

Calcium-dependent phosphatidylinositol-specific phospholipase C from Streptomyces antibioticus (saPLC1)1 employs a unique catalytic mechanism whereby the cleavage of phosphatidylinositol (PI) into inositol 1-phosphate (I-1-P) occurs via a trans-cyclization mechanism involving formation of inositol 1,6-cyclic phosphate intermediate (1,6-IcP, Scheme 1).2 In all other previously studied phospholipases C, this process is known to occur by cis-cyclization to form inositol 1,2-cyclic phosphate intermediate.3
Scheme 1.
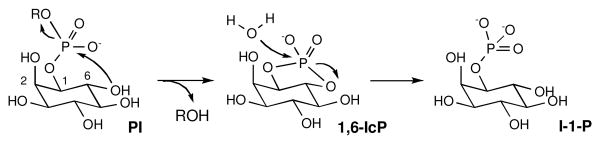
Mechanism of saPLC1-catalyzed hydrolysis of PI.
In this work we examine the kinetics and products of saPLC1 reactions with phosphorothioate and phosphorodithioate analogs of PI.4 To avoid complications related to interfacial interactions between aggregated substrate and saPLC1, we synthesized short-chain (dihexanoyl) analogs of PI (Figure 1), which exist as monomers at the concentrations used for kinetic measurements.5
Figure 1.

Structures of PI, IcP and IP analogs used in this work.
In contrast to the reaction with the natural PI substrate, where the cyclic intermediate is not observable, the cleavage of (Rp+Sp)-DHPsI proceeded with the release of the cyclic phosphorothioate (Figure 2, δ31P = 72.4 ppm). The intermediate was generated only from Rp-DHPsI, and was identified as myo-inositol cis(2-OH,S)-1,6-cyclic phosphorothioate (cis-1,6-IcPs), previously obtained by chemical cyclization of (Sp)-4-nitrophenyl inositol phosphorothioate.6 The cyclic intermediate was gradually hydrolyzed to inositol 1-phosphorothioate (δ31P = 44.1 ppm, I-1-Ps); the identity of the latter was established by comparison of its 1H NMR spectrum with that of the genuine I-1-Ps generated by Bacillus PI-PLC. In agreement with our previous observations,4 the cleavage of Sp-DHPsI was extremely slow (Table 1). In contrast, both Rp and Sp isomers of phosphorodithioate analog, DHsPsI, were readily cleaved, with Sp-isomer being hydrolyzed only ca. 6-times more slowly than Rp (Figure 2 and Table 1). The low Rp/Sp stereoselectivity of saPLC1 in this case is due to a very large increase (4.8×106-fold) in the rate of the cleavage of the Sp-isomer upon introduction of the bridging sulfur (Sp-DHPsI→Sp-DHsPsI). On the other hand, the rates of the cleavage of two other substrates in which pro-S oxygen is left unaltered (Rp-DHPsI and DHPI), were affected only to a small extent by the analogous modification of the bridging position (0.6 and 1.6-fold, respectively, Table 1).
Figure 2.

31P NMR time courses of the cleavage of (Rp+Sp)-DHPsI (A) and (Rp+Sp)-DHsPsI (B) by saPLC1.
Table 1.
Kinetic parameters for the cleavage of PI and its analogs by saPLC1.
| Substrate | Vmax μmol mg-1 min-1 | kO/kS (nonbridging) | kO/kS (bridging) |
|---|---|---|---|
| DHPI | 46.7±1.4 | ||
| DHsPI | 28.7±3.9 | 1.6±0.27 | |
| Rp-DHPsI | 1.21±0.07 | 38.6±3.5 | |
| Rp-DHsPsI | 1.87±0.08 | 15.3±2.7 | 0.65±0.06 |
| Sp-DHPsI | ≤1.41×10-6 | ≥3.0×108 | |
| Sp-DHsPsI | 0.29±0.02 | 99±20 | ≥4.8×10-6 |
In addition to the dramatic increase in the rate of the cleavage of Sp-DHsPsI, this reaction also afforded a product (δ31P = 45.9 ppm) distinct from I-1-Ps. 1H NMR of the isolated product suggested it was inositol 6-phosphorothioate. This conclusion was confirmed by the synthesis of I-6-Ps from myo-inositol, and comparison of its 1H and 31P NMR data with those of the enzyme product.
Our results provide the following new insights into the catalytic mechanism of saPLC1: (A) The release of 1,6-IcPs intermediate during the cleavage of Rp-DHPsI is reminiscent of that in the reaction of H16A mutant of saPLC1 with PI substrate, where formation of 1,6-IcP was detected.2 Both results suggest collectively that interaction of His16 with the pro-R oxygen atom is important for the retention of 1,6-IcP in the active site (Scheme 2). Mutation of His16, or introduction of sulfur at the pro-R position, abolishes this interaction, allowing the cyclic phosphate to escape from the active site. (B) The very high Sp-nonbridging thio-effect7 (≥3.0×108 ) is comparable to that observed previously (1.6×108).4 However, it was not clear previously whether it could arise from either a steric clash of the active site with a bulky sulfur atom in the transition state,8 or a loss of critical catalytic interactions upon O→S modification. The fact that the sterically larger Sp-DHsPsI has a higher turnover rate than Sp-DHPsI indicates that the strong Sp thio-effect arises from a loss of a catalytic interaction, not a steric effect. (C) The very strong inverse bridging thio-effect and the fact that it is observed only for Sp-DHsPsI are intriguing. In general, the magnitude of the bridging thio-effect is determined by the balance of two factors: (i) the loss of general acid catalysis due to poor proton acceptor ability of a mercaptide leaving group, and (ii) greater stability of mercaptide vs alkoxide leaving groups (Lg). The relatively modest inverse effect (1.9×10-3 ) was previously observed for the H55A mutant of saPLC1.4 The much greater effect observed here for the WT enzyme is consistent with a change of mechanism in which the cleavage of Sp-DHsPsI proceeds via a transition state with higher negative charge on Lg. This shift to a more dissociative mechanism likely arises from the lack of stabilization of the negative charge on the nonbridging atom. (D) Even more interesting and unexpected is the formation of I-1-Ps and I-6-Ps by hydrolyses of the cis- and trans-1,6-IcPs, respectively. For hydrolysis of the cyclic phosphate, the replacement of the trans-oxygen of the phosphodiester by sulfur should bring about a very large detriment to catalysis, similar to that for Sp-DHPsI. Thus, a very large (105) nonbridging thio-effect was observed for the hydrolysis of cis-1,2-IcPs by Bacillus PLC.9 saPLC1, however, hydrolyzes trans-1,6-IcPs only one order of magnitude more slowly than cis-1,6-IcPs. To explain this, it is necessary to note that 1,6-IcPs is structurally close to C2-symmetrical scyllo-inositol cyclic phosphate (scyllo-IcP), in which the nonbridging oxygens are homotopic (Scheme 2). Thus, trans-1,6-IcPs could bind in a rotated orientation in which the position of its nonbridging oxygen atom is the same as that in the cis-isomer. The consequence of this rotated binding mode is the change of the leaving group from 6-OH to 1-OH, resulting in the formation of I-6-Ps.
Scheme 2.

Explanation of formation of I-6-Ps from Sp-DHsPsI.
In summary, saPLC1 constitutes a unique enzyme showing an unprecedented strength of interactions as manifested by the highest bridging and nonbridging thio effects ever reported. While the role of saPLC1 in Streptomyces is unknown, this enzyme is capable of cleaving a number of structural analogs of PI, such as scyllo- and L-chiro-PI, as well as 2-O-glycosylated PIs (Bai & Bruzik, unpublished). The latter have been identified as constituents of cell membranes in bacteria, protozoa, fungi and plants.10 Investigation of the range of structures of saPLC1 natural substrates is underway.
Supplementary Material
Acknowledgments
This work was supported by the grant from NIGMS GM 57568.
Footnotes
References
- 1.Iwasaki Y, Tsubouchi Y, Ichihashi A, Nakano H, Kobayashi T, Ikezawa H, Yamane T. Biochim Biophys Acta. 1998;1391:52. doi: 10.1016/s0005-2760(97)00191-4. [DOI] [PubMed] [Google Scholar]
- 2.Bai C, Zhao L, Rebecchi M, Tsai MD, Bruzik KS. J Am Chem Soc. 2009;131:8362. doi: 10.1021/ja902326u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heinz DW, Essen LO, Williams RL. J Mol Biol. 1998;275:635. doi: 10.1006/jmbi.1997.1490. [DOI] [PubMed] [Google Scholar]
- 4.Zhao L, Liu Y, Bruzik KS, Tsai MD. J Am Chem Soc. 2003;125:22. doi: 10.1021/ja029019n. [DOI] [PubMed] [Google Scholar]
- 5.Liu Y, Mihai C, Kubiak RJ, Rebecchi M, Bruzik KS. ChemBiochem. 2007;8:1430. doi: 10.1002/cbic.200700061. [DOI] [PubMed] [Google Scholar]
- 6.Kubiak RJ, Bruzik KS. J Am Chem Soc. 2001;123:1760. doi: 10.1021/ja003675a. [DOI] [PubMed] [Google Scholar]
- 7.Stivers JT, Nagarajan R. Chem Rev. 2006;106:3443. doi: 10.1021/cr050317n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang YL, Hollfelder F, Gordon SJ, Chen L, Keng YF, Wu L, Herschlag D, Zhang ZY. Biochemistry. 1999;38:12111. doi: 10.1021/bi990836i. [DOI] [PubMed] [Google Scholar]
- 9.Hondal RJ, Zhao Z, Riddle SR, Kravchuk AV, Liao H, Bruzik KS, Tsai MD. J Am Chem Soc. 1997;119:9933. [Google Scholar]
- 10.Suzuki E, Tanaka AK, Toledo MS, Levery SB, Straus AH, Takahashi HK. Biochim Biophys Acta. 2008;1780:362. doi: 10.1016/j.bbagen.2007.09.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.