

Abstract
This review summarizes the in vivo assessment--preliminary, preclinical, and clinical--of chemotherapeutics derived from camptothecin or a derivative. Camptothecin is a naturally occurring, pentacyclic quinoline alkaloid that possesses high cytotoxic activity in a variety of cell lines. Major limitations of the drug, including poor solubility and inactivity at physiological conditions, prevent full clinical utilization. Camptothecin remains at equilibrium in an active lactone form and inactive hydrolyzed carboxylate form. The active lactone binds to DNA topoisomerase I cleavage complex, believed to be the single site of activity inhibiting DNA religation, resulting in apoptosis. A series of small molecule camptothecin derivatives have been developed that increase solubility, lactone stability and bioavailability to varying levels of success. A number of macromolecular agents have also been described wherein camptothecin(s) are covalently appended or non-covalently associated with the goal of improving solubility and lactone stability, while taking advantage of the tumor physiology to deliver larger doses of drug to the tumor with lower systemic toxicity. With the increasing interest in drug delivery and polymer therapeutics, additional constructs are anticipated. The goal of this review is to summarize the relevant literature for others interested in the field of camptothecin-based therapeutics, specifically in the context of biodistribution, dosing regimens, and pharmacokinetics with the desire of providing a useful source of comparative data. To this end, only constructs where in vivo data is available are reported. The review includes published reports in English through mid-2009.
Keywords: Camptothecins, topoisomerase I inhibitors, polymer therapeutics, in vivo, cancer therapy
1. INTRODUCTION
Wall and Wani isolated 20-(S)-camptothecin (CPT) in 1966 from the bark of Camptotheca acuminata, but quickly observed that CPT suffers from many limitions including poor stability and solubility.1 A year after the discovery of CPT, Wall and Wani discovered paclitaxel, another anticancer drug, which also showed great promise.2 While both drugs showed powerful anticancer activity,3 CPT’s poor solubility and unpredictable adverse drug interactions favored the development of paclitaxel as a broad spectrum chemotherapeutic.4 However, the CPTs gained much interest in the late 1980’s when the molecular target was identified: DNA topoisomerase I (TOP I) is believed to be the single point of biological activity.5–10 Crystal structures later confirmed the binding pocket for CPT as well as for a series of other compounds.11–13 TOP I is an essential enzyme that relaxes supercoiled DNA prior to transcription through the formation of single strand breaks and religation. Upon binding to TOP I, CPT prevents religation and causes apoptosis. Pommier has reviewed the literature focusing on mechanisms of molecular determinants for response of TOP I inhibitors.14 To overcome the solubility and stability issues associated with camptothecin, various derivatives have been developed. Although a number of small and large molecule compounds are currently in clinical trials only two CPT derivatives, irinotecan and topotecan, are approved for clinical use. Irinotecan is currently used for metastatic colorectal cancer. Topotecan has been approved for ovarian cancer, cervical cancer and small cell lung cancer. These derivatives employ tertiary amine cations to improve solubility and subsequently improve lactone stability. Currently, the CPTs -- notably topotecan,15–21 irinotecan,22–27 9-aminocamptothecin,28, 29 9-nitrocamptothecin30, 31 and belotecan32 -- are being investigated for use as a late-stage therapy either alone or in combination therapies.
Alternatively, polymer technologies may be utilized to afford water-soluble camptothecins. Polymer therapeutics for cancer therapy is a burgeoning field33–36 that combines the therapeutic capacity of small molecule drugs with polymers designed to exploit tumor physiology37, 38 to achieve improved efficacy. These constructs complement the arsenal of self-assembling systems including micelles and liposomes. Both covalent or non-covalent strategies have been applied, and accordingly, macromolecular and supramolecular drug constructs present new possibilities to treat a variety of diseases. Incorporation of these small molecules into any construct is pursued with the intent of surmounting limitations that preclude broad clinical application including poor solubility, rapid clearance, high systemic toxicity and/or poor selectivity toward cancer cells.39
We have organized this review by small molecule and macromolecular agents. Small molecule derivatives are classified according to their site of modification (quinoline ring, lactone or 20-hydroxyl), with a specific section for each drug. Macromolecular agents are subdivided into non-covalent assemblies and covalent constructs with sections dedicated to each architecture and the small molecule agent investigated. As these molecules progressed from benchtop to bedside, new names were utilized to represent the chemical name, the company name, the generic name and the brand name. The situation is further exacerbated when pharmaceutical companies merge or sell their products to other companies. Accordingly, the section heading refers to the most common name with alternate names indicated in parentheses. To simplify our discussion of pharmacokinetics, we focus on the half-life and area under the curve (AUC) values. Available pharmacokinetic parameters such as clearance rates and mean residence times are not included here because it muddies our efforts to providing a concise summary of the field as it applies to future therapeutics.
Furthermore, interpatient variability has been seen in many studies using the CPTs. The inability to accurately determine an optimal dose for all patients has limited the utility of these drugs until better methods for patient specific therapies are developed. For example, one study using irinotecan suggests that body weight and surface area do not accurately determine dosing.40 That is, while many drugs are delivered as a mg/m2 dose in human patients, a flat dose across all patients reported in mg, regardless of patient size, is suggested to be a more appropriate. The current convention for reporting drug doses in animals is mg/kg while human studies use mg/m2. For easiest comparison, we report the published values and convert everything to mg/kg using body weight/surface area conversions found in the Toxicologist’s Pocket Handbook.41 Doses are reported as the dose administered by day rather than the total dose administered over the duration of the experiment. Doses of macromolecular constructs are reported with respect to the amount of CPT rather then total molecular weight of the construct. Toxicity data is included using maximum tolerated doses (MTDs) that are generally classified as the highest dose that does not cause death to the organism or specific organs, does not cause toxic manifestations reducing the life of the animal and does not appreciably decrease the body weight of the animal.42 In most cases described here, the MTD caused mild neutropenia, leucopenia and thrombocytopenia along with other nonhematologic toxicities including fatigue, nausea and diarrhea, but was generally reversible after treatment. Additional details about the pharmacokinetics, pharmacodynamics and toxicity may be found in the appropriate references.
Many of the compounds described in this paper were developed in academic labs providing ample reports of the synthesis, characterization in vitro and in vivo evaluation. Naturally, some new drugs developed and being investigated in the pharmaceutical industry lack publications. One such drug is NKTR-102 from Nektar, which is currently in Phase II trials for second-line colorectal cancer, metastatic breast cancer, platinum-resistant breast cancer and metastatic cervical cancer. We make efforts to mention promising drugs even when publications do not exist, but the lack of publications from the pharmaceutical industry on specific compounds prevents inclusion in other cases.
Herein, we aim to review the relevant literature dealing with in vivo testing of CPT and its derivatives as well as the architectures that incorporate these molecules. In an attempt to deliver a single, clear evaluation and comparison of the in vivo data, we have summarized all of the data collected into tabular format at the end of each section. The data derives from peer-reviewed journals written in English as of September 2009. Other supporting articles leading to the reports of pharmacokinetic data and efficacy are also included. Furthermore, some small molecule derivatives with preliminary efficacy data are included to show the direction of current efforts with small molecules. Macromolecular architectures are only included when pharmacokinetic data of the small molecule drug was available to make the appropriate comparisons.
2. THE CAMPTOTHECINS
2.1 Camptothecin (NSC100880)
The camptothecins are cytotoxic, quinoline alkaloids characterized by the planar pentacyclic ring system.1, 43 While the A-D rings of CPT are necessary to maintain activity, modifications are permissible.44 The E-ring lactone, however, is necessary for activity due to the binding site found in TOP I.45 Hydrolysis or removal of the lactone leads to loss of all activity. The equilibrium between the closed, active lactone and the open, inactive carboxylate form is further influenced by both the affinity of the carboxylate for human serum albumin and the local pH in vivo.46 Originally, CPT was delivered as the sodium salt of the carboxylate to help overcome solubility issues, however, the poor efficacy created a need for new alternatives.47
Even given the hydrolytic sensitivity, the drug remains highly active as an anticancer agent. When delivered in an intralipid formulation through i.m. administration, CPT showed nearly 100% growth inhibition and regression in colon, lung, breast, stomach, ovary and malignant melanoma xenografts.48
Pharmacokinetic studies of CPT in the lactone and carboxylate forms have been performed in rats.49 In various buffers at 37 °C, the carboxylate is the predominant form. In PBS at pH 7.2, 7.4 and 8.0, the half-life of the lactone is 33 min, 22 min and 5.3 min, respectively. Furthermore, equilibrium was achieved between both forms 90 minutes after injection of either 1 mg/kg lactone or carboxylate in rats. The carboxylate was cleared at a much faster rate through the urine and bile as compared to the lactone form. Clearance was also shown to be pH dependent, suggesting that decreasing pH of the urine may reduce bladder toxicity caused by the carboxylate form.50 Additional studies in dogs, monkeys, rats and mice showed toxic effects including emesis, diarrhea, dehydration, coma and death. Intravenous administration of 80 mg/kg or five doses of 0.625 mg/kg/day in dogs showed cumulative toxicity that was entirely reversible in survivors.51 In human subjects, unpredictable toxicity associated with CPT halted clinical trials and opened the door for new antitumor agents.52–56 The preparation and assessment of derivatives through classical structure activity relationships led to increased efficacy and better understanding for the basis for such activity.
Detailed structure activity relationships (SAR) have led to new CPTs with potent antitumor activity.10, 44, 47, 57–63 Many efforts focused on stabilizing the lactone without compromising cytotoxicity. To summarize the SAR studies, the A and B rings are the most tolerant to modification with substitutions at positions 7, 9, 10 and 11 improving or retaining activity. Altering the C and D rings or substituting positions 12 and 14, however, inactivates the molecules. Interestingly, von Hoff has provided evidence that substitutions that increase hydrogen bonding at the 7-position improve binding to TOP I, thus increasing activity over CPT.64 The E ring, where binding to TOP I occurs, tolerates only minor modifications without dramatic, negative effects. For example, enlargement of the ring to form the beta-hydroxy lactone improves stability and drug activity. Additionally, modification of the C20 hydroxyl group through alkylation or acylation has been shown to stabilize the lactone. Acylation is the favored method for linking CPT covalently to macromolecules.
3. CAMPTOTHECIN MODIFICATIONS
Camptothecin modifications have attracted a great deal of research in an effort to increase the therapeutic index of the parent drug. Shortly after the discovery and initial investigations with CPT, new semi-synthetic derivatives were developed that addressed the solubility and stability issues associated with CPT. Certainly, many other derivatives than those described below have been developed including Low’s peptide folate conjugate,65 Chen’s 20-O-linked esters66 and Battaglia’s polyamine conjugates67, but herein we focus only on derivatives with reported in vivo evaluation.
3.1 Quinoline Modifications
The quinoline ring of CPT is the most commonly modified region. These derivatives show increased solubility, lactone stability and antitumor activity. Derivatives include the FDA approved drugs, irinotecan and topotecan among many others. All of the quinoline modified CPTs are shown in Figure 1.
Figure 1.

Quinoline modified camptothecin derivatives.
3.1.1 10-Hydroxycamptothecin
Animal Models
Like CPT, 10-hydroxycamptothecin is naturally occurring.68 Pharmacokinetic studies in rats after i.v. bolus with 10-hydroxyCPT showed a short distribution half-life and long elimination half-life, with a majority of drug excreted in the urine within the first 6 hours while fecal excretion occurred later, dependent on dose.69 Very little systemic toxicity was observed at doses of 1, 3, and 10 mg/kg, including no gastrointestinal toxicity common in many CPTs. Furthermore, it was found that after i.v. administration, the carboxylate form predominated in all organs except in the bone marrow, where the lactone was favored.
3.1.2 Topotecan (Hycamtin, NSC 609669, SK&F 104864)
Animal Models
To improve solubility over CPT and 10-hydroxyCPT, solubilizing groups have been added to the quinoline ring yielding the approved therapeutics topotecan70 and irinotecan.63 Topotecan owes its increased solubility to a tertiary amine at the 9-position, while irinotecan presents solubilizing groups through the 10-hydroxyl moiety. Topotecan was the first topoisomerase I inhibitor approved for clinical trials by the US Food and Drug Administration following CPT. Initial studies using a murine L1210 model showed an MTD in mice of 30 μmol/kg (13.7 mg/kg) as compared to 22 μmol/kg (10.1 mg/kg) for CPT, but the dosing strategy is unclear.70 In a subsequent study topotecan was administered subcutaneously over 72 h to SCID mice with B-lineage acute lymphoblastic leukemia at higher doses of 15.3 mg/m2 (~5.1 mg/kg) as compared to an i.v. dosage of 1.75 mg/m2 (~0.58 mg/kg). The survival rate for treated mice was 57% at 175 days whereas the control mice died at 40 days.71 To further improve the efficacy of topotecan, a mitochondrial inhibitor was given to animals with RIF-1 tumors to decrease the local pH of the tumor to between 6.8 and 6.4 under the hypothesis that the equilibrium of CPT in the open and closed lactone forms could be influenced. However, the improvement in efficacy seen in vitro did not translate to in vivo studies.46
Topotecan was also investigated for activity against both subcutaneous (s.c.) and intracranial (i.c.) xenografts in mice with 1.9 mg/kg (LD10) administered on days 1–5 and 8–12 by i.p. injection.72 Tumor regression was observed in both s.c. and i.c. xenografts, with 39% increase in survival of mice bearing intracranial glioblastoma multiforme xenografts.72 When delivered intraperitoneally (i.p) to mice bearing solid human tumor xenografts once every four days for four total doses, a MTD of 12.5 mg/kg was observed, with modest reduction in tumor volume.73 When 2 mg/kg topotecan was delivered through oral gavage, a maximum plasma concentration was achieved at 0.25 h, with alpha and beta half-lives of 0.55 h and 2.8 h, respectively. These times are significantly longer than those observed for human subjects.73 Similar half-lives were obtained when 1.75 mg/kg topotecan was delivered intraperitoneally, however, AUC values were three times higher than those obtained through oral gavage (0.09 and 0.29 μg•h/mL). An improved response was observed in rhabdomyosarcomas xenografts when delivered at daily doses of up to 2.0 mg/kg for five consecutive days, every three weeks, for up to 20 weeks, with tumor regression and minor toxicity.73 This study was followed with a more thorough investigation of sustained topotecan exposure in Daoy and Rh30 xenografts. The results suggest that sustained exposure is more effective than delivering single, high doses of drug.74
In addition to traditional dosing strategies, oral dosing has been investigated. One study with topotecan compared the modes of delivery for the drug in mice: oral, intravenous, intraperitoneal and subcutaneous.75 When topotecan was delivered as a single dose orally or i.p., the matching MTD values of 10 mg/kg suggested that oral formulations may have clinical relevance. Subcutaneous administration with a MTD of 20 mg/kg suggested lower bioavailability. Correlations between dosing and the route of tumor inoculation were also examined. When mice were inoculated intravenously with L1210 lymphocytic leukemia, oral administration showed 216% increased life span (ILS) as compared to 183% ILS with i.p. administration. Mice with i.v.-inoculated Lewis lung carcinoma showed an ILS of 86–110% for oral administration as compared to complete regression observed when the drug was delivered subcutaneously. Lewis lung tumors implanted subcutaneously showed similar results for oral and i.v. administration, with 90% tumor growth inhibition when topotecan was delivered three times every four days. Mice bearing B16 melanoma through i.v. inoculation experienced increased median survival time through the oral route as compared to i.p. (56% ILS to 49% ILS). Finally, mice with i.p. M5067 reticulum cell sarcoma showed significantly diminished activity for oral administration as compared to i.p. or s.c. administration. These results suggest a potential for oral administration of topotecan.
In a later study, topotecan was orally administered to mice with s.c. NCI-H460 lung tumor xenografts in four doses of 15 mg/kg every four days. No toxicity was observed and 98% tumor growth inhibition was seen for orally administered drug. Upon i.v. administration, 93% inhibition at the same dose was achieved, but lethal toxicity was observed in one of four mice.76 Similar results were also obtained in JCA-1 prostate cancer xenografts. When oral and i.v routes were compared on the same schedule in POVD small cell lung tumor, U87 glioblastoma tumor, COCF colon tumor, SKOV-3 ovarian tumor and A549 non-small-cell lung tumor xenografts, improved tumor growth inhibition was observed in each case regardless of route of administration. Increased weight loss, however, was observed through oral administration on this schedule. An increased half-life was also observed when delivered orally as compared to i.v. (2.77 h vs. 1.95 h), however, AUC values were nearly 5 times lower for oral delivery (0.6 μg•h/mL vs. 2.5 μg•h/mL), suggesting that drug persistence in the plasma may be more important than concentration.
An ideal therapeutic range was later determined to be between 0.2 and 0.7 μM drug for periods greater than 10 hours.77 This window was determined after mice with OVCAR-3 xenografts were treated with a total dose of 12.5 mg/kg topotecan at 1, 5, 10, 20, 40 or 80 daily i.p. injections. Maximal toxicity was observed when the total dose was delivered in the first 5–10 days of treatment and the maximum efficacy was observed when delivered using 20 daily injections of 0.625 mg/kg without any major toxicity.
Human Patients
Preliminary phase I pharmacokinetic studies in which topotecan (MTD of 2.0 mg/m2 (~0.05mg/kg)) was administered for 30 minutes over five consecutive days every three weeks in patients with advanced cancer found AUC values of 4.09 μg•h/mL and alpha and beta half-lives of 0.06 h and 3.5 h, respectively.78 In phase I clinical trials, a dose of 1.5 mg/m2 (~0.041 mg/kg) is the most common MTD, which may be achieved in a variety of ways, including a weekly 24 h i.v. infusion79, 80 or 0.5h i.v. infusion for five consecutive days every three weeks.81, 82 Promising preliminary results with this dosing regime were also observed in phase I trials while a pharmacokinetic profile showed biexponential elimination from the body, with mean alpha and beta half-lives of 0.1 h and 3 h and 39% of the drug excreted in the urine within the first 24 hours.81 Similar results were obtained in a later study.82 Other MTDs and associated dosing strategies for topotecan have been obtained such as 5.5 mg/m2 (~0.18 mg/kg; 24h i.v. infusion every 21 days)83 and 22.5 mg/m2 (~0.61 mg/kg; 0.5h i.v. infusion every 21 days).84 Interestingly, however, MTDs of 0.68 mg/m2 (~0.018 mg/kg) in patients with solid tumors,85 and 10.0 mg/m2 (~0.27 mg/kg) in patients with acute leukemia86 were obtained using a 120 h i.v. infusion every 21 days. Kantarjian attributes the higher MTD to the difference in cancer type.86 Two studies suggest that using higher doses can achieve efficacy with subsequent successful treatment of the toxicities.87, 88 Additionally, each of the clinical trials discussed here were performed to investigate a variety of variables from pediatric patients with solid malignant tumors83, 88 and adult patients with solid tumors79–82, 84, 85, 89 to refractory or relapsed acute leukemia.86, 87
In phase II trials, 30 min i.v. infusions of topotecan at 1.5 mg/m2 (~0.041mg/kg) administered for five consecutive days every three weeks to patients with advanced pancreatic cancer yielded a 122 d median survival time with no significant antitumor response.90 The lack of tumor volume reduction led to recommendation against topotecan for pancreatic cancer treatment. A higher dose of 3.5 mg/m2 given for 30 min five consecutive days every three weeks was also investigated in patients with colorectal cancer. This study relied on co-administration with granulocyte-colony stimulating factor (GCSF) to counteract toxic effects of the higher dose.91 At this dose, a mean AUC of 0.34 μg•h/mL was obtained with alpha and beta half-lives of 0.2 h and 4 h, respectively. While pharmacokinetic parameters remained similar to lower doses reported in the absence of GCSF, an insufficient increase in efficacy did not justify further study.
3.1.3 Irinotecan (CPT-11, Camptosar)
Animal Models
Although topotecan was the first topoisomerase inhibitor approved for clinical trials since CPT, studies with irinotecan entered the clinic only slightly later. Irinotecan (CPT-11) has an ethyl substituent at position 7 and a dipiperidyl carbamate at position 10, which is metabolized to SN38, a 7-ethyl-10-hydroxy derivative that is 100 to 1000 times more cytotoxic than the prodrug.92 Bioactivation of the prodrug has been shown to occur through human carboxylesterase 2 (hCE-2)93 and human hepatic microsomes in the liver, with evidence of participation of the enzyme, P-450 3A, through an oxidized form.94, 95 Rabbit carboxylesterase, however, has been shown to activate CPT-11 more efficiently than the human enzymes.96 This route of activation suggests the opportunity for targeted therapies in cancer cells transfected to overexpress the carboxylesterase proteins. Analysis of human plasma collected from patients receiving CPT-11 infusions showed that the lactone is more stable in the metabolized SN38 (64% lactone) than in parent CPT-11 (37% lactone).97 The area under the curve (AUC) pharmacokinetics of CPT-11 delivered intravenously to mice with L1210 tumors was determined to be 3 μg•h/mL and 23.5 μg•h/mL, corresponding to an 8-fold increase in residence time with only a four-fold increase in dose from 10 to 40 mg/kg.98 Additionally, SN-38 was found to have AUC values between 0.41 and 1.08 μg•h/mL after CPT-11 administration, whereas the AUC value rose to 1.35 μg•h/mL when 10 mg/kg of SN-38 was delivered directly. However, the concentration of SN-38 remained above the ED95 value for 5 h when delivered as CPT-11 as compared to 1 h when given as SN-38.98 This data supports the use of CPT-11 over SN38 due to beneficial solubility and the steady-state kinetics obtained from the prodrug form. However, gastrointestinal toxicity remains a major side effect of CPT-11 therapy.99, 100 Irinotecan was also investigated for oral delivery by administering it as a powder-filled capsule daily for five days every three weeks showing AUC values of 0.65 μg•h/mL and 0.76 μg•h/mL on days 1 and 5, respectively. An MTD of 50 mg/m2 (~1.35 mg/kg) was found in patients with advanced solid tumors.101 Half-lives for irinotecan were determined to be 7 h on day one and 12 h on day five using this strategy. This dosing schedule and pharmacokinetics suggest the potential for further study with orally available irinotecan, however, the gastrointestinal toxicity and myelosuppression remain a drawback.
The antitumor effect of irinotecan was demonstrated in vitro and in vivo in vincristine and adriamycin resistant P388 xenografts.102 Tumor suppression was measured by the percent increase in life span (ILS) compared to control mice after intravenous administration on days 1, 5 and 9 after tumor inoculation. In vincristine resistant tumors, a 130% ILS was observed at total dose of 200 mg/kg of CPT-11 and a 20% ILS was observed with a 4 mg/kg dose of vincristine. Similar results were observed with adriamycin resistant cells.
Human Patients
While irinotecan was able to treat tumors resistant to other therapies, a study with irinotecan as a first line therapy proved interesting. In a phase II trial of 90 patients including those previously treated with 5-fluorouracil and 31 untreated patients, the percentage of partial responses increased from 13.3% to 25.8%, respectively.103 Irinotecan was also investigated in untreated patients with metastatic colorectal cancer with 90 min infusions of 125 mg/m2 (~3.38 mg/kg) weekly for four consecutive weeks every six weeks.104 This study is one of the few studies involving previously untreated patients, thus providing a clear investigation of irinotecan as a first-line therapy. Thirteen patients showed partial response with median survival of 12.1 months. The authors compare this to the commonly used fluorouracil plus leucovorin combination therapy, which offers a 11.5-month median survival time for patients. Side effects of this therapy were observed to be neutropenia in 22% of the patients and diarrhea in 29% of the patients, which was counteracted with diphenhydramine. Similar toxicities and responses were observed at the same dosing schedule in patients with squamous cell carcinoma of the cervix.105
To counteract the gastrointestinal toxicity and resulting diarrhea observed in patients receiving irinotecan, alternative schedules were investigated. With side effects resulting from treatments given weekly, a study of 90 min infusions of irinotecan every three weeks found an MTD of 240 mg/m2 (~6.5mg/kg) with an AUC value of 11.5 μg•h/mL and a mean elimination half-life of 6.7 h.106 At this dose, only 3 out of 72 patients experienced diarrhea who received initial doses higher than the MTD. A later study in patients with metastatic cancer previously treated with surgery, chemotherapy or radiation therapy found a higher MTD of 290 mg/m2 (~7.8 mg/kg) using the same dosing schedule with an AUC value of 18.1 μg•h/mL and a half-life of 13 h.107 This study also showed promising tumor growth inhibition, with four patients experiencing partial response and one with complete response. Furthermore, another study found an MTD of 350 mg/m2 (~9.5 mg/kg) when using the same dosing schedule.108 Due to the success seen with doses up to 350 mg/m2, it is unclear what parameters led to lower MTDs in each study. The effectiveness and length of previous treatments as well as alternative complications associated with the cancer may all contribute to the disparity of reports. To better understand these differences, hepatic function of patients was investigated. The recommended doses between 200 mg/m2 (~5.4 mg/kg) and 350 mg/m2 (~9.5 mg/kg) depend on bilirubin levels, which is a marker for disease states of the liver.109, 110 From the data presented here it is clear that many factors must go into devising a treatment regimen with irinotecan.
While much of the research with irinotecan has focused on solid malignancies and metastases in the lung, liver, pancreas and digestive tract, targeting the brain has been investigated. One phase II trial, built from positive data obtained in preclinical and phase I trials,111–116 tested the efficacy of irinotecan in patients with malignant gliomas.117 This study, however, gave less than desired results. Eighteen patients were treated with 90-min. i.v. infusions once a week for four weeks with two weeks off after treatment to complete a six week cycle. The patients received up to 10 cycles of treatment before stopping the study, with a median of 2 cycles. One patient had complete response, five patients experienced disease stability, five patients progressed, six patients were removed from the study due to toxicity and another refused further therapy. Most recently, a case study reporting the use of irinotecan in combination therapy with 5-fluorouracil and leucovorin acid to treat an ovarian tumor during pregnancy found no adverse side effects observed in the baby up to six months after birth.118
3.1.4 9-Aminocamptothecin (9-AC)
Animal Models
Shortly after the development of irinotecan, a series of CPTs were developed with substitutions on the A-ring.59 One derivative, 9-aminocamptothecin showed the highest activity in cell culture, and later showed antitumor activity in vitro10, 47 and in vivo.119 A pharmacokinetic comparison of 9-aminocamptothecin with CPT in the lactone and sodium carboxylate forms was conducted after i.v. administration in mice.120 Although at different doses, the elimination of 9-AC occurs more rapidly than CPT with elimination half-lives of 1.4 h for 9-AC (5mg/kg) and 24.6 h for CPT (10 mg/kg). The initial plasma lactone concentration was higher after i.v. injection of 9-AC compared CPT at the same doses. Due to fast clearance of 9-AC, the plasma concentration fell below 10 nM at 8 h compared to 48 h for CPT. The promising cytotoxic effects and lactone stability of 9-AC led to the suggestion that continuous i.v. infusion was the ideal method to obtain steady state pharmacokinetics and efficacy.
Human Patients
A pharmacokinetic study of 9-AC in human patients showed dose-dependent half-lives ranging from 4.5 h to 21 h with doses of 0.208 mg/m2 to 1.5 mg/m2 (~0.006–0.041 mg/kg).121 This work was followed by phase I clinical trials of 9-AC administered in a colloidal dispersion as a 72 h continuous i.v. infusion. While there was no evidence of tumor regression, tumor growth did not progress during therapy.122 Various other studies have shown that 9-AC has broad activity in human xenografts including, melanoma, breast, colon and brain tumors,123–126 however, when delivered through continuous intravenous infusion to patients with non-small-cell lung cancer, a response rate of only 9% was observed.127
Similarly, in phase II trials, as second-line therapy for ovarian carcinoma, 18 of 28 patients saw no response to 9-AC, while only 1 had complete response, 2 had partial response and 6 had stable disease.128 While poor efficacy of the drug prevents full scale clinical use, 9-AC has shown potential as a method for sensitizing cells prior to radiation therapy.28
3.1.5 9-Nitrocamptothecin (9-NC, Rubitecan, Orathecin)
Animal Models
After investigations of 9-AC showed little promise, an intermediate in its synthesis, 9-NC, was tested for cytotoxic properties and was found to be converted to 9-AC in vivo.129 Compared with 9-AC, half-life for 9-NC increased from 1.2 h to 10 h in mice given 4.1 mg/kg through i.v. injection in cottonseed oil. Similarly, AUC increased from 63 μg•h/mL to 441μg•h/mL. However, upon oral delivery of 0.1 mg/kg gelatin capsule, a comparison to 9-AC showed that 9-NC had a higher AUC (2.6 μg•h/mL vs. 0.3 μg•h/mL) but lower half-lives (2.5 h vs. 7.1 h) and lower maximum plasma concentrations (3.4 h vs. 10.3 h). Although the drug suffered from poor solubility, oral availability prompted further investigation toward the use of 9-NC clinically.
A pharmacokinetic study between i.v. and oral administration of 9-NC to rats concluded that oral administration of 9-NC may be more effective clincially.130 When delivered through i.v. administration at doses 1.5 mg/kg, 3 mg/kg or 6 mg/kg, half-lives of 0.5 h were obtained for lactone, carboxylate and total drug, regardless of dose. AUC values for the lactone, carboxylate and total drug were 0.25, 0.75 and 1.2 μg•h/mL for each increasing dose. Oral administration of 6 mg/kg of 9-NC provided a slightly longer half-life of 0.8 h with a lower AUC value of 0.25 μg•h/mL for the lactone and carboxylate forms.
Human Patients
Phase I trials with oral 9-NC in patients with metastatic cancer found an MTD of 1.5 mg/m2 (~0.04 mg/kg) on a schedule of five consecutive days weekly.131 At this dose in phase II trials, hematological and gastrointestinal toxicities similar to irinotecan were observed with modest efficacy in patients with ovarian, tubal or peritoneal cancers.132 Pharmacokinetics in these patients showed great variability with AUC values ranging from 0.6 μg•h/mL to 2.8 μg•h/mL and a mean half-life of 11 h. While the results with 9-NC showed moderate promise and the lactone stability improved over CPT, the insolubility and equilibrium of the drug favoring the inactive carboxylate prevented further exploration with this drug without further modification. Similar to 9-AC, however, 9-NC has been investigated as a sensitizer for radiation therapy.133
3.1.6 Lurtotecan (GI47211, GG211)
Animal Models
Lurtotecan is water soluble by virtue of a methylpiperazino group at position 7 and an ethylenedioxy ring bridging positions 10 and 11.134 Initial comparisons to topotecan found that lurtotecan was to be both more soluble (5.8 mg/mL vs. 3.1 mg/mL) and more cytotoxic in vitro.135 Lurtotecan was evaluated in mice with HT-29 and SW48 colon tumor xenografts dosing twice a week for five weeks using a ratio of tumor volume after treatment to tumor volume before treatment (T/B). Success was defined by a T/B ratio <1, meaning that the tumor regressed in size. Lurtotecan provided T/B values of 0.8 and 0.4 at doses of 9 and 12 mg/kg, respectively, in HT-29 xenografts and 0.9 and 0.6 at the same doses in SW48 xenografts. However, body weight loss was observed in both tumor models with two out of six animals dying at the higher dose. Topoecan on the other hand showed T/B values of 4.3 and 2.9 at 9 and 11 mg/kg, respectively in HT-29 cells and 3.1 and 2 in SW48 cells at the same doses with significant body weight loss at all doses.
Human Patients
In phase I clinical trials with doses ranging from 0.3 to 1.75 mg/m2 (~0.008 to 0.047 mg/kg) for five consecutive days every three weeks, an MTD was determined to be 1.5mg/m2 (0.041 mg/kg).136, 137 Lurtotecan was determined to have concentration pharmacokinetic profiles following a three compartment model with total drug alpha, beta and gamma half-lives of 0.095h, 0.91h and 7.1h on day one and 0.062h, 1.2h and 15h on day four of treatment. One and four day AUC values of 0.057 μg•h/mL and 0.064 μg•h/mL were obtained for total drug with 25% corresponding to lactone.
Two different phase I trials investigated the potential of delivering lurtotecan through continuous infusion for 3 days138 or 7, 14 and 21 day continuous infusions.139 When delivered as a 3 day continuous infusion every four weeks to heavily pretreated patients, a MTD of 1.2 mg/m2 (~0.03 mg/kg) was determined, while a slightly higher MTD of 2.0 mg/m2 (~0.05 mg/kg) was found for minimally pretreated patients.138 Over the dose range studied, a mean half-life of 7.5 h was observed for the lactone, with total drug blood concentration four times higher than lactone concentration. Of the 44 patients in this study, only three patients experienced partial responses, while two others observed decreases in hepatic lesions. In the subsequent study, where lurtotecan was administered in 0.3 to 0.5 mg/m2 (~0.008 to 0.013 mg/kg) as a 7, 14 or 21 day continuous infusion, AUC values increased from 0.031 μg•h/mL to 0.18 μg•h/mL when delivered at 0.3 mg/m2 everyday for 7 days and 21 days, respectively. Additionally, only a slight increase to 0.19 μg•h/mL was observed at 0.5 mg/m2 for 21 days. However, these studies and one later study showed significant patient variation between correlation of AUC values and dose, suggesting further investigation into the cause and potential clinical solutions.140
Lurtotecan moved swiftly to phase II trials despite unpredictable pharmacokinetics due to the mild side effects associated with the drug, but only modest antitumor activity was observed.141 Patients with breast cancer (23 patients), colorectal cancer (19 patients) and non-small-cell lung cancer (22 patients) were treated with 30 min i.v. injections of 1.2 mg/m2 (0.03 mg/kg) for five consecutive days every three weeks. No complete responses were obtained in any of the patients, with 13% of breast cancer patients and 9.1% of non-small-cell lung cancer patients experiencing partial responses. In breast cancer patients, 39.1% had stable disease with disease progression in 48%. Colorectal cancer patients showed 37% stable disease and 63% cancer progression. In lung cancer patients, 22.7% had stable disease and 68.2% had progressive cancer. With such modest results, lurtotecan was also investigated as a second-line treatment for small-cell lung cancer, but only 11 out of 66 patients experienced partial response.142
3.1.7 10,11-Methylenedioxy Camptothecins
Animal Models
To overcome the gastrointestinal toxicity of CPT-11,99, 100 a series of fluorinated derivatives were developed. Two fluorinated derivatives, a free hydroxyl and a 20-O-linked ester (BMS422461) showed great promise.143 The compounds showed positive gross log cell kill ability (at MTD) in A2780 (0.06 mg/kg), HT29 (0.13 mg/kg) and HCT116 (0.06 mg/kg) tumors in athymic nude mice when administered i.v. every two days for ten days. Furthermore, BMS422461 showed similar lactone stability as compared to CPT-11 in mouse and human plasma as well as in the presence of mouse or human albumin (between 20 and 34% lactone). The parent compound also possessed a fourfold increase in AUC pharmacokinetics as compared to the prodrug and an eight-fold increase as compared to the β-alanyl intermediate upon intraarterial administration. While the data questions the necessity of the prodrug strategy, the improved solubility of the prodrug over the parent molecule provides clear explanation. A semi-quantitative, histopathological assessment of GI injury after subcutaneous injections of the prodrug was performed with the parent drug and irinotecan dosed every day for five days at the MTD. A relative injury scale of 0 (no injury) to 4 (mucosal atrophy and ulceration) was employed, which provided evidence of diminished toxicity over irinotecan in the new fluorinated molecules with injury values of 0.5, 1.5 and 2.8, respectively.
Wadkins and coworkers explored esters of 10,11-methylenedioxycamptothecins.144 The parent compound, 10,11-methylenedioxycamptothecin showed a 3-fold decrease in half-life and a 5-fold decrease in plasma AUC as compared to CPT after a 10 mg/kg i.v. injection in tumor free mice.120 The poor results prevented further studies until Wadkins and coworkers investigated ester derivatives six years later. All of the compounds were tested in a series of breast cancer cell lines showing nanomolar IC50 values in ZR-75, MDA-231 and BT-20 cells. Two of the derivatives contained an electrophilic chloromethyl group at the 7-position poised for covalent attachment to DNA.144 During in vivo studies with MX-1 and MDA-231 human breast tumor xenografts, the chloromethyl groups did not show cytotoxic enhancement. Furthermore, while the glycinate ester derivatives were more water-soluble than CPT-11, there was no enhanced toxicity observed in either cell line, and could be administered to animals at different doses and different dosing schedules. Success could be achieved using smaller doses over a longer period of time. For example, dosing 0.50 mg/kg every day for five days resulted in eight out of eight complete responses, while 5.0 mg/kg dosed once gave seven out of eight complete responses.144 Additional studies in monolayer cell culture as well as in histocultures provided evidence that the acidic conditions (pH 6.8) of tumor cells increases potency of CPTs including this chloromethyl derivative.145, 146
3.1.8 Morpholino Camptothecins
Animal Models
Kim and coworkers synthesized a library with a variety of A-ring substituents to investigate the effects on the stability of the lactone.147 Initial screening of the compounds found a subgroup that maintained the cytotoxicity of CPT. One compound with a morpholine ring bridging positions 9 and 10 showed retention of TOP I inhibition and increased lactone stability in human serum compared to CPT (but not SN38). However, the additional solubility prompted further in vivo investigations in WiDr xenografts in nude mice. The molecule showed efficacy that was comparable to SN38 at 1/10the the dose delivered i.p. every four days for eight total doses. That is, at an MTD of 10 μmol/kg (4.05 mg/kg), the morpholino compound showed tumor growth inhibition at 98.6% while SN-38 dosed at 100 μmol/kg (39.2 mg/kg) gave a 98.2% inhibitory rate.
3.1.9 Exatecan (DX-8951)
Animal Models
While many of the previously described CPTs have suffered from poor lactone stability, the exatecan equilibrium favors the closed, active lactone form. Exatecan owes its stability and solubility to a six-membered ring containing an exocyclic amine connecting carbons 7 and 9, as well as a methyl at position 10 and fluorine at position 11. Each of the modifications have been shown to increase lactone stability, solubility and in vitro efficacy over CPT and irinotecan without the need for metabolic activation.148 Activity has been noted in pancreatic tumors in vitro149–153 and in subcutaneous xenografts in vivo.154 With such promising activity, Hoffman aimed to investigate the activity of exatecan through the treatment of surgical orthotopic implantation to determine the activity of the drug in normal tissue with metastatic capability as compared to gemcitabine.148 Single doses of the drug delivered to mice with early stage MIA-PaCa-2-GFP tumors provided 93% inhibition at 25 mg/kg and 79% inhibition at 15 mg/kg compared to the control. Gemcitabine gave only 67% for the high dose (300 mg/kg) and 43% low dose (150 mg/kg). Similar results were obtained for the early stage BxPC-3 orthotopic human pancreatic model. Furthermore, when using the same dosing strategy, exatecan was effective in inhibiting lymphatic metastasis and completely eliminating lung metastases in late stage BxPC-3 orthotopic tumors. Little to no effect was observed for gemcitabine, with only 45% and 25% tumor growth inhibition for each tumor, respectively.
When exatecan was investigated for efficacy in mice with SC-6 gastric cancer xenografts, a dosing schedule of four total doses given once every fourth day proved more efficacious than three total doses given once every fourth day or three total doses given once every seventh day.154 Using four doses, between 6.25 mg/kg and 18.75 mg/kg were delivered to mice with greater than 94% tumor growth inhibition and no significant toxicity. Similar potency was observed in a number of cell lines. At 18.75 mg/kg, toxicity manifest in significant loss in body weight and death. Although cell dependent toxicity was observed, exatecan proved to be more potent than irinotecan without the need for metabolic activation while retaining the solubility and improving the lactone stability.
Human Patients
Exatecan was eventually taken into clinical trials, with antitumor activity shown in non-small-cell lung cancer, ovarian cancer, tubal cancer, peritoneal cancer, endometrial cancer, colon cancer, hepatoma, thyoma and small-cell carcinoma of the bladder, as well as patients with platinum, topotecan and taxane resistance.155–158 In patients with advanced solid malignancies on a schedule of 30 min i.v. infusions five days a week every three weeks, MTDs of 0.3 and 0.5 mg/m2 (~0.008 and 0.013 mg/kg) were recommended for heavily pretreated patients and mildly pretreated patients, respectively.155 An average half-life of 8.75 h was determined, with severe myelosuppression experienced at doses above the MTD. Patients with advanced leukemia, however, were treated for 30 min on five consecutive days for three weeks through i.v. infusion, resulting in a recommended dose of 0.9 mg/m2 (~0.024 mg/kg).159 This dose appears to be double the MTD for solid tumors.155
When the dosing strategy was changed from every five days to a single 30 min i.v. infusion every three weeks, the MTD increases to 5.33 mg/m2 (~0.14 mg/kg).160 At this dose, the mean half-life is 7.5 h. While promising pharmacokinetic data was obtained, only six of 11 patients had stable disease, while five showed progressive disease. A subsequent study afforded similar results, with pharmacokinetic analysis showing a lactone AUC value and half-life of 0.663 μg•h/mL and 8h, respectively and total drug values of 2.09 μg•h/mL and 10 h.161 However, by increasing the dosing to consecutive weeks on a schedule of 30 min infusions for three out of four weeks, a recommended dose of 2.1 mg/m2 (~0.057 mg/kg) in heavily pretreated patients and 2.75 mg/m2 (~0.074 mg/kg) in minimally pretreated patients was obtained.162 At the higher dose, an AUC value of 1.095 μg•h/mL and half-life of 8 h was determined, suggesting slight advantage for every three weeks at a higher dose.
While pharmacokinetic data proved promising, poor efficacy prompted investigation of an extended dosing regimen in phase I trials. Exetecan mesylate showed mild toxicity and a mean plasma elimination half-life of 7 h after 24 h i.v. infusions for three consecutive weeks in patients with solid tumors.163 The authors of this study expressed their desire to abandon this route before moving to phase II trials due to the inconvenience associated with the dosing regimen. However, the recommended doses were 0.8 mg/m2 (~0.022 mg/kg) for minimally pretreated patients and 0.53 mg/m2 (~0.014 mg/kg) for heavily pretreated patients. An alternative extended dosing strategy involved a 21 day continuous i.v. infusion at a dose of 0.15 mg/m2 (~0.004 mg/kg) for the first 5 days and incremental increases from days 5 to 21 to reach a steady state plasma concentration.157 Increasing the dose to 0.3 mg/m2 led to AUC values that were significantly higher, 465.8 μg•h/mL, than those obtained when drug is administered over short periods of time. While this dosing schedule was even more cumbersome than a 24 h infusion, the greatly improved pharmacokinetics sets a benchmark for future studies, namely macromolecular delivery of CPTs.
Phase II evaluation of exatecan mesylate on a schedule of 30 min i.v. infusion for five days every three weeks provided 8 h half-lives in patients with metastatic breast cancer. The infusion dose given to minimally pretreated patients was 0.5 mg/m2 (~0.014 mg/kg) while that given to heavily pretreated patients was 0.3 mg/m2 (~0.008 mg/kg).164 Out of 39 patients, no patients experienced a complete response, while three experienced partial and four had minor responses. Sixteen and 14 patients, however, experienced stable and progressive disease, respectively. The authors suggest that although mild toxicity was observed from this dosing strategy, poor efficacy suggests that an alternate schedule be used or that the drug was not effective in treating this tumor. A subsequent study in patients with ovarian, tubal or peritoneal cancer showed slightly higher efficacy at the same dosing schedule, with 7 of 16 patients experiencing stable disease.158 Poor efficacy using this dosing strategy, however, was also observed in patients with non-small cell lung cancer165 and metastatic colorectal adenocarcinoma.166 Poor efficacy was also observed in patients with platinum and taxane resistant ovarian cancer.167 Slight improvement of efficacy was observed when administered at a dose of 0.3 mg/m2 (~0.008 mg/kg) for five consecutive days every three weeks as compared to a dose of 2.1 mg/m2 (~0.057 mg/kg) every week for three consecutive weeks out of four. The modest improvement, however, did not warrant further investigation. Patients with metastatic gastric cancer also experienced poor efficacy, with only 2 out of 41 patients experiencing partial response, 18 exhibiting stable disease and 18 showing progressive disease.168 The median survival time in this cohort of patients was determined to be 197 days with 59% survival at 6 months. Biliary tract cancers treated with this dosing regimen provided similar results and modest survival.169 Modest success was also reported for patients with soft tissue sarcoma, a disease typified by poor survival rates and lack of therapeutic options.170 Phase III studies using exatecan and gemcitabine were also performed and compared to gemcitabine therapy alone showing no extended survival time with co-therapy.171 While initial investigations with exatecan mesylate proved to be promising, results from phase II and phase III clinical trials in a variety of cancers suggest further investigation must be completed to identify the role for exatecan mesylate in cancer therapy.
3.1.10 Belotecan (CKD-602, Camtobell)
Animal Models
More recently a water-soluble CPT derivative with an (isopropylamino)ethyl moiety at position seven, known as belotecan, has been developed. Initial studies in nude mice with human tumor xenografts (CX-1, HT-29, WIDR, LX-1, MX-1, SKOV-3 tumors) showed broad antitumor activity. Potency was three times that of topotecan and slightly higher than CPT.172 The schedule dependence of belotecan was also investigated with doses being administered intraperitoneally to mice bearing L1210 leukemia xenografts on the following dosing schedules: a single dose, 5-daily doses, four total doses every fourth day, two total doses every fourth day and two total doses every seventh days. The antitumor effect and increased life span (ILS) were apparent when four doses were delivered every four days, with very little body weight loss occurring at a dose of 25 mg/kg and 213% ILS.
Significant efforts have focused on the acute toxicity of belotecan in a variety of tumor free animals. Studies have been performed in embryonic and adult rats,173–175 dogs,176 pregnant does and rabbits177 as well as human subjects with small cell lung cancer.178 In general, daily doses of 0.01 mg/kg were well tolerated in both maternal and embryonic subjects depending on the length of administration. Furthermore, the maximum tolerated dose was found to be 0.5 mg/kg when delivered to rats for five consecutive days through i.v. injection.179 At this dose, no deaths were observed, but minor toxicities were found to affect the spleen and thymus. Acute toxicity, with adverse effects on the gastrointestinal, hematopoietic and reproductive systems, occurred at single i.v. doses of 40 mg/kg in male rats and 50 mg/kg in female rats.180 While acute toxicity has been demonstrated, little evidence of pharmacokinetic analysis or efficacy in the literature precludes additional discussion here, but macromolecular constructs containing belotecan will be discussed later.
3.1.11 Silatecans
Animal Models
While much work with the CPTs has focused on improving solubility, a series of molecules with increase lipophilicity have been prepared in an attempt to increase cellular uptake and oral availability. One such library of CPT derivatives, known as “silatecans”, employ silyl substituents at the 7-position to increase lipophilicity in an attempt to improve oral bioavailability and allow the drug to cross the blood-brain barrier. Curran and coworkers developed a library of silyl-modified CPTs with such properties.181 The lactone stability of the silyl modified derivatives exceeded that of CPT and other 10,11-methylenedioxy CPTs, with sustained or increased TOP I activity in various cell lines including a U87 glioma cell line. This activity was highest with 7-t-butyl-dimethylsilyl-10-hydroxycamptothecin (TBDMS-10-hydroxy CPT), which provided promising results in subcutaneous U87 human glioma tumor xenografts. Furthermore, intracranial U87 tumor xenografts were employed to investigate blood brain barrier trafficking of this silatecan. Median survival time in the control group was 58 days, with all animals dead by day 70, whereas all animals treated with subcutaneous injections of the silatecan were alive at 120 days. The pharmacokinetics of DB-67 were later measured in SCID mice and found a 1.4 h plasma half-life of the lactone with a 17 μg•h/mL plasma AUC value.182 Liver showed the highest AUC value of 57 μg•h/mL with the kidney (30 μg•h/mL) and lung (20 μg•h/mL) providing lower AUC values.
A slightly different silatecan, called karenitecin, was developed by Van Hattum and coworkers, which employed a trimethylsilane attached through an ethyl linkage at position 7.183 These derivatives were developed with oral availability in mind due to the increased lipophilicity of the molecule capable of being taken up by cells. The maximum tolerated dose was determined to be 1.0 mg/kg for five consecutive days when administered through i.p. injection, whereas a dose of 1.5 mg/kg administered orally on the same schedule gave equitoxic results. While the in vivo efficacy of karenitecin was the same or slightly better than other CPTs in four colon cancer xenografts, oral bioavailability was a major advantage: 67% of karenitecin is bioavailable compared to 30% for topotecan184 and 49% for 9-AC capsules185.
This study was then expanded into human ovarian cell lines and showed promising effects in vivo.186 When delivered to mice with human tumor xenografts through i.p. administration daily for five days, topotecan showed >75% growth inhibition in only one cell line at doses of 1.5 or 2.0 mg/kg. Conversely, kerenitecin showed >80% growth inhibition in all three cell lines when given at a dose of 1.0 mg/kg. Additional studies showed potent activity against lung, prostate, breast, melanoma, head and neck cancers, medulloblastoma, neuroblastoma and rhabdomyosarcoma.187–189 During pharmacokinetic studies in non-human primates, which best represent a model for cerebrospinal fluid (CSF) uptake, it was determined that the lactone in karenitecin was present at greater than 90% of the measurable drug.190 Furthermore, only 5% of karenitecin was observed in the CSF, with a whole body mean distribution half-life of 0.96 h and an elimination half-life of 7.6 h. Peak CSF distribution was observed between 12 min and 25 min after a 0.1 mg/kg i.v. infusion.
Human Patients
In phase II clinical trials in 41 patients with malignant melanoma, karenitecin was delivered on five consecutive days every three weeks.191 Only one patient showed complete response, while three showed minor response, ten showed stabilized disease during treatment and 27 saw no effect. Clinical trials with karenitecin are still underway.
3.1.12 ST1481
Animal Models
Other lipophilic derivatives of CPT, 7-oxyiminomethyl derivatives, were investigated by Zunino and coworkers.192 From the 37 derivatives synthesized by this group, 27 showed increased activity in cell culture as compared to topotecan and 12 were more active than SN-38. Correlations between drug activity and steric or electronic substituents on the oxime were identified with a tri(t-butyl) compound, ST1481, proving to be the most potent derivative in vitro. In athymic nude mice with NCI-H460 and LX-1 lung tumor xenografts, the MTD of ST1481 was determined to be 3 mg/kg as compared to 15 mg/kg for topotecan when administered on a schedule of every four days for four total doses. A 100% tumor volume inhibition (TVI) was observed when using ST1481at the MTD, with 100% complete responses (CR) in LX-1 tumors as compared to 99% TVI and 50% CR observed with topotecan 10 days after treatment. Furthermore, when topotecan was administered at 2 mg/kg five times a week for 10 weeks, 4 out of 10 tumors had regressed by 30 days but all tumors were present at 100 days. However, at 0.5 mg/kg five times a week for 5 weeks, the ST1481 group had no detectable tumors on day 30. At 100 days, 5 of 8 tumors were not detectable.193 From the data, the oxime derivative proved to be about five times more potent than topotecan, with a five-fold increase in AUC (0.55μg•h/mL; 2.43μg•h/mL) and half-life (2.77 h; 11.8 h) when delivered orally at 15 mg/kg for topotecan and 5mg/kg for the oxime derivative.
3.1.13 Chimmitecan
Animal Models
Ding and coworkers developed a series of 9-alkyl derivatives that inhibited TOP I effectively in vitro. Initial studies concluded that chimmitecan, with an allyl group at position 9 and a hydroxyl at position 10, showed the most promise for in vivo investigation.194 Chimmitecan was delivered every three weeks through i.v. injection at 15 mg/kg in three of four human xenograft nude mouse models with different experimental endpoints (A549 lung cancer, 15 weeks; MDA-MB-435 breast cancer, 12 weeks; BEL-7402 hepatocellular cancer, 12 weeks). HCT-116 colon cancer was treated by dosing every two weeks for 6 total weeks. Antitumor efficacies were reported as percent tumor inhibtion/control for each of the cell lines. Against the four tumor xenograft models, chimmitecan showed efficacies of 23.0%, 24.2%, 28.2% and 17.6%, respectively, while CPT-11 showed efficacies of 34%, 42%, 15% and 21% for each cell line, respectively. When compared to CPT-11 at equivalent doses, chimmitecan was significantly more potent in BEL-7402 and A549 models. When delivered orally to treat A549 tumors every two days for seven total doses, tumor inhibition was observed at low doses of 4.5 mg/kg and antitumor activity was observed at 9.0 mg/kg with 22.2% efficacy.
3.2 E-RING MODIFICATIONS
As previously described, modifications to the A and B rings of CPT improve solubility and lactone stability while often retaining, if not improving, efficacy. The C and D rings are the least common sites for modifications due to complete inactivation of the molecule. Very few modifications to the E-ring have been reported, due to the poor efficacy upon manipulation of the lactone. However, the homocamptothecins shown in Figure 2 have offered promise with E-ring stabilization and anti-tumor activity.
Figure 2.
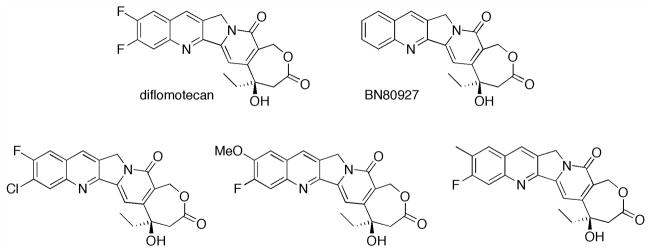
Diflomotecan and related modified camptothecins.
3.2.1 Diflomotecan (BN80915)
Animal Models
Bigg and coworkers developed a series of enlarged E-rings, called homocamptothecins (hCPT), which are characterized by a β-hydroxylactone instead of the natural α-hydroxylactone.195 The addition of a methylene group within the E-ring stabilizes the lactone, leaving 87% in the intact lactone form at pH 7.4 after 24 h compared to only 20% at 1 h for CPT, while retaining TOP I activity comparable to CPT. The in vitro investigation of this series of compounds led to the identification of four lead compounds, which possessed sub-nanomolar IC50 values in one or more cell lines (A427, PC-3, K562adr and MCF7mdr). Interestingly, each of the compounds contains a fluorine substituent at position 10, 11 or both. Each compound was tested in subcutaneous HT-29 tumor xenografts models in nu/nu female athymic nude mice. When administered through i.p. injection 12 times over three weeks using a 4 days on and three days off schedule, a MTD of 0.32 mg/kg was determined. At this MTD, using the methoxy, methyl or difluoro compounds, a tumor growth delay of 12 d, 7 d and 25 d was observed for the derivatives, respectively. The chlorinated compound, however, showed a tumor growth delay of 7 d at 1.25 mg/kg as compared to a 4 d delay at 0.625 mg/kg for CPT. This study was expanded to additional tumor xenografts and compared to topotecan, CPT, and SN-38 with similar results showing higher stability of cleavage complexes and subnanomolar IC50 values.196
Human Patients
In phase I trials, the MTD was determined to be 0.27 mg/day when administered five times orally every three weeks to adults with solid tumors.197 Pharmacokinetics were measured on the fifth consecutive day of treatment showing AUC values of 0.014 μg•h/mL and a half-life of 3.7 h. Furthermore, whil several patients in this study that had been heavily treated prior to this study still showed signs of extended periods of stable disease. When administered through the i.v. route, the MTD of diflomotecan was determined to be 0.15 mg/m2 (~0.0041 mg/kg) as a 20 minute i.v. infusion for five days every three weeks with a recommended dose of 0.125 mg/m2 (~0.0034 mg/kg).198 The treatment also showed very few toxic side effects and either stabilized patients or produced a partial response although this was outside the scope of the study.
A recent phase I study utilizing the flat dosing strategy finds that toxic doses range from 2 mg to 4 mg, due to interpatient variability.199 Upon administration of 2 mg of diflomotecan through 20 min i.v. infusion every three weeks, an AUC value of 0.11 μg•h/mL was found with a half-life of 4 h. When using 3 mg and 4 mg doses, the AUC values increased slightly to 0.12 μg•h/mL and 0.16 μg•h/mL, with half-lives of 3.3 h and 4.6 h, respectively. From the pharmacokinetic data and the toxicities observed, it was determined that the toxic variability was due to drug exposure and not specific dose. With such variability, the authors suggest that further investigation using this strategy for delivery of diflomotecan is not warranted. Interpatient variability with diflomotecan complicates the future utilization of this drug, but future studies at different doses using different schedules may prove advantageous.
3.2.2 Homocamptothecin and BN80927
Animal Models
Although the fluorinated hCPTs showed promising results in vitro and in vivo, Bigg and coworkers investigated a hCPT without a quinoline substituent,200 and a hCPT with a 4-methyl-piperazinomethyl group at position 7, a methyl at position 10 and a chloride at position 11.201 The study of unsubsitituted hCPT provided results similar to the fluorinated compound, showing increased lactone stability and increased TOP I inhibition. While the lactone undergoes slow hydrolysis to the carboxylate form, the seven-membered ring does not spontaneously recyclize. During in vivo studies in athymic mice with HT-29 tumor xenografts, hCPT was administered using a schedule of four days on and three days off for a total of twelve injections at a dose of 1.25 mg/kg as compared to 0.625 mg/kg CPT. Results from this study showed that unsubstituted hCPT inhibited tumor growth as compared to CPT, with tumor volumes of 900, 750, and 400 mm3 for the control, CPT and hCPT, respectively.
The trisubsitituted hCPT showed greater than 90% closed lactone after 3h in human plasma, with 50% of the lactone form still present at 24 h. This new hCPT also showed broad antitumor efficacy in vitro in breast, colon, prostate, ovarian, bladder, leukemia and lung cancers. In vivo efficacy was demonstrated through oral administration to mice with either PC3 or DU145 prostate cancer xenografts. In both models, the preferred schedule was twice a day for 14 days, giving 125% and 175% increase in survival for each cell line, respectively. Only minor toxicity resulted in each model, which rebounded after treatment.
3.3 20-HYDROXY-LINKED MODIFICATIONS
Lactone stability has been shown to increase upon esterification or alkylation of the 20-hydroxyl group. A hypothesis proposed in 1992 implicates the hydroxyl group as a mediator of lactone hydrolysis by activation of water through a hydrogen bond interaction.202 While various ester derivatives have been prepared, only a small number of 20-O-hydroxyl modifications have been tested in vivo. A few examples have already been discussed in the context of A-ring modifications. We address the remaining examples here and discuss utilization of this hydroxyl to covalently append CPT and its derivatives to macromolecular architectures later.
3.3.1 Hydrophobic Esters of CPT
Animal Models
Cao and coworkers reported the synthesis and promise of a series of esters of CPT and 9-nitrocamptothecin (9-NC).203 In human plasma, the lactone of propanoate of CPT diminished to 56% over 6 h as compared to only 0.5% at 2 hours for CPT. Comparatively, the propionate of 9-NC exhibited higher stability in human plasma, with 64.4% lactone present at 6 h and 5.8% present at 51 h, compared to only 7% at one hour for 9-NC. The propanoate of CPT was investigated in CLO-breast tumor and SPA lung tumor xenografts in nude mice, while the propanoate of 9-NC was investigated in SQU colon cancer cells. The breast tumor sizes were measured at 56 days, with average tumor sizes of mice treated with propanoate ranging from ~500–100 mm3 at doses of 5–8 mg/kg, as compared to tumor sizes of ~4500 mm3 in the control animals. Similar results were observed in SPA and SQU tumor xenografts. In a subsequent study with 9-NC, the propanoate ester and butyrate esters showed the greatest toxicity in HL-60 cells and U-937 cells in vitro.204 In vivo data using Doyle lung carcinoma, BRO-melanoma, SPA lung carcinoma and BRE stomach tumor xenografts also suggested promising antitumor activity, however, the poor solubility of the constructs prompted the investigators to dissolve the drugs in cottonseed oil and inject the solutions into the stomach cavity through the anterior wall of the abdomen everyday for five consecutive days each week for the duration of the experiment. While the compounds showed promising lactone stability and antitumor activity, the route of administration was not ideal for prolonged therapy.
3.3.2 Amino Acid Esters of CPT
Animal Models
Lerchen and coworkers at Bayer AG developed a series of 20-hydroxyl linked glycoconjugates of CPT with preferential cellular uptake in cancer cells.152 CPT was acylated with a series of dipeptides, which were then linked to the carbohydrate targeting moiety (p-aminophenyl 3-O-methyl-β-L-fucopyranoside) through a thiourea linkage. Interestingly, when the amino acid adjacent to the CPT was glycine, stability in cell culture was diminished, while valine improved stability. The conjugate with the greatest stability in culture medium and lowest IC50 value in HT29 cells proved to be Sug-HisVal-CPT. When delivered intravenously at the MTD of 32 mg/kg for three consecutive days to mice bearing breast cancer MX-1 xenografts, treated tumor growth/control group growth (T/C%) values were determined to be 1.8%, as compared to 12.7% for topotecan at an MTD dosage of 2.5mg/kg on the same schedule. Fluorescence experiments revealed that cellular uptake in HT29 cells occurs through active transport into the lysosomes. While this study indicates a significant improvement in the design of novel CPTs, further investigation of this conjugate must be completed to determine the pharmacokinetics as compared to the parent molecule.
4. MACROMOLECULAR ARCHITECTURES FOR PASSIVE DRUG DELIVERY
Although a significant library of CPTs has been developed, macromolecular delivery agents have focused on CPT or SN-38. Here, we describe the non-covalent and covalent approaches toward increasing in vivo efficacy using macromolecular constructs. While a large number of architectures have also been developed, many will not be discussed here due to the absence of in vivo data. The compounds described in the literature, that have not yet been investigated in vivo, include non-covalent dendrimer constructs from Ghandehari,205 Grinstaff206 and Simanek207 and covalent dendrimer constructs from Shabat,208–212 and Simanek,213 “clicked” polymers from Emrick,214 micelles from Torchilin215 and Kataoka216, and PEGylated nanoscale graphene oxide from Dai.217
4.1 Non-Covalent Drug Delivery Systems
Various non-covalent drug delivery systems have been developed to improve solubility and lactone stability of CPTs, including micelles, liposomes, dendrimers, nanoparticle drug formulations and hydrogels. Each non-covalent drug delivery vehicle is summarized herein, with comparisons made between the pharmacokinetics and efficacy of the complex to that of the free drug and further summarized in Table 2.
Table 2.
Pharmacokinetic data of macromolecular constructs with non-covalent attachment of camptothecin derivatives.
| Compound (Drug) | Structure | wt% CPT | Subject (Sex) | Tumor Type | Route (time) | Dosing Schedule | Dose (mg/kg) | MTD (mg/kg) | t1/2 (h) | Plasma AUC (μg• h/mL) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PEG-P(bzAsp) (CPT) | Micelle | 40 | ddY mice (M) | None | i.v. | q1d x 1 | 1 | NR | NR | 47 | 218 |
| HSA-DB-L (CPT) | Liposome | 3 | ddY mice | None | i.v. | q1d x 1 | 0.08 | NR | 0.8 | 25 | 225 |
| DSPC-Chol (TPT) | Liposome | 12 | Balb/c mice | None | i.v. | q1d x 1 | 0.6 | NR | α: 2 β: 3 |
360000 | 227 |
| TEA-Pn (CPT-11) | Liposome | 41 | SD rats | None | i.v. | q1d x 1 | 4.1 | >130 | 7 | 1400 | 229 |
| TEA-SOS (CPT-11) | Liposome | 41 | SD rats | None | i.v. | q1d x 1 | 4.1 | >130 | 11 | 2100 | 229 |
| LE-SN38 (SN38) | Liposome | 3 | CD2F1 mice (M) | None | i.v. | q1d x 5 | 0.15 | 0.15 | NR | 3 | 232 |
| LE-SN38 (SN38) | Liposome | 3 | CD2F1 mice (F) | None | i.v. | q1d x 5 | 0.23 | 0.23 | NR | 3 | 232 |
| LE-SN38 (SN38) | Liposome | 3 | Beagle dogs | None | i.v. | q1d x 1 | 0.04 | 0.04 | 3 | 0.5 | 232 |
| OSI-211 (GG211) | Liposome | 14 | SD rats (M) | None | i.v. | q1d x 1 | 1.4 | NR | 21 | 1900 | 233 |
| OSI-211 (GG211) | Liposome | 14 | Athymic mice (F) | None | i.v. | q1d x 1 | 1.26 | NR | 2 | 130 | 234 |
| OSI-211 (GG211) | Liposome | 14 | Adult humans | Solid | i.v. | q1d x 1, q3w | 0.01 | 0.01 | 7 | 12 | 237 |
| OSI-211 (GG211) | Liposome | 14 | Adult humans | Solid | i.v. (0.5 h) | q1d x 3, q3w | 0.01 | 0.01 | 7 | 10 | 238 |
| OSI-211 (GG211) | Liposome | 14 | Adult humans | Solid | i.v. (0.5 h) | q1d x 3, q3w | 0.006 | 0.006 | 5 | 4 | 239 |
| 9NC.NP (9-NC) | NP | 0.6 | Wistar rats (M) | None | i.v. | q1d x 1 | 2 | NR | 2 | 4 | 251 |
| SMEDDS-T (9-NC) | NP | 0.5 | SD rats (M) | None | p.o. (gavage) | q1d x 1 | 0.18 | 0.18 | 4 | 0.36 | 252 |
| SMEDDS-C (9-NC) | NP | 0.5 | SD rats (M) | None | p.o. (gavage) | q1d x 1 | 0.18 | 0.18 | 6 | 0.35 | 252 |
Abbreviations: MTD, maximum tolerated dose; AUC, area under the curve; CPT, camptothecin; TPT, topotecan; CPT-11, irinotecan; GG211, lurtotecan; NP, nanoparticle; M, male; F, female; SD, Sprague-Dawley; i.v., intravenous; p.o., oral; NR, not reported.
Dosing abbreviations: q1d x 1, one dose; q1d x 1, q3w, one dose per week every three weeks.
4.1.1 Micelles
Micelles are macromolecular constructs formed from an aggregation of amphiphilic molecules, which display charged or charge-neutral hydrophilic head groups at the water interface and hydrophobic chains toward the center of the vesicle, commonly forming a spherical structure. The hydrophobic interior of the structure enables efficient encapsulation of hydrophobic molecules, such as the CPTs, for drug delivery.
Camptothecin Micelles
Animal Models
Poly(ethylene glycol) is a polymer commonly used to increase solubility and bioavailability of otherwise insoluble drugs. Through the development of poly(ethylene glycol)-poly(aspartic acid) block copolymers and subsequent partial esterification with benzyl alcohol, the formation of micellar structures containing a benzyl rich core capable of encapsulating CPT and a water soluble PEG corona has been realized.218 Micelles formed from block copolymers containing 5 kDa PEG chains and a poly(aspartic acid) block 25 monomer units long esterified with benzyl groups to 70%, were used to encapsulate CPT for treatment of mice with C26 colon tumor xenografts.219 The micelles were found to have an average diameter of 190 nm and 63% incorporation efficiency. Incorporation efficiency reflects the amount of drug encapsulated in the vesicle after removal of unencapsulated drug. In this example, 2 mg of free CPT were mixed with 5 mg PEG-P(AspBz) resulting in vesicles with approximately 20 wt% CPT. When the micelles were delivered through i.v. injection to tumor bearing mice at doses of 15 mg/kg and 30 mg/kg, 72.5% and 81.5% tumor growth inhibition at 8 days was observed as compared to 51.4% for the solution of free CPT at a dose of 1.5 mg/kg. The micelles released nearly 50% of CPT at 24 h, however, blood plasma levels were 150 times higher at 24 h as compared to free CPT. Furthermore, tumor levels showed an 8-fold increase in CPT when using the micelle as compared to the free drug.
10-Hydroxycamptothecin Micelles
Animal Models
Micelles of poly(ethylene glycol)-poly(γ-benzyl-L-glutamate) were also developed to encapsulate hydroxycamptothecin at 57% efficiency.220 The micelles had an average diameter of 200 nm, with 7.5 wt% drug loading capacity. After i.p. administration of 3 mg/kg for five consecutive days, the micelles showed a slow release of hydroxycamptothecin, with maximum blood concentrations at 1 h as compared to the carboxylate form of the free drug with lower concentrations at 0.25 h. The beta half-lives of the carboxylate and micellar 10-hydroxycamptothecin forms were determined to be 5.8 h and 10.2 h, respectively, with AUC values of 431 μg•h/mL and 1034 μg•h/mL, respectively. Anti-tumor effects in golden hamsters with cheek pouch carcinomas showed a 66% decrease in tumor volume when treated with the micellar formulation and only 50% decrease in tumor volume when treated with the free hydroxycamptothecin. This delivery method, however, suffers from poor loading efficiency and cellular inflammation due to the toxicity of the micelles. Pharmacokinetic data and efficacy data show moderate success, but minimal investigation using non-covalent drug-micelle complexes has been completed due to success with other forms of non-covalent drug delivery. Furthermore, CPT attached covalently to micelles has proven successful as will be discussed later.
4.1.2 Liposomes
Liposomal drug delivery has received much attention for the delivery of a variety of insoluble therapeutics, including the CPTs.221 Burke observed the need for an alternative route to deliver CPTs and investigated liposomal drug delivery with CPT, 9-AC, 9-NC, 10-hydroxyCPT and topotecan. Lactone stability increases when drugs were non-covalently complexed with liposomes.222, 223 Current in vivo efforts with liposomal formulations are summarized below.
Camptothecin Liposomes
Animal Models
A series of lipids were investigated to develop liposomes with high levels of CPT loading. Results showed that cardiolipin and N-glutaryl phosphatidyl ethanolamine (NGPE) had 67% and 97% drug loading, respectively, while other neutral or single, negatively charged head groups provided <5% encapsulation.224 Loading is a measure of the difference between total drug in solution and free drug. At a 12.5:1 wt/wt ratio of lipid to drug, 95% loading is observed, which corresponds to 7 wt% CPT. Antitumor activity in an i.p. injected P388 leukemia mouse model was evaluated using a T/C value, which represents the ratio of median survival in treated mice over control mice. CPT delivered intraperitoneally at a dose of 40 mg/kg resulted in a T/C value of 2.07, whereas toxicity was observed at this dose using liposomal CPT. Decreasing the dose of liposomal CPT to 20 mg/kg resulted in a T/C value of 1.86. A L1210 leukemia model with T/C values between 0.85 and 0.92 for free CPT at doses between 30 and 60 mg/kg, whereas liposomal CPT afforded a T/C of 1.46 at a dose of 20 mg/kg. Biodistribution studies after i.v. administration of 10 mg/kg CPT found high quantities of drug in the lung at 6 h, with decreasing levels at 24 h, while liposomal CPT afforded negligible uptake in all organs.
Alternatively, liposomes developed from bis(dodecyl)benzoic acid and poly(ethylene glycol) with a coating of human serum albumin (HSA) achieved 80% CPT encapsulation efficiency.225 Blood plasma levels increased dramatically from an AUC value of 1.1 μg•h/mL for the CPT solution to an AUC value of 24.8 μg•h/mL for the HSA-coated liposome after a 2.5 mg/kg dose with respect to CPT. When delivered to mice with C26 colon carcinomas through i.v. injection of 15 mg/kg, 84.6% tumor growth inhibition was observed as compared to control mice. However, when delivered at a 10 mg/kg dose on days 1 and 3, significant weight loss (>20%), a common marker for CPT toxicity, was observed. Biodistribution studies with this liposome showed nearly 10-fold increase in tumor accumulation with a 60-fold increase in blood plasma at 8 h as compared to the tumor accumulation seen when using the free drug.
Topotecan Liposomes
Animal Models
The success observed in liposomal formulations of CPT extends to more soluble topotecan. One study, produced a 400-fold increase in plasma AUC when topotecan was encapsulated in sphingomyelin and cholesterol liposomes as compared to free drug.226 Furthermore, lactone stability was enhanced with 84% lactone present at 24h after injection of the liposome compared with only 50% lactone present at five minutes after injection of free drug. Mice bearing L1210 ascites treated with a single i.v. dose of 20 mg/kg free drug or three 4 mg/kg doses on days 1, 5 and 9 provided a 11 day median survival time and 15 day median survival time at three doses of 8 mg/kg. Administration of liposomal topotecan at a single dose of 20 mg/kg or the multiple dosing strategy of 4 mg/kg or 8 mg/kg afforded a median survival time of >60 days. However, liposomal topotecan led to more weight loss as compared to an equivalent dose of free drug. To overcome the toxicity, liposomal doses at half the free drug dose were utilized, resulting in efficacy that was still superior to that seen using the free drug. In a liver metastasis model, similar results were obtained, with liposomal topotecan providing >60 d survival in all animals at all doses half that of free drug. A human breast carcinoma model (MDA-435/LCC6) showed median survival times between 20 and 30 days depending on dose, however, liposomal topotecan provided median survival times between 37 and 52 days.
A subsequent study using DSPC/Chol lipids found that at 48 h greater than 70% topotecan existed in the lactone form when encapsulated in liposomes, while the free drug showed a hydrolysis half-life of 0.33 h.227 Pharmacokinetic data of the DSPC/Chol-topotecan complex show an approximate 40-fold increase in the AUC values of free drug from 9400μg•h/mL to 358400 μg•h/mL with an increase in alpha half-life from 0.1 h to 2.1 h after a single i.v. injected dose of 5 mg/kg. Only a small increase, however, was observed in beta half-life from 2.6 h to 2.9 h. Although liposomal topotecan was found to leak significantly in the presence of plasma, the antitumor efficacy of liposomal topotecan surpassed that of the free drug. When delivered at this dose weekly for two weeks, topotecan showed a 21% tumor growth inhibition at 32 days in mice bearing C26 tumors, while liposomal topotecan exhibited 57% tumor growth inhibition.
Irinotecan Liposomes
Animal Models
Irinotecan was also investigated using DSPC/Chol liposomes which could be loaded with up to 35 wt% drug. This formulation could provide a significant increase in plasma levels when the complex was delivered through i.v. injection at 50 mg/kg to SCID/Rag-2M mice.228 Furthermore, the lactone stability of irinotecan was increased due to liposomal protection, with >95% lactone at 4 h and 80% at 24 h as compared to only 40% for the free drug at 1 hour. Toxicity studies showed that a 100 mg/kg dose of free irinotecan elicited toxic effects within one minute. This toxicity was not observed with the same dosage of the liposomal formulation, but weight loss over time was observed as a toxic marker. When free irinotecan was delivered as a single 50 mg/kg dose to mice inoculated with s.c. LS180 tumors, tumor growth was delayed for 22 days post inoculation as compared to 19 days for control animals. This tumor growth inhibition increased to 30 days when the irinotecan-liposome complex was delivered at 50 mg/kg on days 11, 15 and 19. The liposomal formulation showed delayed growth at 34 days, and no growth at 40 days for the single and triple doses, respectively. Similar results were obtained in LS174T tumors, which exhibit relatively slow-growth and mimic liver metastases secondary to colorectal cancer. The potential for the treatment of liver metastases was suggested by histology wherein accumulation was observed at the tumor’s periphery, while no tumor uptake at 24 h post i.v. injection was observed.
To improve drug loading in liposomes, Drummond and coworkers developed a method of delivery that utilizes polyphosphate or sucrose octasulfate co-encapsulation. This method achieved a loading efficacy of 1.4 mol CPT-11/mol phospholipids or 109,000 drug molecules per liposome, corresponding to a 10- to 20-fold increase in drug loading.229 Blood plasma half-lives of 7 h and 11 h were obtained in Sprague-Dawley rats after i.v. administration of the polyphosphate and sucrose octasulfate liposome complexes, respectively. The sucrose octasulfate formulation showed improved overall pharmacokinetics, with an AUC value of 2134 μg•h/mL and 57 h half-life release of CPT-11 from the liposome. Rats bearing HT29 xenografts received the sucrose encapsulated formulation resulting in four (36.4%) tumor free animals at study end (66 days) as compared to a maximum survival time of 35 days for mice receiving 50 mg/kg CPT-11 every four days for four total i.v. doses of CPT-11. This method of encapsulation has proven optimal for increasing drug loading, plasma half-life and efficacy as compared to the small molecule. The impact that this strategy has on in vivo activity given the need for metabolic activation in the liver remains unclear.
SN-38 Liposomes (LE-SN-38)
Animal Models
To further improve the antitumor activity of liposomal s, SN-38, rather than CPT-11, was encapsulated in liposomes to circumvent the need for metabolic activation of CPT-11. Liposomes were formed from DOPC, cholesterol and cardiolipin with a drug to lipid ratio of 1:18.230 Studies in P388 tumor bearing mice showed median survival of 20 days when CPT-11 was administered at 16 mg/kg over five consecutive days, whereas administration of 4 mg/kg of LE-SN-38 for five consecutive days offered 100% long term survival (>60 days). In mice bearing HT29 xenografts a dose of 8 mg/kg gave 88% tumor growth inhibition for LE-SN-38 as compared to a 36% tumor inhibition observed using free CPT-11. Similar results were observed in mice bearing Capan1 and MX-1 xenografts. Similar tumor growth inhibition was observed when administered as a single dose of 10 mg/kg, 20 mg/kg or 40 mg/kg in the same cell lines, but increased body weight loss at the higher doses suggests an optimal delivery of multiple low doses rather than a single high dose.231
Detailed pharmacokinetics were also reported with this construct, showing plasma AUC values of 3.92 μg•h/mL and a half-life of 6.38 h for SN-38 after a single 10 mg/kg i.v. injection.232 AUC values at 24 h were significant for liver and spleen (~400 μg•h/mL) suggesting recognition by the reticuloendothelial system (RES), which was supported with the presence of extramedullary hematopoesis in dogs. Furthermore, multiple dosing strategies gave MTDs of 5 mg/kg and 7.5 mg/kg for male and female mice, respectively. Single doses afforded a MTD of 37 mg/kg and 46 mg/kg for male and female mice, respectively. Together, these results suggest significant promise for LE-SN38 at multiple low doses, however, accumulation in the RES warrants concern. Recognition by the RES has been overcome through the use of STEALTH liposomes coated in poly(ethylene glycol). Furthermore, the sex dependent variability of pharmacokinetics in small animals also raises interesting questions toward the treatment of cancer in human subjects.
Lurtotecan Liposomes (SPI-355, NX 211, OSI-211)
Animal Models
Liposomal encapsulation of lurtotecan has progressed into phase II clinical trials. PEGylated liposomes, known as STEALTH liposomes, are formed from HSPC/PEG-DSPE with cholesterol resulting in 90% drug encapsulation efficiency in particles of 100 nm diameter.233 Liposomes were determined to have a half-life of 21 h and an AUC of 1852 μg·h/mL after i.v. injection of 10 mg/kg of liposomal drug as compared to 1.58 h and 1.49 μg•h/mL after injection of 8.72 mg/kg free drug. In the first study using mice bearing HT29 colon xenografts, drug doses of 15 mg/kg and 24 mg/kg, given once weekly for three weeks proved toxic, while 6 mg/kg showed minimal toxicity with complete responses in all animals >70 days. The free drug on the same dosing schedule showed less toxicity with only 3 out of 10 complete responses and 1 partial response at 24 mg/kg. In the second study using the same tumor model, lower doses of liposomal lurtotecan were investigated from 0.1 mg/kg to 5 mg/kg as compared to 20 mg/kg for free drug. The MTD for liposomal lurtotecan was determined to be between 3 and 5 mg/kg. Although deaths were observed, complete regression was experienced in 10 out of 10 mice at 5 mg/kg. At a dose of 3 mg/kg, seven out of 10 complete responses were observed with one partial response and one death not associated with the therapy, while 20 mg/kg of free drug afforded no complete responses and only 1 partial response.
A subsequent study with liposomal lurtotecan showed 99.5% tumor growth inhibition with no deaths and 19% body weight loss when 9 mg/kg were delivered through 30 min i.v. infusion to mice with ES-2 tumor xenografts on days 1, 8 and 15. This compares favorably to 95% TGI with 14 mg/kg lurtotecan and 57% TGI with 16 mg/kg topotecan, the respective MTDs.234 Liposomal lurtotecan also proved successful at tumor inhibition when delivered on days 1 and 9 in a KB xenograft model, with 98% TGI at 9 mg/kg. Administration of a 4 mg/kg dose of liposomal drug proved to be as effective at tumor growth inhibition as 16 mg/kg dose of free drug. Pharmacokinetic analysis after administration of 1 mg/kg of liposomal lurtotecan provided an AUC value of 127 μg•h/mL and a 2 h half-life as compared to an AUC value of 0.069 μg•h/mL and a half-life of 0.83 h for the free drug. Biodistribution studies showed that lurtotecan accumulated in tumors 9- to 67-fold more effectively when administered in the liposome as compared to the free drug. However, significantly high splenic uptake was observed for liposomal lurtotecan with maximum concentration at 6 h and a decreased concentration in all organs after that time. Similar pharmacokinetic and biodistribution results were also reported in a later paper from this group.235
Promise was seen in studies comparing different dosing strategies in SCID mice bearing acute myelogenous leukemia and acute lymphocytic leukemia.236 To investigate the effectiveness of liposomal lurtotecan when administered as either an early treatment or as a delayed treatment, mice were injected intravenously with KBM-3B cells followed by an incubation period (6–8 days for early treatment; 15–19 days for delayed treatment). After the predetermined incubation period, treatment began and continued on a schedule of five consecutive days, every two days for three total doses or once a week for two consecutive weeks. When delivered at a dose of 2 mg/kg on days 1, 3 and 5, in either early or delayed therapy, significant toxicity was observed. When the dose was decreased to 1.75 mg/kg, poor efficacy resulted in all cases. Treatment with 6 mg/kg on days 1 and 8 proved to be the most successful in both early and delayed therapy, with an average increased life span of 146% in early and 196% in delayed therapy. In early therapy the lowest and most prolonged dose of 1 mg/kg for five consecutive days provided a survival increase of 174%. Similar results were obtained in HL-60 A5 and Molt-4 A4 leukemia models.
Human Patients
In phase I trials with liposomal lurtotecan, using the same 30 min i.v. infusion once a week for three weeks provided a recommended dose of 3.8 mg/m2 (~0.1 mg/kg).237 At this dose, AUC values were determined to be between 2210 and 28000 μg•h/mL, with a mean value of 12000 μg•h/mL, and a half-life range of 2.5–11 h with a mean of 6.8 h. To further investigate the potential to increase blood plasma concentration of liposomal lurtotecan, 30 min i.v. infusions were administered for three consecutive days to patients with leukemia.238 An MTD similar to what was found previously (3.7 mg/m2) provided an AUC value of 7.1 μg•h/mL after a single injection with a mean half-life of 7.2 h. In patients with refractory solid tumors administered liposomal lurtotecan as a 30 min i.v. infusion on days 1, 2 and 3 for three consectutive weeks found MTDs of 2.1 mg/m2 (~0.057 mg/kg) and 1.8 mg/m2 (~0.049 mg/kg) for minimally pretreated patients and heavily pretreated patients, respectively.239 Pharmacokinetic data obtained from the patients receiving 2.1 mg/m2 gave AUC values of 4.6 μg•h/mL on day one and 7.3 μg•h/mL on day three with half-lives of 6.9 h and 9.3 h, respectively. Significant variability in pharmacokinetic values was once again observed, with AUC ranges from 0.6 to 24 μg•h/mL and half-lives from 3 h to 20 h. Although efficacy was not determined under this set of studies, it was determined that lower doses could be given over extended periods of time with minimal toxicity, however, less than optimal pharmacokinetics were observed.
In phase II trials, liposomal lurtotecan was delivered to patients with topotecan resistant ovarian cancer on days 1 and 8 of a three-week cycle at doses of 2.4 mg/m2 (~0.065 mg/kg). No complete regression was observed. Only 8 of 22 patients had stable disease suggesting further investigation with a different schedule or a different tumor is warranted.240 In a similar study of patients with squamous cell carcinoma of the head and neck using the same dosing regimen, similar results with mild drug toxicity and poor efficacy were found.241 In a comparative study using either this dosing schedule or a 30 min infusion of 1.8 mg/m2 (~0.049 mg/kg) on days 1, 2 and 3 every three weeks found significant advantage using the later dosing schedule.242 In this study, doses on three consecutive days afforded 1 complete response, 5 partial responses, 22 patients with stable disease and 8 patients with progressive disease. No complete responses and only 2 partial responses were observed with the once weekly administration. Interestingly, AUC values in the three daily doses schedule (4.8 μg•h/mL) were slightly lower than the once weekly schedule (5.8 μg•h/mL). Lurtotecan has shown great potential in vitro and in preliminary in vivo studies, surpassing topotecan efficacy. Phase I and II clinical trials, however, have given less than promising results suggesting a need for alternative routes of delivery.
Belotecan Liposomes (CKD-602 Liposomes)
Animal Models
STEALTH® liposomes containing CKD602 were investigated in a series of tumor xenografts in mice to determine the optimal dosing schedules and the MTDs.243 Tumor inhibition was observed when free drug was administered through i.v. administration to mice bearing A375 human melanoma xenografts on a schedule of once a week for three weeks at doses between 10 mg/kg and 20 mg/kg. Higher doses of 30 mg/kg resulted in tumor regression with no observed toxicity in any case. However, when liposomal CKD-602 was administered intravenously, regression was observed at doses as low as 0.15 mg/kg and up to 1.5 mg/kg with only minimal toxicity. Larger doses between 0.3 mg/kg and 3.0 mg/kg administered every two weeks for three total doses resulted in tumor regression with toxicity occurring at 2.5 mg/kg. Similar results were also observed in ES-2 human ovarian xenografts, with a tumor growth delay of >60 days at doses of 2.25 mg/kg and 8 out of 12 mice were cured. Slightly lower efficacy, however, was observed in H82 human small cell lung cancer and HT-29 human colon xenografts.
A pharmacokinetic analysis of liposomal CKD-602 was conducted in mice bearing A375 tumors.244 Liposomal CKD-602 was administered at 1 mg/kg as an i.v. injection, providing a plasma AUC value of 202 μg•h/mL and tumor AUC value of 13 μg•h/mL. The complex showed higher efficacy as compared to a 30 mg/kg injection of free drug, which displayed a plasma AUC value of 9 μg•h/mL and a tumor AUC value of 12 μg•h/mL. Furthermore, uptake in the liver, kidney and spleen showed 2- to 6-fold increase over plasma AUC values when delivered as free drug, whereas liposomal CKD-602 delivery showed an approximate 5-fold decrease. Interestingly, the brain showed a 2.5-fold increase in AUC for liposomal CKD-602 as compared to free drug, suggesting potential utilization of the complex in the treatment of gliomas.
Human Patients
A phase I and pharmacokinetic investigation of liposomal CKD-602 demonstrated a MTD of 2.1 mg/m2 (~0.057 mg/kg) when the complex was administered intravenously once every three weeks.245 This dosage provided a plasma AUC value of 45 μg•h/mL. However, significant interpatient variability was observed, with AUC values ranging from 7 μg•h/mL to 86 μg•h/mL. Partial response seen in 2 out of 5 patients with ovarian cancer. Stable disease was seen in 6 out of 45 patients with sarcoma, hepatocellular, prostate and thyroid cancer. Although the response was not significant in the series of tumors studied here, phase II studies are currently ongoing, with a focus on ovarian, gastric and small cell lung cancers.
DB-67 Liposomes (AR-67 Liposomes)
Animal Models
Liposomal encapsulation of the silatecan, DB-67, was also investigated in SCID mice to monitor plasma and tissue disposition after delivery of the liposomal derivative as compared to the free drug.182 A dose of 10 mg/kg of liposomal DB-67 or non-liposomal DB-67 was administered through tail vein injection to the mice. Interestingly, the AUC values for nonliposomal and liposomal DB-67 were 17.3 μg•h/mL and 8.2 μg•h/mL, respectively. The lactone half-lives of nonliposomal and liposomal DB-67 were 1.4 and 0.9 h, respectively. This data suggest that the liposome releases DB-67 somewhat rapidly after injection. Furthermore, AUC values measured for each organ show that the lactone form of non-liposomal DB-67 has an extended lifetime in the plasma as compared to the liposomal treatment (17 μg•h/mL vs. 7 μg•h/mL). Decreased splenic (14 μg•h/mL vs. 29 μg•h/mL) and lung (20 μg•h/mL vs. 39 μg•h/mL) AUC values were also observed for the non-liposomal treatment. Each of these data suggests that while DB-67 may be a good candidiate for cancer therapy, the use of liposomes drastically change the pharmacokinetic data in a somewhat unexpected fashion.
4.1.3 Nanoparticle Formulations and Emulsions
While CPT has previously been delivered in complex formulations to improve solubility, nanoparticulate structures and emulsions utilizing polymers are described here. Alternative block copolymers including constructs by Onishi246, 247 and Jiang248 have also been developed, however, a lack of complete pharmacological data prevent their inclusion here.
Hydrophobic Chitosan Nanoparticals With Camptothecin
Animal Models
Chitosan was modified with cholanic acid to increase the hydrophobicity of the nanoparticles for encapsulation of CPT.249 An 80% encapsulation efficiency was obtained using this construct. While complete tumor regression was not observed, tumor growth inhibition was apparent. Mice bearing MDA-MB231 human breast cancer xenografts were administered a 30 mg/kg i.v. dose of free CPT, resulting in a 49% tumor growth inhibition. Chitosan nanoparticles containing 10 wt% CPT were given doses of 10 mg/kg and 30 mg/kg resulting in 68% and 77% tumor growth inhibition, respectively. While survival data showed promising results with 75% and 50% of mice alive at 40 days when utilizing 10 mg/kg and 30 mg/kg doses, there were only four mice used in each experiment. Curiously, 25% of mice receiving saline injections were alive at 40 days whereas those treated with free CPT were dead at 35 days. This data suggest that the chitosan nanoparticles slightly improve survival times in mice, however, additional studies with larger test populations are required along with pharmacokinetic investigation to determine their potential for future studies.
Proprietary Nanoparticles With SN-38
Animal Models
In addition to increasing solubility through drug encapsulation, macromolecular constructs often increase lactone stability. When SN-38 is encapsulated in “soft” nanoparticle formulations of 100 to 300 nm in diameter, lactone stability was shown to be 80% at 3 h as compared to 40% with free SN-38.250 Blood plasma levels of 1 μg/mL in the nanoparticle at 24 h compared favorably to free drug at less than 0.01 μg/mL. Mice bearing HT-29 xenografts were treated with the formulations on days 6, 9, 13 and 16 days after implantation and evaluated for drug efficacy by measuring the time for the tumor to reach 1 g in weight. The tumors took 46, 64, 47 and 50 days to reach 1 g using four different formulations as compared to 22 days for no treatment and 35 days with free irinotecan. Although this study offers a brief glimpse into the use of soft nanoparticles for therapy, extensive investigation with these constructs is limited. Challenges of characterization and polydispersity are overshadowed by the positive results suggesting that additional investigation into the pharmacokinetics and efficacy may be warranted.
Poly(Lactic-co-Glycolic Acid) Nanoparticles with 9-Nitrocamptothecin
Animal Models
Nanoparticles measuring 200 nm in diameter with 9-NC at 33% drug loading were prepared from poly(lactic-co-glycolic acid).251 An 0.8 h elimination half-life increased over free drug from 0.8h to 2.45 h at doses of 2 mg/kg through i.v. injection to rats. Similar results were obtained for the half-life of the lactone, suggesting extended lifetime of the active form. The AUC values for the total free drug and lactone were 0.68 μg•h/mL and 0.45 μg•h/mL, respectively as compared to 3.7 μg•h/mL for the nanoparticle. In vitro cytotoxicity assessments showed that the nanoparticles containing 9-NC were 10 times more cytotoxic than the free drug, presumably due to a higher cellular uptake through endocytosis. Additional in vivo studies in tumor bearing mice must be completed to determine the clinical relevance of this construct.
A self-microemulsifying drug delivery system (SMEDDS) was also developed from a mixture of oil (ethyloleat), surfactant (Tween-80 or cremophor EL), cosurfactant (PEG-400) and drug (9-NC).252 The mean particle size was determined to be between 30 and 40 nm depending on whether Tween-80 (T-form) or cremphol EL (C-form) was utilized as the surfactant, respectively. The half-lives of the 9-NC suspension, SMEDDS C-form, SMEDDS T-form and 9-NC solution delivered orally to rats were 3.3 h, 6.3 h, 3.9 h and 3.5 h, respectively. The AUC values obtained for each delivery system were found to be 0.16 μg•h/mL, 0.35 μg•h/mL, 0.36 μg•h/mL and 0.24 μg•h/mL, respectively. Furthermore, oral bioavailability increased from 17% in the free drug suspension to 37% for both SMEDDS. Efficacy studies in nude mice bearing s.c. SKOV-3 ovarian tumors that were treated with 6 mg/kg 9-NC every four days led to 100% tumor growth inhibition at 24 d. This result is significantly better than the 50% growth inhibition observed for the 9-NC suspension. Untreated mice survived between 5 and 10 days.
4.1.4 Hydrogels
Hydrogels are polymers that swell in the presence of water effectively entrapping guest molecules, such as drugs, within the matrix. This technique for drug delivery is attractive due to the biocompatibility, durability, flexibility and ease of injection at the site of interest. Hydrogels have shown great promise in delivering CPTs for cancer therapy.
Camptothecin Hydrogels
Animal Models
The addition of glycerol-2-phosphate to chitosan leads to hydrogel formation at body temperatures due to a lower critical solution temperatures (LCST) of 37 °C.253–255 CPT was loaded into the hydrogel at 4.5 wt% and showed 85% release at 30 days when studied in vitro. When administered to mice bearing RIF-1 tumor xenografts through intratumoral injection at 24 mg/kg, tumor growth delays of 25 days were reported. This compares favorably to the delay of only 8 days when using a 6 mg/kg i.p. injected CPT.256
Alternatively, two biodegradable PLGA-PEG-PLGA copolymers with 5 kDa total polymer molecular weight were prepared that formed gels from room to body temperature.257 These polymers degraded by 50% and 80% over 30 days in vitro. CPT was PEGylated prior to encapsulation due to large pore sizes within the gel that otherwise led to fast release of the unmodified drug. Although incorporation of the modified drug decreased the gel’s LCST, drug release was observed for over one month. Release was dependent on the wt% of copolymer in solution and, to a lesser extent, drug loading. The optimal polymer and drug loading was found to be 18 wt% of a PLGA-PEG-PLGA copolymer with block weights of 1730-1500-1730 Da and 1.5 wt% drug loading. A 65% tumor inhibition rate was observed when this construct was injected subcutaneously into mice with intradermal S-180 sarcomas introduced by injection in the armpits. Higher drug loading (3 wt%) caused toxicity in nearly 50% of animals.
Topotecan Hydrogels
Animal Models
A two-phase system of the PEG hydrogel and liposomes containing topotecan was investigated to exploit the pore size of the parent hydrogel. Free drug entrapped within the liposome-free, one-phase system as well as free drug were used for comparison.258 A 60-fold increase in the release of topotecan entrapped in liposomes within the hydrogel was observed in vitro. Both systems, however, were evaluated for drug release through subcutaneous administration of 5 mg/kg doses, with observed alpha half-lives of 0.84 h, 0.72 h and 2.70 h for i.v. topotecan, s.c. topotecan liposomes and s.c. hydrogels with topotecan liposomes, respectively. Beta half-lives of 6.2 h, 35.1 h and 89.3 h, respectively, were obtained, however, AUC values of 5.8 μg•h/mL, 3.6 μg•h/mL and 3.0 μg•h/mL for s.c. topotecan, s.c. topotecan liposomes and s.c. hydrogels with topotecan liposomes, respectively were observed. The poor area under the curve data for all of the suspensions suggests poor bioavailability in s.c. tissue. The tumor growth suppression in rats with MAT B III tumors, however, gave mixed results, suggesting that free topotecan was the superior in both small and large tumor models.
4.2 Covalent Drug Delivery Systems
The covalent conjugation of CPTs to macromolecular architectures has shown great potential for improving pharmacokinetics and increasing tumor efficacy. Most commonly, CPT is attached to the polymer through an ester bond with the 20-hydroxy moiety. This linkage not only conveys solubility through conjugation with a water-soluble polymer, but also improves lactone stability. Some linkages are chosen as specific substrates for enzymatic cleavage, while others are used due to their pH sensitivity, but may also undergo hydrolysis. The advances with covalently linked CPTs to polymers are discussed here with a table of summarized pharmacokinetics (Table 3) for comparison to the small molecule derivatives in Table 1 and non-covalent pharmacokinetics in Table 2. Covalent constructs offer advantages and disadvantages over non-covalent assemblies. Of the advantages, the opportunity to execute structure-activity studies in a very narrowly defined composition space is attractive. Disadvantages include, in addition to constituting a new drug entity, the burden of characterization. The characterization of covalent macromolecular constructs is oftentimes not trivial, and enthusiasm for biological results need to be tempered with the critical evaluation of the claims on composition. The literature summarized here and the following section clearly contains many well-characterized systems.
Table 3.
Pharmacokinetic data of macromolecular constructs with covalent attachment of camptothecin derivatives.
| Compound (Drug) | Structure | MW (kDa) | wt% CPT | Subject (Sex) | Tumor Type | Route (time) | Dosing Schedule | Dose (mg/kg) | MTD (mg/kg) | t1/2 (h) | Plasma AUC (μg• h/mL) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NK012 (SN-38) | Micelle | 19 | 20 | Balb/c mice (F) | HT-29 (colon) | i.v. | q4d x 3 | 6 | 6 | 31 | 5000 | 259 |
| PEG(CPT)2 (CPT) | Linear Polymer | 40 | 2 | CD1 mice (F) | None | i.v. | q1d x 1 | 6 | NR | α: 0.1 β: 4 |
18300 | 267 |
| PEG(GlyCPT)2 (CPT) | Linear Polymer | 40 | 2 | CD1 mice (F) | None | i.v. | q1d x 1 | 17.5 | NR | α: 0.1 β: 10 |
53300 | 268 |
| Pegamotecan (CPT) | Linear Polymer | 40 | 2 | Adult humans | Solid | i.v. (1h) | q1d x 1, q3w | 3.8 | 3.8 | 94 | 29 | 269 |
| Pegamotecan (CPT) | Linear Polymer | 40 | 2 | Adult humans | Solid + Lymphoma | i.v. (1h) | q1d x 1, q3w | 1.75 | 1.75 | 44 | 27 | 270 |
| IT-101 (CPT) | Linear Polymer | 85 | 5 | SD rats | None | i.v. | q1d x 1 | 1 | 8.8 | α: 0.4 β: 6 γ: 20 |
700 | 273 |
| PLGA (CPT) | Linear Polymer | 33 | 38 | Athymic mice (F) | HT29 (colon) | i.v. | q1d x 1 | 18 | NR | 97 | 240 | 279 |
| PLGA (CPT) | Linear Polymer | Adult humans | Solid | i.v. | q1d x 1, q1w | 25 | 25 | 63 | 27 | 280 | ||
| HPMA (CPT) | Linear Polymer | 28 | 5 | Balb/c mice (M) | None | i.v. | q1d x 1 | 20 | NR | 27 | 1121 | 281 |
| HPMA (CPT) | Linear Polymer | 21 | 10 | Balb/c mice (M) | None | i.v. | q1d x 1 | 20 | NR | 20 | 500 | 281 |
| HPMA (CPT) | Linear Polymer | 20 | 10 | Adult Humans | Solid | i.v. | q1d x 3 | 68 | 68 | 8 | 8700 | 283 |
| T-0128 (SN-38) | Linear Polymer | 130 | 5 | Wistar rats (F) | Walker 256 (sarcoma) | i.v. | q1d x 1 | 1 | 100 | α: 4 β: 17 | 101 | 288 |
| T-0128 (SN-38) | Linear Polymer | 130 | 5 | Balb/c mice (M) | St-4 (gastric) | i.v. | q1d x 1 | 40 | 80 | 30 | 23900 | 292 |
| DE-310 (DX-8951) | Linear Polymer | 300 | 8 | Mice | Meth A (sarcoma) | i.v. | q1d x 1 | 5.7 | 5.7 | NR | 6300 | 293 |
| DE-310 (DX-8951) | Linear Polymer | 300 | 8 | Adult humans | Solid | i.v. (3h) | q1d x 1, q6w | 0.016 | 0.016 | 418 | 1180 | 298 |
| PEG-PLL (CPT) | Branched Polymer | 40 | 6 | Balb/c mice (F) | C26 (colon) | i.v. | q1d x 1 | 10 | NR | 31 | 1400 | 303 |
| DTS-108 (SN-38) | Peptide targeted | 3.6 | 11 | Beagle dogs | None | i.v. (0.8 h) | q1d x 1 | 2.2 | 2.2 | NR | 4.8 | 310 |
Abbreviations: PLGA, poly(L-glutamic acid MTD, maximum tolerated dose; AUC, area under the curve; CPT, camptothecin; TPT, topotecan; CPT-11, irinotecan; GG211, lurtotecan; DX-8951, exatecan; M, male; F, female; SD, Sprague-Dawley; i.v., intravenous; NR, not reported.
Dosing abbreviations: q4d x 3, one dose every four days for three total doses; q1d x 1, one dose; q1d x 1, q3w, once a week every three weeks; q1d x 1, q6w, once a week every six weeks.
Table 1.
Pharmacokinetic data of small molecule camptothecin derivatives.
| Drug | Subject (Sex) | Tumor Type | Route (time) | Dosing Schedule | Dose (mg/kg) | MTD (mg/kg) | t1/2 (h) | Plasma AUC (μg• h/mL) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| CPT | SD Rats (M) | None | i.v. | q1d x 1 | 1.0 | NR | NR | 0.21 | 49 |
| CPT | Balb/c mice | None | i.v. | q1d x 1 | 10 | NR | α: 0.3 β: 4 |
1.80 | 120 |
| Na-CPT | SD Rats (M) | None | i.v. | q1d x 1 | 1.0 | NR | NR | 0.37 | 49 |
| Na-CPT | Balb/c mice | None | i.v. | q1d x 1 | 10 | NR | α: 0.1 β: 2 |
1.47 | 120 |
| TPT | SCID mice | None | s.c. | q1d x 1 | 5.3 | NR | 2 | 0.14 | 71 |
| TPT | Mice | None | p.o. (gavage) | q1d x 1 | 2.0 | 2 | α: 0.6 β: 3 |
0.09 | 73 |
| TPT | Mice | None | i.p. | q1d x 1 | 1.75 | 1.75 | α: 0.3 β: 3 |
0.29 | 73 |
| TPT | Athymic mice (F) | None | i.v. | q1d x 1 | 15 | NR | 2 | 2.5 | 76 |
| TPT | Athymic mice (F) | None | p.o. | q1d x 1 | 15 | NR | 3 | 0.55 | 76 |
| TPT | Adult humans | Solid | i.v. (0.5 h) | q1d x 5, q3w | 0.05 | 0.05 | α: 0.1 β: 4 |
4.1 | 78 |
| TPT | Adult humans | Solid | i.v. (24 h) | q1d x 1, q1w | 0.04 | 0.04 | 4 | 0.12 | 79 |
| TPT | Adult humans | Solid | i.v. (24 h) | q1d x 1, q1w | 0.04 | 0.04 | 3 | 0.06 | 80 |
| TPT | Adult humans | Solid | i.v. (0.5 h) | q1d x 5, q3w | 0.04 | 0.04 | α: 0.2 β: 3 |
3.34 | 82 |
| TPT | Pediatric humans | Solid/Leukemia | i.v. (24 h) | q1d x 1, q3w | 0.15 | 0.15 | α: 0.2 β: 3 |
NR | 83 |
| TPT | Adult humans | Solid | i.v. (0.5 h) | q1d x 1, q3w | 0.61 | 0.61 | 6 | 3.38 | 84 |
| TPT | Adult humans | Solid | i.v. (24 h) | q1d x 3, q3w | 0.03 | 0.03 | 3 | NR | 88 |
| TPT | Adult humans | Solid | i.v. (24 h) | q1d x 5, q3w | 0.06 | 0.06 | NR | 0.11 | 89 |
| TPT | Adult humans | Colorectal | i.v. (0.5 h) | q1d x 5 | 0.10 | 0.10 | 4 | 0.34 | 91 |
| CPT-11 | Balb/c mice (F) | L1210 leukemia | i.v. | q1d x 1 | 10 | NR | β: 1 | 2.96 | 98 |
| CPT-11 | Balb/c mice (F) | L1210 leukemia | i.v. | q1d x 1 | 20 | NR | β: 1 | 7.65 | 98 |
| CPT-11 | Balb/c mice (F) | L1210 leukemia | i.v. | q1d x 1 | 40 | NR | β: 1 | 23.45 | 98 |
| CPT-11 | Adult humans | Solid | p.o. (PFC) | q1d x 5, q 3w | 1.35 | 1.35 | 12 | 0.8 | 101 |
| CPT-11 | Adult humans | Colorectal | i.v. (1.5 h) | q1d x 1, q1w, 4/6 | 3.38 | 3.38 | NR | 3.08 | 104 |
| CPT-11 | Adult humans | Solid | i.v. (1.5 h) | q1d x 1, q3w | 6.49 | 6.49 | 6 | 15.2 | 106 |
| CPT-11 | Adult humans | Solid | i.v. (1.5 h) | q1d x 1, q3w | 7.84 | 7.84 | 13 | 22.3 | 107 |
| CPT-11 | Adult humans | Solid | i.v. (0.5 h) | q1d x 1, q3w | 9.46 | 9.46 | 7 | 25.6 | 109 |
| 9-AC | Balb/c mice | None | i.v. | q1d x 1 | 10 | NR | α: 0.2 β: 1 |
3.58 | 120 |
| 9-AC | Adult humans | Peritoneal | i.p. | q2d x 6, q4w | 0.04 | 0.04 | 21 | 0.46 | 121 |
| 9-AC | Adult humans | Solid | i.v. (24 h) | q1d x 3, q3w | 0.11 | 0.11 | 31 | 0.11 | 122 |
| 9-AC | Mouse | None | p.o. (gavage) | q1d x 1 | 4.1 | NR | 1 | 0.06 | 129 |
| 9-AC | Dog | None | p.o. | q1d x 1 | 1.0 | NR | 21 | 0.31 | 129 |
| 9-AC | Adult human | None | p.o. | q1d x 1 | 0.10 | NR | 7 | 0.31 | 129 |
| 9-AC | Adult human | None | p.o. | q1d x 1 | 1.0 | NR | 13 | 9.12 | 129 |
| 9-NC | Mouse | None | p.o. (gavage) | q1d x 1 | 4.1 | NR | 10 | 0.44 | 129 |
| 9-NC | Dog | None | p.o. | q1d x 1 | 1.0 | NR | 6 | 0.19 | 129 |
| 9-NC | Adult human | None | p.o. | q1d x 1 | 0.1 | NR | 3 | 2.60 | 129 |
| 9-NC | Adult human | None | p.o. | q1d x 1 | 1.0 | NR | 5 | 17.1 | 129 |
| 9-NC | SD rats (M) | None | i.v. | q1d x 1 | 1.5 | NR | 0.5 | 0.55 | 130 |
| 9-NC | SD rats (M) | None | i.v. | q1d x 1 | 3.0 | NR | 0.5 | 1.48 | 130 |
| 9-NC | SD rats (M) | None | i.v. | q1d x 1 | 6.0 | NR | 0.5 | 2.38 | 130 |
| 9-NC | SD rats (M) | None | p.o. (tube) | q1d x 1 | 6.0 | NR | 0.8 | 0.54 | 130 |
| 9-NC | Adult humans | Ovarian, tubal, peritoneal | p.o. | q1d x 4 | 0.04 | NR | 14 | 1.51 | 132 |
| GG211 | Adult humans | Solid | i.v. (0.5 h) | q1d x 5, q3w | 0.03 | 0.03 | 4 | 0.02 | 136 |
| GG211 | Adult humans | Solid | i.v. (0.5 h) | q1d x 5, q3w | 0.03 | 0.03 | α: 0.1 β: 0.9 γ: 9.6 |
0.02 | 137 |
| GG211 | Adult humans | Solid | i.v. (24 h) | q1d x 3, q4w | 0.03 | 0.03 | 6 | NR | 138 |
| GG211 | Adult humans | Solid | i.v. (24 h) | q1d x 21 | 0.01 | 0.01 | NR | 0.15 | 139 |
| GG211 | Adult human | Solid | i.v. (0.5 h) | q1d x 5, q3w | 0.03 | 0.03 | 0.9 | 0.03 | 140 |
| BMS-422461 | SD rats (M) | None | i.a. | q1d x 1 | 1.36 | NR | 0.2 | 0.47 | 143 |
| 10,11-MC | Balb/c mice | None | i.v. | q1d x 1 | 10 | NR | α: 0.1 β: 0.8 | 0.32 | 120 |
| DX-8951 | Adult humans | Solid | i.v. (0.5 h) | q1d x 5, q3w | 0.01 | 0.01 | 11 | 3.2 | 155 |
| DX-8951 | Adult humans | Solid | i.v. (24 h) | q1d x 1, q3w | 0.06 | 0.06 | NR | 1.8 | 156 |
| DX-8951 | Adult humans | Solid | i.v. (24 h) | q1d x 21 | 0.004 | 0.004 | 27 | 0.17 | 157 |
| DX-8951 | Adult humans | Solid | i.v. (0.5 h) | q1d x 1, q3w | 0.14 | 0.19 | 7 | 3.42 | 160 |
| DX-8951 | Adult humans | Solid | i.v. (0.5 h) | q1d x 1, q3w | 0.14 | 0.14 | 10 | 2.1 | 161 |
| DX-8951 | Adult humans | Solid | i.v. (0.5 h) | q1d x 1, q1w | 0.06 | 0.06 | 8 | 1.2 | 162 |
| DX-8951 | Adult humans | Solid | i.v. (24 h) | q1d x 1, q3w | 0.01 | 0.01 | 7 | NR | 163 |
| DX-8951 | Adult humans | Breast | i.v. (0.5 h) | q1d x 5, q3w | 0.01 | 0.01 | 8 | NR | 164 |
| DX-8951 | Adult humans | NSC lung | i.v. (0.5 h) | q1d x 5, q3w | 0.01 | 0.01 | 8 | NR | 165 |
| DB-67 | SCID mice | None | i.v. | q1d x 1 | 10 | NR | 1.4 | 17 | 182 |
| BNP1350 | Rhesus monkeys | None | i.v. (1 h) | q1d x 1 | 0.1 | NR | α: 1 β: 8 |
0.15 | 190 |
| ST1481 | Athymic mice (F) | None | p.o. | q1d x 1 | 5 | NR | 11 | 2.5 | 193 |
| BN80915 | Adult humans | Solid | i.v. (0.3 h) | q1d x 5, q3w | 0.004 | 0.004 | 4 | 0.01 | 198 |
| BN80915 | Adult humans | Solid | p.o. | q1d x 5, q3w | 0.004 | 0.004 | 4 | 0.01 | 197 |
| BN80915 | Adult humans | Solid | i.v. (0.3 h) | q1d x 1, q3w | 0.05 | NR | 3 | 0.16 | 199 |
Abbreviations: MTD, maximum tolerated dose; AUC, area under the curve; CPT, camptothecin; Na-CPT, camptothecin sodium carboxylate; TPT, topotecan; CPT-11, irinotecan; GG211, lurtotecan; DX-8951, exatecan; BNP1350, karenitecin; BN80915, diflomotecan; SD, Sprague-Dawley; SCID, severe combined immunodeficiency; M, male; F, female; NSC, non-small cell; i.v., intravenous; s.c. subcutaneous; p.o., oral; i.p., intraperitoneal; i.a., intrarterial; PFC, powder filled capsule; NR, not reported.
Dosing abbreviations: q1d x 1, one dose; q1d x 5, q3w, five consecutive days every three weeks; q1d x 1, q1w, one dose per week; q1d x 1, q3w, one dose per week every three weeks; q1d x 5, one dose per day for five days; q1d x 1, q1w x 4/6, one dose per week every week for four out of six weeks; q2d x 6, q4w, every two days for six total doses every four weeks; q1d x 21, every day for 21 consecutive days.
4.2.1 Micelles
The individual components in micelles can provide sites for covalent attachment of drugs. Amphiphilic block copolymers with two water-soluble blocks are often times used to obtain micellar structures. In this case, one block is chemically inert while the other is reactive. Covalent attachment of hydrophobic drugs provides an amphiphile that forms micelles. This strategy has seen a great deal of success, particularly for SN-38.
SN-38 Micelles (NK012)
Animal Models
Block copolymers of poly(ethylene glycol)-poly(glutamic acid) were developed for covalent attachment of SN-38 to the carboxylate moieties. Esterification of the phenolate hydroxyl group of SN-38 with acid backbone produces the hydrophobic block for micelle formation.259 Micelles were formed from copolymers 19 kDa in length with a 12 kDa PEG segment, a 7 kDa poly(Glu) segment and incorporation of about 20% SN-38. In nude mice bearing HT-29 colon cancer xenografts, pharmacokinetic studies following i.v. administration of 30 mg/kg micelle or 66.7 mg/kg CPT-11 provided plasma AUC values of 5,010 μg•h/mL for the micelle, 0.022 μg•h/mL for irinotecan and 0.001 μg•h/mL for irinotecan metabolized to SN-38. The half-lives for each of the agents were 31 h, 3 h and 4 h, respectively. Anti-tumor effects were studied in vivo in mice with highly vascularized SBC-3/VEGF tumors as compared to SBC-3/Neo tumors, which do not promote angiogenesis. The micelles showed significant antitumor activity against the highly vascularized tumors with eradication of bulky masses in VEGF positive tumors.
In mice with s.c. Renca renal cell carcinoma xenografts, NK012 was delivered intravenously at 20 mg/kg resulting in complete tumor disappearance by day 21. In contrast, free CPT-11 administered at 30 mg/kg led to only partial response at day 15, followed by progressive disease.260 NK012 treatment led to a 10% decrease in body weight in tumor bearing mice suggesting that there was little toxicity associated with the complex. In lung metastasis models, significant uptake was observed and an overall decrease in metastatic nodules compared to irinotecan and no treatment. Prior to treatment, at day 0, 126 nodules were observed, which increased to 287 nodules at day 21 in untreated mice, as compared to 236 nodules observed after treatment with free irinotecan and only 32 nodules observed after treatment with the NK012 micelles. Furthermore, 6 of 10 mice were alive at 90 days when treated with NK012 as compared to only one remaining mouse at 90 days following treatment with irinotecan. Untreated mice expired by day 65.
Similar results were also observed in orthotopic gastric cancer with peritoneal metastases.261 Mice bearing 44As3Luc tumors were treated with MTDs of either NK012 (30 mg/kg) or CPT-11 (67 mg/kg) for three total doses every four days. NK012 treatment resulted in 80% survival at 150 days using the micelle. Free drug led to no survival at 80 days. Similar results were observed with the 58As tumor model. Tumor uptake of both NK012 and CPT-11 was observed, and the extended half-life of NK012 resulted in increased anti-tumor activity in both the gastric tumor and peritoneal nodules.
While the success observed with NK012 is believed to be due to the enhanced vasculature in solid tumors, investigation of tumor xenografts with highly vascularized (PSN1) and poorly vascularized tumors (Capan1) suggests otherwise.262 When irinotecan was delivered intravenously to mice bearing tumor xenografts at a MTD of 66.7 mg/kg every fourth day for three total doses, a reduction in tumor size was observed from days 4 to 12 in PSN1 tumors but not in Capan1 tumors. Conversely, NK012 caused complete regression of both tumors, regardless of vascularity. Drug distribution monitored with fluorescence distribution showed peak fluorescence intensity at 1 h using CPT-11 as compared to 24 h using NK012, with detection extending past 96 h. One concludes that although differences in vascularization are observed, the extended plasma retention times associated with the increased molecular weight of constructs enable eventual accumulation in tumors, including those with poor blood flow.
Most recently, NK012 was compared to CPT-11 for treatment of malignant gliomas.263 Subcutaneous xenografts treated at MTD (30 mg/kg) every four days for three total doses showed tumor regression beginning on day 5 and reaching complete regression on day 23 until day 80, when relapse was observed. After administration of 30 mg/kg for NK012 and 67 mg/kg CPT-11 to mice bearing orthotopic U87MG/Luc intracranial xenografts, the pharmacokinetic analysis showed 1113 ng/mL SN-38 in the plasma at 2 hours decreasing to 90 ng/mL at 24 hours and 6.88 ng/mL at 72 hours with tumor concentrations of 67.7 ng/mL, 137 ng/mL and 24.6 ng/mL, respectively. Plasma and tumor concentrations were significantly higher using the micelle as compared to free drug, suggesting significant trafficking of SN-38 from the micelle to the tumor. It is unclear what form is transported into the brain. While the antitumor effect was not statistically significant between CPT-11 and NK012, the Kaplan-Meier plot showed all mice had died by 30 days for both the control and CPT-11 treated mice, whereas NK012-treated mice survived for 43 days. While treating gliomas with NK012 was not as successful as treating mice bearing subcutaneous xenografts, this work represents a significant step toward improving treatment of gliomas clinically. Additional studies have shown success in combination therapy with 5-fluoruracil264 and cisplatin265, expanding the therapeutic potential of NK012, which will likely be the goal of future studies using this micellar construct in both mono-therapy and combination therapy.
4.2.2 Linear Polymers
Poly(ethylene glycol)-Camptothecin (PEG(CPT)2, PEG(GlyCPT)2, Prothecan and Pegamotecan)
Animal Models
Poly(ethylene glycol) (PEG) has been used extensively to increase the water solubility of hydrophobic small molecule drugs and macromolecular drug delivery vehicles. PEGylation has been used to increase biocompatibility, masking agents from the reticuloendothelial system while also increasing molecular weight to improve retention in the circulatory system. PEGylation of CPT led to the discovery that CPT esters stabilize the lactone ring.266 Various derivatives of PEGylated camptothecins have been developed, which employ different linkers to attach CPT to 40 kDa PEG. Initially, PEG(CPT)2 was developed through acylation of CPT with PEG-diacarboxylic acid. Glycine was then added as a linker between PEG and CPT to form PEG(GlyCPT)2. Finally, alanine was employed as the linker to form PEG-CPT and later termed Prothecan and Pegamotecan. Each of these derivatives were developed by Enzon Pharmaceuticals Inc.
The antitumor efficacy of PEG(CPT)2 was evaluated in mice bearing either P388/0 leukemia or HT-29 colon xenografts.267 After a 5.2 mg/kg i.v. injection of PEG(CPT)2, alpha and beta half-lives were determined to be 0.07h and 3.5h, respectively with an AUC of 0.018 μg•h/mL. In mice bearing HT-29 colon tumor xenografts, a dose of 2.5 mg/kg CPT afforded a 20% decrease in tumor volume after a dosing schedule of five days a week for five weeks, with 62% increase in tumor volume 2 weeks after treatment. However, using a dose of 3 mg/kg on the same schedule, PEG(CPT)2 afforded an 87% decrease in tumor volume after treatment and a 93% decrease two weeks later. Utilization of PEG(GlyCPT)2 improved the alpha and beta half-lives to 0.1 h and 10 h, respectively, with an increased AUC value of 0.05 μg•h/mL.268 The glycinate ester form appears to be 1.5 times less toxic than free CPT, and show improved pharmacokinetics than the parent PEG dicarboxylate derivative while maintaining similar efficacy. This derivative also showed a significant increase in %ID/g out to 72h in the HT-29 tumor xenografts as compared to other organs, which was not observed using free CPT.
Human Patients
A phase I and pharmacokinetic study of Pegamotecan (Figure 5) found an MTD of 122 mg/m2 (~3.3 mg/kg) as a 1 h i.v. infusion every 3 weeks.269 At this dosing schedule, a plasma AUC value of 29 μg•h/mL was observed, with a 94 h half-life and minimal toxicity seen in patients with solid tumors. While one partial response was noted out of 37 patients and only two minor responses were observed, the promising pharmacokinetics suggested further study. A subsequent study in patients with solid tumors, however, showed a lower MTD of 56 mg/m2 (~1.5 mg/kg), with a similar AUC value of 27 μg•h/mL and a 44 h half-life.270 Minimal toxicity was observed in a later study with two unconfirmed partial responses out of 27 patients. Enzon Pharmaceuticals Inc. halted further phase trials in 2005 due to poor efficacy in Phase II trials and redirected efforts toward alternative antitumor research targets.
Figure 5.
Pegamotecan made using 40 kDa PEG-diacid with two camptothecin moieties attached through alanine linker.
Investigation continued in outside laboratories with phase II trials of patients with gastric and gastro-esophageal adenocarcinoma.271 Using a dosing strategy of 1 h doses every three weeks, an MTD of 122 mg/m2 (~3.3 mg/kg) was determined. Limited efficacy was also noted, with five of 35 patients experiencing a partial response. While this drug alone provided little evidence of efficacy, utilization of this drug in combination therapy may prove more successful.
Cyclodextrin-PEG Polymers (IT-101)
Animal Models
Davis and coworkers synthesized a PEG-containing polymer containing disubstituted β-cyclodextrin moieties and CPT linked through a glycine ester linkage. The final construct had a molecular weight of 97 kDa, with 6.8% drug loading capacity.272 The MTD was determined to be 9 mg/kg, with a drug release half-life of 1.7 h in human plasma. When delivered intravenously to nude mice bearing s.c. LS174T colon carcinoma tumors every four days for three total doses at MTD, a 227% tumor growth delay (TGD) was obtained as compared to only 47% TGD observed when only two doses of 9 mg/kg CPT was delivered on the same schedule. These positive results prompted additional studies toward eventual clinical use. In a later pharmacokinetic study with an 85 kDa polymer-CPT conjugate, a single i.v. injection of 8.8 mg/kg in rats found an elimination half-life of 20 h and an AUC of 693 μg•h/mL.273 Biodistribution studies found that plasma concentrations increased with a significant increase in total CPT. Furthermore, cleavage of CPT from the polymer at the tumor site created a tumor to plasma ratio of 2.5 at 24 h increasing to 21.2 at 48 h, with concentrations of drug higher in the tumor than any other organ. To find an optimal dosing strategy, nude mice with LS174T colon cancer, HT29 colon cancer, H1299 non-small-cell lung cancer, MDA-MB-231 breast cancer, H69 small cell lung cancer or Panc-1 pancreatic cancer xenografts were treated with either a single dose of IT-101 or multiple high and low doses.274 Three doses every week provided increased efficacy over single dose administration, however, efficacy did not increase when delivered at five total doses every four days or five times a week for three weeks due to the extended half-life. In the majority of tumor xenografts, three doses of 16.1 mg/kg over three weeks provided the most promising results, showing the least body weight loss, highest tumor growth delay and highest number of complete regressions. IT-101 has shown significant progress in cancer therapy and clinical trials are currently concluding.
Phthalimide Polymers
Animal Models
Theodorakis and coworkers developed phthalimide-co-acrylic acid polymers synthesized through photopolymerization to afford a 25.5 kDa construct with 21 wt% CPT in one derivative275 or a 15.4 kDa construct with 26 wt% CPT in another.276 In vivo studies with the high molecular weight polymer architectures showed an increase in activity compared to free CPT. Doses of 10 mg/kg of CPT, 21 mg/kg CPT equivalents in the polymer or 2.1 mg/kg CPT equivalents in the polymer afforded ~90 day survival time. The low molecular weight polymer architecture, however, did not provide the same anti-tumor efficacy at low doses.
Poly(L-Glutamic Acid) Polymers (CT-2106)
Animal Models
Li and coworkers developed a poly(L-glutamic acid) polymer containing 7.7 wt% CPT through esterification. In vitro studies of this derivative showed base catalyzed hydrolysis of CPT from the polymer and cytotoxicity in a variety of cell lines.277 Antitumor studies of s.c. H322 tumors in nude mice showed a 32 d tumor growth delay after four 40 mg/kg i.v. injections were administered every four days. Furthermore, when H322 tumors were grown in the lung of nude mice, a dosing schedule of five doses of 10 mg/kg every four days afforded a median survival time of 157 d as compared to the survival time of only 86 d seen for untreated mice and 108 d seen for mice treated with free CPT. When this dose was increased to 40 mg/kg, a median survival of 238 d was obtained as compared to a survival time of only 59 d using one complete dose of 160 mg/kg (less than the LD10 of 177 mg/kg).
Klein and de Vries utilized a similar polymer in vivo, wherein glycine linkers were introduced to investigate the difference in MTD (40 mg/kg) and efficacy.278 While the glycine linkers did not alter efficacy or MTD, drug loading on 50 kDa polymers increased from 15 wt% to 50 wt% when glycine was used. Solubility limited drug loading at ~37 wt%. Increased polymer molecular weights from 33 kDa to 50 kDa increased antitumor effect in C57BL/6 mice with B16 melamona. Similarly, improvement was seen by increasing CPT wt% from 15 wt% to 35 wt%, using the same 40 mg/kg dose. An idealized architecture consisting of a 49 kDa polymer with 37 wt% CPT was used in preliminary in vivo studies of NCI-H460 lung cancer xenografts in athymic mice. Results were successful, showing increased tumor growth delay to 50% after a 30 mg/kg dose. It was later determined that, in nude mice with HT-29 colon carcinoma tumors, this polymer had a 97 h plasma half-life and plasma AUC value of 240 μg•h/mL, as well as a tumor half-life of 84 h and tumor AUC value of 696 μg•h/mL.279
Human Patients
In Phase I trials, CT-2106 was administered intravenously weekly.280 The MTD was determined to be 25 mg/m2 (~0.68 mg/kg) after higher doses of 30 mg/m2 (~0.82 mg/kg) and 35 mg/m2 (~0.95 mg/kg) showed signs of toxicity. Pharmacokinetic data at the MTD provided a 63 h half-life for conjugated CPT and a 36 h half-life for released CPT. At this dose, the plasma AUC value for the conjugate was 27 μg•h/mL, while the plasma AUC for unconjugated CPT was 14 μg•h/mL. While PLGA polymers with CPT offer extended plasma half-lives and slow release of CPT, only 3 of 25 patients experienced stable disease suggesting that additional work need to be completed to determine the clinical relevance of this construct.
Poly[N-(2-Hydroxypropyl) Methacrylamide] Copolymers (MAG-CPT)
Animal Models
Caiolfa and coworkers developed HPMA polymers of 28 kDa and 21 kDa with 5.4 wt% and 10 wt% CPT, respectively.281 CPT was also linked through the tetrapeptide linker GlyPheLeuGly for esterlytic cleavage at the tumor. Pharmacokinetic studies showed 27 h and 20 h half-lives for the high and low molecular weight polymers, respectively, with AUC values of 1023 μg•h/mL and 480 μg•h/mL, respectively. Studies in mice bearing HT-29 colon xenografts showed higher efficacy after the administration of six doses of either 25 mg/kg or 10 mg/kg every four days for the high and low MW polymers, respectively. Each polymer gave 98% tumor growth inhibition one week after the last treatment and 72 d and 62 d tumor growth delay for the high and low MW polymers, respectively. Although a 2-fold increase in AUC for the higher molecular weight polymer was observed, the low molecular weight polymer showed a 2-fold increase in potency, presumably due to an increase in polymer metabolism for the lower molecular weight polymer.
In a subsequent study, Phe-Leu of the tetrapeptide linkage was replaced by 6-aminohexanoic acid.282 The construct, known as MAG-CPT, contains 10 wt% CPT and has a mass of 20 kDa. The polymer with the tetrapeptide linker showed a 2-fold higher potency against a variety of s.c. tumors xenografts in nude mice due to the increased potential for proteolytic cleavage. Although the construct with the Gly-hexanoic acid-Gly linker was less potent, decreased toxicity was also observed resulting in more complete responses when administered at higher doses as compared to the construct with the tetrapeptide linker. This difference in toxicity and efficacy may be attributed to the decreased rate of hydrolysis in the non-peptide linker as compared to the peptide linker.
Human Patients
MAG-CPT has been utilized in Phase I studies, administered as a 30 min. i.v. infusion for three consecutive days every four weeks.283 The MTD of MAG-CPT was determined to 68 mg/m2 (~1.8 mg/kg) with dose limiting cumulative bladder toxicity at higher doses. The plasma AUC value of the construct was found to be 8661 μg•h/mL with an 8 h half-life. Approximately 70% of the dose was excreted through the kidneys within 4 days. The route of excretion, likely leads to the bladder toxicity observed at high doses. Changing this dosing regimen to a once weekly schedule for three weeks in a four week cycle at doses of 80 mg/m2 (~2.2 mg/kg) and 120 mg/m2 (~3.2 mg/kg) saw similar results.284 At the low dose, no adverse toxicities were observed until the second cycle of treatment, whereas cumulative bladder toxicity was observed during the first cycle at the high dose. Carrier bound plasma AUC values for the low dose were 1540 μg•h/mL and 1226 μg•h/mL for the high dose. Alpha and beta plasma half-lives were about 2.5 h and 100 h, respectively, regardless of dose. Unpredictable excretion kinetics and the resulting variable toxicities suggest that this dosing strategy is not practical for clinical development. Changing the dosing regimen once again to a 30 min. infusion once every four weeks at doses between 30 mg/m2 (~0.81 mg/kg) and 240 mg/m2 (~6.5 mg/kg).285 An MTD of 200 mg/m2 (~5.4 mg/kg) was determined, which afforded a 237 h half-life and a plasma AUC value of 9305 μg•h/mL. Once again highly variable urinary excretion proved problematic in determining toxicity and determining an effective dose.
In another study MAG-CPT was administered intravenously at a dose of 60 mg/m2 (~1.6 mg/kg) over 24 h, 3 or 7 days prior to surgery for colorectal cancer.286 After infusion of MAG-CPT patients had mean plasma concentrations or 29,378 ng/mL of polymer bound CPT and 17.3 ng/mL, which decreased to 2588 ng/mL and 12.2 ng/mL at 7 days, respectively. Furthermore, normal tissue had 451 ng/mL MAG-CPT at 7 days as compared to 434 ng/mL in the tumor. The high plasma concentration and poor tumor uptake relative to normal tissue suggest that this construct is perhaps too small to selectively partition into the colorectal tumors through the EPR effect. The variable pharmacokinetics and bladder toxicities associated with this construct prevented further evaluation, however, it is assumed that increasing the size of the construct to bypass glomerular filtration and increase EPR effect may prove to be efficacious.
Carboxymethyl Dextran (Delimotecan, MEN4901, T-0128)
Animal Models
The carbohydrate backbone of carboxymethyl dextran displays acid groups for attaching SN-38. Here, SN-38 is modified with an aminopropyl group and triglycine. The starting polymer has approximately 0.40–0.45 carboxylates per sugar with a molecular weight of about 130 kDa and 3–6 wt% drug loading. This linker has shown 9% release of accumulated polymer in the liver at 6 hours after administration of a 1 mg/kg dose as compared to 1.4% and 23% for diglycine and tetraglycine linkers, respectively.287 Furthermore, it appears that the triglycine linker is a selective substrate for cathepsin B.
A comparison of the polymeric material containing SN-38 and the aminopropyl derivative of SN-38 showed lower potency for the polymeric material in a series of cell lines when compared to topotecan, SN-38 propylamine ether and CPT.288 The ED50 and MTDs of the SN-38 derivative and topotecan were reported as 23 mg/kg (MTD 60 mg/kg) and 5.4 mg/kg (MTD 25 mg/kg), respectively. The ED50 and MTD of the polymer conjugate, however, were 2.3 mg/kg and 100 mg/kg, respectively. Mice bearing MX-1 tumor xenografts experienced 99.8% maximum tumor growth inhibition, with 5 out of 6 mice tumor free at 6 days, after the administration of a 6 mg/kg i.v. dose of T-0128. This inhibition was higher than the 67% maximum tumor growth inhibition observed using the SN-38 derivative at 80 mg/kg. Various tumors and doses were investigated, with significant efficacy enhancement observed using the polymer in all cases. Investigation of the pharmacokinetics provided alpha and beta half-lives of 4 h and 17 h for the carboxymethyl dextran polymer as compared to 0.017 h and 0.88 h for the SN-38 derivative. Plasma AUC values also showed significant enhancement for the polymer, as expected, with 101 μg•h/mL and 0.14 μg•h/mL, respectively. Tumor uptake of the polymeric material was significant when compared to free drug. The liver, spleen and lymph nodes, however, also showed significant uptake, suggesting recognition by the reticuloendothelial system, which was later investigated and confirmed in studies involving macrophage-mediated activation of T-0128.289, 290
To investigate the effects of molecular weight and degree of substitution on the pharmacokinetics of T-0128, a series of carboxymethyl dextran analogues with varying degrees of substitution of FITC dye were developed.291 Using 110 kDa polymer, the optimal degree of substitution of carboxylates of 0.4 provided an AUC value of 6361 μg•h/mL and half-life of 10 h. Varying the molecular weight at a fixed degree of substitution, 0.4 per sugar, established that AUC values increased with molecular weight with 40 kDa < 70 kDa < 250 kDa < 110 kDa. This trend is due to the increased renal excretion low molecular weight polymers (40 kDa and 70 kDa). Significant hepatic uptake is observed for the 250 kDa polymer. Furthermore, low anionic charge, 0.2 to 0.6 carboxylates per sugar, enables decreased hepatic uptake suggesting that the 110 kDa polymer with 0.4 substitutions per sugar would possess the highest tumor accumulation. Pharmacokinetic analysis of polymer bound SN-38 at the highest dose tested (25 mg/kg) gave an AUC of 178,000,000 μg•h/mL and a half-life of 8.2 h in rats bearing Walker-256 tumor xenografts. Non-tumor bearing rats, however, experienced significant increase in AUC and half-life (547,000,000 μg•h/mL; 28h). A subsequent study in mice bearing human tumor xenografts found significant tumor inhibition at one third the MTD of 80 mg/kg once a week for four weeks (H81 gastric, 97.5%; H-110 colon, 98.5%; Mqnu-1 lung, 99.7%; H-74 lung, 90.7%; H-204 esophageal, 78.8%; H-181 liver, 81.2%; H48 pancreatic, 98.8%).292 Pharmacokinetic evaluation of polymer bound SN-38 in nude mice bearing St-4 xenografts found plasma AUC values of 23,900 μg•h/mL and a 30 h half-life after a 40 mg/kg i.v. injection.
Carboxymethyl Dextran Polyalcohol–DX8951 (DE-310)
Animal Models
Exatecan showed significant promise in preliminary studies of antitumor activity with minimal toxicity as compared to other CPT derivatives. The success in preclinical studies did not translate well into human patients and the need for a macromolecular architecture became apparent. Exatecan was incorporated into a carboxymethyl dextran polyalcohol polymer through GlyGlyPheGly spacer with an average molecular weight of 300 kDa and 8 wt% drug loading.293
Mice bearing Meth A fibrosarcoma xenografts were administered a single MTD dose of 11.4 mg/kg resulting in tumor shrinkage out to three weeks.294 This improved efficacy over the free drug was accompanied by a 30% loss in body weight. When a single dose at one quarter of the MTD is given to mice, similar efficacy is observed without the loss in body weight. In a long-term study, mice administered four doses of 5.7 mg/kg every three days or every seven days experienced complete tumor regression at 85 days, with significant body weight loss when delivered every three days. A single dose of 11.4 mg/kg also resulted in complete tumor regression at 85 days, however, weight loss was also observed using this dosing strategy. It should also be noted that body weight decreased significantly in the first 17 days with multiple injections and 8 days with a single injection, but returned to normal within 10 days of reaching a minimum. DE-310 also showed similar efficacy in mice bearing HCT116 colon cancer, PC-6 and PC-12 lung cancers, CDDP liver metastasis and CPA lung metastasis models. A later study showed similar results, but also found evidence that DE-310 was taken up into tumors or macrophages and broken down to release the drugs.295 The release of drug through the amino acid linker cleavage was also determined to be mainly due to the activity of cathepsin B in the tumor.296 Another study shows evidence of meningocele induction in rat fetuses after their mothers received four i.v. doses of 0.3 mg/kg or a single dose of 1 mg/kg.297 Administration between days 7 and 13 of gestation resulted in 100% meningocele formation and suggests significant caution when utilizing DE-310.
Human Patients
In human patients, a dramatic increase in plasma AUC values was obtained when DE-310 was administered using a 3 h i.v. infusion once every six weeks at an MTD of 7.5 mg/m2 (~0.2 mg/kg).298 The plasma AUC was determined to be 1,124 μg•h/mL with a 338 h half-life. Furthermore, a total of 27 patients received doses from 1.0 mg/m2 (~0.03 mg/kg) every two weeks to 9.0 mg/m2 (~0.24 mg/kg) every six weeks. Dose limiting toxicities were observed when 9.0 mg/m2 (~0.24 mg/kg) were delivered every six weeks, but lower doses of 6.0 mg/m2 (~0.16 mg/kg) resulted in no dose limiting toxicity. An intermediary dose of 7.5 mg/m2 (~0.20 mg/kg) was also investigated resulting in only one patient with reversible toxicity suggesting this dose to be used in Phase II clinical trials. Efficacy was also measured in all 27 patients receiving DE-310. One patient experienced complete remission for over 2 years after receiving two doses of 9.0 mg/m2 (~0.24 mg/kg). Another patient had a partial response for three months after one dose of 9.0 mg/m2 and two subsequent doses of 6.0 mg/m2. A third patient had a partial response after seven cycles of 2.0 mg/m2 with progression occurring after 8 months. Furthermore, 1 patient experienced disease stabilization for 6 weeks, another for 8 weeks and another for 10 weeks. Five other patients had disease stabilization for 12 weeks, one for 16 weeks, 2 patients each for 18 weeks and 24 weeks and one patient for 32 weeks. The significant pharmacokinetic improvement and efficacy of DE-310 over the free drug suggested further investigation was warranted. In related studies, however, tumor accumulation is not improved significantly over other organs perhaps posing problems with systemic toxicity if the drug is capable of releasing in normal tissue.299 It remains clear, however, that the significant increase in pharmacokinetics and the convenient dosing schedule with DE-310 will warrant further investigation and may find clinical utility in terminal patients who will benefit from the antitumor efficacy, which may outweigh the toxic side effects.
Carboxymethyl Dextran Polyalcohol–Camptothecin (XMT-1001)
Animal Models
Camptothecin was also employed in a carboxymethyl dextran polyalcohol polymer. The polyalcohol polymer was functionalized with succinic acid, which was later acylated with glycine-camptothecin as shown in Figure 11.300 The starting polymer contained 0.2 carboxylates per sugar, which resulted in a 5–7 wt% CPT after partial acylation to yield a final carboxylate construct of 70 kDa. The polymer was labeled with 111In and CPT was labeled with 3H for dual labeling biodistribution studies in mice. CPT uptake in HT29 colon cancer human tumor xenografts was 2.52% ID/g at 24h with 6% ID/g of carrier in the blood and 7% ID/g of CPT in the liver. XMT-1001 was delivered intravenously to mice bearing LS174T tumors at doses of 59 mg/kg, 44 mg/kg and 22mg/kg resulted in 223%, 196% and 207% tumor growth delay, respectively. Pharmacokinetic studies were also performed in rats and dogs through i.v. administration of 30–300 mg/kg and 5–50 mg/kg, respectively. The conjugated CPT half-life was found to be 3.5–4.0 h in rats and 4.5–5.2h in dogs.
Figure 11.
Carboxymethyl dextran polyalcohol polymers. DE-310 containing 5–7 wt% exatecan in a 360 kDa polymer with 0.4 carboxylates per sugar. XMT-1001 containing 5–7 wt% camptothecin in a 70kDa polymer with 0.2 carboxylates per sugar.
Human Patients
Little animal or human data is available in the literature for this construct, however, Phase 1 studies are currently ongoing. Initial studies in patients bearing solid tumors have not reached an MTD after administration of construct between 1 mg/m2 and 20.5 mg/m2. Release of hydrolysis products in the plasma have also been determined with less than 1% CPT present in the urine. Furthermore, patients with advanced disease prior to treatment have experienced extended periods of stable disease. Although few results with XMT-1001 have been published, this construct has provided some promising data and addition efforts utilizing this construct are warranted.
4.2.3 Branched Polymers
Poly(ethylene glycol) SN-38 (EZN-2208)
Animal Models
Although limited success was observed with PEG-CPT conjugates, SN-38 was incorporated into 4-arm PEG through glycine linkers, increasing the drug loading from 1.7 wt% in PEG-CPT to 3.7 wt% in PEG-SN38.301 In MX-1 tumors, EZN-2208 was administered intravenously at an MTD of 20 mg/kg for six total doses every other day affording 100% tumor growth inhibition with cures in all animals at 16 weeks as compared to cures seen in 44% of animals using CPT-11, regardless of dose. In bulky tumors, both dosing schedules of EZN-2208 resulted in 100% tumor growth inhibition at 10 days and complete regression was observed until the end of the study at 115 days. CPT-11 was slightly more efficacious at a multiple dosing schedule with tumor regrowth occurring at 13 days using the single dosing schedule and at 45 days for the multiple dosing schedule. Similar results were obtained in MiaPaCa-2 and HT-29 cells for CPT-11 regardless of dose. Slightly less efficacy was observed in MiaPaCa-2 xenografts, with 71% tumor growth inhibition on day 69 and 100% animal survival at the termination of the study (day 125) using a single dose. Multiple doses of EZN-2208 resulted in 95% tumor growth inhibition and 66% animals cured at the termination of the study (day 147). In HT-29 xenografts, 68% and 92% tumor growth inhibition was observed using single and multiple doses, respectively. Furthermore, HT-29 xenografts were treated with EZN-2208 upon remission of tumor for up to three cycles, displaying evidence of response to repeated cycles of therapy. Additionally, EZN-2208 showed increased response from CPT-11 resistant cells, with 193% increase in tumor volume 32 days after therapy as compared to 1298% increase when using CPT-11. Pharmacokinetic studies showed that this delivery system displayed a 12 h plasma half-life for SN-38 and an elimination half-life of 26 h as compared to a plasma half-life of 1.7 h and elimination half-life of 2.1 h using CPT-11 as a free drug. Plasma AUC values of EZN-2208 and released SN-38 were 107,065 μg•h/mL and 129 μg•h/mL, respectively. When using CPT-11, AUC values of 194 μg•h/mL and 3 μg•h/mL were obtained. Tumor AUC values were also significantly higher for EZN-2208 with a value of 38825 μg•h/g versus 83 μg•h/g for CPT-11.
Structure activity relationships aimed at the linker were pursued using different amino acids. Hydrolysis half-lives in human plasma depended on amino acid: alanine, 0.21 h; methionine, 0.45 h; sarcosine, 0.32 h; glycine, 0.21 h.302 While the MTDs of the alanine and glycine analogues were determined after a single i.v. injected dose to be 20 mg/kg, antitumor efficacy was investigated using a single MTD dose and six doses of 5 mg/kg. Complete regression was observed in 100% of mice with MX-1 tumor xenografts using both single or multiple dosing schedules in all derivatives except in the single dose with the glycine derivative (83%).
Poly-(L-Lysine) Dendrimer
Animal Models
Dendrimers are hyperbranched polymers synthesized in a controlled fashion to afford a monomolecular entity with dense terminal functionality at the periphery. Peripheral functionalization with drug moieties through covalent attachment affords macromolecular constructs with a large number of drug molecules. Fréchet and Szoka utilized poly(L-lysine) dendrimers as an architecture for peripheral functionalization with CPT and poly(ethylene glycol) to increase size and solubility of the dendrimer to effectively improve the efficacy of CPT.303 CPT was attached to the dendrimer through both glycine and β-alanine linkage to afford ~35 kDa conjugate with 6.5 wt% and 4.5 wt% drug loading, respectively. The glycine linkage proved to be two orders of magnitude more toxic in vitro than the β-alanine linkage due to the increased hydrolysis in the former. In pharmacokinetic studies, a 31 h half-life was observed for the dendrimer construct, with an AUC value of 460% of the injected dose-h/gram tissue (~1380 μg•h/g) after a 10 mg/kg i.v injection. The biodistribution showed 4% ID/g tumor accumulation at 24 h, compared to 0.3% for free drug. Furthermore, free CPT accumulated in the lung, liver and spleen at 24 h, while dendrimeric CPT was observed in the tumor, serum, spleen and to a lesser extent in the lungs at 48 h. Mice with C26 colorectal s.c. tumors were treated with a single dose of 24 mg/kg eight days after inoculation, resulting in 72% tumor growth delay as compared to no significant tumor growth delay observed when using a single 10 mg/kg dose of CPT and 18% tumor growth delay with four 50 mg/kg doses of irinotecan in one week. Lower doses over prolonged periods proved successful with 155% tumor growth delay at 12 mg/kg doses once a week for three weeks. When the same dosing strategy was used in a HT29 mouse model a 122% tumor growth delay with all mice surviving for the length of the experiment while free CPT and irinotecan proved to be less efficacious.
4.2.4 Proteins
Human Serum Albumin
Animal Studies
Blood proteins are another attractive macromolecular architecture for delivery of anticancer drugs to tumors. Human serum albumin (HSA) is the most abundant blood protein and has been shown to accumulate in solid tumors due to the EPR effect. This has led some the link anticancer drugs to the protein for drug delivery. One method used with CPT involves a short poly(ethylene glycol) linker between a camptothecin ester and a maleimide group (Figure 14). The maleimide is then capable of reacting with a cysteine-34 in HSA.304 The final product, HSA-PEG-CPT is well defined and contains a single CPT (0.51 wt% CPT). Conjugation of the drug to albumin also provides a 27-fold increase in water solubility compared to CPT alone depending on the length of the poly(ethylene glycol) linker. When delivered to mice bearing subcutaneous HT-29 human tumor xenografts at four doses of 25 mg/kg (two times the MTD of camptothecin), no adverse effects were observed and an improved T/C % of 47% was achieved as compared to 89% for CPT at 12.5 mg/kg.
Figure 14.
Compounds investigated for human serum albumin linked CPT conjugates.
Alternatively, the PEG-linker was replaced with a peptide spacer (Maleimide-Arg-Arg-Ala-Leu-Ala-Leu-Ala-CPT) susceptible to cleavage by cathepsin B, which is present in lysozomes and overexpressed in various malignant tumors.305 Cleavage studies showed that mice bearing HT-29 human tumor xenografts treated three times with HSA-RRALALA-CPT at the MTD of the free drug (3 × 12.5 mg/kg) gave a T/C% of 17% compared to 40% for the free drug alone. To enhance the cleavage properties of the peptide sequence used, a peptide positional scanning library was developed to determine the optimal sequence for peptidase activity in tumor homogenates.306 From the library of peptide sequences, two sequences were found to show optimal cleavage profiles (Maleimide-Arg-ArgAla-Phe-Met-Ala-CPT and Maleimide-Arg-Arg-Phe-Tyr-Met-Ala-CPT). The sequence containing Arg-Ala-Phe-Met was then investigated in vivo in nude mice with HT-29 human tumor xenografts. The optimized sequences, HSA-RRAFMA-CPT, showed a similar T/C% of 17% at a dose of 2 × 12.5 mg/kg as compared to a 40% T/C% for free drug at a dose of 3 × 12.5 mg/kg.
While the peptide positional scanning technique has shown promise at developing new cleavage sequences, the in vivo results suggest that there is no tumor selectivity. Although tumor homogenates were shown to be active at cleaving this optimized sequences, healthy cells also express the same proteases presenting an issue with potential systemic toxicity. Further optimization of these sequences to target tumor cells while providing higher drug loading may prove beneficial to a range of antitumor therapeutics.
5. MACROMOLECULAR ARCHITECTURES FOR TARGETED DRUG DELIVERY
Few constructs have utilized targeting moieties to localize CPT to tumors through receptor-ligand mediated interactions. While passive tumor targeting has been shown to successfully reach solid tumors with increased vasculature quite efficiently through the EPR effect, actively targeting receptors present on tumor cells allows for tumor localization for both solid tumors and leukemias. Active targeting relies on the presence of a specific receptor overexpressed on cancer cells relative to non-cancerous cells for tumor specific drug delivery. Targeted therapy has proven to be useful in some cases, but significant barriers toward successful clinical implementation are present including the aforementioned challenges to characterization. Some targeted therapies have been reported but have not been investigated in vivo.307
5.1 Luteinizing Hormone-Releasing Hormone-PEG-Camptothecin (LHRH-PEG-CPT)
Animal Models
Breast, ovarian and prostate cancer cells have been shown to overexpress luteinizing hormone-releasing hormone (LHRH) receptors, which are not detected in most other organs. To exploit this tumor targeting potential, LHRH was attached to a 5 kDa PEG chain with CPT attached to the other end.308 Cysteine links CPT through an ester and PEG through a thioether. The molecular weight of the final construct is ~7 kDa, which represents a 5 wt% drug loading. In mice without tumors, tritium labeled PEG and LHRH-PEG showed no detectable uptake in the tumor and limited uptake in other organs with the highest in liver. Mice with tumors showed a significant increase in PEG and LHRH-PEG accumulation. As expected, the ovaries showed an increase in accumulation with targeted PEG in mice without tumors and with tumors. Nude mice with s.c. A2780 ovarian cancer xenografts were treated with 0.5 mg/kg of targeted and non-targeted constructs through i.p. injection. Suprisingly, tumors decreased in size 20 h after treatment. Other macromolecular non-targeted constructs, which have 100% tumor regression >60 d generally do not see statistically significant tumor response until one week after treatment. The non-targeted CPT-PEG-constructs developed by Minko afforded tumor maintenance up to 40 hours and slow increase in tumor size from 40 h to 100 h. Furthermore, a 28-fold increase in relative apoptosis values was observed in tumors compared to the untargeted construct. Apoptosis was measured using ELISA assays for protein expression in tumor homogenates. Additional studies to investigate the physiologic effects of LHRH targeting moieties, showed no change in serum levels of luteinizing hormone and no change in progeny numbers at the next generation.
More complex constructs with citric acid groups installed on the termini of the PEG chain were reported. The resulting six terminal carboxylates were used to attach CPT, LHRH and the BCL2 homology 3 domain (BH3).309 The BH3 peptide is added to suppress the cellular antiapoptotic defense system. A series of derivatives were synthesized with the construct bearing 2 CPT, 2 LHRH and 2 BH3 moieties judged most effective construct. Apoptosis was measured in each construct in mice bearing A2780 tumor xenografts, with the saline control having a relative value of one. The drug construct containing 2 CPT, 2 LHRH and 2 BH3 moieties showed a relative apoptosis unit of 55. A 28% decrease in apoptosis was measured when one of each moiety was present with an approximate 64% decrease in apoptosis when two CPT and 2 BH3 were present without LHRH. Furthermore, the absence of BH3 caused a 55% decrease in apoptosis. Tumor size was also measured 96 h after treatment of 0.4 mg/kg (1 CPT) or 0.7 mg/kg (2 CPT) of construct, showing the most significant response when two of each moiety was present on the PEG construct.
5.2 Vectocell SN-38 (DTS-108)
Animal Models
DTS-108 is a prodrug of SN-38 with a 20-amino acid peptide sequence, known as vectocell, which enables increased cellular trafficking.310 The molecular weight of this construct is 3.2 kDa and contains a highly charged sequence, allowing for and delivery of the topoisomerase I inhibitor directly into the nucleus of the cell. In dogs, the MTD after i.v. infusion of a single dose was determined to be 20 mg/kg or 2.2 mg/kg with respect to SN-38, which is significantly lower than the MTD of irinotecan (30 mg/kg). At this dose, the prodrug AUC values decreased dramatically from 36 μg•h/mL with irinotecan to 4 μg•h/mL with DTS-108 at their respective MTDs. However, the AUC values of SN-38 increase significantly from 0.018 μg•h/mL with irinotecan to 4.8 μg•h/mL for DTS-108. The increase in active drug suggests that more SN-38 is available in the plasma. However, this observation also suggests that a portion is not entering the cell with the aid of the peptide. In nude mice bearing HCT116 tumors, a slight enhancement in antitumor activity was observed when DTS-108 was administered intravenously at a dose of 10.4 mg/kg on days 3, 7 and 11 after tumor implantation, as compared to 20 mg/kg for irinotecan. When delivered on a more frequent schedule (3 times a week for 3 weeks), DTS-108 provides significant tumor growth inhibition, however, this data is not compared with irinotecan. In mice bearing HCT116 colorectal carcinoma, a 3% T/C was observed, with a 23% and 29% T/C in mice bearing NCI-H460 and MDA-MB-231 tumors, respectively. A 44% T/C was observed in rats with LS-174T colon tumor xenografts. Furthermore, efficacy was improved in combination therapy with 5-flurouracil or bevacizumab suggesting further investigation with DTS-108 is warranted. It is unclear from this study whether DTS-108 has an advantage over irinotecan with respect to efficacy, but pharmacokinetics seem to improve and interpatient variability may also improve due to the ability to function without further metabolism to form SN-38. Further studies with DTS-108 are currently ongoing.
6. CONCLUSIONS
Interest in the CPTs has undergone a significant evolution from the initial discovery in the late 1960’s through the investigation of small molecule derivatives to macromolecular constructs and formulations. The initial modifications of the quinoline ring provided increased solubility and cytotoxicity, which led to further structure activity relationships to determine the necessity of the E-ring lactone. The importance of the lactone was confirmed with reports of the TOP I bind site. Further modifications of the E-ring and the 20-(S)-hydroxyl moiety has led to a series of water soluble, highly efficacious CPTs.
While small molecule CPTs have received much attention, macromolecular architectures and supramolecular assemblies have improved pharmacokinetic parameters over the small molecule counterparts. Increasing plasma half-life and AUC values correlate to antitumor efficacy, which continues to improve in a broad series of tumor cell lines in mouse xenograft models as well as clinical trials. Our interest in this field overlaps with our interest in triazine-based dendrimers as drug delivery systems and a synthetic report from our group using a second-generation dendrimer containing CPT.213 Therefore, our motivation for assembling the literature into this review article was selfish in an attempt to better understand the in vivo literature as we move forward into animal studies. A compilation of the in vivo trials for the small molecule drugs suggest that irinotecan and exatecan are the most promising derivatives based on half-life and plasma AUC values. This conclusion may be supported by the number of clinical trials completed and ongoing using irinotecan (>250 according to clinicaltrials.gov). This comparison, however, is not as straightforward when evaluating macromolecular constructs. While each construct has specific half-life and AUC values associated, a number of variables play a role in the selection of the optimal construct. Synthetic ease, linker technology, solubility, drug loading, molecular weight, drug accessibility to esterases and other proteins and polymer degradability must all be taken into account. Furthermore, physiological variables also play a role, which is not as easy to account for.
In selecting a drug to utilize, a number of possible options are available. Implementation is limited by availability, cost and the need for metabolic activation. Antagonistic functional groups, such as the phenolate on 10-hydroxyCPT, and potential side reactions must also be considered. Irinotecan has been successful as a small molecule drug, however, implementation onto a polymer would contradict the benefits of macromolecular delivery to the tumor since drug trafficking to the liver would be necessary for activation. The use of camptothecin then seems to be a logical choice.
The polymers used have also provided evidence of optimal conditions. In most cases a biodegradable polymer backbone is employed such as cyclodextrans, polyamides and polyacetals. The molecular weights of these constructs also provide insight. For example, MAG-CPT, an HPMA based construct with a molecular weight of 20 kDa suffers from renal excretion and bladder toxicity. Higher molecular weight constructs tend to stay in the blood longer and potentially allow more uptake in the tumor. However, DE-310 has a molecular weight of 300 kDa and a plasma half-life of 7 days, which may be beneficial for sustained drug concentrations, but may also present chronic toxicity issues over time. DE-310, however, has had the most significant efficacy data in a Phase I study of any other macromolecular construct with extended periods of remission in slightly more than 50% of the patients. It is unclear, however, what parameter is most indicative of these results. The tetrapeptide linker may be of more importance than the molecular weight of the polymer itself. Each of these parameters may only be corroborated after methodical modifications of each unit in the macromolecular architecture. Interestingly, XMT-1001 has changed the tetrapeptide linker for a Gly-hexanoic acid-Gly linker and decreased the polymer from 300 kDa to 70 kDa. Preliminary data with this construct also suggests disease stabilization, but further studies are needed to support this data.
Furthermore, changes in dose and schedule greatly affect the pharmacokinetics and efficacy of the constructs. These data are summarized in the tables. The doses listed in the tables generally correspond to the MTD unless a recommended dose other than the MTD is mentioned in the specific reference. Some references generated pharmacokinetic data from doses that were not the most efficacious. In general, many different dosing strategies have been evaluated for all species.
The MTD dose with small molecules is generally the dose that offers the greatest therapeutic efficacy, while macromolecular agents were generally tested at doses lower than the MTD of the construct to establish improved efficacy over the small molecules. While macromolecules improve the pharmacokinetic data observed in small molecules, comparisons between different macromolecular constructs cannot be superficially made from the reported data. Data in each report is presented in a slightly different manner. For example, half-lives are sometimes reported using a two-compartment model or a three compartment model, while others just report elimination half-life. Additionally, it is unclear in some cases whether the half-lives are reported with respect to macromolecular drug construct, total drug construct or free drug. Furthermore, such disparity in data may be due to the difference in chemical makeup of the architectures as well as genetic differences in animal and human subjects.
Interpatient variability with both macromolecular constructs and free drug continues to hamper the widespread use of CPTs. Some variability in pharmacokinetics has been shown to occur due to a mutation in ABCG2 when using diflomotecan.311 This protein is believed to be responsible for natural detoxification and has been found to be overexpressed in the placenta, liver and intestine. Allele mutations have shown dramatically increased plasma AUC values for molecule substrates, which include 9-AC,312 SN-38313 and topotecan.314 Although allele variants may provide insight into potential pharmacokinetic outcomes, it is likely that other physiological differences in tumors such as vascularization and expression of other proteins, may also cause variability. Furthermore, a significant number of clinical trials have been completed in patients who have previously been treated with therapy, showing further interpatient variability with improved efficacy in some cases and diminished efficacy in others. One method to decrease interpatient variability and improve efficacy using the CPTs utilizes cellular transfection prior to therapy to overexpress E2F-1 and thus sensitize the tumors to CPT. Promising results have been obtained using this technique, but further assessment of transfection and camptothein cotherapy are needed to verify the clinical relevance of such a technique.
While research aims toward the development of a “magic bullet” capable of treating all cancers in all patients with a single compound, current data suggests the need to tailor therapy for individual patients either through the choice of drugs and dosing schedules or through the use of combination therapy. This review identifies a number of interesting leads for these pursuits as well as comparative data useful for assessing the next generation of candidates.
Figure 3.

Amino acid linked 20-hydroxy ester of camptothecin.
Figure 4.

NK012 showing 12 kDa PEG block and 7 kDa poly(L-glutamic acid) block with 20 wt% overall SN-38.
Figure 6.

IT-101 with a 3.4 kDa PEG chain and overall molecular weight of 85 kDa.
Figure 7.
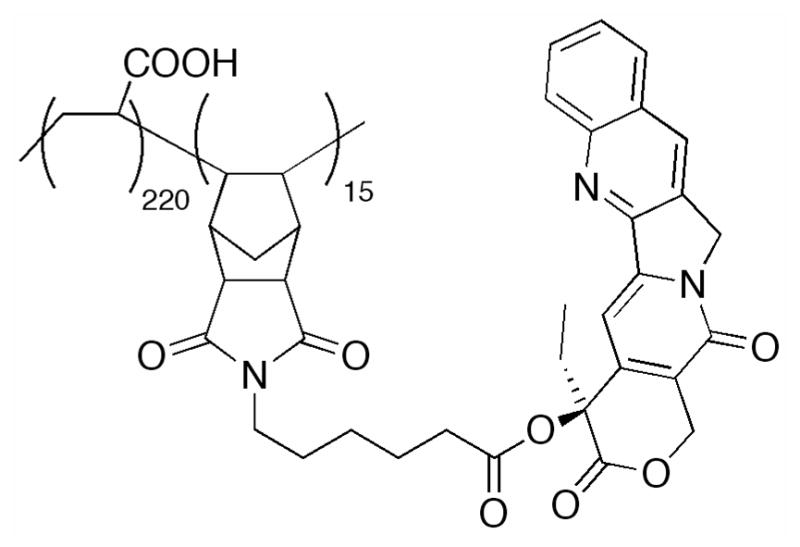
Phthalmide polymers with molecular weight of 25.5 kDa.
Figure 8.

PLGA polymer with glycine linker to CPT containing 37 wt% drug in a 49 kDa polymer.
Figure 9.

HPMA polymer with a GlyPheLeuGly linker or a Gly-hexanoic acid-Gly linker (MAG-CPT) to CPT with 10 wt% drug in a 21 kDa polymer.
Figure 10.

T-0128 polymer conatining SN-38 linked with glycine and propanolamine to a 130 kDa polymer with 3 wt% drug loading and 0.5 carboxylates per sugar.
Figure 12.
EZN-2208 is a 40 kDa four arm PEG with four SN-38 moieties attached.
Figure 13.

Poly(L-lysine) dendrimer containing eight CPT and eight PEG5000 chains.
Figure 15.
Ideal structures of PEG-CPT constructs containing LHRH or LHRH and BH3 antiapoptotic peptide.
Figure 16.
SN-38 linked to Vectocell cell penetrating peptide, through a maleimide linkage.
Scheme 1.
Camptothecin in the lactone form and open carboxylate from.
Acknowledgments
We thank Dr. André Warnecke (Tumor Biology Center, Freiberg) for helpful discussions. Support derives from the National Institutes of Health (R01 NIGMS 64560) and the Robert A. Welch Foundation (A-1439).
References
- 1.Wall ME, Wani MC, Cooke CE, Palmer KH, McPhail AT, Sim GA. Plant antitumor agents. I. The isolation and structure of camptothecin, a novel alkaloidal leukemia and tumor inhibitor from Camptotheca acuminata. J Am Chem Soc. 1966;88:3888–3890. [Google Scholar]
- 2.Wani MC, Taylor HL, Wall ME, Coggon P, McPhail AT. Plant antitumor agentsVI The isolation and structure of taxol, a novel antileukemic and antitumor agent from taxus brevifolia. J Am Chem Soc. 1971;93:2325–2327. doi: 10.1021/ja00738a045. [DOI] [PubMed] [Google Scholar]
- 3.Slichenmyer WJ, Von Hoff DD. New natural products in cancer chemotherapy. J Clin Pharmacol. 1990;30:770–788. doi: 10.1002/j.1552-4604.1990.tb01873.x. [DOI] [PubMed] [Google Scholar]
- 4.Wall ME. Camptothecin and taxol: discovery to clinic. Med Res Rev. 1998;18:299–314. doi: 10.1002/(sici)1098-1128(199809)18:5<299::aid-med2>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 5.Hsiang Y-H, Hertzberg R, Hecht S, Liu LF. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J Biol Chem. 1985;260:14873–14878. [PubMed] [Google Scholar]
- 6.Thomsen B, Mollerup S, Bonven BJ, Frank R, Blocker H, Nielsen OF, Westergaard O. Sequence specificity of DNA topoisomerase I in the presence and absence of camptothecin. EMBO J. 1987;6:1817–1823. doi: 10.1002/j.1460-2075.1987.tb02436.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pommier Y, Covey JM, Kerrigan D, Markovits J, Pham R. DNA unwinding and inhibition of mouse leukemia L1210 DNA topoisomerase I by intercalators. Nucl Acids Res. 1987;15:6713–6731. doi: 10.1093/nar/15.16.6713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jaxel C, Kohn KW, Pommier Y. Topoisomerase I interactions with SV40 DNA in the presence and absence of camptothecin. Nucl Acids Res. 1988;16:11157–11170. doi: 10.1093/nar/16.23.11157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kjeldsen E, Mollerup S, Thomsen B, Bonven BJ, Bolund L, Westergaard O. Sequence-dependent effect of camptothecin on human topoisomerase I DNA cleavage. J Mol Biol. 1988;202:333–342. doi: 10.1016/0022-2836(88)90462-7. [DOI] [PubMed] [Google Scholar]
- 10.Jaxel C, Kohn KW, Wani MC, Wall ME, Pommier Y. Structure-activity study of the actions of camptothecin derivatives on mammalian topoisomerase I: evidence for site specific receptor site and a relation to antitumor activity. Cancer Res. 1989;49:1465–1469. [PubMed] [Google Scholar]
- 11.Redinbo MR, Stewart L, Kuhn P, Champoux JJ, Hol WGJ. Crystal structure of human topoisomerase I in covalent and noncovalent complexes with DNA. Science. 1998;279:1504–1513. doi: 10.1126/science.279.5356.1504. [DOI] [PubMed] [Google Scholar]
- 12.Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin AB, Jr, Stewart L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc Natl Acad Sci USA. 2002;99:15387–15392. doi: 10.1073/pnas.242259599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Staker BL, Feese MD, Cushman M, Pommier Y, Zembower D, Stewart L, Burgin AB., Jr Structures of three classes of anticancer agents bound to the human topoisomerase I-DNA covalent complex. J Med Chem. 2005;48:2336–2345. doi: 10.1021/jm049146p. [DOI] [PubMed] [Google Scholar]
- 14.Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nature Rev. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 15.Hotte SJ, Oza A, Winquist EW, Moore M, Chen EX, Brown S, Pond GR, Dancey JE, Hirte HW. Phase I trial of UCN-01 in combination with topotecan in patients with advanced solid cancers: a Princess Margaret Hospital phase II consortium study. Ann Oncol. 2006;17:334–340. doi: 10.1093/annonc/mdj076. [DOI] [PubMed] [Google Scholar]
- 16.Anzai H, Frost P, Abbruzzese JL. Synergistic cytotoxicity with 2′-deoxy-5-azacytidine and topotecan in vitro and in vivo. Cancer Res. 1992;52:2180–2185. [PubMed] [Google Scholar]
- 17.Romanelli S, Perego P, Graziella P, Cerenini N, Tortoreto M, Zunino F. In vitro and in vivo interaction between cisplatin and topotecan in ovarian carcinoma systems. Cancer Chemother Pharmacol. 1998;41:385–390. doi: 10.1007/s002800050755. [DOI] [PubMed] [Google Scholar]
- 18.Crump M, Lipton J, Hedley D, Sutton D, Shepherd F, Minden M, Stewart K, Beare S, Eisenhauer E. Phase I trial of sequential topotecan followed by etoposide in adults with myeloid leukemia: a National Cancer Institute of Canada Clinical Trials Group Study. Leukemia. 1999;13:343–347. doi: 10.1038/sj.leu.2401308. [DOI] [PubMed] [Google Scholar]
- 19.Schmidt F, Schuster M, Strefer J, Schabet M, Weller M. Topotecan-based combination chemotherapy for human malignant glioma. Anticancer Res. 1999;19:1217–1221. [PubMed] [Google Scholar]
- 20.Raymond E, Burris HA, Rowinsky EK, Eckardt JR, Rodriguez G, Smith L, Weiss G, von Hoff DD. Phase I study of daily times five topotecan and single injection of cisplatin in patients with previously untreated non-small-cell lung carcinoma. Ann Oncol. 1997;8:1003–1008. doi: 10.1023/a:1008253314126. [DOI] [PubMed] [Google Scholar]
- 21.Hammond LA, Eckardt JR, Ganapathi R, Burris HA, Rodriguez GA, Eckhardt SG, Rothenberg ML, Weiss GR, Kuhn JG, Hodges S, von Hoff DD, Rowinsky EK. A phase I and traslational study of sequential administration of the topoisomerase I and II inhibitors topotecan and etoposide. Clin Cancer Res. 1998;4:1459–1467. [PubMed] [Google Scholar]
- 22.Britten CD, Hilsenbeck SG, Eckhardt SG, Marty J, Mangold G, MacDonald JR, Rowinsky EK, von Hoff DD, Weitman SD. Enhanced antitumor activity of 6-hydroxymethylacylfulvene in combination with irinotecan and 5-fluorouracil in the HT29 human colon tumor xenograft model. Cancer Res. 1999;59:1049–1053. [PubMed] [Google Scholar]
- 23.Cao S, Rustum YM. Synergistic antitumor activity of irinotecan in combination with 5-fluorouracil in rats bearing advanced colorectal cancer: role of drug sequence and dose. Cancer Res. 2000;60:3717–3721. [PubMed] [Google Scholar]
- 24.Wasserman E, Sutherland W, Esteban C. Irinotecan plus oxaliplatin: a promising combination for advanced colorectal cancer. Clin Colorectal Cancer. 2001;1:149–153. doi: 10.3816/CCC.2001.n.015. [DOI] [PubMed] [Google Scholar]
- 25.Mayer LD, Janoff AS. Optimizing combination chemotherapy by controlling drug ratios. Mol Interv. 2007;7:216–223. doi: 10.1124/mi.7.4.8. [DOI] [PubMed] [Google Scholar]
- 26.Guichard S, Arnould S, Hennebelle I, Bugat R, Canal P. Combination of oxaliplatin and irinotecan on human colon cancer cell lines: activity in vitro and in vivo. Anti-Cancer Drugs. 2001;12:741–751. doi: 10.1097/00001813-200110000-00006. [DOI] [PubMed] [Google Scholar]
- 27.Harasym TO, Tardi PG, Harasym NL, Harvie P, Johnstone SA, Mayer LD. Increased preclinical efficacy of irinotecan and floxuridine coencapsulated inside liposomes is associated with tumor delivery of synergistic drug ratios. Oncol Res. 2007;16:361–374. doi: 10.3727/000000006783980937. [DOI] [PubMed] [Google Scholar]
- 28.Kirichenko AV, Rich TA, Newman RA, Travis EL. Potentiation of murine MCa-4 carcinoma radioreponse by 9-amino-20-(S)-camptothecin. Cancer Res. 1997;57:1929–1933. [PubMed] [Google Scholar]
- 29.Lamond JP, Mehta MP, Boothman DA. The potential of topoisomerase I inhibitors in the treatment of CNS malignancies: report of a synergistic effect between topotecan and radiation. J Neuro-Oncol. 1996;30:1–6. doi: 10.1007/BF00177437. [DOI] [PubMed] [Google Scholar]
- 30.Karaberis E, Mourelatos D. Enhanced cytogenetic and antitumor effects by 9-nitrocamptothecin and antineoplastics. Teratogen Carcin Mut. 2000;20:141–146. [PubMed] [Google Scholar]
- 31.Lawson KA, Anderson K, Snyder RM, Simmons-Menchaca M, Atkinson J, Sun L-Z, Bandyopadhyay A, Knight V, Gilbert BE, Sanders BG, Kline K. Novel vitamine E analogue and 9-nitro-camptothecin administered as liposome aerosols decrease syngeneic mouse mammary tumor burden and inhibit metastasis. Cancer Chemother Pharmacol. 2004;54:421–431. doi: 10.1007/s00280-004-0817-y. [DOI] [PubMed] [Google Scholar]
- 32.Lee DH, Kim S-W, Bae K-S, Hong J-S, Suh C, Kang Y-K, Lee J-S. A phase I and pharmacologic study of belotecan in combination with cisplatin in patients with previously untreated extensive-stage disease small cell lung cancer. Clin Cancer Res. 2007;13:6182–6186. doi: 10.1158/1078-0432.CCR-07-0534. [DOI] [PubMed] [Google Scholar]
- 33.Duncan R. The dawning era of polymer therapeutics. Nat Rev Drug Discovery. 2003;2:347–360. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 34.Kabanov AV. Polymer genomics: An insight into pharmacology and toxicology of nanomedicines. Adv Drug Delivery Rev. 2006;58:1597–1621. doi: 10.1016/j.addr.2006.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duncan R. Polymer conjugates as anticancer nanomedicines. Nature Rev. 2006;6:688–701. doi: 10.1038/nrc1958. [DOI] [PubMed] [Google Scholar]
- 36.Green JJ, Langer R, Anderson DG. A combinatorial polymer library approach yields insight into nonviral gene delivery. Acc Chem Res. 2008;41:749–759. doi: 10.1021/ar7002336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1985;46:6387–6392. [PubMed] [Google Scholar]
- 38.Greish K, Fang J, Inutsuka T, Nagamitsu A, Maeda H. Macromolecular therapeutics: Advantages and prospects with special emphasis on solid tumor targeting. Clinical Pharmacokinetics. 2003;42:1089–1105. doi: 10.2165/00003088-200342130-00002. [DOI] [PubMed] [Google Scholar]
- 39.Duncan R. Polymer therapeutics for tumour specific delivery. Chem Ind. 1997:262–263. [Google Scholar]
- 40.Mathijssen RHJ, Verweij J, De Jonge MJ, Nooter K, Stoter G, Sparreboom A. Impact of body-size measures on irinotecan clearance: alternative dosing recommendations. J Clin Oncol. 2002;20:81–87. doi: 10.1200/JCO.2002.20.1.81. [DOI] [PubMed] [Google Scholar]
- 41.Derelanko MJ. Toxicologist’s pocket handbook. 2. Informa Healthcare; New York: 2007. [Google Scholar]
- 42.IUPAC Compendium of Chemical Technology. International Union of Pure and Applied Chemistry; NY: 2003. [Google Scholar]
- 43.Gallo RC, Whang-Peng J, Adamson RH. Studies on the antitumor activity, mechanism of action and cell cylce effects of camptothecin. J Nat Cancer Inst. 1971;46:789–795. [PubMed] [Google Scholar]
- 44.Wani MC, Ronman PE, Lindley JT, Wall ME. Plant antitumor agents. 18. Synthesis and biological activity of camptothecin analogues. J Med Chem. 1980;23:554–560. doi: 10.1021/jm00179a016. [DOI] [PubMed] [Google Scholar]
- 45.Adamovics JA, Hutchinson CR. Prodrug analogs of the antitumor alkaloid camptothecin. J Med Chem. 1979;22:310–314. doi: 10.1021/jm00189a018. [DOI] [PubMed] [Google Scholar]
- 46.Gabr A, Kuin A, Aalders M, El-Gawly H, Smets LA. Cellular pharmacokinetics and cytotoxicity of camptothecin and topotecan at normal and acidic pH. Cancer Res. 1997;57:4811–4816. [PubMed] [Google Scholar]
- 47.Hsiang Y-H, Liu LF, Wall ME, Wani MC, Nicholas AW, Manikumar G, Kirshenbaum S, Silber R, Potmesil M. DNA topoisomerase I-mediated DNA cleavage and cytotoxicity of camptothecin analogues. Cancer Res. 1989;49:4385–4389. [PubMed] [Google Scholar]
- 48.Giovanella BC, Hinz HR, Kozielski AJ, Stehlin JS, Silber R, Potmesil M. Complete growth inhibition of human cancer xenografts in nude mice by treatment with 20-(S)-camptothecin. Cancer Res. 1991;51:3052–3055. [PubMed] [Google Scholar]
- 49.Scott DO, Bindra DS, Stella VJ. Plasma pharmacokinetics of the lactone and carboxylate forms of 20(S)-camptothecin in anesthetized rats. Pharm Res. 1993;10:1451–1457. doi: 10.1023/a:1018919224450. [DOI] [PubMed] [Google Scholar]
- 50.Scott DO, Bindra DS, Sutton SC, Stella VJ. Urinary and biliary disposition of the lactone and carboxylate forms of 20(S)-camptothecin in rats. Drug Metabolism and Disposition. 1994;22:438–442. [PubMed] [Google Scholar]
- 51.Schaeppi U, Fleischman RW, Cooney DA. Toxicity of Camptothecin (NSC-100880) Cancer Chemother Rep. 1974;5:25–35. [PubMed] [Google Scholar]
- 52.Gottlieb JA, Guarino AM, Call JB, Oliverio VT, Block JB. Preliminary pharmacological and clinical evaluation of camptothecin sodium (NSC 100880) Cancer Chemother Rep. 1970;54:461–470. [PubMed] [Google Scholar]
- 53.Creaven PJ, Allen LM, Muggia FM. Plasma camptothecin (NSC-100880) levels during a 5-day course of treatment: relation to dose and toxicity. Cancer Chemother Rep. 1972;56:573–578. [PubMed] [Google Scholar]
- 54.Muggia FM, Creaven PJ, Hansen HH, Cohen MN, Selawry DS. Phase I clinical trials of weekly and daily treatment with camptothecin (NSC100880). Correlation with clincial studies. Cancer Chemother Rep. 1972;56:515–521. [PubMed] [Google Scholar]
- 55.Gottlieb JA, Luce JK. Treatment of malignant melanoma with camptothecin (NSC 100880) Cancer Chemother Rep. 1972;56:103–105. [PubMed] [Google Scholar]
- 56.Moertel CG, Schutt AJ, Reitmeier RJ, Hahn RG. Phase II study of camptothecin (NSC 100880) in the treatment of advanced gastrointestinal cancer. Cancer Chemother Rep. 1972;56:95–101. [PubMed] [Google Scholar]
- 57.Wall ME, Wani MC, Cook CE, Palmer KH, McPhail AT, Sim GA. Plant antitumor agents. I. Isolation and structure of camptothecin, a novel alkaloidal leukemia and tumor inhibitor from Camptotheca acuminata. J Am Chem Soc. 1966;88:3888–3890. [Google Scholar]
- 58.Wall ME, Wani MC, Natschke SM, Nicholas AW. Plant antitumor agents. 22. Isolation of 11-hydroxycamptothecin from camptotheca acuminata decne: total synthesis and biological activity. J Med Chem. 1986;29:1553–1555. doi: 10.1021/jm00158a044. [DOI] [PubMed] [Google Scholar]
- 59.Wani MC, Nicholas AW, Wall ME. Plant antitumor agents. 23. Synthesis and antileukemic activity of camptothecin analogs. J Med Chem. 1986;29:2358–2363. doi: 10.1021/jm00161a035. [DOI] [PubMed] [Google Scholar]
- 60.Wani MC, Nicholas AW, Manikumar G, Wall ME. Plant antitumor agents. 25. Total synthesis and antileukemic activity of ring A substituted camptothecin analogs. Structure-activity correlations. J Med Chem. 1987;30:1774–1779. doi: 10.1021/jm00393a016. [DOI] [PubMed] [Google Scholar]
- 61.Wani MC, Nicholas AW, Wall ME. Plant antitumor agents. 28. Resolution of a key tricyclic synthon, 5′(RS)-1,5-dioxo-5′-hydroxy-2′H,5′H,6′H-6′-oxopyrano[3′,4′-f]-6,8-tetrahydroindolizine: total synthesis and antitumor activity of 20(S)- and 20(R)-camptothecin. J Med Chem. 1987;30:2317–2319. doi: 10.1021/jm00395a024. [DOI] [PubMed] [Google Scholar]
- 62.Nicholas AW, Wani MC, Manikumar G, Wall ME, Kohn KW, Pommier Y. Plant antitumor agents. 29. Synthesis and biological activity of ring D and ring E modified analogs of camptothecin. J Med Chem. 1990;33:972–978. doi: 10.1021/jm00165a014. [DOI] [PubMed] [Google Scholar]
- 63.Sawada S, Matsuoka S, Nokata K, Nagata H, Furuta T, Yokokura T, Miyasaka T. Synthesis and antitumor activity of 20(S)-camptothecin derivatives: A-ring modified and 7,10-disubstituted camptothecins. Chem Pharm Bull. 1991;39:3183–3188. doi: 10.1248/cpb.39.3183. [DOI] [PubMed] [Google Scholar]
- 64.Wadkins RM, Bearss D, Manikumar G, Wani MC, Wall ME, von Hoff DD. Hydrophilic camptothecin analogs that form extremely stable cleavable complexes with DNA and topoisomerase I. Cancer Res. 2004;64:6679–6683. doi: 10.1158/0008-5472.CAN-04-1885. [DOI] [PubMed] [Google Scholar]
- 65.Henne WA, Doorneweerd DD, Hilgebrink AR, Kularatne SA, Low PS. Synthesis and of a folate peptide camptothecin prodrug. Bioorg Med Chem Lett. 2006;16:5350–5355. doi: 10.1016/j.bmcl.2006.07.076. [DOI] [PubMed] [Google Scholar]
- 66.Lu H, Lin H, Jiang Y, Zhou X, Wu B, Chen J. Synthesis and antitumor activity of 20-O-linked succinate-based camptothecin ester derivatives. Lett Drug Design Disc. 2006;3:83–86. [Google Scholar]
- 67.Samor C, Guerrini A, Varchi G, Beretta GL, Fontana G, Bombardelli E, Carenini N, Zunino F, Bertucci C, Fiori J, Battaglia A. The role of polyamine architecture on the pharmacological activity of open lactone camptothecin-polyamine conjugates. Bioconjugate Chem. 2008;19:2270–2279. doi: 10.1021/bc800033r. [DOI] [PubMed] [Google Scholar]
- 68.Wani MC, Wall ME. Plant antitumor agents. II. Structure of two new alkaloids from Camptotheca acuminata. J Org Chem. 1969;34:1364–1367. [Google Scholar]
- 69.Zhang R, Li Y, Cai Q, Liu T, Sun H, Chambless B. Preclinical pharmacology of the natural product anticancer agent 10-hydroxycamptothecin, an inhibitor of topoisomerase I. Cancer Chemother Pharmacol. 1998;41:257–267. doi: 10.1007/s002800050738. [DOI] [PubMed] [Google Scholar]
- 70.Kingsbury WD, Boehm JC, Jakas DR, Holden KG, Hecht SM, Gallagher G, Caranfa MJ, McCabe FL, Faucette LF, Johnson RK. Synthesis of water soluble (aminoalkyl) camptothecin analogues: inhibition of topoisomerase I and antitumor activity. J Med Chem. 1991;34:98–107. doi: 10.1021/jm00105a017. [DOI] [PubMed] [Google Scholar]
- 71.Uckun FM, Stewart CF, Reaman G, Chelstrom LM, Jin J, Chandran-Langlie M, Waddick KG, White J, Evans WE. In vitro and in vivo activity of topotecan against human B-lineage acute lymphoblastic leukemia cells. Blood. 1995;85:2817–2828. [PubMed] [Google Scholar]
- 72.Friedman HS, Houghton PJ, Schold SC, Keir S, Bigner DD. Activity of 9-dimethylaminomethyl-10-hydroxycamptothecin against pediatric and adult central nervous system tumor xenografts. Cancer Chemother Pharmacol. 1994;34:171–174. doi: 10.1007/BF00685936. [DOI] [PubMed] [Google Scholar]
- 73.Houghton PJ, Cheshire PJ, Myers L, Stewart CF, Synold TW, Houghton JA. Evaluation of 9-dimethylaminomethyl-10-hydroxycamptothecin against xenografts derived from adult and childhood solid tumors. Cancer Chemother Pharmacol. 1992;31:229–239. doi: 10.1007/BF00685553. [DOI] [PubMed] [Google Scholar]
- 74.Pawlik CA, Houghton PJ, Stewart CF, Cheshire PJ, Richmond LB, Danks MK. Effective schedules of exposure of medulloblastoma and rhabdomyosarcoma xenografts to topotecan correlate with in vitro assays. Clin Cancer Res. 1998;4:1995–2002. [PubMed] [Google Scholar]
- 75.McCabe FL, Johnson RK. Comparative activity of oral and parenteral topotecan in murine tumor models: efficacy of oral topotecan. Cancer Invest. 1994;12:308–313. doi: 10.3109/07357909409023029. [DOI] [PubMed] [Google Scholar]
- 76.De Cesare M, Zunino F, Pace S, Pisano C, Pratesi G. Efficacy and toxicity profile of oral topotecan in a panel of human tumour xenografts. Eur J Cancer. 2000;36:1558–1564. doi: 10.1016/s0959-8049(00)00141-6. [DOI] [PubMed] [Google Scholar]
- 77.Guichard S, Montazeri A, Chatelut E, Hennebelle I, Bugat R, Canal P. Schedule-dependent activity o topotecan in OVCAR-3 ovarian carcinoma xenograft: pharmacokinetic and pharmacodynamic evaluation. Clin Cancer Res. 2001;7:3222–3228. [PubMed] [Google Scholar]
- 78.Grochow LB, Rowinsky EK, Johnson R, Ludeman S, Kaufmann SH, McCabe FL, Smith BR, Hurowitz L, DeLisa A, Donehower RC, Noe DA. Pharmacokinetics and pharmacodynamics of topotecan in patients with advanced cancer. Drug Metabolism and Disposition. 1992;20:706–713. [PubMed] [Google Scholar]
- 79.Haas NB, LaCreta FP, Walczak J, Hudes GR, Brennan JM, Ozols RF, O’Dwyer PJ. Phase I/pharmacokinetic study of topotecan by 24-hour continuous infusion weekly. Cancer Res. 1994;54:1220–1226. [PubMed] [Google Scholar]
- 80.O’Dwyer PJ, LaCreta FP, Haas NB, Halbherr T, Frucht H, Goosenberg E, Yao KS. Clinical, pharmacokinetic and biological studies of topotecan. Cancer Chemother Pharmacol. 1994;34(Suppl):S46–S52. doi: 10.1007/BF00684863. [DOI] [PubMed] [Google Scholar]
- 81.Rowinsky EK, Grochow LB, Hendricks CB, Ettinger DS, Forasteire AA, Hurowitz LA, McGuire WP, Sartorius SE, Lubejko BG, Kaufmann SH, Donehower RC. Phase I and pharmacologic study of topotecan: a novel topoisomerase I inhibitor. J Clin Oncol. 1992;10:647–656. doi: 10.1200/JCO.1992.10.4.647. [DOI] [PubMed] [Google Scholar]
- 82.Verweij J, Lund B, Beijnen JH, Planting A, De Boer-Dennert M, Koier I, Rosing H, Hansen HH. Phase I and pharmacokinetics study of topotecan, a new topoisomerase I inhibitor. Ann Oncol. 1993;4:673–678. doi: 10.1093/oxfordjournals.annonc.a058623. [DOI] [PubMed] [Google Scholar]
- 83.Blaney SM, Balis FM, Cole DE, Craig C, Reid JM, Ames MM, Krailo M, Reaman G, Hammond D, Poplack DG. Pediatric phase I trial and pharmacokinetic study of topotecan administered as a 24-hour continuous infusion. Cancer Res. 1993;53:1032–1036. [PubMed] [Google Scholar]
- 84.Wall JG, Burris HAI, von Hoff DD, Rodriguez G, Kneuper-Hall R, Shaffer D, O’Rourke T, Brown T, Weiss G, Clark G, McVea S, Brown J, Johnson R, Friedman C, Smith B, Mann WS, Kuhn J. A phase I clinical and pharmacokinetic study of the topoisomerase I inhibitor topotecan (SK&F 104864) given as an intravenous bolus every 21 days. Anti-Cancer Drugs. 1992;3:337–345. doi: 10.1097/00001813-199208000-00004. [DOI] [PubMed] [Google Scholar]
- 85.Burris HAI, Awads A, Kuhn JG, Eckardt JR, Cobb PW, Rinaldi DA, Fields S, Smith L, von Hoff DD. Phase I and pharmacokinetic studies of topotecan administered as a 72 or 120 h continuous infusion. Anti-Cancer Drugs. 1994;5:394–402. doi: 10.1097/00001813-199408000-00002. [DOI] [PubMed] [Google Scholar]
- 86.Kantarjian HM, Beran M, Ellis A, Zwelling L, O’Brien S, Cazenave L, Koller C, Rios MB, Plunkett W, Keating MJ, Estey EH. Phase I study of topotecan, a new topoisomerase I inhibitor, in patients with refractory or relapsed acute leukemia. Blood. 1993;81:1146–1151. [PubMed] [Google Scholar]
- 87.Rowinsky EK, Adjei A, Donehower RC, Gore SD, Jones RJ, Burke PJ, Cheng Y-C, Grochow LB, Kaufmann SH. Phase I and pharmacodynamic study of the topoisomerase I-inhibitor topotecan in patients with refractory acute leukemia. J Clin Oncol. 1994;12:2193–2203. doi: 10.1200/JCO.1994.12.10.2193. [DOI] [PubMed] [Google Scholar]
- 88.Pratt CB, Stewart C, Santana VM, Bowman L, Furman W, Ochs J, Marina N, Kuttesch JF, Heideman R, Sandlund JT, Avery L, Meyer WH. Phase I study of topotecan for pediatric patients with malignant solid tumors. J Clin Oncol. 1994;12:539–543. doi: 10.1200/JCO.1994.12.3.539. [DOI] [PubMed] [Google Scholar]
- 89.Gerrits CJH, Schellens JHM, Burris H, Eckardt JR, Planting AST, van der Burg MEL, Rodriguez GI, Loos WJ, van Beurden V, Hudson I, von Hoff DD, Verweij J. A comparison of clinical pharmacodynamics of different administration schedules of oral topotecan (hycamtin) Clin Cancer Res. 1999;5:69–75. [PubMed] [Google Scholar]
- 90.O’Reilly E, Donehower RC, Rowinsky EK, Ord S, Grochow LB. A phase II trial of topotecan in patients with previously untreated pancreatic cancer. Anti-Cancer Drugs. 1996;7:410–414. doi: 10.1097/00001813-199606000-00006. [DOI] [PubMed] [Google Scholar]
- 91.Rowinsky EK, Baker SD, Burks K, O’Reilly S, Donehower RC, Grochow LB. High-dose topotecan with granulocyte-colony stimulating factor in fluoropyrimidine-refractory colorectal cancer: a phase II and pharmacodynamic study. Ann Oncol. 1998;9:173–180. doi: 10.1023/a:1008266630701. [DOI] [PubMed] [Google Scholar]
- 92.Hertzberg R, Caranfa MJ, Holden KG, Jakas DR, Gallagher G, Mattern MR, Mong SM, Bartus JO, Johnson RK, Kingsbury WD. Modification of the hydroxlactone ring of camptothecin: inhibition of mammalian topoisomerase I and biological activity. J Med Chem. 1989;32:715–720. doi: 10.1021/jm00123a038. [DOI] [PubMed] [Google Scholar]
- 93.Yano H, Kayukawa S, Iida S, Nakagawa C, Oguri T, Sanda T, Ding J, Mori F, Ito A, Ri M, Inagaki A, Kusumoto S, Ishida T, Komatsu H, Inagaki H, Suzuki A, Ueda R. Overexpression of caboxylesterase-2 results in enhanced efficacy of topoisomerase I inhibitor, irinotecan (CPT-11), for multiple myeloma. Cancer Sci. 2008;99:2309–2314. doi: 10.1111/j.1349-7006.2008.00936.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dodds HM, Haaz MC, Riou JF, Robert J, Rivory LP. Identification of a new metabolite of CPT-11 (Irinotecan): pharmacological properties and activation to SN-38. J Pharmacol Exp Ther. 1998;286:578–583. [PubMed] [Google Scholar]
- 95.Haaz MC, Rivory LP, Riche C, Vernillet L, Robert J. Metabolism of irinotecan (CPT-11) by human hepatic microsomes: participation of cytochrome P-450 3A and drug interactions. Cancer Res. 1998;58:468–472. [PubMed] [Google Scholar]
- 96.Danks MK, Yoon KJ, Bush RA, Remack JS, Wierdl M, Tsurkan L, Kim SU, Garcia E, Metz MZ, Najbauer J, Potter PM, Aboody KS. Tumor-targeted enzyme/prodrug therapy mediates long-term disease-free survival of mice bearing disseminated neuroblastoma. Cancer Res. 2007;67:22–25. doi: 10.1158/0008-5472.CAN-06-3607. [DOI] [PubMed] [Google Scholar]
- 97.Rivory LP, Chatelut E, Canal P, Mathieu B, Robert AJ. Kinetics of the in vivo interconversion of the carboxylate and lactone forms of irinotecan (CPT-11) and of its metabolite SN-38 in patients. Cancer Res. 1994;54:6330–6333. [PubMed] [Google Scholar]
- 98.Kaneda N, Nagata H, Furuta T, Yokokura T. Metabolism and pharmacokinetics of the camptothecin analogue CPT-11 in the mouse. Cancer Res. 1990;50:1715–1720. [PubMed] [Google Scholar]
- 99.Ohno R, Okada K, Masaoka T, Kuramoto A, Arima T, Yoshida Y. An early phase II study of CPT-11: a new derivative of camptothecin for the treatment of leukemia and lymphoma. J Clin Oncol. 1990;8:1907–1912. doi: 10.1200/JCO.1990.8.11.1907. [DOI] [PubMed] [Google Scholar]
- 100.Masuda N, Fukuoka M, Kusunoki Y, Matsui K, Takifuji N, Kudoh S, Negoro S, Nishioka M, Nakagawa K, Takada M. CPT-11: a new derivative of camptothecin for the treatment of refractory or relapsed small-cell lung cancer. J Clin Oncol. 1992;10:1225–1229. doi: 10.1200/JCO.1992.10.8.1225. [DOI] [PubMed] [Google Scholar]
- 101.Pitot HC, Adjei AA, Reid JM, Sloan JA, Atherton PJ, Rubin J, Alberts SR, Duncan BA, Denis L, Schaaf LJ, Yin D, Sharma A, McGovern P, Miller LL, Erlichman C. A phase I and pharmacokinetic study of a powder-filled capsule formulation of oral irinotecan (CPT-11) given daily for 5 days every 3 weeks in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2006;58:165–172. doi: 10.1007/s00280-005-0138-9. [DOI] [PubMed] [Google Scholar]
- 102.Tsuruo T, Matsuzaki T, Matsshita M, Saito H, Yokokura T. Antitumor effect of CPT-11, a new derivative of camptothecin, against pleiotropic drug-resistant tumors in vitro and in vivo. Cancer Chemother Pharmacol. 1988;21:71–74. doi: 10.1007/BF00262744. [DOI] [PubMed] [Google Scholar]
- 103.Pitot HC, Wender D, O’Connell MJ, Schroeder G, Goldberg RM, Rubin J, Mailliard JA, Knost JA, Ghosh C, Kirshling RJ, Levitt R, Windshitl HE. Phase II trial of irinotecan in patients with metastatic colorectal carcinoma. J Clin Oncol. 1994;15:2910–2919. doi: 10.1200/JCO.1997.15.8.2910. [DOI] [PubMed] [Google Scholar]
- 104.Conti JA, Kemeny NE, Saltz LB, Huang Y, Tong WP, Chou T-C, Sun M, Pulliam S, Gonzalez C. Irinotecan is an active agent in untreated patients with metastatic colorectal cancer. J Clin Oncol. 1996;14:709–715. doi: 10.1200/JCO.1996.14.3.709. [DOI] [PubMed] [Google Scholar]
- 105.Verschraegen CF, Levy T, Kudelka AP, Llerena E, Ende K, Freedman RS, Edwards CL, Hord M, Steger M, Kaplan AL, Kieback D, Fishman A, Kavanaugh JJ. Phase II study of irinotecan in prior chemotherapy-treated squamous cell carcinoma of the cervix. J Clin Oncol. 1997;15:625–631. doi: 10.1200/JCO.1997.15.2.625. [DOI] [PubMed] [Google Scholar]
- 106.Rowinsky EK, Grochow LB, Ettinger DS, Sartorius SE, Lubejko BG, Chen T-L, Rock MK, Donehower RC. Phase I and pharmacological study of the novel topoisomerase I inhibitor 7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecin (CPT-11) administered as a ninety-minute infusion every 3 weeks. Cancer Res. 1994;54:427–436. [PubMed] [Google Scholar]
- 107.Pitot HC, Goldberg RM, Reid JM, Sloan JA, Skaff PA, Erlichman C, Rubin J, Burch PA, Adjei AA, Alberts SA, Schaaf LJ, Elfring G, Miller LL. Phase I dose-finding and pharmacokinetic trial of irinotecan hydrochloride (CPT-11) using a once-every-three-week dosing schedule for patients with advanced solif tumor malignancy. Clin Cancer Res. 2000;6:2236–2244. [PubMed] [Google Scholar]
- 108.Rougier P, Bugat R, Douillard JY, Culine S, Suc E, Brunet P, Becouarn Y, Ychou M, Marty M, Extra JM, Bonneterre J, Adenis A, Seitz JF, Ganem G, Namer M, Conroy T, Negrier S, Merrouche Y, Burki F, Mousseau M, Herait P, Mahjoubi M. Phase II study of irinotecan in the treatment of advanced colorectal cancer in chemotherapy-naive patients and patients pre-treated with fluorouracil-based chemotherapy. J Clin Oncol. 1997;15:251–260. doi: 10.1200/JCO.1997.15.1.251. [DOI] [PubMed] [Google Scholar]
- 109.Raymond E, Boige V, Faivre S, Sanderink G-J, Rixe O, Vernillet L, Jacques C, Gatineau M, Ducreux M, Armand JP. Dosage adjustment and pharmacokinetic profile of irinotecan in cancer patients with hepatic dysfunction. J Clin Oncol. 2002;20:4303–4312. doi: 10.1200/JCO.2002.03.123. [DOI] [PubMed] [Google Scholar]
- 110.Boige V, Taïeb J, Hebbar M, Malka D, Debaere T, Hannoun L, Magherini E, Mignard D, Poynard T, Ducreux M. Irinotecan as first-line chemotherapy in patients with advanced hepatocellular carcinoma: a multicenter phase II study with dose adjustment according to baseline serum bilirubin level. Eur J Cancer. 2006;42:456–459. doi: 10.1016/j.ejca.2005.09.034. [DOI] [PubMed] [Google Scholar]
- 111.Vassal G, Morizet J, Bissery M-C, Boland I, Ardouin P, Mathieu-Boue A, Gouyette A. Activity of the camptothecin analog CPT-11 (irinotecan) against medulloblastoma xenografts. Proc Am Assoc Cancer Res. 1994;35:366. [Google Scholar]
- 112.Savaraj N, Xu R, Wu CJ, Landy H, Chua L, Solomon J, Freun L. Correlation of in vitro antitumor activity of irinotecan and topoisomerase I activity an dlevels in brain tumors. Proc Am Assoc Cancer Res. 1995;14:492. [Google Scholar]
- 113.Armand JP, Extra YM, Catimel G, Abigerges D, Marty M, Clavel M. Rationale for the dosage and schedule of CPT-11 (irinotecan) selected for phase II studies, as determined by European phase I studies. Ann Oncol. 1996;7:837–842. doi: 10.1093/oxfordjournals.annonc.a010763. [DOI] [PubMed] [Google Scholar]
- 114.Masuda N, Kudoh S, Fukuoka M. Irinotecan (CPT-11): Pharmacology and clinical applications. Crit Rev Oncol Hematol. 1996;24:3–26. doi: 10.1016/1040-8428(96)00201-6. [DOI] [PubMed] [Google Scholar]
- 115.O’Reilly S, Rowinshy EK. The clinical status of irinotecan (CPT-11), a novel water soluble camptothecin analogue: 1996. Crit Rev Oncol Hematol. 1996;24:47–70. doi: 10.1016/1040-8428(96)00211-9. [DOI] [PubMed] [Google Scholar]
- 116.Hare CB, Elion GB, Houghton PJ, Houghton JA, Keir S, Marcelli SL, Bigner DD, Friedman HS. Therapeutic efficacy of the topoisomerase I inhibitor 7-ethyl-10-(4-[1-piperidino]-1-piperidino)-carbonyloxy-camptothecin against pediatric and adult central nervous system tumor xenografts. Cancer Chemother Pharmacol. 1997;39:187–191. doi: 10.1007/s002800050558. [DOI] [PubMed] [Google Scholar]
- 117.Batchelor TT, Gilbert MR, Supko JG, Carson KA, Nabors LB, Grossman SA, Lesser GJ, Mikkelsen T, Phuphanich S. Phase 2 study of weekly irinotecan in adults with recurrent malignant glioma: Final report of NABTT 97–11. Neuro-Oncology. 2004;6:21–27. doi: 10.1215/S1152851703000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Taylor J, Amanze A, Di Federico E, Verschraegen CF. Irinotecan use during pregnancy. Obstet Gynecol. 2009;114:451–452. doi: 10.1097/AOG.0b013e3181a1d478. [DOI] [PubMed] [Google Scholar]
- 119.Potmesil M, Giovanella BC, Liu LF, Wall ME, Silber R, Stehlin JS, Hsiang Y-H, Wani MC. Preclinical studies of DNA topoisomerase I-targeted 9-amino and 10,11-methylenedioxy camptothecins. In: Potmesil M, Kohn KW, Liu LF, Muggia FM, Ross WE, Silber R, editors. DNA topoisomerases in cancer. Oxford University Press; 1991. pp. 299–311. [Google Scholar]
- 120.Supko JG, Malspeis L. Pharmacokinetics of the 9-amino and 10,11-methylenedioxy derivatives of camptothecin in mice. Cancer Res. 1993;53:3062–3069. [PubMed] [Google Scholar]
- 121.Muggia FM, Liebes l, Hazarika M, Wadler S, Hamilton A, Hornreich G, Sorich J, Chiang C, Newman E, Potmesil M, Hochster H. Phase I and pharmacologic study of i.p. 9-aminocamptothecin given as six fractions over 14 days. Anti-Cancer Drugs. 2002;13:819–825. doi: 10.1097/00001813-200209000-00006. [DOI] [PubMed] [Google Scholar]
- 122.Eder JP, Jr, Supko JG, Lynch T, Bryant M, Vosburgh E, Shulman LN, Xu G, Kufe DW. Phase I trial of the colloidal dispersion formulation of 9-amino-20(S)-camptothecin administered as a 72-hour continuous intravenous infusion. Clin Cancer Res. 1998;4:317–324. [PubMed] [Google Scholar]
- 123.Giovanella BC, Stehlin JS, Wall ME, Wani MC, Nicholas AW, Liu LF, Silber R, Potmesil M. DNA topoisomerase I-targeted chemotherapy of human colon cancer in xenografts. Science. 1989;246:1046–1048. doi: 10.1126/science.2555920. [DOI] [PubMed] [Google Scholar]
- 124.Pantazis P, Hinz HR, Mendoza JT, Kozielski AJ, Williams LJ, Jr, Stehlin JS, Giovanella BC. Complete inhibition of growth followed by death of human malignant melanoma cells in vitro and regression of human xenografts in immunodeficient mice induced by camptothecins. Cancer Res. 1992;52:3980–3987. [PubMed] [Google Scholar]
- 125.Mattern MR, Hofmann GA, Polsky RM, Funk LR, McCabe FL, Johnson RK. In vitro and in vivo effects of clinically important camptothecin analogs on multidrug-resistant cells. Oncol Res. 1993;5:467–474. [PubMed] [Google Scholar]
- 126.Pantazis P, Kozielski AJ, Vardeman DM, Petry ER, Giovanella BC. Efficacy of camptothecin congeners in the treatment of human breast carcinoma xenografts. Oncol Res. 1994;5:273–281. [PubMed] [Google Scholar]
- 127.Vokes EE, Ansari RH, Masters GA, Hoffman PC, Klepsch A, Ratain MJ, Sciortino DF, Lad TE, Krauss S, Fishkin PAS, Golomb HM. A phase II study of 9-aminocamptothecin in advanced non-small-cell lung cancer. Ann Oncol. 1998;9:1085–1090. doi: 10.1023/a:1008432729754. [DOI] [PubMed] [Google Scholar]
- 128.Hochster H, Plimack ER, Runowicz CD, Speyer J, Wallach RC, Sorich J, Mandeli J, Wadler S, Wright J, Muggia FM. Biweekly 72-hour 9-aminocamptothecin infusion as second-line therapy for ovarian carcinoma: Phase II study of the new york gynecologic oncology group and the eastern cooperative oncology group. J Clin Oncol. 2004;22:120–126. doi: 10.1200/JCO.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 129.Hinz HR, Harris NJ, Natelson EA, Giovanella BC. Pharmacokinetics of the in vivo and in vitro conversion of 9-nitro-20(S)-camptothecin to 9-amino-20(S)-camptothecin in humans, dogs and mice. Cancer Res. 1994;54:3096–3100. [PubMed] [Google Scholar]
- 130.Chen J, Ping Q, Guo J, Chu X, Song M. Pharmacokinetics of lactone, carboxylate and total 9-nitro-camptothecin with different doses and administration routes in rats. Biopharm Drug Dispos. 2006;27:53–59. doi: 10.1002/bdd.480. [DOI] [PubMed] [Google Scholar]
- 131.Verschraegen CF, Natelson EA, Giovanella BC, Kavanaugh JJ, Kudelka AP, Freedman RS, Edwards CL, Ende K, Stehlin JS. A phase I clinical and pharmacological study of oral 9-nitrocamptothecin, a novel water-insoluble topoisomerase I inhibitor. Anti-Cancer Drugs. 1998;9:36–44. doi: 10.1097/00001813-199801000-00004. [DOI] [PubMed] [Google Scholar]
- 132.Verschraegen CF, Gupta A, Loyer E, Kavanaugh JJ, Kudelka AP, Freedman RS, Edwards CL, Harris N, Steger M, Steltz V, Giovanella BC, Stehlin JS. A phase II clinical and pharmacological study of oral 9-nitrocamptothecin in patients with refractory epithelial ovarian, tubal or peritoneal cancer. Anti-Cancer Drugs. 1999;10:375–383. doi: 10.1097/00001813-199904000-00005. [DOI] [PubMed] [Google Scholar]
- 133.Amorino GP, Hercules SK, Mohr PJ, Pyo H, Choy H. Preclinical evaluation of the orally active camptothecin analog, RFS-2000 (9-nitro-20(S)-camptothecin) as a radiation enhancer. Int J Radiation Oncology Biol Phys. 2000;47:503–509. doi: 10.1016/s0360-3016(00)00461-2. [DOI] [PubMed] [Google Scholar]
- 134.Luzzio MJ, Besterman JM, Emerson DL, Evans MG, Lackey K, Leitner PL, McIntyre G, Morton B, Myers PL, Peel M, Sisco JM, Sternbach DD, Tong W-Q, Truesdale A, Uehling DE, Vuong A, Yates J. Synthesis and antitumor activity of novel water soluble derivatives of camptothecin as specific inhibitors of topoisomerase I. J Med Chem. 1995;38:395–401. doi: 10.1021/jm00003a001. [DOI] [PubMed] [Google Scholar]
- 135.Emerson DL, Besterman JM, Brown HR, Evans MG, Leitner PP, Luzzio MJ, Shafffer JE, Sternbach DD, Uehling DE, Vuong A. In Vivo antitumor activity of two new seven-substituted water-soluble camptothecin analogues. Cancer Res. 1995;55:603–609. [PubMed] [Google Scholar]
- 136.Gerrits CJH, Creemers GJ, Schellens JH, Wissel PS, Planting AST, Kunka R, Selinger K, De Boer-Dennert M, Marijnen Y, Harteveld M, Verweij J. Phase I and pharmacological study of the new topoisomerase I inhibitor GI47211, using a daily x5 intravenous administration. Brit J Cancer. 1996;73:744–750. doi: 10.1038/bjc.1996.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Eckhardt SG, Baker SD, Eckardt J, Burke TG, Warner DL, Kuhn JG, Rodriguez G, Fields S, Thurman A, Smith L, Rothenberg ML, White L, Wissel PS, Kunka R, DePee S, Littlefield D, Burris HAI, von Hoff DD, Rowinsky EK. Phase I and pharmacokinetic study of GI147211, a water-soluble camptothecin analogue, administered for five consecutive days every three weeks. Clin Cancer Res. 1998;4:595–604. [PubMed] [Google Scholar]
- 138.Paz-Ares L, Kunka R, DeMaria D, Cassidy J, Alden M, Beranek P, Kaye S, Littlefield D, Reilley D, DePee S, Wissel PS, Twelves C, O’Dwyer PJ. A phase I clinical and pharmackinetic study of the new topoisomerase inhibitor GI147211 given as a 72-h continuous infusion. Br J Cancer. 1998;78:1329–1336. doi: 10.1038/bjc.1998.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Stevenson JP, DeMaria D, Sludden J, Kaye SB, Paz-Ares L, Grochow LB, McDonald A, Selinger K, Wissel PS, O’Dwyer PJ, Twelves C. Phase I/pharmacokinetic study of the topoisomerase I inhibitor GG211 administered as a 21-day continuous infusion. Ann Oncol. 1999;10:339–344. doi: 10.1023/a:1008313011289. [DOI] [PubMed] [Google Scholar]
- 140.Schellens JH, Heinrich B, Lehnert M, Gore ME, Kaye SB, Dombernowsky P, Paridaens R, van Oosterom AT, Verweij J, Loos WJ, Calvert H, Pavlidis N, Cortes-Funes H, Wanders J, Roelvink M, Sessa C, Selinger K, Wissel PS, Gamucci T, Hanauske A. Population pharmacokinetic and dynamic analysis of the topoisomerase I inhibitor lurtotecan in phase II studies. Invest New Drugs. 2002;20:83–93. doi: 10.1023/a:1014454821885. [DOI] [PubMed] [Google Scholar]
- 141.Gamucci T, Paridaens R, Heinrich B, Schellens JH, Pavlidis N, Verweij J, Sessa C, Kaye S, Roelvink M, Wanders J, Hanauske A. Activity and toxicity of GI47211 in breast, colorectal and non-small-cell lung cancer patients: an EORTC-ECSG phase II clinical study. Ann Oncol. 2000;11:793–797. doi: 10.1023/a:1008373031714. [DOI] [PubMed] [Google Scholar]
- 142.Sessa C, Wanders J, Roelvink M, Dombernowsky P, Nielsen D, Morant R, Drings P, Wissel PS, Hanauske A-R. Second-line treatment of small-cell lung cancer with the camptothecin-derivative GI147211: A study of the EORTC early clinical studies group (ECSG) Ann Oncol. 2000;11:207–210. doi: 10.1023/a:1008372404504. [DOI] [PubMed] [Google Scholar]
- 143.Rose WC, Marathe PH, Jang GR, Monticello TM, Balasubramanian BN, Long B, Fairchild CR, Wall ME, Wani MC. Novel fluoro-substituted camptothecins: in vivo antitumor activity, reduced gastrointestinal toxicity and pharmacokinetic characterization. Cancer Chemother Pharmacol. 2006;58:73–85. doi: 10.1007/s00280-005-0128-y. [DOI] [PubMed] [Google Scholar]
- 144.Wadkins RM, Potter P, Vladu B, Marty J, Mangold G, Weitman SD, Manikumar G, Wani MC, Wall ME, von Hoff DD. Water soluble 20(S)-glycinate esters of 10,11-methylenedioxycamptothecins are highly active against human breast cancer xenografts. Cancer Res. 1999;59:3424–3428. [PubMed] [Google Scholar]
- 145.Adams DJ, Dewhirst MW, Flowers JL, Gamcsik MP, Colvin OM, Manikumar G, Wani MC, Wall ME. Camptothecin analogues with enhanced antitumor activity at acidic pH. Cancer Chemother Pharmacol. 2000;46:263–271. doi: 10.1007/s002800000157. [DOI] [PubMed] [Google Scholar]
- 146.Flowers JL, Hoffman RM, Driscoll TA, Wall ME, Wani MC, Manikumar G, Friedman HS, Dewhirst MW, Colvin OM, Adams DJ. The activity of camptothecin analogues is enhanced in histocultures of human tumors and human tumor xenografts by modulation of extracellular pH. Cancer Chemother Pharmacol. 2000;52:253–361. doi: 10.1007/s00280-003-0635-7. [DOI] [PubMed] [Google Scholar]
- 147.Kim D-K, Ryu DH, Lee JY, Lee N, Kim Y-K, Kim J-S, Chang K, Im G-J, Kim T-K, Choi W-S. Synthesis and biological evaluation of novel A-ring modified hexacyclic camptothecin analogues. J Med Chem. 2001;44:1594–1602. doi: 10.1021/jm0004751. [DOI] [PubMed] [Google Scholar]
- 148.Sun F-X, Tohgo A, Bouvet M, Yagi S, Nassirpour R, Moossa AR, Hoffman RM. Efficacy of camptothecin analog DX-8951f (exatecan mesylate) on human pancreatic cancer in an orthotopic metastatic model. Cancer Res. 2003;63:80–85. [PubMed] [Google Scholar]
- 149.Mitsui I, Kumazawa E, Hirota Y, Aonuma M, Sugimori M, Ohsuki S, Uoto K, Ejima A, Terasawa H, Sato K. A new water-soluble camptothecin derivative. DX-8951f, exhibits potent antitumor activity against human tumors in vitro and in vivo. Jpn J Cancer Res. 1995;86:776–782. doi: 10.1111/j.1349-7006.1995.tb02468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Takiguchi S, Kumazawa E, Shimazoe T, Tohgo A, Kono A. Antitumor effect of DX-8951, a novel camptothecin analog, on human pancreatic tumor cells and their CPT-1-resistant variants cultured in vitro and xenografted into nude mice. Jpn J Cancer Res. 1997;88:760–769. doi: 10.1111/j.1349-7006.1997.tb00448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Joto N, Ishii M, Minami M, Kuga H, Mitsui I, Tohgo A. DX8951f, a water-soluble camptothecin analog, exhibits potent antitumor activity against a human lung cancer cell line and its SN-38-resistant variant. Int J Cancer. 1997;72:680–686. doi: 10.1002/(sici)1097-0215(19970807)72:4<680::aid-ijc21>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 152.Lerchen H-G, Baumgarten J, von dem Bruch K, Lehmann TE, Sperzel M, Kempka G, Fiebig H-H. Design and optimization of 20-O-linked camptothecin glycoconjugates as anticancer agents. J Med Chem. 2001;44:4186–4195. doi: 10.1021/jm010893l. [DOI] [PubMed] [Google Scholar]
- 153.Ishii M, Iwahana M, Mitsui I, Minami M, Imagawa S, Tohgo A, Ejima A. Growth inhibitory effect of a new camptothecin analog. DX8951f, on various drug-resistant sublines including BCRP-mediated camptothecin derivative-resistant variants derived from the human lung cancer cell line PC-6. Anticancer Drugs. 2000;11:353–362. doi: 10.1097/00001813-200006000-00005. [DOI] [PubMed] [Google Scholar]
- 154.Kumazawa E, Jimbo T, Ochi Y, Tohgo A. Potent and broad antitumor effects of DX-8951f, a water soluble camptothecin derivative, against various human tumors xenografted in nude mice. Cancer Chemother Pharmacol. 1998;42:210–220. doi: 10.1007/s002800050807. [DOI] [PubMed] [Google Scholar]
- 155.Rowinsky EK, Johnson TR, Geyer CEJ, Hammond LA, Eckhardt SG, Drengler R, Smetzer L, Coyle J, Rizzo J, Schwartz G, Tolcher A, von Hoff DD, De Jager RL. DX-8951f, a hexacyclic camptothecin analog, on a daily-times-five schedule: a phase I and pharmacokinetic study in patients with advanced solid malignancies. J Clin Oncol. 2000;18:3151–3163. doi: 10.1200/JCO.2000.18.17.3151. [DOI] [PubMed] [Google Scholar]
- 156.Royce ME, Hoff PM, Dumas P, Lassere Y, Lee JJ, Coyle J, Ducharme M, De Jager RL, Pazdur R. Phase I and pharmacokinetic study of exatecan mesylate (DX-8951f): a novel camptothecin analog. J Clin Oncol. 2001;19:1493–1500. doi: 10.1200/JCO.2001.19.5.1493. [DOI] [PubMed] [Google Scholar]
- 157.Garrison M, Hammond LA, Geyer CEJ, Schwartz G, Tolcher A, Smetzer L, Figueroa JA, Ducharme M, Coyle J, Takimoto CH, De Jager RL, Rowinshy EK. A phase I and pharmacokinetic (PK) study of exatecan mesylate administered as a protratced 21-day infusion in patients with advanced solid malignancies. Clin Cancer Res. 2003;9:2527–2537. [PubMed] [Google Scholar]
- 158.Verschraegen CF, Kudelka AP, Hu W, Vincent M, Kavanaugh JJ, Loyer E, Bastien L, Duggal A, De Jager RL. A phase II study of intravenous exatecan mesylate (DX-8951f) administered daily for 5 days every 3 weeks to patients with advanced ovarian, tubal or peritoneal cancer resistant to platinum, taxane and topotecan. Cancer Chemother Pharmacol. 2004;53:1–7. doi: 10.1007/s00280-003-0696-7. [DOI] [PubMed] [Google Scholar]
- 159.Giles FJ, Cortes JE, Thomas DA, Garcia-Manero G, Faderl S, Jeha S, De Jager RL, Kantarjian HM. Phase I and pharmacokinetic study of DX-8951f (exatecan mesylate), a hexacyclic camptothecin, on a daily-times-five schedule in patients with advanced leukemia. Clin Cancer Res. 2002;8:2134–2141. [PubMed] [Google Scholar]
- 160.Boige V, Raymond E, Faivre S, Gatineau M, Meely K, Mekhaldi S, Pautier P, Ducreux M, Rixe O, Armand JP. Phase I and pharmacokinetic study of the camptothecin analog DX-8951f administered as a 30-minute infusion every 3 weeks in patients with advanced cancer. J Clin Oncol. 2000;18:3986–3992. doi: 10.1200/JCO.2000.18.23.3986. [DOI] [PubMed] [Google Scholar]
- 161.Minami H, Fujii H, Igarashi T, Itoh K, Tamanoi K, Oguma T, Sasaki Y. Phase I and pharmacological study of a new camptothecin derivative, exatecan mesylate (DX-8951f), infused over 30 minutes every three weeks. Clin Cancer Res. 2001;7:3056–3064. [PubMed] [Google Scholar]
- 162.Braybrooke JP, Boven E, Bates NP, Ruijter R, Dobbs N, Cheverton PD, Pinedo HM, Talbot DC. Phase I and pharmacokinetic study of the topoisomerase I inhibitor, exatecan mesylate (DX-8951f), using a weekly 30-minute intravenous infusion, in patients with advanced solid malignancies. Ann Oncol. 2003;14:913–921. doi: 10.1093/annonc/mdg243. [DOI] [PubMed] [Google Scholar]
- 163.Sharma S, Kemeny N, Schwartz GK, Kelsen D, O’Reilly E, Ilson D, Coyle J, De Jager RL, Ducharme MP, Kleban S, Hollywood E, Saltz LB. Phase I study of topoisomerase I inhibitor exatecan mesylate (DX-8951f) given as weekly 24-hour infusions three of every four weeks. Clin Cancer Res. 2001;7:3963–3970. [PubMed] [Google Scholar]
- 164.Esteva FJ, Rivera E, Valero MCV, Royce M, Duggal A, Colucci P, De Jager R, Hortobagyi GN. A phase II study of intravenous exatecan mesylate (DX-8951f) administered daily for 5 days every 3 weeks to patients with metastatic breast carcinoma. Cancer. 2003;98:900–907. doi: 10.1002/cncr.11557. [DOI] [PubMed] [Google Scholar]
- 165.Braybrooke JP, Ranson M, Manegold C, Mattson K, Thatcher N, Cheverton P, Sekiguchi M, Suzuki M, Oyama R, Talbot DC. Phase II study of exatecan mesylate (DX8951f) as first line therapy for advanced non-small cell lung cancer. Lung Cancer. 2003;41:215–219. doi: 10.1016/s0169-5002(03)00190-9. [DOI] [PubMed] [Google Scholar]
- 166.Royce M, Rowinsky EK, Hoff PM, Coyle J, De Jager R, Pazdur R, Saltz LB. A phase II study of intravenous exatecan mesylate (DX8951-f) administered daily for five days every three weeks to patients with metastatic adenocarcinoma of the colon or rectum. Invest New Drugs. 2004;22:53–61. doi: 10.1023/b:drug.0000006174.87869.6b. [DOI] [PubMed] [Google Scholar]
- 167.Clamp A, Adams M, Atkinson R, Boven E, Calvert AH, Cervantes A, Lotz TGJ, Vasey P, Cheverton P, Jayson GC. A phase IIA study of the topoisomerase I inhibitor, exatecan mesylate (DX-8951f), adminitsered at two different dose schedules in patients with platinum- and taxane-resistant/refractory ovarian cancer. Gynecol Oncol. 2004;95:114–119. doi: 10.1016/j.ygyno.2004.06.047. [DOI] [PubMed] [Google Scholar]
- 168.Ajani JA, Takimoto C, Becerra CR, Silva A, Baez L, Cohn A, Major P, Kamida M, Feit K, De Jager R. A phase II clinical and pharmacokinetic study of intravenous exatecan mesylate (DX-8951f) in patients with untreated metastatic gastric cancer. Invest New Drugs. 2005;23:479–484. doi: 10.1007/s10637-005-2907-z. [DOI] [PubMed] [Google Scholar]
- 169.Abou-Alfa G, Rowinsky EK, Patt YZ, Schwartz GK, Kelsen DP, Sharma S, Siegel E, Becerra CR, Eckhardt SG, Feit K, De Jager R, O’Reilly EM. A phase II study of intravenous exatecan administered daily for 5 days, every 3 weeks to patients with biliary tract cancers. Am J Clin Oncol. 2005;28:334–339. doi: 10.1097/01.coc.0000158829.63542.2c. [DOI] [PubMed] [Google Scholar]
- 170.Reichardt P, Nielsen OS, Bauer S, Hartmann JT, Schöffski P, Christensen TB, Pink D, Daugaard S, Marreaud S, Van Glabbeke M, Blay JY. Exatecan in pretreated adult patients with advanced soft tissue sarcoma: results of a phase II study of the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer. 2007;43:1017–1022. doi: 10.1016/j.ejca.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 171.Abou-Alfa G, Letourneau R, Harker G, Modiano M, Hurwitz H, Tchekmedyian NS, Feit K, Ackerman J, De Jager RL, Eckhardt SG, O’Reilly EM. Randomized phase III study of exatecan and gemcitabine compared with gemcitabine alone in untreated advanced pancreatic cancer. J Clin Oncol. 2006;24:4441–4447. doi: 10.1200/JCO.2006.07.0201. [DOI] [PubMed] [Google Scholar]
- 172.Lee J-H, Lee J-M, Kim J-K, Ahn S-K, Lee S-J, Kim M-Y, Jew S-S, Park J-G, Hong CI. Antitumor activity of 7-[2-(N-Isopropylamino)ethyl]-(20S)-camptothecin, CKD602, as a potent DNA topoisomerase I inhibitor. Arch Pharm Res. 1998;21:581–590. doi: 10.1007/BF02975379. [DOI] [PubMed] [Google Scholar]
- 173.Kim JC, Shin DH, Kim SH, Kim JK, Park SC, Son WC, Lee HS, Suh JE, Kim CY, Ha CS, Chung MK. Subacute toxicity evaluation of a new camptothecin anticancer agent CKD-602 administered by intravenous injection to rats. Reg Toxicol Pharmacol. 2004;40:356–369. doi: 10.1016/j.yrtph.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 174.Chung M-K, Kim C-Y, Kim J-C. Reproductive toxicity evaluation of a new camptothecin anticancer agent, CKD-602, in pregnant/lactating female rats and their offspring. Cancer Chemother Pharmacol. 2007;59:383–395. doi: 10.1007/s00280-006-0290-x. [DOI] [PubMed] [Google Scholar]
- 175.Chung M-K, Kim J-C, Han S-S. Embryonic effects of CKD-602, a new camptothecin anticancer agent, in rats. Reprod Toxicol. 2005;20:165–173. doi: 10.1016/j.reprotox.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 176.Kim J-C, Shin D-H, Park S-H, Park S-C, Kim Y-B, Kim H-C, Cha S-W, Cho K-H, Kang B-H, Chung M-K. 4-Week repeated intravenous dose toxicity study of a new camptothecin anticancer agent CKD-602 in dogs. Food Chem Toxicol. 2005;43:699–706. doi: 10.1016/j.fct.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 177.Chung M-K, Kim J-C, Han S-S. Effects of CKD-602, a new camptothecin anticancer agent, on pregnant does and embryo-fetal development in rabbits. Drug Chem Toxicol. 2005;1:35–49. doi: 10.1081/dct-39685. [DOI] [PubMed] [Google Scholar]
- 178.Lee DH, Kim S-W, Suh C, Lee J-S, Lee J-H, Lee S-J, Ryoo BY, Park K, Kim JS, Heo DS, Kim NK. Belotecan, new camptothecin analogue, is active in patients with small-cell lung cancer: results of a multicenter early phase II study. Ann Oncol. 2008;19:123–127. doi: 10.1093/annonc/mdm437. [DOI] [PubMed] [Google Scholar]
- 179.Han J, Cha S-W, Kim C-Y, Lee G-S, Suh J-E, Kim J-K, Kim J-C, Kang B-H. Toxicity study of CKD-602, a camptothecin anticancer agent: 5-day repeated intravenous administration in rats. J Appl Pharmacol. 2004;12:49–54. [Google Scholar]
- 180.Kim JC, Shin D-H, Kim SH, Kim JK, Cha SW, Han J, Suh JE, Chung M-K. Acute toxicity of CKD-602, a new anticancer agent, in rats. J Appl Pharmacol. 2004;12:43–48. [Google Scholar]
- 181.Pollack IF, Erff M, Bom D, Burke T, Strode JT, Curran DP. Potent topoisomerase I inhibition by novel silatecans eliminates glioma proliferation in vitro and in vivo. Cancer Res. 1999;59:4898–4905. [PubMed] [Google Scholar]
- 182.Zamboni WC, Jung LL, Strychor S, Joseph E, Zamboni BA, Fetterman SA, Sidone BJ, Burke TG, Curran DP, Eiseman JL. Plasma and tissue disposition of non-liposomal DB-67 and liposomal DB-67 in C.B-17 SCID mice. Invest New Drugs. 2008;26:399–406. doi: 10.1007/s10637-007-9109-9. [DOI] [PubMed] [Google Scholar]
- 183.Van Hattum AH, Pinedo HM, Schluper HMM, Hausheer FH, Boven E. New highly lipophilic camptothecin BNP1350 is an effective drug in experimental human cancer. Int J Cancer. 2000;88:260–266. doi: 10.1002/1097-0215(20001015)88:2<260::aid-ijc18>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 184.Schellens JH, Creemers GJ, Beijnen JH, Rosing H, De Boer-Dennert M, McDonald M, Davies B, Verweij J. Bioavailability and pharmacokinetics of oral topotecan: a new topoisomerase I inhibitor. Br J Cancer. 1996;73:1268–1271. doi: 10.1038/bjc.1996.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Sparreboom A, De Jonge MJ, Punt CJ, Nooter K, Loos WJ, Porro MG, Verweij J. Pharmacokinetics and bioavailability of oral 9-aminocamptothecin capsules in adult patients with solid tumors. Clin Cancer Res. 1998;4:1915–1919. [PubMed] [Google Scholar]
- 186.Van Hattum AH, Schluper HMM, Hausheer FH, Pinedo HM, Boven E. Novel camptothecin derivative BNP1350 in experimental human ovarian cancer: determination of efficacy and possible mechanisms of resistance. Int J Cancer. 2002;100:22–29. doi: 10.1002/ijc.10434. [DOI] [PubMed] [Google Scholar]
- 187.Hausheer FH, Haridas K, Zhao M, Murali D, Seetharamulu P, Yao S, Reddy D, Pavankumar P, Saxe J, Qiuli H, Rustum YM. Karenitecins: a novel class of orally active highly lipophilic topoisomerase I inhibitors. Proc Am Assoc Cancer Res. 1997;38:227. [Google Scholar]
- 188.Hausheer FH, Haridas K, Zhao M, Murali D, Seetharamulu P, Yao S, Reddy D, Pavankumar P, Wu M, Saxe J, Huang Q, Rustum YM. Kerenitecins (part II): a novel class of orally active highly lipophilic topoisomerase I inhibitors. Proc Am Assoc Cancer Res. 1998;39:420. [Google Scholar]
- 189.Kerr JZ, Berg SL, Hausheer FH, Blaney SM. Karenitecins: cytotoxicity studies in pediatric tumor cell lines. Proc Am Assoc Cancer Res. 1999;40:112. [Google Scholar]
- 190.Thompson PA, Berg SL, Aleksic A, Kerr JZ, McGuffey L, Dauser R, Nuchtern JG, Hausheer F, Blaney SM. Plasma and cerebrospinal fluid pharmacokinetic study of BNP1350 in nonhuman primates. Cancer Chemother Pharmacol. 2004;53:527–532. doi: 10.1007/s00280-004-0765-6. [DOI] [PubMed] [Google Scholar]
- 191.Daud A, Valkov N, Centeno B, Derderian J, Sullivan P, Munster P, Urbas P, DeConti RC, Berghorn E, Liu Z, Hausheer FH, Sullivan D. Phase II trial of kerenitecin in patients with malignant melanoma: clinical and translational study. Clin Cancer Res. 2005;11:3009–3016. doi: 10.1158/1078-0432.CCR-04-1722. [DOI] [PubMed] [Google Scholar]
- 192.Dallavalle S, Ferrari A, Biasotti B, Merlini L, Penco S, Gallo G, Marzi M, Tinti MO, Martinalli R, Pisano C, Carminati P, Carenini N, Beretta G, Perego P, De Cesare M, Pratesi G, Zunino F. Novel 7-oxyiminomethyl derivatives of camptothecin with potent in vitro and in vivo antitumor activity. J Med Chem. 2001;44:3264–3274. doi: 10.1021/jm0108092. [DOI] [PubMed] [Google Scholar]
- 193.De Cesare M, Pratesi G, Perego P, Carenini N, Tinelli S, Merlini L, Penco S, Pisano C, Bucci F, Vesci L, Pace S, Capocasa F, Carminati P, Zunino F. Potent antitumor activity and improved pharmacological profile of ST1481, a novel 7-substituted camptothecin. Cancer Res. 2001;61:7189–7195. [PubMed] [Google Scholar]
- 194.Huang M, Gao H, Chen Y, Zhu H, Cai Y, Zhang X, Miao Z, Jiang H, Zhang J, Shen H, Lin L, Wei L, Ding J. Chimmitecan, a novel 9-substituted camptothecin, with improved anticancer pharmacologic profiles in vitro and in vivo. Clin Cancer Res. 2007;13(4):1298–1307. doi: 10.1158/1078-0432.CCR-06-1277. [DOI] [PubMed] [Google Scholar]
- 195.Lavergne O, Lesueur-Ginot L, Pla Rodas F, Kasprzyk PG, Pommier J, Demarquay D, Prevost G, Ulibarri G, Rolland A, Schiano-Liberatore A-M, Harnett J, Pons D, Camara J, Bigg DCH. Homocamptothecins: Synthesis and antitumor activity of novel E-ring modified camptothecin analogues. J Med Chem. 1998;41:5410–5419. doi: 10.1021/jm980400l. [DOI] [PubMed] [Google Scholar]
- 196.Larsen A, Gilbert C, Chyzak G, Plisov SY, Naguibneva I, Lavergne O, Lesueur-Ginot L, Bigg DCH. Unusual potency of BN 80915, a novel fluoriated E-ring modified camptothecin, toward human colon carcinoma cells. Cancer Res. 2001;61:2961–2967. [PubMed] [Google Scholar]
- 197.Gelderbolm H, Salazar R, Verweij J, Pentheroudakis G, De Jonge MJ, Devlin M, van Hooije C, Seguy F, Obach R, Prunonosa J, Principe P, Twelves C. Phase I and pharmacological and bioavailability study of oral diflomotecan (BN80915), a novel E-ring-modified camptothecin analogue in adults with solid tumors. Clin Cancer Res. 2003;9:4101–4107. [PubMed] [Google Scholar]
- 198.Scott L, Soepenberg O, Verweij J, De Jonge MJ, Th Planting AS, McGovern D, Principe P, Obach R, Twelves C. A multicentre phase I and pharmacokinetic study of BN80915 (diflomotecan) administered daily as a 20-min intravenous infusion for 5 days every 3 weeks to patients with advanced solid tumors. Ann Oncol. 2007;18:569–575. doi: 10.1093/annonc/mdl439. [DOI] [PubMed] [Google Scholar]
- 199.Graham JS, Falk S, Samuel LM, Cendros JM, Evans TRJ. A multi-centre dose-escalation and pharmacokinetic study of diflomotecan in patients with advanced malignancy. Cancer Chemother Pharmacol. 2009;63:945–952. doi: 10.1007/s00280-008-0795-6. [DOI] [PubMed] [Google Scholar]
- 200.Lesueur-Ginot L, Demarquay D, Kiss R, Kasprzyk PG, Dassonneville L, Bailly C, Camara J, Lavergne O, Bigg DCH. Homocamptothecin, an E-ring modified camptothecin with enhanced lactone stability, retains topoisomerase I-targeted activity and antitumor properties. Cancer Res. 1999;59:2939–2943. [PubMed] [Google Scholar]
- 201.Demarquay D, Huchet M, Coulomb H, Lesueur-Ginot L, Lavergne O, Camara J, Kasprzyk PG, Prevost G, Bigg DCH. BN89027: A novel homocamptothecin that inhibits proliferation of human tumor cells in vitro and in vivo. Cancer Res. 2004;64:4942–4949. doi: 10.1158/0008-5472.CAN-03-3872. [DOI] [PubMed] [Google Scholar]
- 202.Fassberg J, Stella JV. A kinetic and mechanistic study of the hydrolysis of camptothecin and some analogues. J Pharm Sci. 1992;81:676–684. doi: 10.1002/jps.2600810718. [DOI] [PubMed] [Google Scholar]
- 203.Cao Z, Harris N, Kozielski A, Vardeman D, Stehlin JS, Giovanella B. Alkyl esters of camptothecin and 9-nitrocamptothecin: Synthesis, in vivo pharmacokinetics, toxicity and antitumor activity. J Med Chem. 1998;41:31–37. doi: 10.1021/jm9607562. [DOI] [PubMed] [Google Scholar]
- 204.Cao Z, Pantazis P, Mendoza JT, Early J, Kozielski A, Harris N, Liehr J, Stehlin JS, Giovanella B. Structure-activity relationship of alkyl 9-nitrocamptothecin esters. Recent Res Novel Medicinal Chem. 2001;1:157–171. [Google Scholar]
- 205.Kolhatkar RB, Swaan P, Ghandehari H. Potential oral delivery of 7-ethyl-10-hydroxy-camptothecin (SN-38) using poly(amidoamine) dendrimers. Pharmaceutical Res. 2008;25:1723–1729. doi: 10.1007/s11095-008-9572-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Morgan MT, Nakanishi Y, Kroll DJ, Griset AP, Carnahan MA, Wathier M, Oberlies NH, Manikumar G, Wani MC, Grinstaff MW. Dendrimer-encapsulated camptothecins: increased solubility, cellular uptake, and cellular retention affords enhanced anticancer activity in vitro. Cancer Res. 2006;66:11913–11921. doi: 10.1158/0008-5472.CAN-06-2066. [DOI] [PubMed] [Google Scholar]
- 207.Zhang W, Jiang J, Qin C, Perez LM, Parrish AR, Safe SH, Simanek EE. Triazine dendrimers for drug delivery: evaluation of solubilization properties, activity in cell culture, and in vivo toxicity of a candidate vehicle. Supramol Chem. 2003;15:607–616. [Google Scholar]
- 208.Gopin A, Rader C, Shabat D. New chemical adaptor unit designed to release a drug from a tumor targeting davice by enzymetic triggering. Bioorg Med Chem. 2004;12:1853–1858. doi: 10.1016/j.bmc.2004.01.041. [DOI] [PubMed] [Google Scholar]
- 209.Pessah N, Reznik M, Shamis M, Yantiri F, Xin H, Bowdish K, Shomron N, Ast G, Shabat D. Bioactivation of carbamate-based 20(S)-camptothecin prodrugs. Bioorg Med Chem. 2004;12:1859–1866. doi: 10.1016/j.bmc.2004.01.039. [DOI] [PubMed] [Google Scholar]
- 210.Shamis M, Lode HN, Shabat D. Bioactivation of self-immolative dendritic prodrugs by catalytic antibody 38C2. J Am Chem Soc. 2004;126:1726–1731. doi: 10.1021/ja039052p. [DOI] [PubMed] [Google Scholar]
- 211.Gopin A, Ebner S, Attali B, Shabat D. Enzymatic activation of second-generation denritic prodrugs: canjugation of self-immolative dendrimers with poly(ethylene glycol) via click chemistry. Bioconjugate Chem. 2006;17:1432–1440. doi: 10.1021/bc060180n. [DOI] [PubMed] [Google Scholar]
- 212.Erez R, Ebner S, Attali B, Shabat D. Chemotherapeutic bone-targeted bisphophonate prodrugs with hydrolytic mode of activation. Bioorg Med Chem Lett. 2008;18:816–820. doi: 10.1016/j.bmcl.2007.11.029. [DOI] [PubMed] [Google Scholar]
- 213.Venditto VJ, Allred K, Allred CD, Simanek EE. Intercepting the synthesis of triazine dendrimers with nucleophilic pharmacophores: a general strategy toward drug delivery vehicles. Chem Commun. 2009 doi: 10.1039/b911353c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Parrish B, Emrick T. Soluble camptothecin derivatives prepared by click cycloaddition chemistry on functional aliphatic polyesters. Bioconjugate Chem. 2007;18:263–267. doi: 10.1021/bc060201d. [DOI] [PubMed] [Google Scholar]
- 215.Mu L, Chrastina A, Levchenko T, Torchilin VP. Micelles from Poly(ethylene glycol)-phosphatidyl ethanolamine conjugates (PEG-PE) as pharmaceutical nanocarriers for poorly soluble drug camptothecin. J Biomed Nanotechnol. 2005;1:190–195. [Google Scholar]
- 216.Cabral H, Nakanishi M, Kumagai M, Jang W-D, Nishiyama N, Kataoka K. A photo-activated targeting chemotherapy using glutathione sensitive camptothecin-loaded polymeric micelles. Pharm Res. 2009;26:82–92. doi: 10.1007/s11095-008-9712-2. [DOI] [PubMed] [Google Scholar]
- 217.Liu Z, Robinson JT, Sun X, Dai H. PEGylated nanographene oxide for delivery of water-insoluble cancer drugs. J Am Chem Soc. 2008;130:10876–10877. doi: 10.1021/ja803688x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Watanabe M, Kawano K, Yokoyama M, Opanasopit P, Okano T, Maitani Y. Preparation of camptothecin-loaded polymeric micelles and evaluation of their incorporation and circulation stability. Int J Pharm. 2006;308:183–189. doi: 10.1016/j.ijpharm.2005.10.030. [DOI] [PubMed] [Google Scholar]
- 219.Kawano K, Watanabe M, Yamamoto T, Yokoyama M, Opanasopit P, Okano T, Maitani Y. Enhanced antitumor effect of camptothecin loaded in long-circulating polymeric micelles. J Control Release. 2006;112:329–332. doi: 10.1016/j.jconrel.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 220.Ding X-Q, Chen D, Wang A-X, Li S, Chen Y, Wang J. Antitumor effects of hydroxycamptothecin-loaded poly[ethylene glycol]-poly[3-benzyl-L-glutamate] micelles against oral squamous cell carcinoma. Oncol Res. 2007;16:313–323. doi: 10.3727/000000006783980991. [DOI] [PubMed] [Google Scholar]
- 221.Emerson DL. Liposomal delivery of camptothecins. Pharm Sci Technol To. 2000;3:205–209. doi: 10.1016/s1461-5347(00)00268-6. [DOI] [PubMed] [Google Scholar]
- 222.Burke TG, Staubus AE, Mishra AK, Malak H. Liposomal stabilization of camptothecin’s lactone ring. J Am Chem Soc. 1992;114:8318–8319. [Google Scholar]
- 223.Burke TG, Mishra AK, Wani MC, Wall ME. Lipid bilayer partitioning and stability of camptothecin drugs. Biochemistry. 1993;32:5352–5364. doi: 10.1021/bi00071a010. [DOI] [PubMed] [Google Scholar]
- 224.Sugarman SM, Zou Y, Wasan K, Poirot K, Kumi R, Reddy S, Perez-Soler R. Lipid-complexed camptothecin: formulation and initial biodistribution and antitumor activity studies. Cancer Chemother Pharmacol. 1996;37:531–538. doi: 10.1007/s002800050425. [DOI] [PubMed] [Google Scholar]
- 225.Watanabe M, Kawano K, Toma K, Hatori Y, Maitani Y. In vivo antitumor activity of camptothecin incorporated in liposomes formulated with an artificial lipid and human serum albumin. J Control Release. 2008;127:231–238. doi: 10.1016/j.jconrel.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 226.Tardi P, Choice E, Masin D, Redelmeier T, Bally MB, Madden TD. Liposomal encapsulation of topotecan enhances anticancer efficacy in murine and human xenograft models. Cancer Res. 2000;60:3389–3393. [PubMed] [Google Scholar]
- 227.Liu J-J, Hong R-L, Cheng W-F, Hong K, Chang F-H, Tseng Y-L. Simple and efficient liposomal encapsulation of topotecan by ammonium sulfate gradient: stability, pharmacokinetic and therapeutic evaluation. Anti-Cancer Drugs. 2002;13:709–717. doi: 10.1097/00001813-200208000-00005. [DOI] [PubMed] [Google Scholar]
- 228.Messerer CL, Ramsay EC, Waterhouse D, Ng R, Simms EM, Harasym N, Tardi P, Mayer LD, Bally MB. Liposomal irinotecan: formulation deelopment and therapeutic assessment in murine xenograft models of colorectal cancer. Clin Cancer Res. 2004;10:6638–6649. doi: 10.1158/1078-0432.CCR-04-0221. [DOI] [PubMed] [Google Scholar]
- 229.Drummond DC, Noble CO, Guo Z, Hong K, Park JW, Kirpotin DB. Development of a highly active nanoliposomal irinotecan using a novel intraliposomal stabilization strategy. Cancer Res. 2006;66:3271–3277. doi: 10.1158/0008-5472.CAN-05-4007. [DOI] [PubMed] [Google Scholar]
- 230.Zhang JA, Xuan T, Parmar M, Ma L, Ugwu S, Ali S, Ahmad I. Development and characterization of a novel liposome-based formulation of SN-38. Int J Pharm. 2004;270:93–107. doi: 10.1016/j.ijpharm.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 231.Lei S, Chien P-Y, Sheikh S, Zhang A, Ali S, Ahmad I. Enhanced therapeutic efficacu of a novel liposome-based formulation of SN-38 against human tumor models in SCID mice. Anti-Cancer Drugs. 2004;15:773–778. doi: 10.1097/00001813-200409000-00006. [DOI] [PubMed] [Google Scholar]
- 232.Pal A, Khan S, Wang Y-F, Kamath N, Sarkar AK, Ahmad A, Sheikh S, Ali S, Carbnaro D, Zhang A, Ahmad I. Preclinical safety, pharmacokinetics and antitumor efficacy profile of liposome-entrapped SN-38 formulation. Anticancer Res. 2005;25:331–342. [PubMed] [Google Scholar]
- 233.Colbern GT, Dykes DJ, Engbers C, Musterer R, Hiller A, Pegg E, Saville R, Weng S, Luzzio MJ, Uster P, Amantea M, Working PK. Encapsulation of the topoisomerase I inhibitor GL147211C in PEGylated (STEALTH) liposomes: pharmacokinetics and antitumor activity in HT29 colon tumor xenografts. Clin Cancer Res. 1998;4:3077–3082. [PubMed] [Google Scholar]
- 234.Emerson DL, Bendele R, Brown EB, Chiang S, Desjardins JP, Dihel LC, Gill SC, Hamilton M, LeRay JD, Moon-McDermott L, Moynihan K, Richardson FC, Tomkinson B, Luzzio MJ, Baccanari D. Antitumor efficacy, pharmacokinetics and biodistribution of NX211: a low-clearance liposomal formulation of lurtotecan. Clin Cancer Res. 2000;6:2903–2912. [PubMed] [Google Scholar]
- 235.Desjardins JP, Abbott EA, Emerson DL, Tomkinson BE, LeRay JD, Brown EN, Hamilton M, Dihel LC, Ptaszynski M, Bendele RA, Richardson FC. Biodistribution of NX211, liposomal lurtotecan, in tumor-bearing mice. Anti-Cancer Drugs. 2001;12:235–245. doi: 10.1097/00001813-200103000-00009. [DOI] [PubMed] [Google Scholar]
- 236.Tomkinson B, Bendele R, Giles FJ, Brown EB, Gray A, Hart K, LeRay JD, Meyer D, Pelanne M, Emerson DL. OSI-211, a novel liposomal topoisomerase I inhibitor, is active in SCID mouse models of human AML and ALL. Leukemia Res. 2003;27:1039–1050. doi: 10.1016/s0145-2126(03)00092-4. [DOI] [PubMed] [Google Scholar]
- 237.Kehrer DFS, Bos AM, Verweij J, Groen HJ, Loos WJ, Sarreboom A, de Jonge MJA, Hamilton M, Cameron T, de Vries EGE. Phase I and pharmacologic study of liposomal lurtotecan, NX211: Urinary excretion predicts hematologic toxicity. J Clin Oncol. 2002;20:1222–1231. doi: 10.1200/JCO.2002.20.5.1222. [DOI] [PubMed] [Google Scholar]
- 238.Giles FJ, Tallman MS, Garcia-Manero G, Cortes JE, Thomas DA, Wierda WG, Verstovek S, Hamilton M, Barrett E, Albitar M, Kantarjian HM. Phase I and pharmacokinetic study of a low-clearance, unilamellar liposomal formulation of lurtotecan, a topoisomerase 1 inhibitor, in patients with advanced leukemia. Cancer. 2004;100:1449–1458. doi: 10.1002/cncr.20132. [DOI] [PubMed] [Google Scholar]
- 239.Gelmon K, Hirte HW, Fisher B, Walsh W, Ptaszynski M, Hamilton M, Onetto N, Eisenhauer E. A phase 1 study of OSI-211 given as an intravenous infusion days 1, 2, and 3 every three weeks with solid cancers. Invest New Drugs. 2004;22:263–275. doi: 10.1023/B:DRUG.0000026252.86842.e2. [DOI] [PubMed] [Google Scholar]
- 240.Seiden MV, Muggia FM, Astrow A, Matulonis U, Campos S, Roche M, Sivret J, Rusk J, Barrett E. A phase II study of liposomal lurtotecan (OSI-211) in patients with topotecan resistant ovarian cancer. Gynecol Oncol. 2004;93:229–232. doi: 10.1016/j.ygyno.2003.12.037. [DOI] [PubMed] [Google Scholar]
- 241.Duffaud F, Borner M, Chollet P, Vermorken JB, Bloch J, Degardin M, Rolland F, CD, Baron B, Lacombe D, Fumoleau P. Phase II study of OSI-211 (liposomal lurtotecan) in patients with metastatic or loco-regional recurrent squamous cell carcinoma of the head and neck. An EORTC New Drug Development Group Study. Eur J Cancer. 2004;40:2748–2752. doi: 10.1016/j.ejca.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 242.Dark GG, Calvert AH, Grimshaw R, Poole C, Swenerton K, Kaye S, Coleman R, Jayson G, Le T, Ellard S, Trudeau M, Vasey P, Hamilton M, Cameron T, Barrett E, Walsh W, McIntosh L, Eisenhauer E. Randomized trial of two intravenous schedules of the topoisomerase I ihibitor liposomal lurtotecan in women with relapsed epithelial ovarian cancer: a trial of the National Cancer Institute of Canada clinical trials group. J Clin Oncol. 2005;23:1859–1866. doi: 10.1200/JCO.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 243.Yu NY, Conway C, Pena RLS, Chen JY. STEALTH Liposomal CKD-602, a topoisomerase I inhibitor, improves teh therapeutic index in human tumor xenograft models. Anticancer Res. 2007;27:2541–2546. [PubMed] [Google Scholar]
- 244.Zamboni WC, Strychor S, Joseph E, Walsh DR, Zamboni BA, Parise RA, Tonda ME, Yu NY, Engbers C, Eiseman JL. Plasma, tumor and tissue disposition of STEALTH liposomal CKD-602 (S-CKD602) and nonliposomal CKD-602 in mice bearing A375 human melanoma xenografts. Clin Cancer Res. 2007;13:7217–7223. doi: 10.1158/1078-0432.CCR-07-1035. [DOI] [PubMed] [Google Scholar]
- 245.Zamboni WC, Ramalingam S, Friedland DM, Edwards RP, Stoller RG, Strychor S, Maruca L, Zamboni BA, Belani CP, Ramanathan RK. Phase I and pharmacokinetic study of pegylated liposomal CKD-602 in patients with advanced malignancies. Clin Cancer Res. 2009;15:1466–1472. doi: 10.1158/1078-0432.CCR-08-1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Kunii R, Onishi H, Machida Y. Preparation and antitumor characteristics of PLA/(PEG-PPG-PEG) nanoparticles loaded with camptothecin. Eur J Pharm Biopharm. 2007;67:9–17. doi: 10.1016/j.ejpb.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 247.Miura H, Onishi H, Sasatsu M, Machida Y. Antitumor characteristics of methoxypolyethylene glycol-poly(DL-lactic acid) nanoparticles containing camptothecin. J Control Release. 2004;97:101–113. doi: 10.1016/j.jconrel.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 248.Zhang L, Hu Y, Jiang X, Yang C, Lu W, Yang YH. Camptothecin derivative-loaded poly(caprolactone-co-lactide)-b-PEG-b-poly(caprolactone-co-lactide) nanoparticles and their biodisribution in mice. J Control Release. 2004;96:135–148. doi: 10.1016/j.jconrel.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 249.Min KH, Park K, Kim Y-S, Bae SM, Lee S, Jo HG, Park RW, Kim I-S, Jeong SY, Kim K, Kwon IC. Hydrophobically modified glycol chitosan nanoparticles-encapsulated camptothecin enhance the drug stability and tumor targeting in cancer therapy. J Control Release. 2008;127:208–218. doi: 10.1016/j.jconrel.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 250.Williams J, Lansdown R, Sweitzer R, Romanowski M, LaBell R, Ramaswami R, Unger E. Nanoparticle drug delivery system for intravenous delivery of topoisomerase inhibitors. J Control Release. 2003;91:167–172. doi: 10.1016/s0168-3659(03)00241-4. [DOI] [PubMed] [Google Scholar]
- 251.Dadashzadeh S, Derakhshandeh K, Shirazi FH. 9-Nitrocamptothecin polymeric nanoparticles: cytotoxicity and pharmacokinetic studies of lactone and total forms of drug in rats. Anti-Cancer Drugs. 2008;19:805–811. doi: 10.1097/CAD.0b013e3283099e5c. [DOI] [PubMed] [Google Scholar]
- 252.Lu J-L, Wang J-C, Zhao S-X, Liu X-Y, Zhao H, Zhang X, Zhou S-F, Zhang Q. Self-microemulsifying drug delivery system (SMEDDS) improves anticancer effect of oral 9-nitrocamptothecin on human cancer xenografts in nude mice. Eur J Pharm Biopharm. 2008;69:899–907. doi: 10.1016/j.ejpb.2008.02.023. [DOI] [PubMed] [Google Scholar]
- 253.Chenite A, Chaput C, Wang D, Combes C, Buschmann M, Hoemann CD, Leroux JC, Atkinson BL, Binette F, Selmani A. Novel injectable neutral solutions of chitosan fom biodegradable gels in situ. Biomaterials. 2000;21:2155–2161. doi: 10.1016/s0142-9612(00)00116-2. [DOI] [PubMed] [Google Scholar]
- 254.Chenite A, Buschmann M, Wang D, Chaput C, Kndani N. Rheological characterization of thermogelling chitosan/glycerol-phosphate solutions. Carbohydr Polym. 2001;46:39–47. [Google Scholar]
- 255.Jarry C, Chaput C, Chenite A, Renaud MA, Buschmann M, Leroux JC. Effects of steam sterilization on thermogelling chitosan-based gels. J Biomed Mater Res. 2001;58:127–135. doi: 10.1002/1097-4636(2001)58:1<127::aid-jbm190>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 256.Berrada M, Serreqi A, Dabbarh F, Owusu A, Gupta A, Lehnert S. A novel non-toxic camptothecin formulation ofr cancer chemotherapy. Biomaterials. 2005;26:2115–2120. doi: 10.1016/j.biomaterials.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 257.Yu L, Chang GT, Zhang H, Ding JD. Injectable block copolymer hydrogels for sustained release of a PEGylated drug. Int J Pharm. 2007;348:95–106. doi: 10.1016/j.ijpharm.2007.07.026. [DOI] [PubMed] [Google Scholar]
- 258.Lalloo A, Chao P, Hu P, Stein S, Sinko PJ. Pharmacokinetic and pharmacodynamic evaluation of a novel in situ forming poly(ethylene glycol)-based hydrogel for the controlled delivery of the camptothecins. J Control Release. 2006;112:333–342. doi: 10.1016/j.jconrel.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 259.Koizumi F, Kitagawa M, Negishi T, Onda T, Matsumoto S-I, Hamaguchi T, Matsumura Y. Novel SN-38-incorporating polymeric micelles, NK012, eradicate vascular endothelial growth factor-secreting bulky tumors. Cancer Res. 2006;66:10048–10056. doi: 10.1158/0008-5472.CAN-06-1605. [DOI] [PubMed] [Google Scholar]
- 260.Sumitomo M, Koizumi F, Asano T, Horiguchi A, Ito K, Asano T, Kakizoe T, Hayakawa M, Matsumura Y. Novel SN-38-incorporated polymeric micelle, NK012, strongly suppresses renal cancer progression. Cancer Res. 2008;68:1631–1635. doi: 10.1158/0008-5472.CAN-07-6532. [DOI] [PubMed] [Google Scholar]
- 261.Nakajima TE, Yanagihara K, Takigahira M, Yasunaga M, Kato K, Hamaguchi T, Yamada Y, Shimada Y, Mihara K, Ochiya T, Matsumura Y. Antitumor effect of SN-38-releasing polymeric micelles, NK012, on spontaneous peritoneal metastases from orthotopic gastric cancer in mice compared with irinotecan. Cancer Res. 2008;68:9318–9322. doi: 10.1158/0008-5472.CAN-08-2822. [DOI] [PubMed] [Google Scholar]
- 262.Saito Y, Yasunaga M, Kuroda J, Koga Y, Matsumura Y. Enhanced distribution of NK012, a polymeric micelle-encapsulated SN-38, and sustained release of SN-38 within tumors can beat a hypovascular tumor. Cancer Sci. 2008;99:1258–1264. doi: 10.1111/j.1349-7006.2008.00806.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Kuroda J, Kuratsu J-I, Yasunaga M, Koga Y, Saito Y, Matsumura Y. Potent antitumor effect of SN-38-incorporating polymeric micelle, NK012, against malignant glioma. Int J Cancer. 2009;124:2505–2511. doi: 10.1002/ijc.24171. [DOI] [PubMed] [Google Scholar]
- 264.Nakajima TE, Yasunaga M, Kano Y, Koizumi F, Kato K, Hamaguchi T, Yamada Y, Shirao K, Shimada Y, Matsumura Y. Synergistic antitumor activity of the novel SN-38-incorporating polymeric micelles, NK012, combined with 5-fluorouracil in a mouse model of colorectal cancer, as compared with that of irinotecan plus 5-flurouracil. Int J Cancer. 2008;122:2148–2153. doi: 10.1002/ijc.23381. [DOI] [PubMed] [Google Scholar]
- 265.Nagano T, Yasunaga M, Goto K, Kenmotsu H, Koga Y, Kuroda J, Nishimura Y, Sugino T, Matsumura Y. Antitumor activity of NK012 combined with cisplatin against small cell lung cancer and intestinal mucosal changes in tumor-bearing mouse after treatment. Clin Cancer Res. 2009;15:4348–4355. doi: 10.1158/1078-0432.CCR-08-3334. [DOI] [PubMed] [Google Scholar]
- 266.Greenwald R, Pendri A, Conover C, Gilbert C, Yang R, Xia J. Drug delivery systems. 2.Camptothecin-20-O-poly(ethylene glycol) ester transport forms. J Med Chem. 1996;39:1938–1940. doi: 10.1021/jm9600555. [DOI] [PubMed] [Google Scholar]
- 267.Conover CD, Pendri A, Lee C, Gilbert CW, Shum KL, Greenwald RB. Camptothecin delivery systems: the antitumor activity of a camptothecin-20-O-polyethylene glycol ester transport form. Anticancer Res. 1997;17:3361–3368. [PubMed] [Google Scholar]
- 268.Conover CD, Greenwald RB, Pendri A, Gilbert C, Shum KL. Camptothecin delivery systems: enhanced efficacy and tumor accumulation of camptothecin following its conjugation to polyethylene glycol via a glycine linker. Cancer Chemother Pharmacol. 1998;42:407–414. doi: 10.1007/s002800050837. [DOI] [PubMed] [Google Scholar]
- 269.Rowinsky EK, Rizzo J, Ochoa L, Takimoto CH, Forouzesh B, Schwartz G, Hammond LA, Patnaik A, Kwiatek J, Goetz A, Denis L, McGuire J, Tolcher AW. A phase I and pharmacokinetic study of pegylated camptothecin as a 1-hour infusion every 3 weeks in patients with advanced solid malignancies. J Clin Oncol. 2003;21:148–157. doi: 10.1200/JCO.2003.03.143. [DOI] [PubMed] [Google Scholar]
- 270.Posey JA, III, Saif MW, Carlisle R, Goetz A, Rizzo J, Stevenson S, Rudoltz MS, Kwiatek J, Simmons P, Rowinsky EK, Takimoto CH, Tolcher AW. Phase I study of weekly polyethylene glycol-camptothecin in patients with advanced solid tumors and lymphomas. Clin Cancer Res. 2005;11:7866–7871. doi: 10.1158/1078-0432.CCR-05-0783. [DOI] [PubMed] [Google Scholar]
- 271.Scott LC, Yao JC, Benson AB, III, Thomas AL, Falk S, Mena RR, Picus J, Wright J, Mulcahy MF, Ajani JA, Evans TRJ. A phase II study of pegylated-camptothecin (pegamotecan) in the treatment of locally advanced and metastatic gastric and gastro-oesophageal junction adenocarcinoma. Cancer Chemother Pharmacol. 2009;63:363–370. doi: 10.1007/s00280-008-0746-2. [DOI] [PubMed] [Google Scholar]
- 272.Cheng J, Khin KT, Davis ME. Antitumor activity of 3-cyclodextrin polymer-camptothecin conjugates. Molec Pharmaceut. 2004;1:183–193. doi: 10.1021/mp049966y. [DOI] [PubMed] [Google Scholar]
- 273.Schluep T, Cheng J, Khin KT, Davis ME. Pharmacokinetics and biodistribution of the camptothecin-polymer conjugate IT-101 in rats and tumor-bearing mice. Cancer Chemother Pharmacol. 2006;57:654–662. doi: 10.1007/s00280-005-0091-7. [DOI] [PubMed] [Google Scholar]
- 274.Schluep T, Hwang J, Cheng J, Heidel JD, Bartlett DW, Hollister B, Davis ME. Preclinical efficacy of the camptothecin-polymer conjugate IT-101 in multiple cancer models. Clin Cancer Res. 2006;12:1606–1614. doi: 10.1158/1078-0432.CCR-05-1566. [DOI] [PubMed] [Google Scholar]
- 275.Lee N-J, Ju S-S, Cho W-J, Kim S-H, Kang K-T, Brady T, Theodorakis EA. Synthesis and biological activity of medium molecular weight polymers of camptothecin. Eur Poly J. 2003;39:367–374. [Google Scholar]
- 276.Lee N-J, Ju S-S, Cho W-J, Kim S-H, Kang K-T, Brady T, Theodorakis EA. Synthesis and antitumor activity of medium molecular weight phthalimide polymers of camptothecin. Polym Int. 2003;52:1339–1345. [Google Scholar]
- 277.Zou Y, Wu Q-P, Tansey W, Chow D, Hung M-C, Charnsangavej C, Wallace S, Li C. Effectiveness of water soluble poly(L-glutamic acid)-camptothecin conjugate against resistant human lung cancer xenografted in nude mice. Int J Oncol. 2001;18:331–336. doi: 10.3892/ijo.18.2.331. [DOI] [PubMed] [Google Scholar]
- 278.Singer JW, Bhatt R, Tulinsky J, Buhler KR, Heasley E, Klein P, de Vries P. Water-soluble poly-(L-glutamic acid)-Gly-camptothecin conjugates enhance camptothecin stability and efficacy in vivo. J Control Release. 2001;74:243–247. doi: 10.1016/s0168-3659(01)00323-6. [DOI] [PubMed] [Google Scholar]
- 279.Bhatt R, de Vries P, Tulinsky J, Bellamy G, Baker B, Singer JW, Klein P. Synthesis and in vivo antitumor activity of poly(L-glutamic acid) conjugates of 20(S)-camptothecin. J Med Chem. 2003;46:190–193. doi: 10.1021/jm020022r. [DOI] [PubMed] [Google Scholar]
- 280.Homsi J, Simon GR, Garrett CR, Springett G, De Conti R, Chiappori AA, Munster PN, Burton MK, Stromatt S, Allievi C, Angiuli P, Eisenfeld A, Sullivan DM, Daud AI. Phase I trial of poly-L-glutamate camptothecin (CT-2106) administered weekly in patients with advanced solid malignancies. Clin Cancer Res. 2007;13:5855–5861. doi: 10.1158/1078-0432.CCR-06-2821. [DOI] [PubMed] [Google Scholar]
- 281.Caiolfa VR, Zamai M, Fiorino A, Frigerio E, Pellizzoni C, d’Argy R, Ghigleri A, Castelli MG, Farao M, Pesenti E, Gigli M, Angelucci F, Suarato A. Polymer-bound camptothecin: initial biodistribution and antitumour activity studies. J Control Release. 2000;65:105–119. doi: 10.1016/s0168-3659(99)00243-6. [DOI] [PubMed] [Google Scholar]
- 282.Zamai M, vande Ven M, Farao M, Gratton E, Ghigleri A, Castelli MG, Fontana E, d’Argy R, Fiorino A, Pesenti E, Suarato A, Caiolfa VR. Camptothecin poly[N-(2-hydroxypropyl) methacrylamide] copolymers in antitopoisomerase-I tumor therapy: intratumor release and antitumor efficacy. Mol Cancer Ther. 2003;2:29–40. [PubMed] [Google Scholar]
- 283.Schoemaker NE, van Kesteren C, Rosing H, Jansen S, Swart M, Lieverst J, Fraier D, Breda M, Pellizzoni C, Spinelli R, Grazia Porro M, Beijnen JH, Schellens JHM, ten Bokkel Huinink WW. A phase I and pharmacokinetic study of MAG-CPT, a water-soluble polymer conjugate of camptothecin. Brit J Cancer. 2002;87:608–614. doi: 10.1038/sj.bjc.6600516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Wachters FM, Groen HJM, Maring JG, Gietema JA, Porro MG, Dumez H, de Vries EGE, van Oosterom AT. A phase I study with MAG-camptothecin intravenously administered weekly for 3 weeks in a 4 week cycle in adult patients with solid tumours. Brit J Cancer. 2004;90:2261–2267. doi: 10.1038/sj.bjc.6601811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Bissett D, Cassidy J, de Bono JS, Muirhead F, Main M, Robson L, Fraier D, Magne ML, Pellizzoni C, Porro MG, Spinelli R, Speed W, Twelves C. Phase I and pharmacokinetic (PK) study of MAG-CPT (PNU 166148): a polymeric derivative of camptothecin (CPT) Brit J Cancer. 2004;91:50–55. doi: 10.1038/sj.bjc.6601922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Sarapa N, Britto MR, Speed W, Jannuzzo MG, Breda M, James CA, Porro MG, Rocchetti M, Wanders A, Mahteme H, Nygren P. Assessment of normal and tumor tissue uptake of MAG-CPT, a polymer-bound prodrug of camptothecin, in patients undergoing elective surgery for colorectal carcinoma. Cancer Chemother Pharmacol. 2003;52:424–430. doi: 10.1007/s00280-003-0685-x. [DOI] [PubMed] [Google Scholar]
- 287.Harada M, Sakakibara H, Yano T, Suzuki T, Okuno S. Determinants for the drug release from T-0128, camptothecin analogue-carboxymethyl dextran conjugate. J Control Release. 2000;69:399–412. doi: 10.1016/s0168-3659(00)00321-7. [DOI] [PubMed] [Google Scholar]
- 288.Okuno S, Harada M, Yano T, Yano S, Kiuchi S, Tsuda N, Sakamura Y, Imai J, Kawaguchi T, Tsujihara K. Complete regression of xenografted human carcinomas by camptothecin analogue-carboxymethyl dextran conjugate (T-0128) Cancer Res. 2000;60:2988–2995. [PubMed] [Google Scholar]
- 289.Harada M, Imai J, Okuno S, Suzuki T. Macrophage-mediated activation of camptothecin analogue T-2513-carboxymethyl dextran conjugate (T-0128): possible cellular mechanism for antitumor activity. J Control Release. 2000;69:389–397. doi: 10.1016/s0168-3659(00)00320-5. [DOI] [PubMed] [Google Scholar]
- 290.Binaschi M, Parlani M, Bellarosa D, Bigioni M, Salvatore C, Palma C, Crea A, Maggi CA, Manzini S, Goso C. Human and murine macrophages mediate activation of MEN 4901/T-0128: a new promising camptothecin analogue-polysaccharide conjugate. Anti-Cancer Drugs. 2006;17:1119–1126. doi: 10.1097/01.cad.0000236307.20339.b4. [DOI] [PubMed] [Google Scholar]
- 291.Harada M, Murata J-I, Sakamura Y, Sakakibara H, Okuno S, Suzuki T. Carrier and dose effects on the pharmacokinetics of T-0128, a camptothecin analogue-carboxymethyl dextran conjugate, in non-tumor- and tumor-bearing rats. J Control Release. 2001;71:71–86. doi: 10.1016/s0168-3659(00)00372-2. [DOI] [PubMed] [Google Scholar]
- 292.Fujita F, Koike M, Fujita M, Sakamoto Y, Okuno S, Kawaguchi T, Yano S, Yano T, Kiuchi S, Fujiwara T, Kudoh S, Kakushima M. MEN4901/T-0128, a new camptothecin derivative-carboxymethyldextran conjugate, has potent antitumor activities in a panel of human tumor xenografts in nude mice. Clin Cancer Res. 2005;11:1650–1657. doi: 10.1158/1078-0432.CCR-04-1756. [DOI] [PubMed] [Google Scholar]
- 293.Inoue K, Kumazawa E, Kuga H, Susaki H, Masubuchi N, Kajimura T. CM-dextran-polyalcohol-camptothecin conjugate. In: Maeda H, editor. Polymer Drugs in the Clinical Stage. Kluwer Academic/Plenum Publishers; New York: 2003. [Google Scholar]
- 294.Kumazawa E, Ochi Y. DE-310, a novel macromolecular carrier system for the camptothecin analog DX-8951f: Potent antitumor activities in various murine models. Cancer Sci. 2004;95:168–175. doi: 10.1111/j.1349-7006.2004.tb03199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Ochi Y, Shiose Y, Kuga H, Kumazawa E. A possible mechanism for the long-lasting antitumor effect of the macromolecular conjugate DE-310: mediation by cellular uptake and drug release of its active camptothecin analog DX-8951. Cancer Chemother Pharmacol. 2005;55:323–332. doi: 10.1007/s00280-004-0911-1. [DOI] [PubMed] [Google Scholar]
- 296.Shiose Y, Ochi Y, Kuga H, Yamashita F, Hashida M. Relationship between drug release of DE-310, macromolecular prodrug of DX-8951f, and cathepsins activity in several tumors. Biol Pharm Bull. 2007;30:2365–2370. doi: 10.1248/bpb.30.2365. [DOI] [PubMed] [Google Scholar]
- 297.Kato M, Matsuhashi K, Shimomura K, Shimada M, Hagiwara M, Fujikawa K, Furuhama K. Examination of meningocele induced by the antitumor agent DE-310 in rat fetuses. Reprod Toxicol. 2005;20:495–502. doi: 10.1016/j.reprotox.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 298.Soepenberg O, De Jonge MJ, Sparrenboom A, de Bruin P, Eskens FALM, de Heus G, Wanders J, Cheverton P, Ducharme MP, Verweij J. Phase I and pharmacokinetic study of DE-310 in patients with advanced solid tumors. Clin Cancer Res. 2005;11:703–711. [PubMed] [Google Scholar]
- 299.Wente MN, Kleeff J, Buchler MW, Wanders J, Cheverton P, Langman S, Friess H. DE-310, a macromolecular prodrug of the topoisomerase-I-inhibitor exatecan (DX-8951), in patients with operable solid tumors. Invest New Drugs. 2005;23:339–347. doi: 10.1007/s10637-005-1442-2. [DOI] [PubMed] [Google Scholar]
- 300.Yurkovetskiy AV, Fram RJ. XMT-1001, a novel polymeric camptothecin pro-drug in clinical development for patients with advanced cancer. Adv Drug Delivery Rev. 2009;61:1193–1202. doi: 10.1016/j.addr.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 301.Sapra P, Zhao H, Mehling M, Malaby J, Kraft P, Longley C, Greenberger LM, Horak ID. Novel delivery of SN38 markedly inhibits tumor growth in xenografts, including a camptothecin-11- refractory model. Clin Cancer Res. 2008;14:1888–1896. doi: 10.1158/1078-0432.CCR-07-4456. [DOI] [PubMed] [Google Scholar]
- 302.Zhao H, Rubio B, Sapra P, Wu D, Reddy P, Sai P, Martinez A, Gao Y, Lozanguiez Y, Longley C, Greenberger LM, Horak ID. Novel prodrugs of SN38 using multiarm poly(etylene glycol) linkers. Bioconjugate Chem. 2008;19:849–859. doi: 10.1021/bc700333s. [DOI] [PubMed] [Google Scholar]
- 303.Fox ME, Guillaudeu S, Fréchet JMJ, Jerger K, Macaraeg N, Szoka FC. Synthesis and in vivo antitumor efficacy of PEGylated poly(L-lysine) dendrimer-camptothecin conjugates. Molec Pharmaceut. 2009 doi: 10.1021/mp9001206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Warnecke A, Kratz F. Maleimide-oligo(ethylene glycol) derivatives of camptothecin as albumin-binding prodrugs: synthesis and antitumor efficacy. Bioconjugate Chem. 2003;14:377–387. doi: 10.1021/bc0256289. [DOI] [PubMed] [Google Scholar]
- 305.Schmid B, Chung D-E, Warnecke A, Fichtner I, Kratz F. Albumin-binding prodrugs of camptothecin and doxorubicin with an Ala-Leu-Ala-Leu-linker that are cleaved by cathepsin B: synthesis and antitumor efficacy. Bioconjugate Chem. 2007;18:702–716. doi: 10.1021/bc0602735. [DOI] [PubMed] [Google Scholar]
- 306.Schmid B, Warnecke A, Fichtner I, Jung M, Kratz F. Development of albumin-binding comptothecin prodrugs using a peptide positional scanning library. Bioconjugate Chem. 2007;18:1786–1799. doi: 10.1021/bc0700842. [DOI] [PubMed] [Google Scholar]
- 307.Moon S-J, Govindan SV, Cardillo TM, D’Souza CA, Hansen HJ, Goldenberg DM. Antibody conjugates of 7-ethyl-10-hydoxycamptothecin (SN-38) for targeted cancer chemotherapy. J Med Chem. 2008;51:6916–6929. doi: 10.1021/jm800719t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Dharap SS, Wang Y, Chandna P, Khandare JJ, Qiu B, Gunaseelan S, Sinko PJ, Stein S, Farmanfarmaian A, Minko T. Tumor-specific targeting of an anticancer drug delivery system by LHRH peptide. Proc Natl Acad Sci USA. 2005;102:12962–12967. doi: 10.1073/pnas.0504274102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Chandna P, Saad M, Wang Y, Ber E, Khandare JJ, Vetcher AA, Soldatenkov VA, Minko T. Targeted proapoptotic anticancer drug delivery system. Molec Pharmaceut. 2007;4:668–678. doi: 10.1021/mp070053o. [DOI] [PubMed] [Google Scholar]
- 310.Meyer-Losic F, Nicolazzi C, Quinnero J, Ribes F, Michel M, Dubois V, de Coupade C, Boukaissi M, Chene A-S, Tranchant I, Arranz V, Zoubaa I, Fruchart J-S, Ravel D, Kearsey J. DTS-108, a novel peptidic prodrug of SN38: In vivo efficacy and toxicokinetic studies. Clin Cancer Res. 2008;14:2145–2153. doi: 10.1158/1078-0432.CCR-07-4580. [DOI] [PubMed] [Google Scholar]
- 311.Sparrenboom A, Gelderbolm H, Marsh S, Ahluwalia R, Obach R, Principe P, Twelves C, Verweij J, McLeod HL. Diflomotecan pharmacokinetics in relation to ABCG2 421>CA genotype. Clin Pharmacol Ther. 2004;76:38–44. doi: 10.1016/j.clpt.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 312.Rajendra R, Grounder MK, Saleem A, Schellens JH, Ross DD, Bates SE, Sinko PJ, Rubin EH. Differential effects of the breat cancer resistance protein on the cellular accumulation and cytotoxicity of 9-aminocamptothecin and 9-nitrocamptothecin. Cancer Res. 2003;63:3228–3233. [PubMed] [Google Scholar]
- 313.Nakatomi K, Yoshikawa M, Oka M, YI, Hayasaka S, Sano K, Shiozawa K, Kawabata S, Soda H, Ishikawa T, Tanabe S, Kohno S. Transport of 7-ethyl-10-hydroxycamptothecin (SN-38) by breast cancer resistance protein ABCG2 in human lung cancer cells. Biochem Biophys Res Commmun. 2001;288:827–832. doi: 10.1006/bbrc.2001.5850. [DOI] [PubMed] [Google Scholar]
- 314.Maliepaard M, van Gastelen MA, de Jong LA, Pluim D, van Waardenburg RC, Ruevekamp-Helmers MC, Floot BG, Schellens JH. Overexpression of the BCRP/MXR/ABCP gene in a topotecan-selected ovarian tumor cell line. Cancer Res. 1999;59:4559–4563. [PubMed] [Google Scholar]