLS) protein] resulted in undetectable nuclear fluorescence (Fig. 2J), indicating that residual nuclear localization seen with Cdc17(1–175/M20A) is due to the presence of the intact NLS.
Functional assays
Next, the part played by the NTDs of Cdc17 in the in vivo function of the protein was investigated. To do this, PCR-mediated gene targeting was used to construct a diploid S.pombe strain in which the cdc17+ gene had been deleted from one chromosome and replaced with the kanMX6 marker cassette (see Materials and Methods for details). Following meiosis and sporulation, tetrad dissection demonstrated that the cdc17::kanMX6 spores were capable of germination but not colony formation. Thus, as expected, cdc17+ is an essential gene (data not shown).
To examine the role of the NTD of Cdc17, plasmid vectors were constructed to express various mutant forms of Cdc17 under the control of the thiamine-repressible nmt41 promoter (29–31). The choice of these expression conditions was based on quantitation of Cdc17 protein levels in transformed cells by western blotting using affinity purified anti-Cdc17 C-terminal peptide antisera (see Materials and Methods). These results indicated that the level of Cdc17 protein resulting from expression from the nmt41 promoter, in the presence of thiamine (i.e. with the promoter in its repressed state), was at most 2-fold higher than the endogeneous Cdc17 level (Fig. 5A, lanes 5 and 6). Consistent with this, expression of Cdc17 under these conditions was found to be able to fully rescue cdc17::kanMX6 cells without causing the cell elongation seen at higher or lower expression levels (cdc17::kanMX6 cells carrying plasmid pREP41X-Cdc17 divided at 15.1 ± 1.4 µm when grown in thiamine-containing minimal medium at 32°C, whereas cdc17::kanMX6 cells carrying plasmid pREP81X-Cdc17 divided at 18.8 ± 5.3 µm under the same conditions; expression levels from the nmt81 promoter in pREP81X are generally 10-fold lower than from the nmt41 promoter in pREP41X).
In all, 10 Cdc17 expression constructs were generated, including the full-length wild-type gene (diagrammed in Fig. 5B). To test the ability of the mutant proteins to rescue cdc17::kanMX6 cells, a plasmid shuffle strategy was used. First, a cdc17::kanMX6 haploid strain containing plasmid pUR19-Cdc17 was isolated. This plasmid, which carries the entire cdc17+ gene region as well as the ura4+ selectable marker, was originally isolated from a genomic DNA library on the basis of its ability to rescue the temperature-sensitive cdc17-K42 mutant (see Materials and Methods). Next, each of the pREP41X-Cdc17 plasmids was transformed into this strain. Transformants obtained carrying both pUR19-Cdc17 and the relevant pREP41X-Cdc17 plasmid were then grown up in supplemented EMMT medium (EMM containing 5 µM thiamine) and ∼1000 cells per plate plated on EMMT medium containing uracil and 5-FOA, to select for clones in which the ura+ pUR19-Cdc17 plasmid had been lost. Following 4–6 days incubation at 32°C, the results were scored by comparing colony number and size, and by microscopic examination. The results, summarized in Table 1, are discussed below.
Table 1. In vivo function of truncated Cdc17 proteins.
| Plasmid | Off | On |
|---|---|---|
| pREP41X |
– |
– |
| pREP41X-Cdc17 |
+++ |
+++ |
| pREP41X-Cdc17-K416A |
– |
– |
| pREP41X-Cdc17-NΔ19 |
– |
+++ |
| pREP41X-Cdc17-NΔ31 |
– |
+++ |
| pREP41X-Cdc17-NΔ65 |
– |
+++ |
| pREP41X-Cdc17-NΔ96 |
– |
+++ |
| pREP41X-Cdc17-NΔ125 |
– |
+++ |
| pREP41X-Cdc17-NΔ390 |
– |
– |
| pREP41X-Cdc17-M20A |
– |
+++ |
| pREP81X-Cdc17 | ++ | +++ |
Plasmids were tested for their ability to rescue cdc17Δ cells by the plasmid shuffle assay described in the text. Off, nmt promoter in plasmid pREP41X in repressed state. On, nmt promoter derepressed. Growth was scored subjectively after 4 days at 32°C as follows: +++, indistinguishable from wild type; ++, good growth but poorer than wild type; +, reduced growth; –, no growth.
In the absence of wild-type Cdc17 expression from plasmid pUR19-Cdc17, plasmid pREP41X was unable to support growth of the cdc17::kanMX6 haploid: no colonies were formed on the 5-FOA plates (Table 1 and Fig. 5C, bottom). Plasmid pREP41X-Cdc17 was able to fully support growth of the cdc17::kanMX6 (Table 1 and Fig. 5C, top). The cdc17::kanMX6 cells carrying pREP41X-Cdc17 grew well and underwent cell division at a cell length indistinguishable from wild type (with cell size at division of ∼15.1 µm, as above). In contrast, none of the mutant constructs tested was able to rescue in cells grown in the presence of thiamine, to repress the nmt41 promoter (see Fig. 5C, middle, for an example and Table 1 for summary). In all but one case, the cells formed abortive microcolonies of elongated cells. The exception to this were those cells expressing Cdc17-NΔ19. In this case, elongated cells were not seen; instead the microcolonies comprised a small number of swollen, almost spherical cells.
The ability of the mutant proteins to rescue the cdc17::kanMX6 strain when grown in the absence of thiamine was also tested. Under these conditions, expression from the nmt41 promoter is induced, leading to significant overproduction of the Cdc17 protein: Figure 5A (lanes 1–4) shows that the level of wild-type Cdc17 protein present in cells grown in the absence of thiamine is 100–200-fold greater than endogenous Cdc17 levels. Consistent with results obtained in other organisms, overproduction of all but two of the mutant Cdc17 proteins was sufficient to restore viability to cdc17::kanMX6 haploid cells (Table 1). The exceptions were the catalytically inactive Cdc17-K416A protein and Cdc17-NΔ390 (Table 1).
Separate expression of nuclear and mitochondrial Cdc17 proteins
Next, whether co-expressed nuclear and mitochondrial forms of the Cdc17 would support growth of cdc17::kanMX6 cells was tested (Fig. 6A). To achieve this, a cdc17::kanMX6 haploid strain carrying plasmid pREP41X-Cdc17-NΔ19 was created by transforming this plasmid into cdc17::kanMX6 cells carrying pUR19-Cdc17 and counter-selecting using 5-FOA on medium lacking thiamine to lose pUR19-Cdc17. Under these conditions (i.e. with the nmt promoter fully derepressed due to the absence of thiamine in the growth medium), the substantial overproduction of the nuclear Cdc17-NΔ19 protein is sufficient to support growth of the cdc17::kanMX6 cells. Next, this strain was transformed with plasmids pREP42X, pREP42X-Cdc17-M20A and pREP42X-Cdc17-M20AΔNLS expressing mitochondrial Cdc17 proteins. (Note that, in contrast to the LEU2-containing pREP41X, pREP42X carries the ura4+ marker, allowing both this plasmid and pREP41X to be selected independently in the same cell.) Transformants obtained in the absence of thiamine were then grown in liquid medium and plated onto EMM medium containing thiamine, to repress the nmt41 promoters in both plasmids.
Figure 6.

Role of PIP box in Cdc17 function. (A) Schematic outline of strategy to test requirement for the Cdc17 PIP box for mitotic growth. Haploid cdc17::kanMX6 cells containing pUR19-Cdc17 were (1) transformed with either plasmid pREP41X-Cdc17-NΔ19 or -Cdc17-NΔ31 (expressing nuclear Cdc17, indicated as Nu-Cdc17, with/without the PIP box), (2) plated on 5-FOA on medium lacking thiamine to allow for loss of pUR19-Cdc17, then (3) transformed with either pREP42X-Cdc17-M20A or pREP42X-Cdc17-M20AΔNLS (expressing mitochondrial Cdc17, indicated as Mt-Cdc17, with/without the internal NLS), before finally being plated on medium containing thiamine (gray box) to repress expression from the nmt41/42 promoters. (B) Results of assay using pREP42X-Cdc17-M20A at the final stage. Cells co-expressing Cdc17-M20A and Cdc17-NΔ19 were ∼95% viable and divided at near normal cell size (left), whereas cells co-expressing Cdc17-M20A and Cdc17-NΔ31 were only ∼15% viable and divided at a significantly elongated cell size (right). Phenotypes were scored after 5 days incubation at 32°C. See text for details and discussion.
The results of this experiment were as follows. As noted above, in cells grown in the presence of thiamine, plasmid pREP41X-Cdc17-NΔ19 alone is unable to rescue the cdc17::kanMX6 defect (Table 1). Predictably, when the cdc17::kanMX6 cells also contain pREP42X, the same result is seen. However, when pREP42X is replaced by either pREP42X-Cdc17-M20A or pREP42X-Cdc17-M20AΔNLS, growth on thiamine is restored (Fig. 6B, left). Cells carrying both plasmids displayed normal cell morphology and underwent cell division at a cell length (14.4 ± 3.3 µm) close to wild type. These results indicate that co-expression of nuclear and mitochondrial forms of Cdc17 is able to substitute for loss of cdc17+ function.
Role of PCNA binding motif (PIP box) in vivo
Next, the same assay was used to test whether expression of the PIP-box mutant Cdc17-NΔ31 would be sufficient to rescue cdc17::kanMX6 when co-expressed with either of the mitochondrial proteins Cdc17-M20A or Cdc17-M20AΔNLS (Fig. 6A). It was found that, in both cases, co-expression of the nuclear Cdc17-NΔ31 protein alongside the mitochondrial Cdc17 resulted in a significant reduction in colony forming ability, to only ∼15% of the numbers of the Cdc17-NΔ19 control (see Fig. 6B, right, for Cdc17-M20A). In addition, the cells were highly elongated compared with the control (32.2 versus 14.4 µm). It is possible to conclude from this that the PIP box makes a significant contribution to nuclear Cdc17 function but that it is not essential for function. Interestingly, when the viable colonies from the first plates were grown up in liquid medium and replated onto the same medium, viability was restored to near control levels (∼80% compared with >95% for similarly treated control cells) and cell size at cell division was only marginally increased over the control. In short, prolonged growth results in improved cell viability, with the viable cells dividing at a reduced cell size compared with the initial plating on thiamine-containing minimal medium. The most likely explanation for this is that cells with elevated Cdc17 levels are being selected for. However, immunoblotting showed that no significant increases in Cdc17 protein level could be detected over time in cells growing on thiamine-containing medium (data not shown).
Overproduction of Cdc17 proteins
Although the PIP box is not absolutely required for nuclear Cdc17 function, it was found that gross overproduction of Cdc17 proteins containing the PIP box interfered with cell cycle progression. Specifically, overproduction, in wild-type cells, of full-length Cdc17 from the strong nmt1 promoter in plasmid pREP3X caused cell elongation, with cells undergoing division at ∼22.3 µm. This effect was also seen when the overproduced protein lacked the MTS at the N-terminus (Cdc17-NΔ19; cell size at cell division of ∼21.4 µm, compared with wild-type cells at ∼15 µm), or when the entire C-terminal catalytic domain was deleted (Cdc17-CΔ391; ∼21.1 µm) but was reduced when the PIP box was deleted (Cdc17-NΔ31; ∼17.6 µm). A construct in which sequences encoding the Cdc17 PIP box were replaced with those encoding the PIP box of the large subunit of fission yeast RF-C (Cdc17-NΔ31-R, see Materials and Methods for details) was also tested. Interestingly, Cdc17-NΔ31-R was a more potent inhibitor of cell cycle progression than Cdc17-NΔ19; cells carrying plasmid pREP41X-Cdc17-NΔ31-R underwent division at ∼21 µm in the absence of thiamine, compared with ∼15 µm for cells carrying pREP41X-Cdc17-NΔ19.
Role of the conserved non-catalytic domain (NCD)
In addition to analysing the function of the NTD of Cdc17, the role of the adjacent domain, the conserved NCD, was also examined. The NCD is a conserved feature of all eukaryotic and archaeal ATP-dependent DNA ligases, although the degree of conservation between proteins from different species varies (1). To test whether the NCD was required for Cdc17 function in fission yeast, a plasmid expressing a truncated form of Cdc17, called Cdc17-NΔ390, lacking both the NTD and the NCD, was constructed. pREP41X-Cdc17-NΔ390 was transformed into cdc17::kanMX6 cells containing pUR19-Cdc17, and transformants plated on medium containing 5-FOA but lacking thiamine, to ensure high level expression of Cdc17-NΔ390. No colonies were obtained by this method (Fig. 7A), suggesting that Cdc17-NΔ390, unlike Cdc17-NΔ125, is unable to compensate for loss of the wild-type Cdc17 protein when expressed from pREP41X. However, the difference observed between Cdc17-NΔ125 and Cdc17-NΔ390 may simply be due the relative level of the two proteins present in cells, as western blotting of wild-type cells transformed with either pREP41X-Cdc17-NΔ125 or pREP41X-Cdc17-NΔ390 showed that the Cdc17-NΔ390 protein was present at low levels only (Fig. 7B). However, when the Cdc17-NΔ390 protein was expressed under the control of the full-strength nmt1 promoter in plasmid pREP3X to levels considerably higher than those of the endogenous Cdc17 protein (Fig. 7B, compare lanes 1 and 6), it was still unable to rescue either cdc17::kanMX6 or the temperature-sensitive cdc17-K42 and cdc17-L16 alleles under restrictive conditions (data not shown).
Figure 7.
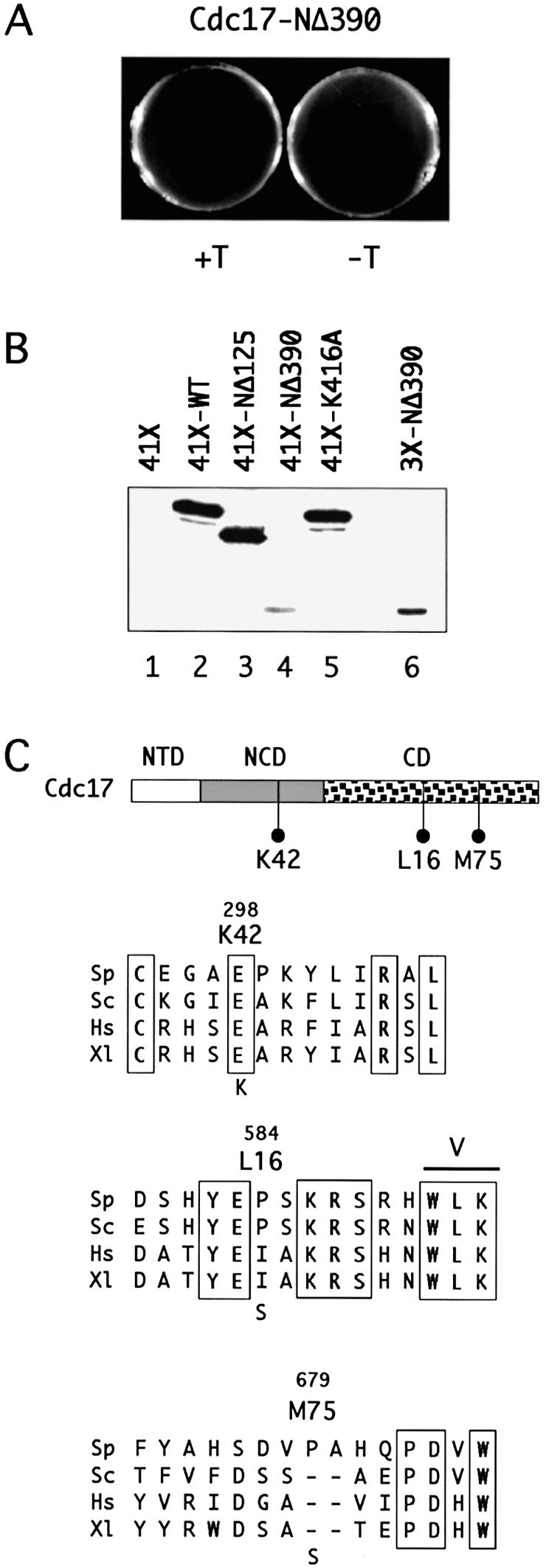
Role of NCD in Cdc17 function. (A) Expression of Cdc17-NΔ390 from nmt41 promoter fails to rescue haploid cdc17::kanMX6. (B) Immunoblot of protein extracts prepared from wild-type cells (grown in the absence of thiamine) expressing Cdc17, Cdc17-NΔ125, Cdc17-NΔ390 and Cdc17-K416A from the nmt41 promoter in plasmid pREP41X (lanes 2–5, respectively) or Cdc17-NΔ390 from the full-strength nmt1 promoter in pREP3X (lane 6). Lane 1 contains protein prepared from cells carrying the empty vector pREP41X. Note that under these conditions, detection of the endogenous Cdc17 protein was not possible. (C) Location of mutations in temperature-sensitive Cdc17 proteins, together with protein sequence alignments showing context of mutations. Sp, S.pombe. Sc, S.cerevisiae. Hs, human. Xl, Xenopus. See legend to Figure 1 for Swiss-Prot accession numbers.
These experiments suggest that the NCD plays an important, indeed indispensable, role in Cdc17 function in vivo. In support of this, the mutation in the temperature-sensitive cdc17-K42 allele was found to map to the NCD: an invariant glutamate at position 298 in the wild-type protein is changed to lysine in Cdc17-K42 (Table 2 and Fig. 7C, see Materials and Methods). The mutations in the other extant cdc17 alleles, cdc17-L16 and cdc17-M75, map within the catalytic domain (Table 2 and Fig. 7C). Both are proline to serine changes, at amino acids 584 and 679, respectively. Proline 584 is located between DNA ligase conserved motifs IV and V in a region of the protein that is well conserved across a variety of eukaryotic species including yeast, human, Xenopus and Drosophila (Fig. 7C), as well as archaeal species (data not shown). Proline 584 itself is conserved between the fission yeast and budding yeast Lig I proteins, but not in higher eukaryotes. In contrast, proline 679, the site of the Cdc17-M75 mutation, is located in a region of relatively low sequence similarity between species, between conserved motifs V and VI. The fission yeast protein possesses a two amino insertion in this region (Fig. 7C).
Table 2. Location of temperature-sensitive mutants in Cdc17.
| Mutant protein |
K42 |
L16 |
M75 |
| Wild-type sequence |
Glu298 |
Pro584 |
Pro679 |
| Mutant sequence |
Lys298 |
Ser584 |
Ser679 |
| Location |
NCD |
CD |
CD |
| Sub-domain | IV–V | V–VI |
NCD, non-catalytic conserved domain; CD, catalytic domain.
DISCUSSION
Lig I plays key roles in DNA replication and repair in eukaryotic cells (1,2). In budding yeast, the Cdc9 protein is targeted to both nuclear and mitochondrial compartments by virtue of the utilization of alternative in-frame start codons (22). In the nucleus, Cdc9 functions in S-phase to ligate together Okazaki fragments formed during discontinuous synthesis of the lagging strand (3–6), and also in a range of DNA repair pathways vital for maintaining genome integrity throughout the cell cycle (7,8). In mitochondria, Cdc9 has been shown to play a vital role in the repair of DNA damage and most likely also functions in mtDNA replication (22,23). In this paper, Cdc17, the essential Lig I protein of fission yeast (3,5,24,25), is shown to be targeted to both the nucleus and mitochondria (Fig. 2). Mitochondrial targeting is dependent upon the presence of an N-terminal MTS, a putative amphipathic α-helix, and is essential for cell viability (Figs 1–4).
Nuclear localization of Lig I in fission yeast is also dependent upon sequences located in the NTD of the protein (Figs 1–4). Replacement of amino acids lysine 62, arginine 63 and lysine 64 with three consecutive alanine residues abolishes nuclear Cdc17-GFP targeting, suggesting that these form part of the Cdc17 NLS. Since amino acids 32–65 alone were unable to localize GFP, sequences beyond this must also be required. Previous studies on conserved features of NLSs have identified two major groups (45). Canonical NLSs comprise a single stretch of basic amino acids. The NLS of SV40 large T-antigen (PKKKRK) is one such sequence. Bipartite NLSs comprise two stretches of basic amino acids, generally separated by a spacer of ∼10 residues. In addition to these two groups, other types of NLSs that show no obvious resemblance to the above and which are heterogeneous in nature have been identified experimentally (46,47). With respect to Cdc17, inspection of the NTD sequence identifies a second cluster of basic amino acids (KKQK) spanning residues 97–100 that could form part of a non-standard bipartitite NLS but further mutational analysis will be required to determine if this is the case.
The sliding clamp PCNA plays an important role in many aspects of DNA metabolism in eukaryotic cells, and a large number of PCNA binding proteins have been identified. Many, though not all, of these proteins interact with PCNA via a short protein sequence motif called a PIP box (17) that was first identified in studies of the mammalian DNA replication and cell cycle inhibitor p21Cip1 (41). In the case of Lig I, the mammalian and yeast proteins possess a PIP box at or near their N-termini that is distinct from their NLS sequences. Experiments with the human enzyme have clearly demonstrated a role for the PIP box in directing localization of Lig I to sites of ongoing DNA replication, so-called replication foci or replication factories (20). The results presented here demonstrate that the presence of the PIP box is non-essential for fission yeast Cdc17 function, although the viability of cells expressing nuclear Cdc17 proteins lacking the PIP box is somewhat compromised (Fig. 6). Visualization of Cdc17 lacking the PIP box by indirect immunofluorescence (Fig. 3B) reveals a nuclear staining pattern that is indistinguishable from that seen with the full-length protein, although the latter also localizes to the mitochondria (Fig. 3A). However, at the level of resolution used in this study, it is not possible to conclude that the PIP box plays no role in the intranuclear localization of Cdc17. It is unclear, for example, whether the punctate intranuclear staining patterns observed (see Fig. 3A and B) represent sites of ongoing replication or whether they present an artifact of the fixation procedure.
The importance of the conserved NCD for Cdc17 function is also highlighted by this study. Comparative sequence analysis identifies the NCD as a conserved feature of all three eukaryotic ATP-dependent cellular ligases (I, III and IV) as well as viral and archaeal enzymes (1). The level of sequence similarity between NCDs from ligases from different groups (e.g. human Lig III and Lig IV) is not great but can be detected by reiterative ψ-BLAST searching irrespective of the input query sequence. Within a single group, the level of sequence identity between NCDs is lower than the level of sequence identity displayed by the catalytic domains from the same enzymes. Deleting the NCD abolishes Cdc17 function even when the truncated protein is present at levels higher than endogenous Cdc17 in wild-type cells. Deletion of the NCD also results in significant reduction in protein stability (Fig. 7). One of the three existing temperature-sensitive mutations in Cdc17 (Cdc17-K42) maps to the NCD, the other two being located in the catalytic domain. Taken together these results provide clear evidence that the NCD plays an essential role in Cdc17 function, consistent with the degree of sequence conservation shown across species.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank our friends and colleagues, past and present, for their help in the course of this work, particularly Dr L. Johnston (NIMR, London) and Dr K. Hardwick (WTCCB, University of Edinburgh) for providing plasmids, Dr H. Ohkura (WTCCB, University of Edinburgh) for providing secondary antibodies for immunofluorescence and for technical advice, Dr P. Fantes, Dr H. Tanaka and F. Gray for critical reading of the manuscript, and Professor S. Linn (UC Berkeley) for his editorial comments. I.V.M. was supported by a studentship from the Darwin Trust of Edinburgh. S.A.M. is a Wellcome Trust Senior Research Fellow in Basic Biomedical Science.
REFERENCES
- 1.Martin I.V. and MacNeill,S.A. (2002) ATP-dependent DNA ligases. Genome Biol., 3, REVIEWS3005 [Epub 2002 Mar 19]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tomkinson A.E. and Mackey,Z.B. (1998) Structure and function of mammalian DNA ligases. Mutat. Res., 407, 1–9. [DOI] [PubMed] [Google Scholar]
- 3.Nasmyth K.A. (1977) Temperature-sensitive lethal mutants in the structural gene for DNA ligase in the yeast Schizosaccharomyces pombe. Cell, 12, 1109–1120. [DOI] [PubMed] [Google Scholar]
- 4.Johnston L.H. and Nasmyth,K.A. (1978) Saccharomyces cerevisiae cell cycle mutant cdc9 is defective in DNA ligase. Nature, 274, 891–893. [DOI] [PubMed] [Google Scholar]
- 5.Nasmyth K.A. (1979) Genetic and enzymatic characterization of conditional lethal mutants of the yeast Schizosaccharomyces pombe with a temperature-sensitive DNA ligase. J. Mol. Biol., 130, 273–284. [DOI] [PubMed] [Google Scholar]
- 6.Bielinsky A.K. and Gerbi,S.A. (1999) Chromosomal ARS1 has a single leading strand start site. Mol. Cell, 3, 477–486. [DOI] [PubMed] [Google Scholar]
- 7.Johnston L.H. (1979) The DNA repair capability of cdc9, the Saccharomyces cerevisiae mutant defective in DNA ligase. Mol. Gen. Genet., 170, 89–92. [DOI] [PubMed] [Google Scholar]
- 8.Moore C.W. (1982) cdc9 ligase-defective mutants of Saccharomyces cerevisiae exhibit lowered resistance to lethal effects of bleomycin. J. Bacteriol., 151, 1617–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barnes D.E., Tomkinson,A.E., Lehmann,A.R., Webster,A.D. and Lindahl,T. (1992) Mutations in the DNA ligase I gene of an individual with immunodeficiencies and cellular hypersensitivity to DNA-damaging agents. Cell, 69, 495–503. [DOI] [PubMed] [Google Scholar]
- 10.Prigent C., Satoh,M.S., Daly,G., Barnes,D.E. and Lindahl,T. (1994) Aberrant DNA repair and DNA replication due to an inherited enzymatic defect in human DNA ligase I. Mol. Cell. Biol., 14, 310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bentley D., Selfridge,J., Millar,J.K., Samuel,K., Hole,N., Ansell,J.D. and Melton,D.W. (1996) DNA ligase I is required for fetal liver erythropoiesis but is not essential for mammalian cell viability. Nature Genet., 13, 489–491. [DOI] [PubMed] [Google Scholar]
- 12.Bentley D.J., Harrison,C., Ketchen,A.M., Redhead,N.J., Samuel,K., Waterfall,M., Ansell,J.D. and Melton,D.W. (2002) DNA ligase I null mouse cells show normal DNA repair activity but altered DNA replication and reduced genome stability. J. Cell Sci., 115, 1551–1561. [DOI] [PubMed] [Google Scholar]
- 13.Subramanya H.S., Doherty,A.J., Ashford,S.R. and Wigley,D.B. (1996) Crystal structure of an ATP-dependent DNA ligase from bacteriophage T7. Cell, 85, 607–615. [DOI] [PubMed] [Google Scholar]
- 14.Odell M., Sriskanda,V., Shuman,S. and Nikolov,D.B. (2000) Crystal structure of eukaryotic DNA ligase-adenylate illuminates the mechanism of nick sensing and strand joining. Mol. Cell, 6, 1183–1193. [DOI] [PubMed] [Google Scholar]
- 15.Sriskanda V., Schwer,B., Ho,C.K. and Shuman,S. (1999) Mutational analysis of Escherichia coli DNA ligase identifies amino acids required for nick-ligation in vitro and for in vivo complementation of the growth of yeast cells deleted for CDC9 and LIG4. Nucleic Acids Res., 27, 3953–3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levin D.S., Bai,W., Yao,N., O’Donnell,M. and Tomkinson,A.E. (1997) An interaction between DNA ligase I and proliferating cell nuclear antigen: implications for Okazaki fragment synthesis and joining. Proc. Natl Acad. Sci. USA, 94, 12863–12868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Warbrick E. (1998) PCNA binding through a conserved motif. Bioessays, 20, 195–199. [DOI] [PubMed] [Google Scholar]
- 18.Cardoso M.C., Joseph,C., Rahn,H.P., Reusch,R., Nadal-Ginard,B. and Leonhardt,H. (1997) Mapping and use of a sequence that targets DNA ligase I to sites of DNA replication in vivo. J. Cell Biol., 139, 579–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Montecucco A., Savini,E., Weighardt,F., Rossi,R., Ciarrocchi,G., Villa,A. and Biamonti,G. (1995) The N-terminal domain of human DNA ligase I contains the nuclear localization signal and directs the enzyme to sites of DNA replication. EMBO J., 14, 5379–5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Montecucco A., Rossi,R., Levin,D.S., Gary,R., Park,M.S., Motycka,T.A., Ciarrocchi,G., Villa,A., Biamonti,G. and Tomkinson,A.E. (1998) DNA ligase I is recruited to sites of DNA replication by an interaction with proliferating cell nuclear antigen: identification of a common targeting mechanism for the assembly of replication factories. EMBO J., 17, 3786–3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levin D.S., McKenna,A.E., Motycka,T.A., Matsumoto,Y. and Tomkinson,A.E. (2000) Interaction between PCNA and DNA ligase I is critical for joining of Okazaki fragments and long-patch base-excision repair. Curr. Biol., 10, 919–922. [DOI] [PubMed] [Google Scholar]
- 22.Willer M., Rainey,M., Pullen,T. and Stirling,C.J. (1999) The yeast CDC9 gene encodes both a nuclear and a mitochondrial form of DNA ligase I. Curr. Biol., 9, 1085–1094. [DOI] [PubMed] [Google Scholar]
- 23.Donahue S.L., Corner,B.E., Bordone,L. and Campbell,C. (2001) Mitochondrial DNA ligase function in Saccharomyces cerevisiae. Nucleic Acids Res., 29, 1582–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnston L.H., Barker,D.G. and Nurse,P. (1986) Cloning and characterization of the Schizosaccharomyces pombe DNA ligase gene cdc17. Gene, 41, 321–325. [DOI] [PubMed] [Google Scholar]
- 25.Barker D.G., White,J.H.M. and Johnston,L.H. (1987) Molecular characterization of the DNA ligase gene, cdc17, from the fission yeast Schizosaccharomyces pombe. Eur. J. Biochem., 162, 659–667. [DOI] [PubMed] [Google Scholar]
- 26.Schäfer B. and Wolf,K. (2004) Mitochondrial genetics in a petite-negative yeast. In Egel,R. (ed.), The Molecular Biology of Schizosaccharomyces pombe. Springer, Berlin, pp. 415–443. [Google Scholar]
- 27.Nasmyth K.A. (1977) Temperature-sensitive lethal mutants in the structural gene for DNA ligase in the yeast Schizosaccharomyces pombe. Cell, 12, 1109–1120. [DOI] [PubMed] [Google Scholar]
- 28.Moreno S., Klar,A. and Nurse,P. (1991) Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol., 194, 795–823. [DOI] [PubMed] [Google Scholar]
- 29.Maundrell K. (1990) nmt1 of fission yeast. A highly transcribed gene completely repressed by thiamine. J. Biol. Chem., 265, 10857–10864. [PubMed] [Google Scholar]
- 30.Maundrell K. (1993) Thiamine-repressible expression vectors pREP and pRIP for fission yeast. Gene, 123, 127–130. [DOI] [PubMed] [Google Scholar]
- 31.Basi G., Schmid,E. and Maundrell,K. (1993) TATA box mutations in the Schizosaccharomyces pombe nmt1 promoter affect transcription efficiency but not the transcription start point or thiamine repressibility. Gene, 123, 131–136. [DOI] [PubMed] [Google Scholar]
- 32.Forsburg S.L. (1993) Comparison of Schizosaccharomyces pombe expression systems. Nucleic Acids Res., 21, 2955–2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bähler J., Wu,J.Q., Longtine,M.S., Shah,N.G., McKenzie,A.,III, Steever,A.B., Wach,A., Philippsen,P. and Pringle,J.R. (1998) Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast, 14, 943–951. [DOI] [PubMed] [Google Scholar]
- 34.Barbet N., Muriel,W.J. and Carr,A.M. (1992) Versatile shuttle vectors and genomic libraries for use with Schizosaccharomyces pombe. Gene, 114, 59–66. [DOI] [PubMed] [Google Scholar]
- 35.Reynolds N., Warbrick,E., Fantes,P.A. and MacNeill,S.A. (2000) Essential interaction between the fission yeast DNA polymerase δ subunit Cdc27 and Pcn1 (PCNA) mediated through a C-terminal p21Cip1-like PCNA binding motif. EMBO J., 19, 1108–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alfa C.E., Gallagher,I.M. and Hyams,J.S. (1993) Antigen localization in fission yeast. Methods Cell Biol., 37, 201–222. [DOI] [PubMed] [Google Scholar]
- 37.Nash R. and Lindahl,T. (1996) DNA ligases. In DePamphilis,M.L. (ed.), DNA Replication in Eukaryotic Cells. Cold Spring Harbor Laboratory Press, New York, pp. 575–586. [Google Scholar]
- 38.von Heijne G. (1986) Mitochondrial targeting sequences may form amphiphilic helices. EMBO J., 5, 1335–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bannai H., Tamada,Y., Maruyama,O., Nakai,K. and Miyano,S. (2002) Extensive feature detection of N-terminal protein sorting signals. Bioinformatics, 18, 298–305. [DOI] [PubMed] [Google Scholar]
- 40.Claros M.G. and Vincens,P. (1996) Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur. J. Biochem., 241, 779–786. [DOI] [PubMed] [Google Scholar]
- 41.Warbrick E., Lane,D.P., Glover,D.M. and Cox,L.S. (1995) A small peptide inhibitor of DNA replication defines the site of interaction between the cyclin-dependent kinase inhibitor p21(waf1) and proliferating cell nuclear antigen. Curr. Biol., 5, 275–282. [DOI] [PubMed] [Google Scholar]
- 42.Robinow C.F. and Hyams,J.S. (1989) General cytology of fission yeasts. In Nasim,A., Young,P. and Johnson,B.F. (eds), Molecular Biology of the Fission Yeast. Academic Press, San Diego, pp. 273–330. [Google Scholar]
- 43.Yaffe M.P., Harata,D., Verde,F., Eddison,M., Toda,T. and Nurse,P. (1996) Microtubules mediate mitochondrial distribution in fission yeast. Proc. Natl Acad. Sci. USA, 93, 11664–11668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pelloquin L., Belenguer,P., Menon,Y., Gas,N. and Ducommun,B. (1999) Fission yeast Msp1 is a mitochondrial dynamin-related protein. J. Cell Sci., 112, 4151–4161. [DOI] [PubMed] [Google Scholar]
- 45.Kaffman A. and O’Shea,E.K. (1999) Regulation of nuclear localization: a key to a door. Annu. Rev. Cell. Dev. Biol., 15, 291–339. [DOI] [PubMed] [Google Scholar]
- 46.Christophe D., Christophe-Hobertus,C. and Pichon,B. (2000) Nuclear targeting of proteins: how many different signals? Cell Signal, 12, 337–341. [DOI] [PubMed] [Google Scholar]
- 47.Pichon B., Mercan,D., Pouillon,V., Christophe-Hobertus,C. and Christophe,D. (2000) A method for the large-scale cloning of nuclear proteins and nuclear targeting sequences on a functional basis. Anal. Biochem., 284, 231–239. [DOI] [PubMed] [Google Scholar]