

Summary
Rat glucose transporter isoform 1 or rGLUT1, which is expressed in neonatal heart and the epithelial cells that form the blood-brain barrier, facilitates uptake of the trivalent arsenicals arsenite as As(OH)3 and methylarsenite as CH3As(OH)2. GLUT1 may be the major pathway for arsenic uptake into heart and brain, where the metalloid causes cardiotoxicity and neurotoxicity. In this paper, we compare the translocation properties of GLUT1 for trivalent CH3As(OH)2 and glucose. Substitution of Ser66, Arg126 and Thr310, residues critical for glucose uptake, led to decreased uptake of glucose but increased uptake of CH3As(OH)2. The Km for uptake of CH3As(OH)2 of three clinically identified mutants, S66F, R126K and T310I, were decreased 4–10 fold compared to native GLUT1. The osmotic water permeability coefficient (Pf) of GLUT1 and the three clinical isolates increased in parallel with the rate of CH3As(OH)2 uptake. GLUT1 inhibitors Hg(II), cytochalasin B and forskolin reduced uptake of glucose but not CH3As(OH)2. These results indicate that CH3As(OH)2 and water use a common translocation pathway in GLUT1 that is different than that of glucose transport.
Keywords: GLUT1, glucose permease, arsenite, monomethylarsenous acid, water translocation pathway, GLUT1 deficiency syndrome (GLUT1-DS)
Introduction
Arsenic is an environmental toxin that ranks first on the EPA’s list for toxic substances (http://www.atsdr.cdc.gov/cercla/05list.html). It enters the environment primarily from geochemical sources, although it is introduced anthropogenically as well 1. The major source of human arsenic exposure is in the food supply and drinking water. Chronic arsenic poisoning via drinking water has been reported in many countries, including Bangladesh, India, China, Chile and United States 2. In Bangladesh, over 46 million people drink arsenic-contaminated well water that contains as much as 1000-fold more arsenic than the World Health Organization (WHO) limit of 10 ppb 3. Chronic arsenic exposure has been associated with multiple diseases, including neurological disorders, diabetes mellitus, various cancers and cardiovascular diseases such as coronary artery disease, stroke and peripheral arterial disease 4. However, the mechanisms by which arsenic causes human disease are far from clear.
The initial step in arsenic toxicity is uptake into cells. We have identified two uptake pathways: aquaglyceroporin channels, in particular the liver isoform AQP9, and the glucose permease GLUT1 conduct trivalent As(OH)3 and CH3As(OH)2 5, 6, which both have oxidative status of +3. GLUT1, which belongs to the major facilitator superfamily (MFS) 7, facilitates uptake of glucose in many cell types, including neonatal cardiomyocytes, erythrocytes and the endothelial cells that form the blood-brain barrier 8. We propose that GLUT1 is the major pathway for movement of trivalent inorganic and methylated arsenic into heart and brain, where aquaglyceroporins are not abundantly expressed, and this uptake may contribute to cardiovascular disease and neurotoxicity 9.
In the absence of crystal structure data, most structure/function studies on GLUT1 have been proposed using homology modeling and site-directed mutagenesis. Homology modeling of human GLUT1 based on glycerol phosphate transporter (GlpT) 10 has provided useful predictions on the GLUT1 translocation mechanism. Here we used another GLUT1 homologue, the E. coli lactose permease (LacY) 11 to predict an open form model of rGLUT1 to better explain the observation of differential transport of arsenic and glucose (Fig. 1). This model can fit the secondary structure of 12 transmembrane segments (TMs) which have been proposed based on solvent accessibility by cysteine-scanning mutagenesis and affinity labeling assays 12–23. In addition, GLUT1 was found to have weak water transport activity 24, possibly through a water permeation pathway that is different than the glucose transport pathway 25. A clinically identified mutation, T310I, which causes a GLUT1-deficiency syndrome (GLUT1-DS) 26, decreases glucose transport but increases water permeation 25, supporting the idea of separate glucose and water pathways. We previously suggested that As(OH)3 could go through the glucose pathway as a cyclic trimer that resembles the glucose molecule 27. CH3As(OH)2 and glucose reciprocally inhibited uptake of each other noncompetitively 9, suggesting different binding sites for the two molecules. Here we propose that that As(OH)3 and CH3As(OH)2 go through the water pathway in GLUT1, and we present data on uptake of CH3As(OH)2 by wild type and mutant rat GLUT1s expressed in Xenopus laevis oocytes that support this proposal. Rat GLUT1 was chosen for this study because it has a higher rate of CH3As(OH)2 transport than human GLUT1 9. Substitutions in residues Ser66, Arg126 and Thr310 led to a dramatic decrease in glucose transport but a significant increase in permeation of both water and CH3As(OH)2. Hg(II), cytochalasin B (cytB) and forskolin, inhibitors of glucose transport via GLUTs, reduced glucose transport but had little inhibition on CH3As(OH)2 transport. These data support our hypothesis that trivalent arsenicals and water share a common translocation pathway that is different from the main glucose pathway.
Fig. 1. GLUT1 homology model.
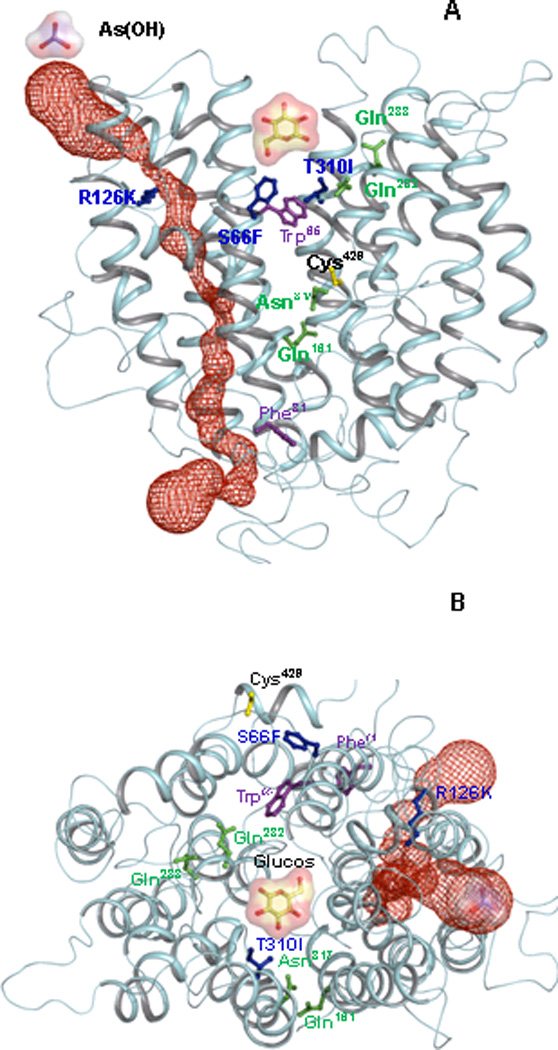
Helices are rendered as Cα worms, colored cyan. (A) View from within the plane of the membrane, with the exofacial side at top and cytosollic side at bottom. In this open form, the glucose binding site is shown as a cavity, (B) Exofacial view of the open form of GLUT1. Selected residues are shown in ball and stick: Gln161, Gln282, Gln283 and Asn317 are shown in green. The GLUT1-DS mutants S66F, R126K, T310I are shown in blue. Cys429, the site of Hg(II) binding, is shown in yellow. Trp65 and Phe81, which are the extra- and intracellular forskolin binding sites, respectively, are shown in magenta. The auxiliary (red) channel is shown as space filling representation. Glucose and As(OH)3 molecules are illustrated as ball-and-stick with molecular surface representations.
Experimental Procedures
Oligonucleotide-directed mutagenesis
Mutations in rat GLUT1 were introduced in the plasmid pL2-5-rGLUT1 by site-directed mutagenesis (Stratagene, La Jolla, CA). The sequences of the mutagenic oligonucleotides and amino acid changes are described in Table 1. The mutation was confirmed by sequencing the entire gene using a CEQ2000 DNA sequencer (Beckman Coulter, Fullerton, CA).
Table 1.
Mutagenic oligonucleotides for construction of Ser66, Arg126, and Thr310 mutants of rGLUT1.
| Residues | Amino Acid Change | Mutagenic Oligonucleotide |
|---|---|---|
| Ser66 | Ser→Ala | CTCACCACACTCTGGGCGCTCTCCGTGGCCATC |
| Ser→Cys | CTCACCACACTCTGGTGCCTCTCCGTGGCCATC | |
| Ser→Phe | CTCACCACACTCTGGTTTCTCTCCGTGGCCATC | |
| Ser→Thr | CTCACCACACTCTGGACCCTCTCCGTGGCCATC | |
| Ser→Tyr | CTCACCACACTCTGGTATCTCTCCGTGGCCATC | |
| Arg126 | Arg→Ala | ATGCTGATCCTGGGCGCGTTCATCATTGGAGTG |
| Arg→His | ATGCTGATCCTGGGCCATTTCATCATTGGAGTG | |
| Arg→Lys | ATGCTGATCCTGGGCAAATTCATCATTGGAGTG | |
| Arg→Leu | ATGCTGATCCTGGGCCTGTTCATCATTGGAGTG | |
| Thr310 | Thr→Ala | CAGCCTGTGTATGCCGCGATCGGCTCGGGTATC |
| Thr→Phe | CAGCCTGTGTATGCCTTTATCGGCTCGGGTATC | |
| Thr→Ile | CAGCCTGTGTATGCCATTATCGGCTCGGGTATC | |
| Thr→Arg | CAGCCTGTGTATGCCCGTATCGGCTCGGGTATC | |
| Thr→Ser | CAGCCTGTGTATGCCTCTATCGGCTCGGGTATC | |
| Thr→Val | CAGCCTGTGTATGCCGTGATCGGCTCGGGTATC |
Expression of rGLUT1 and mutants in oocytes
Plasmid pL2-5-rGLUT1 and its mutants were linearized with NotI. Capped cRNAs were synthesized in vitro (mMESSAGE mMACHINE ultra kit, Applied Biosystems, Austin, TX), and dissolved in distilled water at a final concentration of 0.4 to 0.5 ng/nl. State V and VI defolliculated X. laevis oocytes were injected with 50 ng of cRNA of wild type rGLUT1 or its mutants. Control oocytes were injected with 50 nl of water. The oocytes were incubated at 16°C for 3 days in complete ND96 buffer containing 5 mM HEPES, pH 7.5, 96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 2.5 mM sodium pyruvate, 0.5 mM theophylline, and 2 µg/ml of gentamicin sulphate.
Transport assays
Transport assays for arsenic and glucose in oocytes were performed as described 9. For assay of CH3As(OH)2 (a gift from William Cullen, University of British Columbia, Canada) accumulation, groups of 5 to 10 oocytes were incubated in 0.5 ml incomplete ND96 buffer (5 mM HEPES, pH 6.5, 96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2) with the indicated concentrations of CH3As(OH)2 at room temperature. After either 30 or 60 min, as indicated, the reaction was terminated by washing the oocytes 3 times with 0.75 ml of incomplete ND96 buffer. Oocytes were then transferred to 1.5 ml microcentrifuge tube and completely digested with 50 µl 70% nitric acid. Each sample was diluted with 2 ml HPLC grade water (Sigma, St. Louis, MO). Arsenic concentrations were quantified by inductively coupled plasma-mass spectrometry (ICP-MS) (ELAN 9000, PerkinElmer, Norwalk, CT).
For the assays of glucose transport, the oocytes were incubated in 0.5 ml of incomplete ND96 buffer containing 0.5 µCi/ml D-[U-14C] glucose (Perkin-Elmer, Wellesley, MA) and 0.1 mM unlabelled glucose (Sigma, St. Louis, MO) at room temperature for 1 hr. Oocytes injected with rGLUT1 or water (control) were pretreated in the presence or absence of the indicated concentrations of cytB, forskolin, dimethyl sulfoxide (DMSO) or HgCl2 at room temperature for 25 min before initiation of the transport assay. CytB and forskolin were prepared in DMSO. Uptake was terminated by washing the oocytes three times with 0.5 ml incomplete ND96 buffer containing 0.1 mM unlabelled glucose. Individual oocyte was then solubilized with 0.1 ml of 10% SDS, and radioactivity was quantified using a liquid scintillation counter. Each set of transport experiment was repeated at least twice with different batches of oocytes.
Immunological detection of expression of rGLUT1 and its mutants
Oocyte membranes were isolated as described with minor modifications 28. Briefly, six oocytes were homogenized together in incomplete ND96 buffer by vortexing, followed by centrifugation. The membranes were washed twice with incomplete ND96 and suspended in 0.2 ml the same buffer, solubilized with 5X SDS loading buffer for 10 min at room temperature and separated by SDS-PAGE. Immunoblotting was performed using a commercial anti-GLUT1 primary antibody (Alpha Diagnostic, San Antonio).
Measurement of water osmolarity
Oocytes were incubated for 3 days at 16°C in 205 mosM ND96 medium. Pf was determined by placing oocytes in 1:5 diluted incomplete ND96, with swelling recorded using a microscope (Nikon LABOPHOT-2) and camera system (SPOT PURSUIT Diagnostic Instruments Inc.) at 10 sec intervals continuously for 1 min. The water permeation coefficient (Pf) was calculated as reported 29.
Statistical Analysis
Quantitative results are shown as the mean ± standard deviation. Statistical analyses were performed by Student's t test for paired data between control and mutants. P values <0.05 were considered significant. For each assay, at least two animals and totally eight samples were used. One group of assay (four samples) is presented.
Structural modeling of rGLUT1
A rat GLUT1 homology inward-facing model was constructed using the three dimensional structure of lactose permease of Escherichia coli (LacY and PDB code: 1PV6) as a template using Modeller 9v6 30. The outward-facing model was constructed based on the LacY model of Smirnova et al. 11. The final model was subject to energy minimization using CHARMM 31. Initial energy minimization was done with a steep descent algorithm (100 steps) and followed by adopted basis Newton-Raphson method (1000 steps). The Cα-Cα distance in the rGLUT1 outward model was comparable to LacY outward model proposed by Irina Smirnova et al. (Table 2). This rGLUT1 outwardly-facing model is also consistent with the secondary model proposed by Mueckler and Makepeace 23. The channel was calculated using CAVER 32, and graphical images were rendered with PyMOL 33.
Table 2.
The Cα-Cα distance compared between the rat GLUT1 outward model and LacY outward model
| LacY model (ref) |
GLUT1 model (Current Study) |
||||
|---|---|---|---|---|---|
| Pair of Residues |
Inward-facing crystal structure (1PV6) Cα-Cα,Å |
Outward-facing model Cα-Cα, Å |
Pair of Residues |
Inward-facing model Cα-Cα, Å |
Outward-facing model Cα-Cα, Å |
| 73–401 | 41 | 27 | 86–475 | 41 | 32 |
| 73–340 | 36 | 21 | 86–401 | 36 | 25 |
| 136–340 | 34 | 17 | 152–401 | 35 | 17 |
| 137–340 | 32 | 16 | 153–401 | 35 | 17 |
| 136–401 | 40 | 24 | 152–475 | 41 | 23 |
| 137–401 | 38 | 22 | 153–475 | 37 | 22 |
| 105–310 | 34 | 41 | 116–357 | 34 | 46 |
| 164–310 | 27 | 43 | 180–357 | 30 | 43 |
| 164–375 | 33 | 49 | 180–447 | 33 | 48 |
Results
Glucose and arsenic transport in wild type and mutant rGLUT1
Structure-function relationships in GLUT1 have been studied by site-directed mutagenesis 12–23. Five residues, including Gln161, Gln282, Gln283, Thr310 and Asn317, have been predicted to form hydrogen bonds with glucose in the pyranose form 23. All five are highly conserved in human GLUT1 through human GLUT5, indicating that they might play a similar role in formation of hydrogen bonds. We examined the effect of substitutions of these five residues and eight missense clinical GLUT1-DS mutations: S66F 34, G91D 35, R126H 36, R126L 34, E146K 34, K256V 34, T310I 37 and R333W 34 on glucose and arsenic uptake in rGLUT1. GLUT1-DS causes a decrease rate of glucose transport through the blood-brain barrier, resulting in an inadequate energy supply to brain and delayed neurological development 38, 39.
Mutations of each of five putative hydrogen bond donors (Gln161, Gln282, Gln283, Thr310, and Asn317) to alanine had no significant effect on either glucose or CH3As(OH)2 transport (data not shown), suggesting that elimination of a single hydrogen bond may not be sufficient to influence the initial binding of glucose 40, 41 or CH3As(OH)2. All eight GLUT1-DS mutants decreased glucose transport. The G91D, E146K, K256V and R333W mutants exhibited reduced uptake of both CH3As(OH)2 and glucose (data was not shown). In contrast, the S66F and T310I mutants exhibited a 2-fold increase in CH3As(OH)2 uptake (Fig. 2C and 4C), and the Arg126 substitutions R126H and R126L had no effect on CH3As(OH)2 transport even though they reduced glucose uptake (Fig. 3C). Thus mutations in Ser66, Arg126 and Thr310 differentially affect glucose and arsenical transport.
Fig. 2. Transport of glucose and CH3As(OH)2 via GLUT1 and Ser66 mutants.
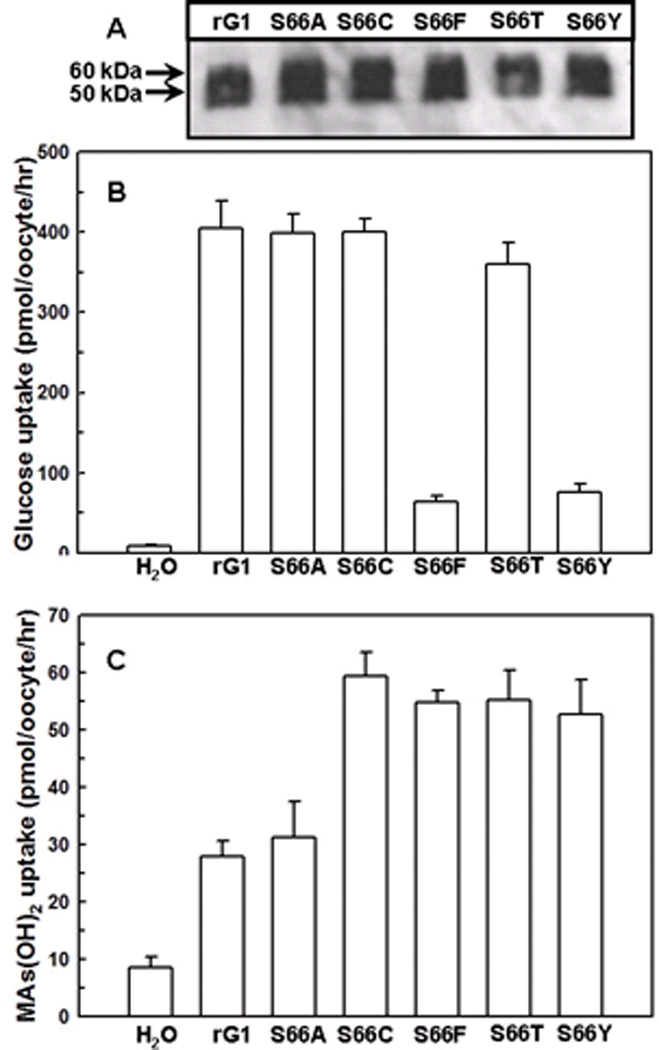
(A) Western blotting results of GLUT1 and Ser66 mutants. Membranes of oocytes were prepared as described under Methods. (B) Transport of D-[U-14C] glucose in oocytes expressing GLUT1 and Ser66 mutants, along with a water injected control. Glucose was used at a final concentration of 0.1 mM. (C) Transport of CH3As(OH)2 in oocytes expressing GLUT1 and Ser66 mutants, along with a water injected control. CH3As(OH)2 was used at a final concentration of 0.1 mM. The values in each plot are means of two independent assays.
Fig. 4. Transport of glucose and CH3As(OH)2 via GLUT1 and Thr310 mutants.
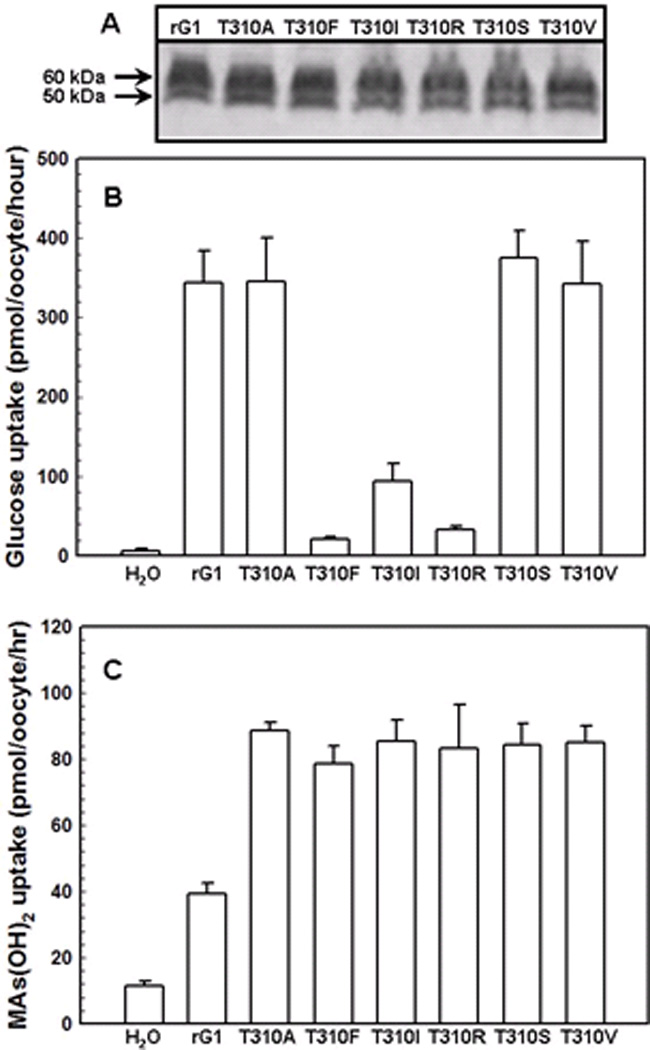
(A) Western blot analysis of rGLUT1 and Thr310 mutants expressed in oocyte membranes was probed with an anti-GLUT1 primary antibody. (B) Transport of 0.1 mM D-[U-14C]glucose in oocytes expressing GLUT1 and Thr310 mutants, along with a water injected control. (C) Transport of 0.1 mM CH3As(OH)2 in oocytes expressing GLUT1 and Thr310 mutants, along with a water injected control. The values in each plot are means of two independent assays.
Fig. 3. Transport of glucose and CH3As(OH)2 via GLUT1 and Arg126 mutants.
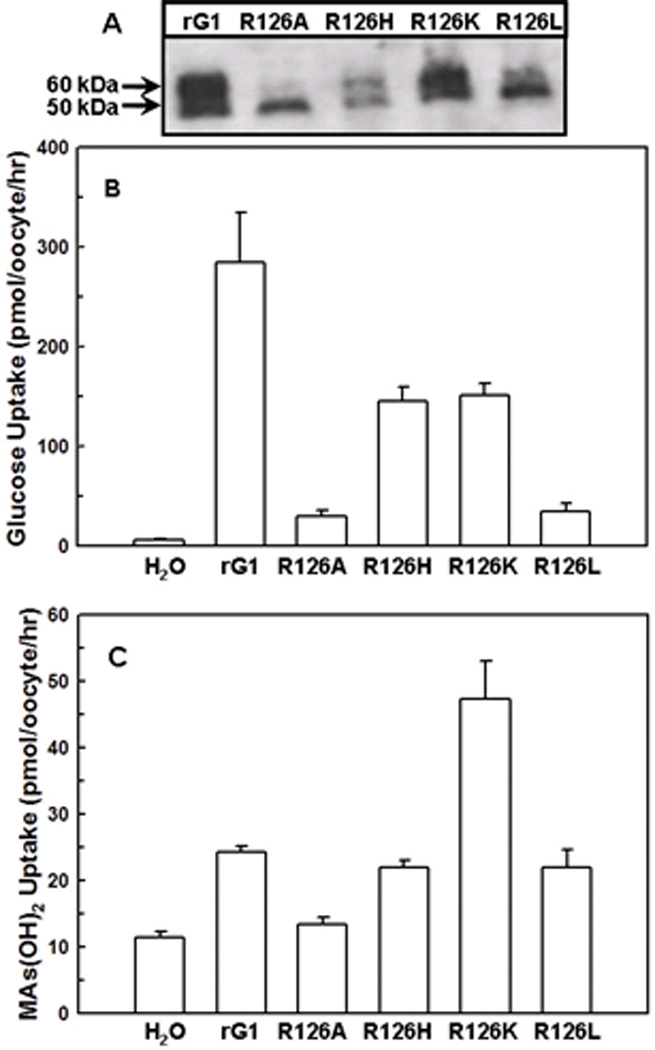
(A) Western blot analysis of rGLUT1 and Arg126 mutants expressed in oocyte membranes was probed with an anti-GLUT1 primary antibody. (B) Transport of 0.1 mM D-[U-14C]glucose in oocytes expressing GLUT1 and Arg126 mutants, along with a water injected control. (C) Transport of 0.1 mM CH3As(OH)2 in oocytes expressing GLUT1 and Arg126 mutants, along with a water injected control. The values in each plot are means of two independent assays.
To examine their roles in more details, the three residues were replaced by residues of different sizes and hydrophobicity (Table 1). From immunoblotting, in each case the oocyte membrane contained similar amounts of the mutant GLUT1 proteins (Fig. 2A and also found with other mutants in Fig. 3A, 4A, and 8A). Ser66 is predicted to be the last amino acid of a large extracellular loop connecting transmembrane sector 1 (TM1) and TM2 23. Substitution of Ser66 with larger aromatic residues (S66F or S66Y) decreased glucose transport (Fig. 2B), while replacement with residues with larger or smaller sizes (S66C, F, T, Y) resulted in a significant increase in uptake of CH3As(OH)2 (Fig. 2C).
Fig. 8. Correlation between CH3As(OH)2 uptake (■) and osmotic water permeability (Pf) (□) for oocytes expressing rGLUT1 and its mutants.
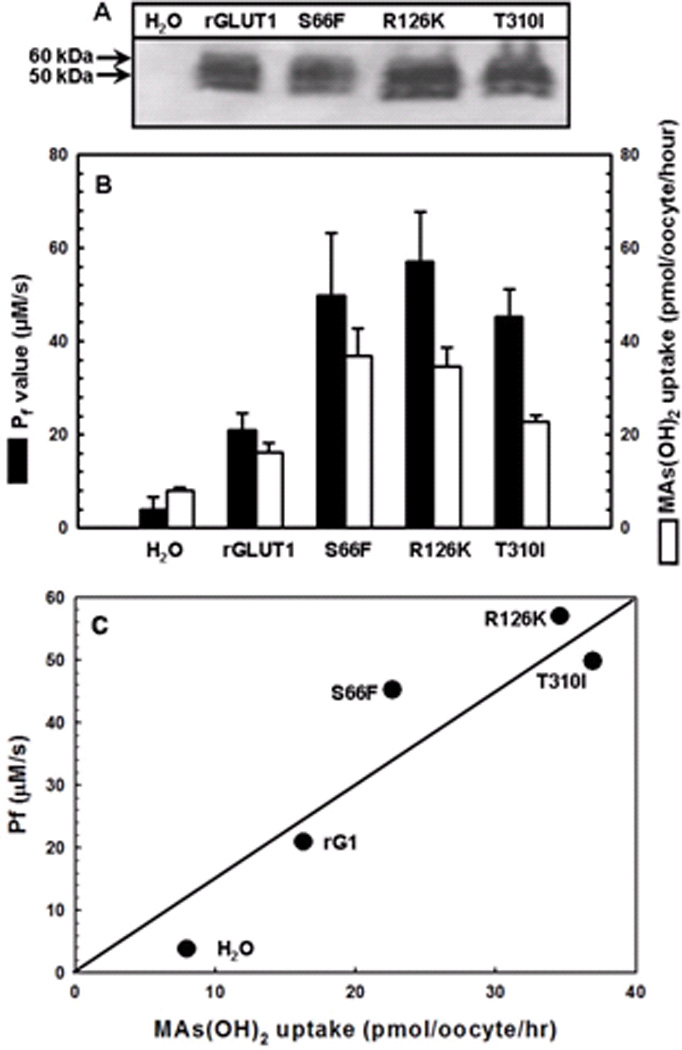
(A) Western blot analysis of rGLUT1 and its mutants expressed in the oocyte membrane was probed with an anti-GLUT1 primary antibody. (B) Pf values were measured by transferring oocytes into 1:5 diluted ND96 buffer. Swelling assays were performed at room temperature for 1 min. CH3As(OH)2 transport was assayed with the same batch of oocytes. The values in each plot are means of two independent assays. (C) Correlation of Pf values with CH3As(OH)2 uptake using the data from (B).
Arg126, the mutation most frequently found in GLUT1-DS 42, is the last amino acid of the extracellular loop connecting TM3 and TM4 23, and is predicted to be located near the opening of the primary glucose translocation pathway 42. Altering Arg126 to alanine, histidine, lysine or leucine considerably decreased glucose uptake (Fig. 3B). However, the efficiency of CH3As(OH)2 uptake by Arg126 mutants was found to be Ala < His = Leu = wild type < Lys. Importantly, while the R126K mutant showed a two-fold decrease in glucose accumulation compared with wild type, it exhibited a dramatic increase in CH3As(OH)2 transport (Fig. 3C). R126A and R126L were expressed in high amounts, but the amounts of the fully glycosylated form (Fig. 3A, upper band) were reduced, possibly resulting from aberrant trafficking to the plasma membrane, as has been observed with human SGLT1 mutants 43. R126H was present in lower amounts in the oocyte membrane, perhaps due to improper folding.
Thr310 is highly conserved throughout the GLUT family, from GLUT1 to GLUT5. It is predicted to be at or near the exofacial end of TM8 and to participate in formation of the water-accessible glucose translocation pathway 23. Six different Thr310 mutants (T310A, F, I, R, S, V) exhibited increased CH3As(OH)2 uptake, while three of them, T310F, T310I and T310R, showed decreased glucose uptake (Fig. 4B). The Km for CH3As(OH)2 uptake in wild type, S66F, R126K and T310I was determined (Fig. 5). In these assays, background uptake of water-injected oocytes was subtracted from all groups. While the Vmax values for the wild type and mutants were similar, wild type affinity for CH3As(OH)2 (Km = 3.9 mM) is lower than in S66F, R126K or T310I (Km values of 0.52 mM, 0.4 mM and 1.0 mM, respectively).
Fig. 5. Kinetic analysis of CH3As(OH)2 uptake by (A) rGLUT1, (B) S66F, (C) R126K, and (D) T310I.
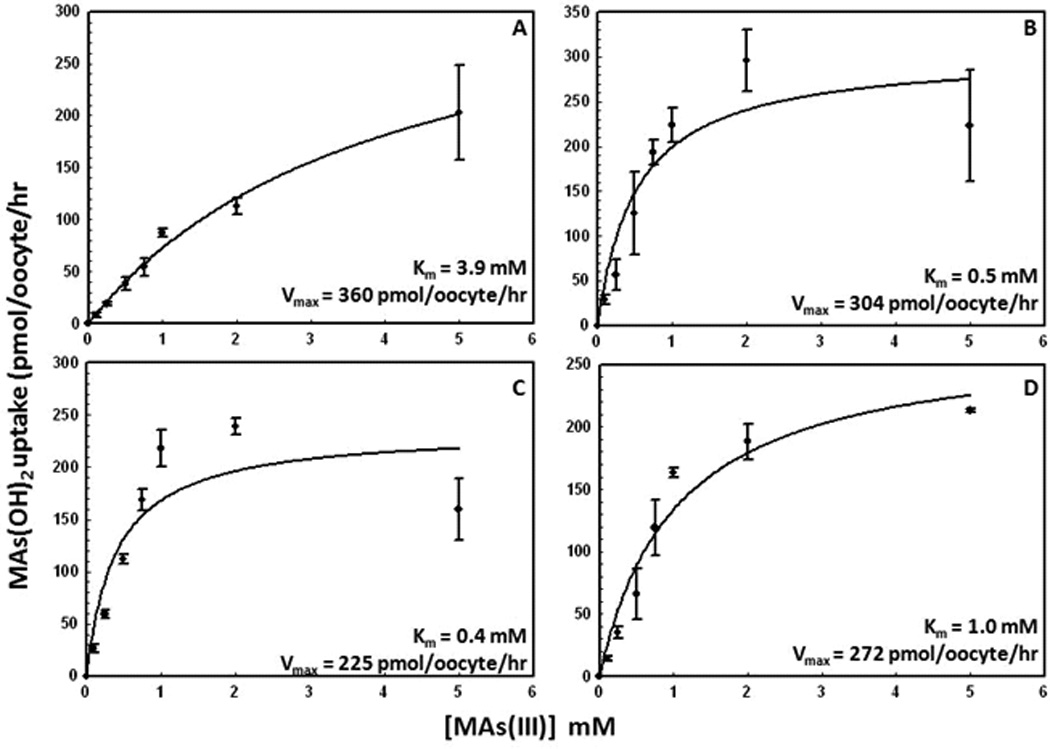
Kinetic parameters were calculated from CH3As(OH)2 uptake in oocytes expressing rGLUT1 or its mutants after subtraction of uptake in oocytes injected with water. Uptake assays in oocytes were assayed after 30 min of incubation with the indicated concentrations of CH3As(OH)2. The values in each plot are means of two independent assays. The kinetic parameters were calculated using Sigma Plot 9.0.
GLUT1 inhibitors do not affect CH3As(OH)2 transport
The docking sites for β-D-glucose and several well-characterized inhibitors have been predicted in the hGLUT1 homology model 40. CytB and forskolin are inhibitors of glucose permeases. In this model, glucose and forskolin dock near the glucose binding site (Trp65) on the extracellular face of GLUT1. Forskolin can also penetrate the plasma membrane and bind to another site (Phe81) at the intracellular face of the translocation pathway. CytB docks at the intracellular face of the pathway but further toward the membrane than the binding site for forskolin. The effect of these inhibitors on CH3As(OH)2 uptake was examined (Fig. 6). CytB and forskolin abolished glucose transport compared with the water-injected control (Fig. 6A), but neither inhibitor had an effect on CH3As(OH)2 transport (Fig. 6B). These results are consistent with our previous observation that cytB and foskolin inhibit glucose but not CH3As(OH)2 uptake when rGLUT1 was expressed in S. cerevisiae 9.
Fig. 6. Inhibition of glucose and CH3As(OH)2 via rGLUT1 by GLUT inhibitors.
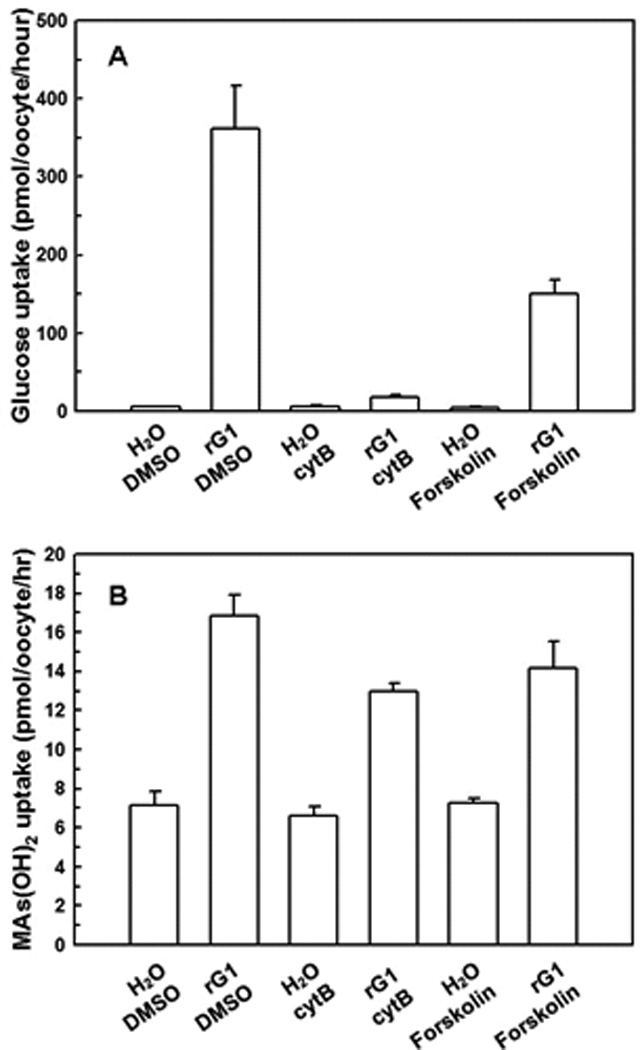
(A) Transport of glucose was inhibited by cytB and forskolin in oocytes injected with rGLUT1 cRNA. (B) Transport of MAs(III) was barely affected by cytB and forskolin in oocytes injected with rGLUT1 cRNA. The values in each plot are means of three independent assays.
Mercurials bind to cysteine residues (Cys429 in GLUT1 44) and occlude transport pathways 45. The effect of 0.2 mM HgCl2 on glucose and CH3As(OH)2 uptake via rGLUT1 was examined (Fig. 7). Transport of glucose was inhibited by Hg(II) (Fig. 7A), while CH3As(OH)2 uptake decreased largely unaffected (Fig. 7B). Again, these results indicate the glucose pathway in GLUT1 is different from the pathway for CH3As(OH)2 translocation.
Fig. 7. Inhibition of glucose and CH3As(OH)2 via rGLUT1 by HgCl2.
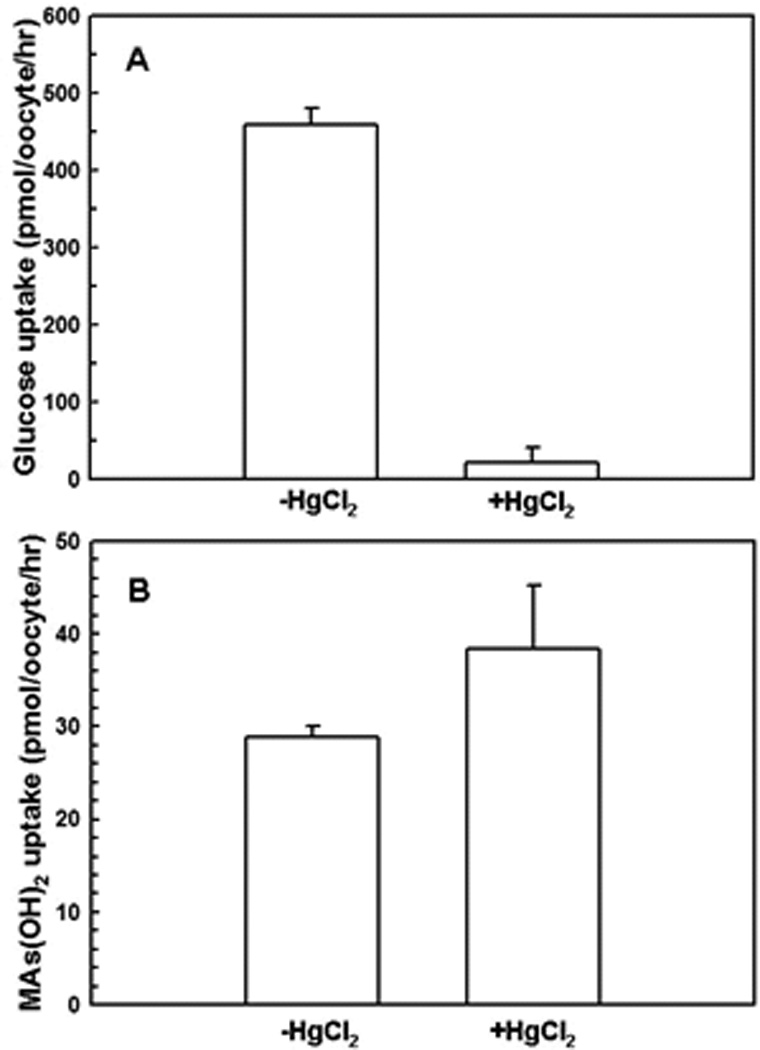
(A) Transport of glucose by oocytes expressing rGLUT1 after 20-min incubation with HgCl2 was calculated after subtraction of the rates from water-injected oocytes. (B) Transport of CH3As(OH)2 by oocytes expressing rGLUT1 after 20-min incubation with HgCl2 was calculated after subtraction of the rates from water-injected oocytes. The values in each plot are means of three independent assays.
Water permeability and CH3As(OH)2 uptake are affected in parallel in GLUT1 mutants
Rat GLUT1 exhibits moderate water permeability 24. Water permeability by wild type and mutant rGLUT1s was examined using an oocyte swelling assay. The Pf values were normalized relative to GLUT1 expression in the plasma membrane, as determined by quantitative immunoblotting (Fig. 8A). The rates of CH3As(OH)2 uptake were measured in the same batch of oocytes (Fig. 8B) The Pf values of each mutant increased in direct proportion with the rate of CH3As(OH)2 uptake (Fig. 8C), consistent with our hypothesis that CH3As(OH)2 and water use the same translocation pathway in GLUT1.
Discussion
Glucose permeases are essential plasma membrane transporters that facilitate the majority of glucose uptake in nearly every mammalian tissue 46. The GLUT1 isoform also catalyzes the adventitious uptake of CH3As(OH)2, and, to a less extent, As(OH)3, when expressed in yeast or X. laevis oocytes 9, which highlights its important roles in both nutrient uptake and arsenic toxicity. It is reasonable to predict that other mammalian glucose permeases would have a similar function in uptake of the metalloid arsenic. GLUT1 also serves as a weak water transporter 24. However, the nature of the pathways for transport of multiple substrates in a single permease is not well understood.
The transport results reported here demonstrate that glucose and CH3As(OH)2 have different translocation properties via GLUT1. The drastic decrease in glucose uptake in mutants S66F and S66Y indicates that a bulky side chain at Ser66 may block glucose binding (Fig. 2B), and a hydrophilic environment is not required at this position since replacement of the serine hydroxyl with a hydrophobic side chain did not affect glucose transport (Fig. 2B). However, all mutations in this position increased uptake of arsenic (Fig. 2C). Substitution of other residues for Arg126 each decreased glucose uptake, suggesting that an arginine at this position is involved in glucose transport (Fig. 3B). At the same time, substitution of Arg126 with a lysine residue produced a dramatic increase in CH3As(OH)2 transport (Fig. 3C), suggesting that a smaller positive residue could increase the size of the permeation pathway, for water and arsenicals. Thr310 has been proposed to form hydrogen bond with glucose at the exofacial side of the binding site 19. Replacement of Thr310 with amino acids containing larger side chains such as phenylalanine, isoleucine or arginine led to decreased transport of glucose, while alterations to small residues such as alanine, serine or valine did not affect glucose transport (Fig. 4B), indicating that the size of this residue alone affects glucose transport. In contrast, all substitutions of Thr310 resulted in significant increases in CH3As(OH)2 uptake (Fig. 4C), quite different from the glucose uptake results, suggesting that conformational changes in the glucose pathway are transmitted to the separate water and arsenical pathway in GLUT1.
Sugar transporters such as the lactose permease (LacY) 47 and Na+-coupled glucose permease (vSGLT1) 48 use conformational changes to reorient the substrates from one side of the membrane to the other during the transport reaction. Binding of substrates lead to changes from outward- to inward-facing conformations. GLUT1 would similarly be expected to couple conformational change to sugar or arsenical transport. Logically, glucose would be expected to bind to an outward-facing cavity, inducing a conformational change that drives the reorientation of the site to inward-facing. Therefore, an open conformation of GLUT1 is modeled, and the five residues predicted to form hydrogen bonds with glucose are located in a large open cavity to which glucose molecules initially bind (Fig. 1). The Cα-Cα distances between the rGLUT1 and the LacY outwardly-facing models 11 were reasonably similar (Table 2). The arrangement of the helices in the rGLUT1 outwardly-facing model was consistent with the hGLUT1 model proposed by Mueckler and Makepeace 23. On the other hand, arsenicals appear to bind to a different location than glucose to a site that may be similar to the entrance of the putative water channel. In this model, this secondary pathway is proposed to function as an auxiliary channel for both water and arsenic (Fig. 1). In rGLUT1 model, Ser66 and Thr310 are located in the putative glucose binding pocket. Therefore, replacement of these two residues to larger amino acids would be predicted to sterically reduce glucose binding and transport, while arsenic transport might not be expected to be affected, as was experimentally observed (Fig. 2 and 4). The S66F and T310I mutations have been shown to increase water permeation, and also increase arsenical uptake (Fig. 8B). Although a homology model can only provide limited accuracy, it provides testable predictions for further exploration. For examples, the functions of residues that line the putative auxiliary channel, including residues from helix 2, 3, and 4, in arsenical and water transport in future experiments, which may also give insight into ways to decrease uptake of arsenicals without reducing normal sugar transport function.
In solution, trivalent arsenic has tetrahedral structure that mimics water and glycerol 49 but is much smaller than glucose, so it is reasonable to speculate that it binds to a different site than glucose. If they do not compete for the same initial binding site, arsenic and glucose should inhibit each other non-competitively, as has been observed 9. In further support of this idea, transport of glucose and arsenic are affected differently by inhibitors such as Hg(II), cytB or forskolin. Hg(II) is proposed to bind Cys429 in GLUT1, which is the only cysteine with a thiolate exposed from the exofacial side of the permease 44 (Fig. 1). Binding of this impermeable thiol-reactive reagent at the extracellular surface inhibits glucose transport, but not CH3As(OH)2 uptake (Fig. 7). Forskolin and glucose are predicted to dock at near each other on the extracellular face of the GLUT1. Forskolin binds near Trp65 in the exofacial cavity but also penetrates the plasma membrane and binds to an alternate site near Phe81 on the intracellular face of the permease 40. The precise site of cytB binding is not known but is proposed to dock at the outer face further down the large open cavity than forskolin 40. None of these inhibitors of glucose uptake affected CH3As(OH)2 uptake (Fig. 6B).
In summary, preponderance of evidence point to different routes for these two through GLUT1.Adventitious transport of arsenicals via a nutrient transporter makes accumulation of these toxic substances unavoidable. However, identifying the differences in their translocation mechanisms offers the possibility of designing ways to prevent entry of this environmental toxin while allowing uptake of glucose.
Acknowledgments
This work is supported by National Institutes of Health Grant GM55425 to B.P.R. and ES016856 to Z.L.
Abbreviations
- GLUT1
glucose transporter isoform 1
- As(OH)3
inorganic arsenite
- CH3As(OH)2
methylarsenite
- Pf
osmotic water permeability
- cytB
cytochalasin B
- DMSO
dimethyl sulfoxide
- GLUT1-DS
GLUT1-deficiency syndrome
Contributor Information
Xuan Jiang, Email: xjiang@med.wayne.edu.
Joseph R. McDermott, Email: jmcdermo@oakland.edu.
A. Abdul Ajees, Email: ajees@fiu.edu.
Barry P. Rosen, Email: brosen@fiu.edu.
Zijuan Liu, Email: liu2345@oakland.edu.
References
- 1.Abernathy CO, Thomas DJ, Calderon RL. J Nutr. 2003;133:1536S–1538S. doi: 10.1093/jn/133.5.1536S. [DOI] [PubMed] [Google Scholar]
- 2.Nordstrom DK. Science. 2002;296:2143–2145. doi: 10.1126/science.1072375. [DOI] [PubMed] [Google Scholar]
- 3.Alam MG, Allinson G, Stagnitti F, Tanaka A, Westbrooke M. Int J Environ Health Res. 2002;12:235–253. doi: 10.1080/0960312021000000998. [DOI] [PubMed] [Google Scholar]
- 4.Weinhold B. Environ Health Perspect. 2004;112:A880–A887. doi: 10.1289/ehp.112-a880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Z, Styblo M, Rosen BP. Environ Health Perspect. 2006;114:527–531. doi: 10.1289/ehp.8600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu Z, Shen J, Carbrey JM, Mukhopadhyay R, Agre P, Rosen BP. Proc Natl Acad Sci U S A. 2002;99:6053–6058. doi: 10.1073/pnas.092131899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marger MD, Saier MH., Jr Trends in biochemical sciences. 1993;18:13–20. doi: 10.1016/0968-0004(93)90081-w. [DOI] [PubMed] [Google Scholar]
- 8.Vannucci SJ, Maher F, Simpson IA. Glia. 1997;21:2–21. doi: 10.1002/(sici)1098-1136(199709)21:1<2::aid-glia2>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 9.Liu Z, Sanchez MA, Jiang X, Boles E, Landfear SM, Rosen BP. Biochem Biophys Res Commun. 2006;351:424–430. doi: 10.1016/j.bbrc.2006.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang Y, Lemieux MJ, Song J, Auer M, Wang DN. Science. 2003;301:616–620. doi: 10.1126/science.1087619. [DOI] [PubMed] [Google Scholar]
- 11.Smirnova I, Kasho V, Choe JY, Altenbach C, Hubbell WL, Kaback HR. Proc Natl Acad Sci U S A. 2007;104:16504–16509. doi: 10.1073/pnas.0708258104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heinze M, Monden I, Keller K. Biochemistry. 2004;43:931–936. doi: 10.1021/bi030175w. [DOI] [PubMed] [Google Scholar]
- 13.Olsowski A, Monden I, Krause G, Keller K. Biochemistry. 2000;39:2469–2474. doi: 10.1021/bi992160x. [DOI] [PubMed] [Google Scholar]
- 14.Mueckler M, Roach W, Makepeace C. J Biol Chem. 2004;279:46876–46881. doi: 10.1074/jbc.M408632200. [DOI] [PubMed] [Google Scholar]
- 15.Mueckler M, Makepeace C. J Biol Chem. 2005;280:39562–39568. doi: 10.1074/jbc.M509050200. [DOI] [PubMed] [Google Scholar]
- 16.Mueckler M, Makepeace C. J Biol Chem. 1999;274:10923–10926. doi: 10.1074/jbc.274.16.10923. [DOI] [PubMed] [Google Scholar]
- 17.Mueckler M, Makepeace C. J Biol Chem. 2008;283:11550–11555. doi: 10.1074/jbc.M708896200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hruz PW, Mueckler MM. J Biol Chem. 1999;274:36176–36180. doi: 10.1074/jbc.274.51.36176. [DOI] [PubMed] [Google Scholar]
- 19.Mueckler M, Makepeace C. J Biol Chem. 2004;279:10494–10499. doi: 10.1074/jbc.M310786200. [DOI] [PubMed] [Google Scholar]
- 20.Mueckler M, Makepeace C. J Biol Chem. 2002;277:3498–3503. doi: 10.1074/jbc.M109157200. [DOI] [PubMed] [Google Scholar]
- 21.Hruz PW, Mueckler MM. Biochemistry. 2000;39:9367–9372. doi: 10.1021/bi000821g. [DOI] [PubMed] [Google Scholar]
- 22.Mueckler M, Makepeace C. J Biol Chem. 2006;281:36993–36998. doi: 10.1074/jbc.M608158200. [DOI] [PubMed] [Google Scholar]
- 23.Mueckler M, Makepeace C. Biochemistry. 2009;48:5934–5942. doi: 10.1021/bi900521n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fischbarg J, Kuang KY, Vera JC, Arant S, Silverstein SC, Loike J, Rosen OM. Proc Natl Acad Sci U S A. 1990;87:3244–3247. doi: 10.1073/pnas.87.8.3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iserovich P, Wang D, Ma L, Yang H, Zuniga FA, Pascual JM, Kuang K, De Vivo DC, Fischbarg J. J Biol Chem. 2002;277:30991–30997. doi: 10.1074/jbc.M202763200. [DOI] [PubMed] [Google Scholar]
- 26.De Vivo DC, Trifiletti RR, Jacobson RI, Ronen GM, Behmand RA, Harik SI. N Engl J Med. 1991;325:703–709. doi: 10.1056/NEJM199109053251006. [DOI] [PubMed] [Google Scholar]
- 27.Liu Z, Boles E, Rosen BP. J Biol Chem. 2004;279:17312–17318. doi: 10.1074/jbc.M314006200. [DOI] [PubMed] [Google Scholar]
- 28.Preston GM, Carroll TP, Guggino WB, Agre P. Science. 1992;256:385–387. doi: 10.1126/science.256.5055.385. [DOI] [PubMed] [Google Scholar]
- 29.Carbrey JM, Cormack BP, Agre P. Yeast. 2001;18:1391–1396. doi: 10.1002/yea.782. [DOI] [PubMed] [Google Scholar]
- 30.Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. Curr Protoc Protein Sci. 2007;Chapter 2(Unit 29) doi: 10.1002/0471140864.ps0209s50. [DOI] [PubMed] [Google Scholar]
- 31.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. J. Comp. Chem. 1983;4:187–217. [Google Scholar]
- 32.Petrek M, Otyepka M, Banas P, Kosinova P, Koca J, Damborsky J. BMC Bioinformatics. 2006;7:316. doi: 10.1186/1471-2105-7-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DeLano WL. http://www.pymol.org. [Google Scholar]
- 34.Wang D, Kranz-Eble P, De Vivo DC. Hum Mutat. 2000;16:224–231. doi: 10.1002/1098-1004(200009)16:3<224::AID-HUMU5>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 35.Klepper J, Willemsen M, Verrips A, Guertsen E, Herrmann R, Kutzick C, Florcken A, Voit T. Hum Mol Genet. 2001;10:63–68. doi: 10.1093/hmg/10.1.63. [DOI] [PubMed] [Google Scholar]
- 36.Brockmann K, Wang D, Korenke CG, von Moers A, Ho YY, Pascual JM, Kuang K, Yang H, Ma L, Kranz-Eble P, Fischbarg J, Hanefeld F, De Vivo DC. Ann Neurol. 2001;50:476–485. doi: 10.1002/ana.1222. [DOI] [PubMed] [Google Scholar]
- 37.Klepper J, Wang D, Fischbarg J, Vera JC, Jarjour IT, O'Driscoll KR, De Vivo DC. Neurochem Res. 1999;24:587–594. doi: 10.1023/a:1022544131826. [DOI] [PubMed] [Google Scholar]
- 38.Klepper J, Voit T. Eur J Pediatr. 2002;161:295–304. doi: 10.1007/s00431-002-0939-3. [DOI] [PubMed] [Google Scholar]
- 39.Takata K, Hirano H, Kasahara M. Int Rev Cytol. 1997;172:1–53. doi: 10.1016/s0074-7696(08)62357-8. [DOI] [PubMed] [Google Scholar]
- 40.Salas-Burgos A, Iserovich P, Zuniga F, Vera JC, Fischbarg J. Biophys J. 2004;87:2990–2999. doi: 10.1529/biophysj.104.047886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dwyer DS. Proteins. 2001;42:531–541. [PubMed] [Google Scholar]
- 42.Pascual JM, Wang D, Yang R, Shi L, Yang H, De Vivo DC. J Biol Chem. 2008;283:16732–16742. doi: 10.1074/jbc.M801403200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lostao MP, Hirayama BA, Panayotova-Heiermann M, Sampogna SL, Bok D, Wright EM. FEBS Lett. 1995;377:181–184. doi: 10.1016/0014-5793(95)01339-3. [DOI] [PubMed] [Google Scholar]
- 44.Wellner M, Monden I, Keller K. Biochem J. 1994;299(Pt 3):813–817. doi: 10.1042/bj2990813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Savage DF, Stroud RM. J. Mol. Biol. 2007;368:607–617. doi: 10.1016/j.jmb.2007.02.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Joost HG, Thorens B. Mol Membr Biol. 2001;18:247–256. doi: 10.1080/09687680110090456. [DOI] [PubMed] [Google Scholar]
- 47.Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S. Science. 2003;301:610–615. doi: 10.1126/science.1088196. [DOI] [PubMed] [Google Scholar]
- 48.Faham S, Watanabe A, Besserer GM, Cascio D, Specht A, Hirayama BA, Wright EM, Abramson J. Science. 2008;321:810–814. doi: 10.1126/science.1160406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ramírez-Solis A, Mukopadhyay R, Rosen BP, Stemmler TL. Inorg Chem. 2004;43:2954–2959. doi: 10.1021/ic0351592. [DOI] [PMC free article] [PubMed] [Google Scholar]