

Abstract
Cancer cells demonstrate deviant behavior that induces apoptotic signaling. In order to survive, cancer cells typically acquire changes enabling evasion of death signals. One way they do this is by increasing the expression of anti-apoptotic BCL-2 proteins. Anti-apoptotic BCL-2 family proteins antagonize death signaling by forming heterodimers with pro-death proteins. Heterodimer formation occurs via binding of the pro-apoptotic protein’s BH3 domain into the hydrophobic cleft of anti-apoptotic proteins. The BH3 mimetics are small molecule antagonists of the anti-apoptotic BCL-2 members that function as competitive inhibitors by binding to the hydrophobic cleft. Under certain conditions, antagonism of anti-apoptotic BCL-2 family proteins can unleash pro-death molecules in cancer cells. Thus, the BH3 mimetics are a new class of cancer drugs that specifically target a mechanism of cancer cell survival, to selectively kill cancer cells.
Introduction
For decades, cytotoxic chemotherapy, along with surgery and radiation, has been one of the three important modalities used to treat cancer. As most chemotherapeutics were discovered by empirical screens, the molecular mechanisms of how they kill cells are poorly understood. An ideal chemotherapeutic drug would target only neoplastic cells, signaling their removal from the body without damaging adjacent cells. However, conventional chemotherapy is associated with many toxicities, largely due to the presence in normal cells of the targets of most conventional agents, ie DNA and microtubules. A truly cancer-selective therapy must target a molecule or property that is selectively present in cancer cells to avoid toxicity to normal cells.
In order for a cancerous cell to develop it must obtain the ability to surmount essential checkpoints that would normally send a deregulated cell to its demise. Cancer cells attain the capacity to evade the body’s own immune response and to grow in a stressful environment, where both oxygen and nutrients are limited. In order to maintain a high proliferation rate, tumor cells generate their own growth signal, while, simultaneously becoming insensitive to growth inhibitory effects (Hanahan and Weinberg, 2000; Johnstone et al., 2002). The cells obtain mutations in key cellular proteins that allow the developing cancer to grow, albeit breaking certain cellular rules, such as genomic instability, oncogene activation, loss of adhesion, along with breach of cell cycle checkpoints. Each of the breaches mentioned can cause activation of the mitochondrial (intrinsic) apoptotic pathway. The oncogene c-myc, has been shown to activate apoptosis (Evan et al., 1992), similarly anoikis or loss of adhesion leads to the activation of pro-apoptotic factors (Puthalakath et al., 1999; Puthalakath et al., 2001). Cancer cells have a unique requirement to overcome the death signaling engendered by these behaviors. Targeting the mechanisms cancers use to escape apoptosis offers the possibility, therefore, of a wide therapeutic index.
The BCL-2 family
The BCL-2 family of proteins governs whether a cell continues to live or instead commits to death via the mitochondrial apoptotic pathway. Interactions between pro- and anti-apoptotic BCL-2 family proteins control the decision making process at the mitochondrion, modulating the cells sensitivity to death (Kim et al., 2006; Letai et al., 2002; Wei et al., 2000). BCL-2, BCL-XL, MCL-1, BCL-W and BFL-1 are the anti-apoptotic members of the family (Boise et al., 1993; Choi et al., 1995; Cleary and Sklar, 1985; Gibson et al., 1996; Kozopas et al., 1993; Tsujimoto et al., 1985). They share sequence homology in four α– helical BCL-2 homology domains (BH) BH1-4. While the multidomain pro-apoptotic proteins BAX, BAK and BOK, the promoters of apoptosis, share homology in the domains BH1 through BH3 (Chittenden et al., 1995; Oltvai et al., 1993). BOK expression is largely limited to reproductive tissues, and less is known about its function, though it is widely supposed that it behaves much like BAX and BAK (Hsu et al., 1997). Lastly, the group sharing the least homology is the BH3-only pro-death group, which includes PUMA, NOXA, BIM, BID, HRK, BMF, BAD and BIK (Boyd et al., 1995; Inohara et al., 1997; Nakano and Vousden, 2001; O’Connor et al., 1998; Oda et al., 2000; Wang et al., 1996; Yang et al., 1995). They are so named because they demonstrate homology only in the BH3 domain. The BH3 domain, along with the presence of BAX and BAK, are absolutely essential for the death function of the BH3-only proteins (Cheng et al., 2001; Wei et al., 2001; Zha and Reed, 1997).
Death signals emanating from diverse cellular locations caused by DNA damage, growth factor deprivation and oncogene activation are directed through the mitochondrial pathway by activation (transcriptional or post-translational modifications) of BH3-only proteins (Evan et al., 1992; Nakano and Vousden, 2001; Oda et al., 2000). In viable cells BAX and BAK exist as monomers, however, upon receiving death signals from the activated BH3-only proteins, they homooligomerize and insert into the mitochondrial membrane (Eskes et al., 2000; Korsmeyer et al., 2000; Wei et al., 2001). Insertion of homooligomerized BAX and BAK causes outer mitochondrial membrane permeabilization (MOMP). Permeabilization is followed by release of cytochrome c and other pro-apoptotic factors from the mitochondria. Peremabilization of the mitochondrial outer membrane is the key step in commitment to death via the mitochondrial apoptotic pathway (Fig 1).
Fig 1. The mitochondrial apoptotic pathway.

Cellular stress activates both sensitizer and activator BH3-only proteins. The sensitizer BH3-only proteins inhibit the anti-apoptotic BCL-2 family, while the direct activators cause the activation leading to oligomerization and insertion of BAX and BAK into the mitochondrial membrane. Mitochondrial outer membrane permeabilization (MOMP) causes the release of Cytochrome c, which forms a complex with caspase-9, APAF-1 and dATP/dADP triggering downstream apoptotic events.
There is general agreement that BAX and BAK require activation prior to effecting permeabilization of the mitochondrion. While, some reports emphasize the importance of activator BH3 proteins in facilitating this transition (Kim et al., 2006; Kuwana et al., 2002; Letai et al., 2002; Wei et al., 2000) other reports stress the neutralization of the anti-apoptotic members (Willis et al., 2005; Willis et al., 2007). The former describes a select few of the BH3-only proteins termed activators (BIM, tBID and perhaps PUMA) that have the ability to bind directly to BAX and BAK causing their activation (Kim et al., 2006; Kuwana et al., 2005; Letai et al., 2002). In the latter case, BH3-only proteins are sensitizers that cause death by binding the anti-apoptotic BCL-2 proteins causing release of proapoptotic proteins (Fig 2) (Willis et al., 2005; Willis et al., 2007).
Fig 2. The binding affinity of BH3-only peptides to the anti-apoptotic Bcl-2 family members.
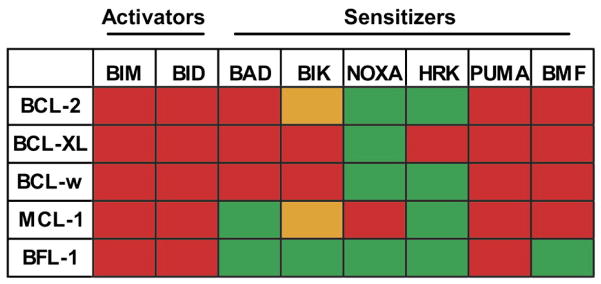
BH3-only peptides (Top) demonstrate a distinct binding pattern to the anti-apoptotic Bcl-2 family members (Left). Red depicts high affinity binding, orange depicts low- affinity binding and green depicts non-detectable binding. The BH3-only proteins are subdivided into activators and sensitizers. Modified with permission from (Deng et al., 2007).
Following its release, cytochrome c (Korsmeyer et al., 2000) binds in a complex with APAF-1, caspase-9 and dATP/dADP to form the apoptosome (Zou et al., 1999). From the apoptosome platform the downstream effectors of death, the caspases, are activated (Acehan et al., 2002; Zou et al., 1997; Zou et al., 1999).
Apoptotic road blocks in cancer
Delineation of the mitochondrial apoptotic pathway leads to an important question, how does a cancer cell survive the apoptotic signaling events activated by its aberrant behavior? One mechanism is to select for a block in apoptotic signaling at the mitochondria by increasing the expression of anti-apoptotic BCL-2 members. There are many mechanisms known by which BCL-2 is expressed at high levels in cancer cells. BCL-2 was originally cloned from the breakpoint of the chromosomal translocation t(14; 18) in patients with follicular lymphoma (Cleary and Sklar, 1985; Tsujimoto et al., 1985). The translocation placed BCL-2 next to enhancer elements of the immunoglobulin promoter causing an increased expression of BCL-2. The BCL-2 translocation occurs in approximately 90% of follicular lymphomas and in about one third of diffuse large B cell lymphomas (Weiss et al., 1987). However, BCL-2 overexpression has also been linked to gene amplification (Rao et al., 1998), hypermethylation of the BCL-2 gene (Hanada et al., 1993) or chromosomal deletions causing the loss of micro-RNAs involved in the silencing of BCL-2 (Cimmino et al., 2005). A series of genetic studies in mice really emphasized the oncogenic potential of BCL-2 (Letai et al., 2004; Strasser et al., 1990; Vaux et al., 1988). Work examining the combined action of c-myc and BCL-2 in mice showed that for neoplastic progression there is a requirement for the deregulated cellular proliferation to be coupled with a compensatory inhibition of apoptosis.
Reciprocally, cancer cells can evade death through mutations that inactivate the effector arm of apoptosis, such as, the mutations of BAX observed in some solid tumors and hematopoietic malignancies (Meijerink et al., 1998; Meijerink et al., 1995; Rampino et al., 1997). An especially common defect of cancers is a mutation in the tumor suppressor gene p53 (Greenblatt et al., 1994) which in turn has a negative effect on activation of apoptosis, as the BH3-only proteins NOXA and PUMA are transcriptional targets of p53 (Jeffers et al., 2003; Nakano and Vousden, 2001; Oda et al., 2000; Shibue et al., 2003; Villunger et al., 2003). However, there are also indirect mechanisms for thwarting p53 function including mutations in ATM, a kinase involved in the phosphorylation and activation of p53 (Banin et al., 1998).
A recent paper segregated each of these blocks in apoptosis into three classes. In a Class A block, there is a loss of activator BH3-only proteins. A Class B block is caused by a failure to activate the effector arm of apoptosis through loss or inactivation of BAX or BAK. Finally, increased expression of an inhibitor protein such as BCL-2/MCL-1 causes a class C block (Deng et al., 2007) (Fig 3). A novel technique called BH3 profiling can detect the class of apoptotic block a cancer cell has evolved in order to evade apoptosis. BH3 profiling exploits the binding pattern of sensitizer BH3 proteins to the anti-apoptotic proteins (Certo et al., 2006; Letai et al., 2002) (Fig 2). An example of how this works is if the cancer cell has selected for upregulation of MCL-1 to avoid apoptosis, the sensitizer BH3 peptide NOXA would cause release of cytochrome c from the isolated mitochondria, while the BAD peptide would be ineffective, diagnosing a MCL-1 dependent Class C block. BH3 profiling has been used to categorize diffuse large cell lymphoma cell lines into each of the three classes of apoptotic blocks and shown that membership in the class C block correlates with increased drug sensitivity (Del Gaizo Moore et al., 2007; Deng et al., 2007).
Fig 3. The apoptotic blocks utilized by cancer cells.
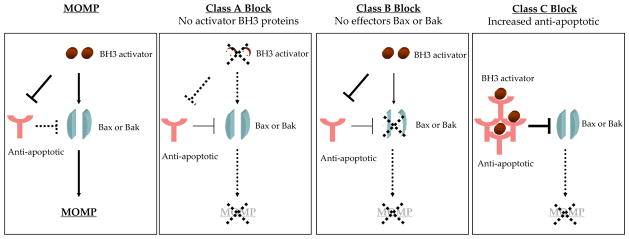
In order to evade apoptosis cancer cells select for blocks in apoptotic signaling. These blocks have been categorized based on which group of the BCL-2 family is altered. A class A block is caused by a loss of BH3-only proteins, a Class B block is due to loss or inactivation of BAX and BAK and a class C block is caused by enhanced expression of anti-apoptotic BCL-2 proteins. Following activation and insertion of BAX and BAK into the mitochondrial memberane, the outer mitochondrial membrane is permeabilized (MOMP) enabling release of cytochrome c and activation of apoptosis.
How does increased expression of an anti-apoptotic correlate with enhanced drug sensitivity? From results generated by overexpression studies in cell lines, it is easy to envision that increased expression of BCL-2 would provide additional opposition against chemotherapy (Martinou et al., 1994; Miyashita and Reed, 1993; Takahashi et al., 2003; Zhang and Insel, 2001). However, as mentioned previously, during oncogenesis the abnormal phenotype of oncogene activation and genomic instability acquired for deregulated growth initiates apoptotic signals, in the form of activated BH3-only proteins. A compensatory increased level of an anti-apoptotic protein is selected to buffer the death signals generated. Therefore, the anti-apoptotic proteins are filled with death initiator signals or are “primed” for death and the cancer cell is often dependent or addicted to the anti-apoptotic protein (Certo et al., 2006; Del Gaizo Moore et al., 2008; Letai et al., 2004). The relatively “full” state of these selected anti-apoptotic proteins does not afford for the binding of many subsequent pro-apoptotic proteins. This is in contrast to the case of forced overexpression in cell lines, in which most of the overexpressed anti-apoptotic proteins are “empty” and therefore competent to sequester subsequent pro-death signaling molecules.
Design of BCL-2 targeting drugs
Once it was understood that cancer cells depended on BCL-2 or related anti-apoptotic proteins for survival - it became apparent that this could be of great practical significance, as a mechanism to specifically target cancer cells (Letai et al., 2004). One of the first strategies employed to target the BCL-2 family, was the use of antisense oligonucleotides to knockdown the mRNA expression of BCL-2 (Cotter et al., 1994). Oblimersen (Genasense) from Genta is an 18-mer phosphorothioated antisense oligonucleotide directed against the open reading frame of BCL-2 (Klasa et al., 2002). The clinical effect of the drug has been modest in clinical trials. (O’Brien et al., 2005), and it is not clear how well the drug lowers BCL-2 protein levels in cells in vivo.
The next strategy that emerged was to antagonize the function of BCL-2 rather than reduce its levels. This was approached mechanistically following delineation of the crystal structure of BCL-XL, which revealed that the BH1- BH3 domains formed a hydrophobic groove (Muchmore et al., 1996), where the α-helix of the BH3-only proteins bind (Sattler et al., 1997). The structural analysis of BCL-XL bound to the BAK BH3 peptide (Sattler et al., 1997) was a proof of concept experiment indicating that it could be possible to create small molecules that bound to the hydrophobic groove of BCL-XL, inhibiting its anti-apoptotic function. Since this preliminary experiment, numerous small molecules with varying degree of specificity and effectiveness have been synthesized and the mechanism by which they induce apoptosis is depicted in (Fig 4).
Fig 4. Model of BH3 mimetic- induced apoptosis.
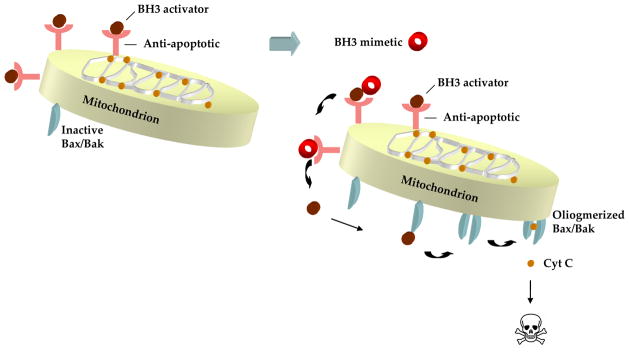
Anti-apoptotic Bcl-2 family members buffer the death signals activated by the deregulated growth of cancer cells. BH3 mimetics induce apoptosis by binding to the hydrophobic groove of the anti-apoptotic BCL-2 members displacing bound activator BH3-only proteins, enabling activation of BAX and BAK and the downstream apoptotic events.
A number of natural compounds that function as cell permeable, small molecule mimics of the BH3 domain were found using a library screening process including Tetrocarcin A, Antimycin and gossypol (Kitada et al., 2003; Nakashima et al., 2000; Tzung et al., 2001). Gossypol is found in cottonseeds, it is a natural polyphenol capable of displacing BH3 peptides from BCL-XL with a dissociation constant in the low micromolar range (Becattini et al., 2004; Kitada et al., 2003). An altered version of gossypol without the two reactive aldehyde groups has been synthesized. However, the new compound termed apogossypol has actually reduced affinity for BCL-XL (Becattini et al., 2004). TW-37 is another gossypol derivative that was designed using the structure of the BIM BH3 domain as a model; it binds with high affinity to BCL-2, BCL-XL and MCL-1 (Verhaegen et al., 2006). Two molecules were discovered from a chemical library screen, tested for the ability to disrupt the BCL-XL/BAK BH3 complex and they were named BH3I-1 and BH3I-2 (Degterev et al., 2001). In contrast to random screening of compounds another approach to drug discovery is computer aided design of a specific ligand molecule based on the structure of the recipient receptor. This method enabled the discovery of the organic compound HA14-1 (Wang et al., 2000). In general, most of these molecules display a biochemical affinity for certain anti-apoptotic BCL-2 proteins, linked to their ability to kill certain cells, and even in some cases improve survival of xenografts (Degterev et al., 2001; Kitada et al., 2003; Mohammad et al., 2007). However, the mechanism of cell killing is often unclear, and it is not clear that the primary event is antagonism of anti-apoptotic BCL-2 proteins (van Delft et al., 2006).
ABT-737
Of the compounds discovered to date, there is the most evidence to support killing via direct interaction with the BCL-2 family for the BH3 mimetic ABT-737 (Abbott Laboratories). ABT-737 was ineffective at activating apoptosis in cells doubly deficient of BAX and BAK, demonstrating that the mechanism of action is solely through the BCL-2 family (van Delft et al., 2006). ABT-737 was discovered by Abott using a strategy of combining screening using nuclear magnetic resonance, structure based design and combinatory chemical synthesis. The strategy was named “SAR by NMR” (Oltersdorf et al., 2005). The lead compound ABT-737 mimicked the BH3 domain of BAD and bound selectively to BCL-2, BCL-XL and BCL-W. Of importance the binding efficiency for ABT-737 to the subset of the anti-apoptotic members was is in the nanomolar range, hence making in vivo therapeutically significant concentrations attainable. The crystal structure of ABT-737 bound to BCL-XL shows the chloro-biphenyl and thio-phenyl moieties bind to the p2 and p4 pockets of the hydrophobic groove of BCL-XL (Lee et al., 2007). ABT-737, similar to the BAD peptide, binds with poor affinity to MCL-1 and BFL-1 with a dissociation constant in the micro-molar range (Fig 2) (Oltersdorf et al., 2005).
Obatoclax is a synthetic derivative of prodiginines (GX015-070) from Gemin X Biotech (Nguyen et al., 2007; Reed, 2003; Trudel et al., 2007a). However, the affinity for binding to the BCL-2 family is inferior to ABT-737, again with dissociation constants in hundreds of nanomolar range. TW-37 has also been shown to bind to BCL-2, BCL-XL and MCL-1 so it could potentially be a pan-BCL-2 mimetic (Verhaegen et al., 2006). Similarly, to many other putative BH3 mimetics, obatoclax is not entirely dependent on BAX and BAK for its method of apoptosis induction (Konopleva et al., 2008). A compound that binds with high affinity to all anti-apoptotic proteins is almost certainly going to be more toxic to cancer cells. Furthermore, such a drug will not stimulate selection for expression of alternative antiapoptotic BCL-2 family proteins, as none would escape inhibition. However, the concern is that there would be greater toxicity to normal cells, so that it is not clear that an improved therapeutic index would be obtained by a pan-BCL-2 family inhibitor. Our observation that cancer cells are far more likely to be “primed” than normal cells lends some hope to such an approach, but true confirmation would require extensive in vivo testing.
Clinical use of BH3 mimetics
Upfront therapy
Of the small molecule inhibitors of BCL-2 described thus far, four are at present in clinical trials – Genasense, TW-37, obatoclax, and ABT-263. In in vitro experiments, ABT-737 has mono-therapy toxicity to leukemia, lymphoma, and at higher concentrations is also able to induce apoptosis in multiple myeloma, glioma and small cell lung cancer cell lines (Chauhan et al., 2007; Oltersdorf et al., 2005; Tagscherer et al., 2008; Trudel et al., 2007b). Numerous studies including work carried out by our laboratory have demonstrated that primary cells from patients with chronic lymphocytic leukemia (Del Gaizo Moore et al., 2007), acute myeloid leukemia (Konopleva et al., 2006), acute lymphocytic leukemia (Del Gaizo Moore et al., 2008), along with B-cell neoplasms like follicular lymphoma and marginal zone lymphoma (Vogler et al., 2008) are extremely sensitive to ABT-737, with widespread apoptosis when treated with concentrations in the nanomolar range. Similarly, obatoclax the pan-BCL-2 inhibitor shows single agent efficiency in the killing of multiple myeloma (Trudel et al., 2007a), AML cell lines and primary samples. However, it also causes cell cycle arrest independent of apoptosis (Konopleva et al., 2008; Trudel et al., 2007a). Although, TW-37 has been shown to bind BCL-2, BCL-XL and MCL-1, the mechanism by which it induces apoptosis has not yet been elucidated. It causes a reduction in proliferation in pancreatic and B-cell lymphomas along with reducing tumor size in xenograft models, however, whether this is BCL-2 dependent needs to be clarified (Mohammad et al., 2007; Wang et al., 2008).
In order to enhance its clinical potential ABT-737 has been modified to make the drug orally available. ABT-263 binds to serum proteins resulting in a longer oral half life. The tumoricidal action of ABT-263 was demonstrated in xenograft models of small cell lung cancer and acute lymphoblastic leukemia cancer cells, where ABT-263 caused complete regression of the tumors (Shoemaker et al., 2008; Tse et al., 2008). The drug is now in clinical trials for chronic lymphocytic leukemia, lymphoma and small cell lung cancer (Wilson et. al., 2008). Notably, ABT-263 binds to BCL-2 with affinity <100 picomolar, making it several logs more potent than the other small molecules detailed above.
Intrinsic and acquired resistance therapy
The BH3 mimetics are often not very effective as single killing agents against some of the epithelial cancers, such as, the pancreatic, ovarian and breast cancers (Huang and Sinicrope, 2008; Kutuk and Letai, 2008; Witham et al., 2007). However, the other niche for these drugs is in combination therapy, where the BCL-2 antagonist serves to inhibit BCL-2 mediated resistance, enabling killing by conventional chemotherapy. Numerous examples exist in the literature of an enhanced apoptotic response when the BH3 mimetics are combined with traditional therapies to treat various cancers such as melanoma (Verhaegen et al., 2006), pancreatic (Huang and Sinicrope, 2008), glioma (Tagscherer et al., 2008), breast (Kutuk and Letai, 2008), multiple myeloma (Trudel et al., 2007b), and B-cell malignant models (Mohammad et al., 2007; Tse et al., 2008). Thus, it appears that the conventional therapies provide the additional priming event required to enable the Bcl-2 antagonists to kill the above examples of intrinsically resistant cancers. Resistance to conventional chemotherapy may also develop over time (acquired resistance). Upon initial treatment there is a dramatic response, which tapers over time due to the acquisition of resistance through various mechanisms, including alteration of the expression of Bcl-2 family members. A recent report, demonstrated the use of the BH3 mimetic ABT-737 in the treatment of acquired resistance in cancer cells, it was effective at resensitizing drug resistant breast cancer cell lines to apoptosis via paclitaxel. This study highlighted an important factor, during the acquisition of resistance against the chemotherapeutic drug, albeit in vitro, the breast cancer cell lines selected for increasing the expression of BCL-2 family members to gain resistance. ABT-737 in combination with other chemotherapeutics was efficient in overcoming this altered BCL-2 balance to reinstate killing (Kutuk and Letai, 2008), emphasizing the potential effectiveness of the use of BH3 mimetics in combination therapy.
Why are some cells resistant and some cells sensitive to ABT-737? Two conditions must be met for ABT-737 to kill cells. Firstly, BCL-2 (or BCL-XL and BCL-W) need to be primed with death initiating signals. These signals may be BH3 activators such as BIM, or perhaps activated monomeric BAX or BAK. These pro-death proteins are then released upon ABT-737 binding to the hydrophobic groove of BCL-2, enabling activation of apoptosis (Fig 4) (Del Gaizo Moore et al., 2007). Secondly, the displaced death initiating signal (eg BIM or BAX)) needs to exceed the quantity of empty MCL-1 and BFL-1, which are not targeted by ABT-737. Excess empty MCL-1 or BFL-1 would otherwise sequester displaced pro-death proteins so that apoptosis would not be activated. High levels of MCL-1 or BFL-1 have been shown to correlate with resistance to the ABT-737 (Konopleva et al., 2006; Lin et al., 2007; Tagscherer et al., 2008; van Delft et al., 2006). The resistance caused by MCL-1 can be overcome by inactivation via knockdown (Lin et al., 2007) or through addition of agents that decrease MCL-1, such as seliciclib (Chauhan et al., 2007; Chen et al., 2007; Nguyen et al., 2007). However, it is worthy of note that cells which have either lost or mutated BAX or BAK that fall into a class B block of apoptosis are also resistant to BH3 mimetics. Similarly, cells with reduced levels of BH3-only activator proteins (Class A) will also be unable to respond BH3 mimetics (Deng et al., 2007). Thus, resistance to the BH3 mimetics is not solely dependent on the expression levels of MCL-1/BFl-1. Utilization of the BH3 profiling tool facilitates identification of the apoptotic block employed by cancer cells or the block acquired upon resistance to chemotherapy. Therefore, this tool would aid in the recognition of cancers that are likely to be susceptible to either pan- or selective BH3 mimetics, enabling a personalized approach to treatment (Del Gaizo Moore et al., 2007; Del Gaizo Moore et al., 2008; Deng et al., 2007).
Conclusion
Huge inroads have been made in delineating the apoptotic pathway over the last two decades, in particular the mechanism by which the BCL-2 family functions through selective protein-protein interactions to control mitochondrial apoptosis. Simultaneously, an understanding of how cancer cells evade death at the molecular level has been achieved with the BCL-2 family playing a starring role in death avoidance. The stage is set for the next generation of therapeutics, the small-molecule inhibitors of anti-apoptotic BCL-2 proteins, to be tested and fully exploited clinically, either as single agents or in combination therapy.
References
- Acehan D, Jiang X, Morgan DG, Heuser JE, Wang X, Akey CW. Three-dimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation. Mol Cell. 2002;9:423–32. doi: 10.1016/s1097-2765(02)00442-2. [DOI] [PubMed] [Google Scholar]
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–7. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- Becattini B, Kitada S, Leone M, Monosov E, Chandler S, Zhai D, et al. Rational design and real time, in-cell detection of the proapoptotic activity of a novel compound targeting Bcl-X(L) Chem Biol. 2004;11:389–95. doi: 10.1016/j.chembiol.2004.02.020. [DOI] [PubMed] [Google Scholar]
- Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA, et al. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- Boyd JM, Gallo GJ, Elangovan B, Houghton AB, Malstrom S, Avery BJ, et al. Bik, a novel death-inducing protein shares a distinct sequence motif with Bcl-2 family proteins and interacts with viral and cellular survival-promoting proteins. Oncogene. 1995;11:1921–8. [PubMed] [Google Scholar]
- Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–65. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- Chauhan D, Velankar M, Brahmandam M, Hideshima T, Podar K, Richardson P, et al. A novel Bcl-2/Bcl-X(L)/Bcl-w inhibitor ABT-737 as therapy in multiple myeloma. Oncogene. 2007;26:2374–80. doi: 10.1038/sj.onc.1210028. [DOI] [PubMed] [Google Scholar]
- Chen S, Dai Y, Harada H, Dent P, Grant S. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res. 2007;67:782–91. doi: 10.1158/0008-5472.CAN-06-3964. [DOI] [PubMed] [Google Scholar]
- Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, et al. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell. 2001;8:705–11. doi: 10.1016/s1097-2765(01)00320-3. [DOI] [PubMed] [Google Scholar]
- Chittenden T, Harrington EA, O’Connor R, Flemington C, Lutz RJ, Evan GI, et al. Induction of apoptosis by the Bcl-2 homologue Bak. Nature. 1995;374:733–6. doi: 10.1038/374733a0. [DOI] [PubMed] [Google Scholar]
- Choi SS, Park IC, Yun JW, Sung YC, Hong SI, Shin HS. A novel Bcl-2 related gene, Bfl-1, is overexpressed in stomach cancer and preferentially expressed in bone marrow. Oncogene. 1995;11:1693–8. [PubMed] [Google Scholar]
- Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102:13944–9. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary ML, Sklar J. Nucleotide sequence of a t(14;18) chromosomal breakpoint in follicular lymphoma and demonstration of a breakpoint-cluster region near a transcriptionally active locus on chromosome 18. Proc Natl Acad Sci U S A. 1985;82:7439–43. doi: 10.1073/pnas.82.21.7439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotter FE, Johnson P, Hall P, Pocock C, al Mahdi N, Cowell JK, et al. Antisense oligonucleotides suppress B-cell lymphoma growth in a SCID-hu mouse model. Oncogene. 1994;9:3049–55. [PubMed] [Google Scholar]
- Degterev A, Lugovskoy A, Cardone M, Mulley B, Wagner G, Mitchison T, et al. Identification of small-molecule inhibitors of interaction between the BH3 domain and Bcl-xL. Nat Cell Biol. 2001;3:173–82. doi: 10.1038/35055085. [DOI] [PubMed] [Google Scholar]
- Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD, Letai A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest. 2007;117:112–21. doi: 10.1172/JCI28281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Gaizo Moore V, Schlis KD, Sallan SE, Armstrong SA, Letai A. BCL-2 dependence and ABT-737 sensitivity in acute lymphoblastic leukemia. Blood. 2008;111:2300–9. doi: 10.1182/blood-2007-06-098012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell. 2007;12:171–85. doi: 10.1016/j.ccr.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Eskes R, Desagher S, Antonsson B, Martinou JC. Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol Cell Biol. 2000;20:929–35. doi: 10.1128/mcb.20.3.929-935.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, et al. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–28. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- Gibson L, Holmgreen SP, Huang DC, Bernard O, Copeland NG, Jenkins NA, et al. bcl-w, a novel member of the bcl-2 family, promotes cell survival. Oncogene. 1996;13:665–75. [PubMed] [Google Scholar]
- Greenblatt MS, Bennett WP, Hollstein M, Harris CC. Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res. 1994;54:4855–78. [PubMed] [Google Scholar]
- Hanada M, Delia D, Aiello A, Stadtmauer E, Reed JC. bcl-2 gene hypomethylation and high-level expression in B-cell chronic lymphocytic leukemia. Blood. 1993;82:1820–8. [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hsu SY, Kaipia A, McGee E, Lomeli M, Hsueh AJ. Bok is a pro-apoptotic Bcl-2 protein with restricted expression in reproductive tissues and heterodimerizes with selective anti-apoptotic Bcl-2 family members. Proc Natl Acad Sci U S A. 1997;94:12401–6. doi: 10.1073/pnas.94.23.12401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Sinicrope FA. BH3 mimetic ABT-737 potentiates TRAIL-mediated apoptotic signaling by unsequestering Bim and Bak in human pancreatic cancer cells. Cancer Res. 2008;68:2944–51. doi: 10.1158/0008-5472.CAN-07-2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inohara N, Ding L, Chen S, Nunez G. harakiri, a novel regulator of cell death, encodes a protein that activates apoptosis and interacts selectively with survival-promoting proteins Bcl-2 and Bcl-X(L) EMBO J. 1997;16:1686–94. doi: 10.1093/emboj/16.7.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4:321–8. doi: 10.1016/s1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002;108:153–64. doi: 10.1016/s0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- Kim H, Rafiuddin-Shah M, Tu HC, Jeffers JR, Zambetti GP, Hsieh JJ, et al. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol. 2006;8:1348–58. doi: 10.1038/ncb1499. [DOI] [PubMed] [Google Scholar]
- Kitada S, Leone M, Sareth S, Zhai D, Reed JC, Pellecchia M. Discovery, characterization, and structure-activity relationships studies of proapoptotic polyphenols targeting B-cell lymphocyte/leukemia-2 proteins. J Med Chem. 2003;46:4259–64. doi: 10.1021/jm030190z. [DOI] [PubMed] [Google Scholar]
- Klasa RJ, Gillum AM, Klem RE, Frankel SR. Oblimersen Bcl-2 antisense: facilitating apoptosis in anticancer treatment. Antisense Nucleic Acid Drug Dev. 2002;12:193–213. doi: 10.1089/108729002760220798. [DOI] [PubMed] [Google Scholar]
- Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10:375–88. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Konopleva M, Watt J, Contractor R, Tsao T, Harris D, Estrov Z, et al. Mechanisms of antileukemic activity of the novel Bcl-2 homology domain-3 mimetic GX15–070 (obatoclax) Cancer Res. 2008;68:3413–20. doi: 10.1158/0008-5472.CAN-07-1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsmeyer SJ, Wei MC, Saito M, Weiler S, Oh KJ, Schlesinger PH. Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 2000;7:1166–73. doi: 10.1038/sj.cdd.4400783. [DOI] [PubMed] [Google Scholar]
- Kozopas KM, Yang T, Buchan HL, Zhou P, Craig RW. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc Natl Acad Sci U S A. 1993;90:3516–20. doi: 10.1073/pnas.90.8.3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutuk O, Letai A. Alteration of the mitochondrial apoptotic pathway is key to acquired paclitaxel resistance and can be reversed by ABT-737. Cancer Res. 2008;68:7985–94. doi: 10.1158/0008-5472.CAN-08-1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, et al. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell. 2005;17:525–35. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, et al. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–42. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- Lee EF, Czabotar PE, Smith BJ, Deshayes K, Zobel K, Colman PM, et al. Crystal structure of ABT-737 complexed with Bcl-xL: implications for selectivity of antagonists of the Bcl-2 family. Cell Death Differ. 2007;14:1711–3. doi: 10.1038/sj.cdd.4402178. [DOI] [PubMed] [Google Scholar]
- Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–92. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- Letai A, Sorcinelli MD, Beard C, Korsmeyer SJ. Antiapoptotic BCL-2 is required for maintenance of a model leukemia. Cancer Cell. 2004;6:241–9. doi: 10.1016/j.ccr.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Lin X, Morgan-Lappe S, Huang X, Li L, Zakula DM, Vernetti LA, et al. ‘Seed’ analysis of off-target siRNAs reveals an essential role of Mcl-1 in resistance to the small-molecule Bcl-2/Bcl-XL inhibitor ABT-737. Oncogene. 2007;26:3972–9. doi: 10.1038/sj.onc.1210166. [DOI] [PubMed] [Google Scholar]
- Martinou JC, Dubois-Dauphin M, Staple JK, Rodriguez I, Frankowski H, Missotten M, et al. Overexpression of BCL-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron. 1994;13:1017–30. doi: 10.1016/0896-6273(94)90266-6. [DOI] [PubMed] [Google Scholar]
- Meijerink JP, Mensink EJ, Wang K, Sedlak TW, Sloetjes AW, de Witte T, et al. Hematopoietic malignancies demonstrate loss-of-function mutations of BAX. Blood. 1998;91:2991–7. [PubMed] [Google Scholar]
- Meijerink JP, Smetsers TF, Sloetjes AW, Linders EH, Mensink EJ. Bax mutations in cell lines derived from hematological malignancies. Leukemia. 1995;9:1828–32. [PubMed] [Google Scholar]
- Miyashita T, Reed JC. Bcl-2 oncoprotein blocks chemotherapy-induced apoptosis in a human leukemia cell line. Blood. 1993;81:151–7. [PubMed] [Google Scholar]
- Mohammad RM, Goustin AS, Aboukameel A, Chen B, Banerjee S, Wang G, et al. Preclinical studies of TW-37, a new nonpeptidic small-molecule inhibitor of Bcl-2, in diffuse large cell lymphoma xenograft model reveal drug action on both Bcl-2 and Mcl-1. Clin Cancer Res. 2007;13:2226–35. doi: 10.1158/1078-0432.CCR-06-1574. [DOI] [PubMed] [Google Scholar]
- Muchmore SW, Sattler M, Liang H, Meadows RP, Harlan JE, Yoon HS, et al. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature. 1996;381:335–41. doi: 10.1038/381335a0. [DOI] [PubMed] [Google Scholar]
- Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7:683–94. doi: 10.1016/s1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- Nakashima T, Miura M, Hara M. Tetrocarcin A inhibits mitochondrial functions of Bcl-2 and suppresses its anti-apoptotic activity. Cancer Res. 2000;60:1229–35. [PubMed] [Google Scholar]
- Nguyen M, Marcellus RC, Roulston A, Watson M, Serfass L, Murthy Madiraju SR, et al. Small molecule obatoclax (GX15–070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc Natl Acad Sci U S A. 2007;104:19512–7. doi: 10.1073/pnas.0709443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien SM, Cunningham CC, Golenkov AK, Turkina AG, Novick SC, Rai KR. Phase I to II multicenter study of oblimersen sodium, a Bcl-2 antisense oligonucleotide, in patients with advanced chronic lymphocytic leukemia. J Clin Oncol. 2005;23:7697–702. doi: 10.1200/JCO.2005.02.4364. [DOI] [PubMed] [Google Scholar]
- O’Connor L, Strasser A, O’Reilly LA, Hausmann G, Adams JM, Cory S, et al. Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 1998;17:384–95. doi: 10.1093/emboj/17.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288:1053–8. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–81. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–19. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- Puthalakath H, Huang DC, O’Reilly LA, King SM, Strasser A. The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol Cell. 1999;3:287–96. doi: 10.1016/s1097-2765(00)80456-6. [DOI] [PubMed] [Google Scholar]
- Puthalakath H, Villunger A, O’Reilly LA, Beaumont JG, Coultas L, Cheney RE, et al. Bmf: a proapoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science. 2001;293:1829–32. doi: 10.1126/science.1062257. [DOI] [PubMed] [Google Scholar]
- Rampino N, Yamamoto H, Ionov Y, Li Y, Sawai H, Reed JC, et al. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science. 1997;275:967–9. doi: 10.1126/science.275.5302.967. [DOI] [PubMed] [Google Scholar]
- Rao PH, Houldsworth J, Dyomina K, Parsa NZ, Cigudosa JC, Louie DC, et al. Chromosomal and gene amplification in diffuse large B-cell lymphoma. Blood. 1998;92:234–40. [PubMed] [Google Scholar]
- Reed JC. Apoptosis-targeted therapies for cancer. Cancer Cell. 2003;3:17–22. doi: 10.1016/s1535-6108(02)00241-6. [DOI] [PubMed] [Google Scholar]
- Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, et al. Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science. 1997;275:983–6. doi: 10.1126/science.275.5302.983. [DOI] [PubMed] [Google Scholar]
- Shibue T, Takeda K, Oda E, Tanaka H, Murasawa H, Takaoka A, et al. Integral role of Noxa in p53-mediated apoptotic response. Genes Dev. 2003;17:2233–8. doi: 10.1101/gad.1103603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker AR, Mitten MJ, Adickes J, Ackler S, Refici M, Ferguson D, et al. Activity of the Bcl-2 family inhibitor ABT-263 in a panel of small cell lung cancer xenograft models. Clin Cancer Res. 2008;14:3268–77. doi: 10.1158/1078-0432.CCR-07-4622. [DOI] [PubMed] [Google Scholar]
- Strasser A, Harris AW, Bath ML, Cory S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature. 1990;348:331–3. doi: 10.1038/348331a0. [DOI] [PubMed] [Google Scholar]
- Tagscherer KE, Fassl A, Campos B, Farhadi M, Kraemer A, Bock BC, et al. Apoptosis-based treatment of glioblastomas with ABT-737, a novel small molecule inhibitor of Bcl-2 family proteins. Oncogene. 2008 doi: 10.1038/onc.2008.259. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Saito H, Atsukawa K, Ebinuma H, Okuyama T, Ishii H. Bcl-2 prevents doxorubicin-induced apoptosis of human liver cancer cells. Hepatol Res. 2003;25:192–201. doi: 10.1016/s1386-6346(02)00244-9. [DOI] [PubMed] [Google Scholar]
- Trudel S, Li ZH, Rauw J, Tiedemann RE, Wen XY, Stewart AK. Preclinical studies of the pan-Bcl inhibitor obatoclax (GX015–070) in multiple myeloma. Blood. 2007a;109:5430–8. doi: 10.1182/blood-2006-10-047951. [DOI] [PubMed] [Google Scholar]
- Trudel S, Stewart AK, Li Z, Shu Y, Liang SB, Trieu Y, et al. The Bcl-2 family protein inhibitor, ABT-737, has substantial antimyeloma activity and shows synergistic effect with dexamethasone and melphalan. Clin Cancer Res. 2007b;13:621–9. doi: 10.1158/1078-0432.CCR-06-1526. [DOI] [PubMed] [Google Scholar]
- Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–8. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- Tsujimoto Y, Gorham J, Cossman J, Jaffe E, Croce CM. The t(14;18) chromosome translocations involved in B-cell neoplasms result from mistakes in VDJ joining. Science. 1985;229:1390–3. doi: 10.1126/science.3929382. [DOI] [PubMed] [Google Scholar]
- Tzung SP, Kim KM, Basanez G, Giedt CD, Simon J, Zimmerberg J, et al. Antimycin A mimics a cell-death-inducing Bcl-2 homology domain 3. Nat Cell Biol. 2001;3:183–91. doi: 10.1038/35055095. [DOI] [PubMed] [Google Scholar]
- van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–99. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–2. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- Verhaegen M, Bauer JA, Martin de la Vega C, Wang G, Wolter KG, Brenner JC, et al. A novel BH3 mimetic reveals a mitogen-activated protein kinase-dependent mechanism of melanoma cell death controlled by p53 and reactive oxygen species. Cancer Res. 2006;66:11348–59. doi: 10.1158/0008-5472.CAN-06-1748. [DOI] [PubMed] [Google Scholar]
- Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–8. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- Vogler M, Dinsdale D, Sun XM, Young KW, Butterworth M, Nicotera P, et al. A novel paradigm for rapid ABT-737-induced apoptosis involving outer mitochondrial membrane rupture in primary leukemia and lymphoma cells. Cell Death Differ. 2008;15:820–30. doi: 10.1038/cdd.2008.25. [DOI] [PubMed] [Google Scholar]
- Wang JL, Liu D, Zhang ZJ, Shan S, Han X, Srinivasula SM, et al. Structure-based discovery of an organic compound that binds Bcl-2 protein and induces apoptosis of tumor cells. Proc Natl Acad Sci U S A. 2000;97:7124–9. doi: 10.1073/pnas.97.13.7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Yin XM, Chao DT, Milliman CL, Korsmeyer SJ. BID: a novel BH3 domain-only death agonist. Genes Dev. 1996;10:2859–69. doi: 10.1101/gad.10.22.2859. [DOI] [PubMed] [Google Scholar]
- Wang Z, Song W, Aboukameel A, Mohammad M, Wang G, Banerjee S, et al. TW-37, a small-molecule inhibitor of Bcl-2, inhibits cell growth and invasion in pancreatic cancer. Int J Cancer. 2008;123:958–66. doi: 10.1002/ijc.23610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M, et al. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 2000;14:2060–71. [PMC free article] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–30. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss LM, Warnke RA, Sklar J, Cleary ML. Molecular analysis of the t(14;18) chromosomal translocation in malignant lymphomas. N Engl J Med. 1987;317:1185–9. doi: 10.1056/NEJM198711053171904. [DOI] [PubMed] [Google Scholar]
- Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, et al. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19:1294–305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE, et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315:856–9. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- Witham J, Valenti MR, De-Haven-Brandon AK, Vidot S, Eccles SA, Kaye SB, et al. The Bcl-2/Bcl-XL family inhibitor ABT-737 sensitizes ovarian cancer cells to carboplatin. Clin Cancer Res. 2007;13:7191–8. doi: 10.1158/1078-0432.CCR-07-0362. [DOI] [PubMed] [Google Scholar]
- Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80:285–91. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- Zha H, Reed JC. Heterodimerization-independent functions of cell death regulatory proteins Bax and Bcl-2 in yeast and mammalian cells. J Biol Chem. 1997;272:31482–8. doi: 10.1074/jbc.272.50.31482. [DOI] [PubMed] [Google Scholar]
- Zhang L, Insel PA. Bcl-2 protects lymphoma cells from apoptosis but not growth arrest promoted by cAMP and dexamethasone. Am J Physiol Cell Physiol. 2001;281:C1642–7. doi: 10.1152/ajpcell.2001.281.5.C1642. [DOI] [PubMed] [Google Scholar]
- Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–13. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- Zou H, Li Y, Liu X, Wang X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem. 1999;274:11549–56. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]