

Abstract
An efficient stereocontrolled construction of the fully substituted isoindolone subunit of muironolide A is described. The approach is centered on the intramolecular Diels-Alder reaction between the enol form of the β-keto amide and an α β γ δ-unsaturated ester, followed by the installation of the cyclohexene double bond.
Muironolide A is a tetrachlorinated marine metabolite isolated in minute quantities from a specimen of Phorbas1 that is also known to produce phorboxazoles A and B.2 The halogenation in muironolide A is present in the form of a trichloromethyl group attached to a carbinol stereocenter, with an additional chloro substituent at the cyclopropyl group (Figure 1). Another structural element characteristic of muironolide A is the 16-membered bislactone that incorporates a conformationally intriguing hexahydro-1H-isoindolone subunit, which is the focus of this study. Remarkably, using microcryoprobe NMR spectroscopy,3 Molinski and co-workers fully elucidated the structure of muironolide A with only 90 µg (152 nmol) of the material available. This exceedingly small amount was insufficient to investigate biological activity of the natural product, although an initial screen indicated a notable activity against the HCT116 cancer cell line.1 In a sense, muironolide A can be categorized as a “nearly extinct” natural product; considering that its reisolation from the natural source is not feasible, the chemical synthesis is the only alternative to provide material for elucidation of its biological function. Accordingly, our goal is to develop a robust and scalable synthesis of muironolide A.
Figure 1.

Structure of muironolide A
In the preliminary synthetic study we concentrated on the development of an efficient, concise entry to the isoindolinone subunit of muironolide A by a suitable intramolecular Diels-Alder (IMDA) cycloaddition, identifying compound 1 as a key target of the study (Scheme 1).4,5 Isoindolinone 1 is closely related to the heterocyclic subunit of the natural product and contains most of its structural information. At the onset, our synthetic planning has been informed by conformational analysis of the bicyclic ring system. The pattern of substitution in isoindolinone 1 forces it to adopt a rather strained conformation in the ground state. A similar conformational preference was found for muironolide A by NMR spectroscopic studies.1 In contrast, a potential precursor to 1, intermediate 2, adopts a less strained half-chair conformation, suggesting that an IMDA approach based on isomerization of 2 into conjugation is likely to be thermodynamically unfavorable. The prediction based on this analysis is also supported by experimental observations reported recently by Molinski and co-workers in their elegant synthetic approach to muironolide A. The attempted rearrangement of the double bond in an intermediate similar to 2 into conjugation with the lactam carbonyl group was found to be problematic.5
Scheme 1.

As a tactical solution to this problem, we considered β-keto amide 3a as a precursor to 1, which by way of its enol form 3b enables an IMDA or double Michael disconnection to simple acyclic amide 4a, along with its enol form 4b that is properly functionalized for the cycloaddition process. Another benefit of this design is the potential for Lewis-acid complexation to the β-keto amide 4a/4b that could eventually enable a catalytic asymmetric variant of the IMDA approach to the isoindolinone core of muironolide A.6
To gauge the feasibility of this design in greater detail, we investigated the Diels-Alder reaction with both the 4E- and 4Z-isomers of 4a. The common carboxylic acid counterpart was used in the form of dioxinone 8, and its three-step synthesis from ethyl (2E)-2,4-dimethyl-2- pentenoate is shown in Scheme 2a. Reduction of the substrate with i-Bu2AlH and Swern oxidation afforded 2,4-dimethyl-2-pentenal (6), and its olefination with reagent 7 delivered requisite dioxinone 8.7 For the (4E)-amide precursor (Scheme 2b), oxidation of (E)-4-bromo-3-methyl-2-butene-1-ol8 followed by a Wittig 2-carbon elaboration provided ester 11, and its direct reaction with 4-methoxybenzylamine completed the short synthesis of the E-amine counterpart. The synthesis of the amine counterpart of (4Z)-4a required a late-stage ester installation; therefore, Z-amine 15 was prepared in three steps from propargyl alcohol as shown in Scheme 2c. Copper-catalyzed methylmagnesation of propargyl alcohol followed by the iodine quench afforded (Z)-3-iodo-2-methyl-propen-1-ol.9 The synthesis was completed by the Kumada coupling with vinylmagnesium bromide10 and a one-pot elaboration of the hydroxy group to PMBprotected amine 15.
Scheme 2.

Investigation of the IMDA reaction began with amide (4E)-4a, which was readily accessed by an efficient thermal coupling between dioxinone 8 and amine 12 with pyridinium p-toluenesulfonate in toluene at reflux (Scheme 3).11 Early experimentation revealed that heating the substrate in toluene at reflux results in a clean, highly endo-selective cyclization to 16, reaching 90% conversion after 18 h (see Figure S1 in the Supporting Information for the kinetic profile). No additive was required. Performing the reaction under basic conditions (Cs2CO3, EtOAc, 25 °C; LiN(SiMe3)2, THF, 0 °C; n-Bu4NF, THF, 0 °C)6,12 generally offered no advantages and resulted in either low reactivity or decomposition. Carrying out the original thermal IMDA reaction in the presence of triethylamine also had no effect on the rate of the reaction (Figure 2). Collectively, these observations are indicative of a pericyclic process rather than a stepwise anionic double Michael addition pathway.6,12
Scheme 3.

Reduction of 16 with NaBH4 in the presence of CeCl3 in methanol selectively afforded 17, whose relative stereochemistry was corroborated by comprehensive NOE experiments. Further treatment of 17 with i-Bu2AlH and MnO2 produced unsaturated aldehyde, which could be effectively epimerized at the C4 position upon exposure to piperidinium trifluoroacetate at 75 °C.13 The relative configuration of aldehyde 19, which has the same stereochemistry at C4 as muironolide A, was also established by NOE experiments.
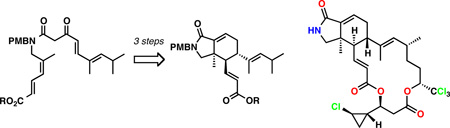
The IMDA reaction of (4Z)-4a was investigated next (Scheme 4). The substrate was prepared in two steps from dioxinone 8 and amine 15, which included the direct amide formation followed by site-selective cross-metathesis with methyl acrylate,14 providing (4Z)-4a in 77% yield over 2 steps. Heating of this compound in toluene at reflux again resulted in smooth intramolecular cycloaddition, albeit at a somewhat slower rate than that of the corresponding isomer (4E)-4a (see Figure S1 in the Supporting Information for the kinetic profile). In contrast to the cyclization of the E-isomer, the product of exocyclization was favored, and similarly high level of diastereocontrol exceeding 30:1 was observed. The stereochemistry of 3a was ascertained through chemical correlation by its 3-step conversion to aldehyde 19.
Scheme 4.

The significance of the IMDA cyclization of (4Z)-4a is that it directly provides the ring system of muironolide A with the correct stereochemistry. The fully assembled isoindolinone ring system was reached by the installation of the C8,C9-double bond in three straightforward transformations: 1) reduction of the ketone (NaBH4, CeCl3, MeOH; 2) methanesulfonation (CH3SO2Cl, Et3N); and 3) elimination (DBU, PhMe, 85 °C), which afforded 21 in 64% yield after the three steps.
Finally, the stereochemical aspects of the IMDA reactions of isomers (4E)-4b and (4Z)-4b, the reactive enol forms of (4E)-4a and (4Z)-4a, deserve a comment. The alternative transition structures (TS) for the exo and endo Diels-Alder cycloaddition of (4E)-4b and (4Z)-4b are depicted in Scheme 5. Upon consideration of these structures, we hypothesize that the effect of secondary orbital interactions is countered by the hydrogen bond stabilizing the reactive enol form of the substrate. This hydrogen bonding is not possible for the exo TS of (4E)-4b and the endo TS of (4Z)-4b. This is expected to substantially destabilize these conformations, resulting in the observed mode of high diastereoselectivty for the two isomers. We observed a notably lower diastereoselectivity of ~5:1 for the IMDA reaction of the corresponding trimethylsilyl enol ethers15 favoring the same diastereomer (PhMe, reflux, 60 h), suggesting that the secondary orbital interactions do not influence the stereochemical course of the reaction, and the conformational preferences appear to be reinforced by hydrogen bonding in the enol form.
Scheme 5.

Proposed transition structures for the IMDA reactions of enol tautomers (4E)-4b and (4Z)-4b.
In closing, we validated an IMDA approach to the core isoindolinone subunit of muironolide A, an exceedingly scarce marine natural product characterized by Molinski in 2009 using state-of-the-art nanoscale NMR technology. This approach potentially capitalizes on the hydrogenbond stabilization of the reactive enol form of the β-keto amide substrate to achieve the desired high diastereoselectivity. The product can be rapidly advanced to the fully functionalized indolinone ring system of the natural product with the correct stereochemical relationship of the substituents. The reported process paves the way for the develoment of an enantioselective variant based on Lewis-acid catalysis, which is currently under active investigation in our laboratory.
Supplementary Material
Acknowledgment
Dr. Hongjun Zhou (UC Santa Barbara) is thanked for continued assistance with NMR spectroscopy. This work is supported by the NIH/NIGMS (R01 GM077379) and kind additional funding from Amgen and Eli Lilly.
Footnotes
Supporting Information Available General experimental procedures, characterization data, and copies of 1H and 13C NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Dalisay DS, Morinaka BI, Skepper CK, Molinski TF. J. Am. Chem. Soc. 2009;131:7552–7553. doi: 10.1021/ja9024929. [DOI] [PubMed] [Google Scholar]
- 2.(a) Searle PA, Molinski TF. J. Am. Chem. Soc. 1995;117:8126–8131. [Google Scholar]; (b) Searle PA, Molinski TF, Brzezinski LJ, Leahy JW. J. Am. Chem. Soc. 1996;118:9422–9423. [Google Scholar]
- 3.Molinski TF. Nat. Prod. Rep. 2010;27:321–329. doi: 10.1039/b920545b. [DOI] [PubMed] [Google Scholar]
- 4.For selected reviews on the intramolecular Diels-Alder reaction, see: Funk RL, Vollhardt KPC. Chem. Soc. Rev. 1980;9:41–61. Brieger G, Bennet JN. Chem. Rev. 1980;80:63–97. Ciganek E. Org. React. 1984;32:1–374. Roush WR. In: Intramolecular Diels-Alder Reactions. In Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 5. New York: Pergamon Press; 1991. pp. 513–550. Winkler JD. Chem. Rev. 1996;96:167–176. doi: 10.1021/cr950029z. Bear BR, Sparks SM, Shea KJ. Angew. Chem. Int. Ed. 2001;40:820–849. Nicolaou KC, Snyder SA, Montagnon T, Vassilikogiannakis G. Angew. Chem. Int. Ed. 2002;41:1668–1698. doi: 10.1002/1521-3773(20020517)41:10<1668::aid-anie1668>3.0.co;2-z. Takao K-i, Munakata R, Tadano K-i. Chem. Rev. 2005;105:4779–4807. doi: 10.1021/cr040632u. Tadano K-i. Eur. J. Org. Chem. 2009:4381–4394. Juhl M, Tanner D. Chem. Soc. Rev. 2009;38:2983–2992. doi: 10.1039/b816703f.
- 5.Flores B, Molinski TF. Org. Lett. 2011;13:3932–3935. doi: 10.1021/ol201461n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For an example of a strategic application of an IMDA reaction involving an enol derived from a β-keto carbonyl precursor as a diene counterpart see: Scheerer JR, Lawrence JF, Wang GC, Evans DA. J. Am. Chem. Soc. 2007;129:8968–8969. doi: 10.1021/ja073590a. See also refs. 12c, 15.
- 7.Boeckman RK, Jr, Thomas AJ. J. Org. Chem. 1982;47:2823–2824. [Google Scholar]
- 8.(a) Yamashita MY, Yasumoto T, Rawal VH. Heterocycles. 1998;48:79–93. [Google Scholar]; (b) Babler JM, Buttner WJ. Tetrahedron Lett. 1976;17:239–241. [Google Scholar]; (c) Van TN, De Kimpe N. Tetrahedron. 2000;56:7969–7973. [Google Scholar]
- 9.(a) Duboudin JG, Jousseaume B, Saux A. J. Organomet. Chem. 1979;168:1–11. [Google Scholar]; (b) Liu F, Negishi E. J. Org. Chem. 1997;62:8591–8594. doi: 10.1021/jo971360g. [DOI] [PubMed] [Google Scholar]
- 10.(a) Liu P, Jacobsen EN. J. Am. Chem. Soc. 2001;123:10772–10773. doi: 10.1021/ja016893s. [DOI] [PubMed] [Google Scholar]; (b) Clausen DJ, Wan S, Floreancig PE. Angew. Chem. Int. Ed. 2011;50:5178–5181. doi: 10.1002/anie.201007757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Reber KP, Tilley SD, Sorensen EJ. Chem. Soc. Rev. 2009;38:3022–3034. doi: 10.1039/b912599j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Clemens RJ, Witzeman JS. J. Am. Chem. Soc. 1989;111:2186–2193. [Google Scholar]
- 12.Eanples of related intermolecular cyclizations: Yang Z, Shannon D, Truong V-L, Deslongchamps P. Org. Lett. 2002;4:4693–4696. doi: 10.1021/ol027125o. Petrovic D, Brückner R. Org. Lett. 2011;13:6524–6527. doi: 10.1021/ol202809y. Intramolecular: Xue H, Yang J, Gopal P. Org. Lett. 2011;13:5696–5699. doi: 10.1021/ol2024554.
- 13.(a) Stivala CE, Zakarian A. J. Am. Chem. Soc. 2008;130:3774–3776. doi: 10.1021/ja800435j. [DOI] [PubMed] [Google Scholar]; (b) Stivala CE, Zakarian A. Org. Lett. 2009;11:839–842. doi: 10.1021/ol8027797. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Araoz R, Servent D, Molgo J, Iorga BI, Fruchart-Gaillard C, Benoit E, Gu Z, Stivala C, Zakarian A. J. Am. Chem. Soc. 2011;133:10499–10511. doi: 10.1021/ja201254c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Stivala CE, Zakarian A. Tetrahedron Lett. 2007;48:6845–6848. [Google Scholar]
- 14.(a) Chatterjee AK, Morgan JP, Scholl M, Grubbs RH. J. Am. Chem. Soc. 2000;122:3783–3784. [Google Scholar]; (b) Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. J. Am. Chem. Soc. 2000;122:8168–8179. [Google Scholar]; (c) Dewi P, Randl S, Blechert S. Tetrahedron Lett. 2005;46:577–580. [Google Scholar]
- 15.For an example of an IMDA reaction with silyl enol ethers derived from β-keto carbonyl compounds see: Grimaud L, Ferezou J-P, Prunet J, Lallemand J-Y. Tetrahedron. 1997;53:9253–9268.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.