

SUMMARY
The epidermal growth factor receptor (EGFR) is implicated in human cancers and is the target of several classes of therapeutic agents, including antibody-based drugs. Here, we describe X-ray crystal structures of the extracellular region of EGFR in complex with three inhibitory VHH domains or nanobodies. VHH domains, the smallest natural antigen-binding modules, are readily engineered for diagnostic and therapeutic applications. All three VHH domains prevent ligand-induced EGFR activation, but use two distinct mechanisms. 7D12 sterically blocks ligand binding to EGFR in a manner similar to cetuximab. EgA1 and 9G8 bind an epitope near the EGFR domain II/III junction preventing receptor conformational changes required for high-affinity ligand binding and dimerization. This epitope is accessible to the convex VHH paratope, but inaccessible to the flatter paratope of monoclonal antibodies. Appreciating the modes of binding and inhibition of these VHH domains will aid in their development for tumor imaging and/or cancer therapy.
INTRODUCTION
Aberrant activation of the epidermal growth factor receptor (EGFR) is implicated in a number of human cancers, including colorectal, lung, brain, and head and neck tumors (Baselga and Arteaga, 2005; Gullick, 1991; Huang et al., 2009). It is well established that antibody binding to the extracellular region of EGFR can inhibit ligand-induced receptor activation and tumor growth (Gill et al., 1984; Sato et al., 1983). Several antibodies with these properties, including cetuximab/Erbitux®, are in current use or development in the clinic (Schmitz and Ferguson, 2009; You and Chen, 2011; Zhang et al., 2007).
Whereas antibodies that bind EGFR and other targets have shown promise in the clinic, there are impediments to their effective application and future development (Beck et al., 2010). The large size of monoclonal antibodies (mAbs) limits tumor penetration, restricting their effectiveness, and generation of new or modified mAbs is costly and laborious. Both problems can be mitigated by exploiting heavy chain only antibodies (HCAbs) from camelids (Hamers-Casterman et al., 1993; Muyldermans et al., 1994). Whereas the antigen recognition region in conventional antibodies comprises the variable regions of both the heavy and the light chains (VH and VL respectively), the antigen recognition region of HCAbs comprises a single variable domain, referred to as a VHH domain or nanobody. This single Ig domain is stable and can be generated rapidly and cheaply with simple expression systems (Harmsen and De Haard, 2007). Single VHH domains can be powerful diagnostic imaging tools, and are being developed for a range of research applications (Steyaert and Kobilka, 2011; Vaneycken et al., 2011). For therapeutic use, VHH domains (monomeric or multivalent) can be modified to extend serum half-life and/or functionality (Saerens et al., 2008).
The clinical success of EGFR-targeted mAbs has prompted significant interest in developing VHH domains that bind to and inhibit this receptor. Several EGFR-specific VHH domains have been reported (Roovers et al., 2007; Roovers et al., 2011) that have the potential to reproduce the clinical efficacy of mAbs such as cetuximab in an agent that is more stable and far less costly to produce. Moreover, potent multivalent VHH molecules can be generated that bind a number of targets (Emmerson et al., 2011; Jahnichen et al., 2010; Roovers et al., 2011), offering the potential to engineer multivalent agents that combine cetuximab-like EGFR inhibition with other modes of binding to EGFR or to other cancer targets. Fusing the targeted VHH domain (or domains) to one that recognizes serum albumin, can also dramatically increase serum half-life (Tijink et al., 2008).
We previously described the structural basis of EGFR inhibition by Fab fragments from three different mAbs: cetuximab, necitumumab and matuzumab (Li et al., 2008; Li et al., 2005; Schmiedel et al., 2008). Each sterically blocks a large conformational transition from an unactivated or “tethered” extracellular EGFR configuration to one that is dimerization-competent. In the tethered configuration, two of the four domains in the EGFR extracellular region (domains II and IV) make intramolecular autoinhibitory contacts, occluding the dimerization interface and separating the two halves of the EGF binding site (in domains I and III). Ligand binding stabilizes a conformation in which domains I and III are brought close together and domain II/IV intramolecular interactions are broken (Burgess et al., 2003). All three EGFR-targeted mAbs bind to domain III (Schmitz and Ferguson, 2009). The epitopes of cetuximab and necitumumab overlap with the domain III ligand binding region, whereas the matuzumab epitope does not. Cetuximab and necitumumab inhibit EGFR by directly interfering with ligand binding and blocking the activating conformational transition (Li et al., 2008; Li et al., 2005). Matuzumab inhibits EGFR exclusively by preventing the activating conformational transition. (Schmiedel et al., 2008)
In this report, we describe the structural basis for EGFR inhibition by three VHH domains. In multivalent formats, each of these VHH domains block ligand induced EGFR activation and cellular proliferation (Roovers et al., 2007; Roovers et al., 2011). Our structural analysis reveals modes of conformational constraint of EGFR by these VHH domains that have not been seen with inhibitory mAbs. The three VHH domains were isolated from an “immune” phage library generated from lymphocytes of Llama glama that had been immunized with A431 epidermoid carcinoma cells and A431 membrane preparations (Gainkam et al., 2008; Hofman et al., 2008; Roovers et al., 2007). One, VHH domain (7D12), was selected for its ability to compete with cetuximab for EGFR binding (Roovers et al., 2011). We show how the much smaller VHH domain can block both cetuximab and ligand binding. The other two VHH domains (EgA1 and 9G8) arose from a screen for molecules that inhibit ligand binding to EGFR (Hofman et al., 2008; Roovers et al., 2007) – the same strategy used to select the originator of cetuximab from a panel of mouse monoclonal antibodies (Sato et al., 1983). Interestingly, EgA1 and 9G8 do not compete with cetuximab for binding to EGFR (Roovers et al., 2011). Instead, these VHH domains bind to an epitope that is inaccessible to cetuximab and that undergoes large conformational changes during EGFR activation – sterically inhibiting the receptor.
RESULTS
VHH domain binding to the EGFR extracellular region
We first determined the affinity constants for interaction of each VHH domain with the EGFR extracellular region using surface plasmon resonance (SPR). Soluble EGFR extracellular region (sEGFR; amino acids 1-618 of the mature protein (Ferguson et al., 2000)) was passed over sensor chips to which VHH domains 7D12, EgA1, or 9G8 had been immobilized by amine-coupling. The equilibrium SPR response was measured over a range of sEGFR concentrations, and the resulting data were fit to a single site Langmuir binding equation. To evaluate possible bias due to covalent immobilization of VHH domains on the sensor surface, we also conducted binding assays in which biotinylated VHH domains were bound to streptavidin-coated sensor chips. Similar binding constants were determined by both methods (Table 1).
Table 1.
Equilibrium binding constants of sEGFR binding to inhibitory VHH domains
| Immobilized VHH | KD value (nM) for binding of indicated analyte | |||
|---|---|---|---|---|
| SEGFR1 | sEGFR501 | sEGFRd3 | SEGFRvIII | |
| 7D12 | 219 ± 20 (279 ± 19) | 143 ± 18 | 47 ± 3.6 | 263 ± 33 |
| EgA1 | 276 ± 7.0 (238 ± 42) | 356 ± 19 | > 2,500 | 822 ± 57 |
| 9G8 | 166 ± 1.2 (263 ± 76) | 317 ± 14 | > 5,000 | 525 ± 51 |
Numbers in brackets are for binding to exogenously biotinylated VHH immobilized to streptavidin coated SA sensor chips. All other KD values were determined for binding to VHH-AVIs that were amine-coupled to CM5 sensor chips.
The dissociation constants (KD) values for binding of sEGFR to immobilized VHH domains were between 166 and 276nM (Table 1). These affinities are 50 – 100 fold weaker than those for sEGFR binding to the structurally characterized Fab fragments of cetuximab (2.3±0.5nM) (Li et al., 2005) and of necitumumab (3.3±0.5nM) (Li et al., 2008), but are similar to those reported for the matuzumab Fab fragment (113±25nM) (Schmiedel et al., 2008). The apparent KD value for the binding of 7D12 to cell surface EGFR has been reported in the low nanomolar range (Oliveira et al., 2012), suggesting that additional factors that are only manifest at the cell surface may contribute to binding of this VHH to EGFR. Similar observations were made for matuzumab (Schmiedel et al., 2008).
As an initial approach for assigning epitopes to domains in sEGFR, we measured VHH domain binding to sEGFR truncation variants. Deleting most of domain IV (with sEGFR501) has little effect on binding (< 2-fold) for all three VHH domains (Table 1). Studies with isolated domain III (sEGFRd3: amino acids 311-514) revealed a complete loss of EgA1 and 9G8 binding, whereas 7D12 binding was retained – and increased by approximately 4 fold (suggesting that its epitope lies entirely on domain III). Interestingly, all three VHH domains bound to the truncated extracellular region of the oncogenic EGFR variant III (sEGFRvIII), which lacks domain I and much of domain II, but retains residues 273-311 of domain II (Sugawa et al., 1990; Wong et al., 1992). The fact that EgA1 and 9G8 bind sEGFRvIII but not sEGFRd3 suggests that a significant part of their epitopes may lie in the C-terminal part of domain II.
Antibody cross-competition
To compare the location of the epitopes for the three VHH domains with those of cetuximab and mAb425 (the murine version of matuzumab) and the EGF binding site, we used SPR-based competition assays. Fab fragments from cetuximab (Fab225) and mAb425 (Fab425), and EGF were amine coupled to CM5 sensor chips. The equilibrium SPR responses obtained for injections of 100 nM sEGFR with and without 5 μM of each VHH were measured (Figure 1A). All three VHH domains dramatically reduced EGF binding by sEGFR, which could arise through direct competition for the ligand binding site, and/or indirect conformational effects. VHH 7D12 reduces the SPR response to a level similar to that observed for competition with cetuximab, as expected, whereas EgA1 and 9G8 compete more effectively than mAb 425 for sEGFR binding to EGF. Only 7D12 reduces sEGFR binding to FabC225, suggesting that its binding site on domain III may overlap with the cetuximab epitope. VHH domains 9G8 and EgA1 both abolished sEGFR binding to immobilized Fab425. Addition of 9G8 and EgA1 to sEGFR enhanced the SPR response for immobilized FabC225, suggesting that these VHH:sEGFR complexes can bind to FabC225. These data confirm and extend our previous results using phage-displayed versions of these VHH domains to compete with mAbs for binding to an immobilized Fc fusion of sEGFR (Roovers et al., 2011).
Figure 1. Cross competition of VHH, Fab and EGF binding to sEGFR.
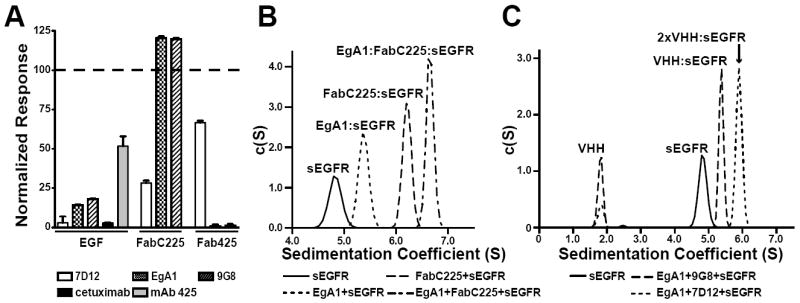
A. BIAcore analysis of the effect of added competitor upon binding of sEGFR to immobilized EGF or Fab. For CM5 sensor chips to which EGF, FabC225 or Fab425 had been amine coupled, the SPR responses for 100 nM sEGFR plus 5 μM VHH (7D12, EgA1 or 9G8) or 10 μM mAb (cetuximab or mAb 425) are shown, normalized to the SPR response for 100 nM sEGFR alone. Error bars indicate the standard deviation on at least three independent measurements. B. Sample of 5 μM sEGFR alone and mixtures of 5 μM sEGFR with (i) 5 μM EgA1, (ii) 5 μM FabC225, or (iii) 5 μM EgA1 plus 5 μM FabC225 were subject to velocity ultracentrifugation. Sedimentation velocity c(S) species analysis shows that sEGFR forms 1:1 complexes with EgA1 (5.4 S) and FabC225 (6.2S), and a ternary EgA1:FabC225:sEGFR (6.6S) when VHH and Fab are added. C. Similar velocity centrifugation shows a ternary 2xVHH:sEGFR complex (5.9S) for mixtures of sEGFR with EgA1 and 7D12, whereas only 1:1 VHH:sEGFR complex (5.4S) is seen for samples containing sEGFR, EgA1 and 9G8. See also Figure S1.
To further investigate the simultaneous binding of EgA1 and FabC225 to sEGFR we used sedimentation velocity analytical ultracentrifugation (SV-AUC). Whereas sEGFR alone (at 5 μM) sediments 4.8S, adding 5 μM EgA1 or 5 μM FabC225 yields species that sediment at 5.4S (EgA1:sEGFR) or 6.2S (FabC225:sEGFR) respectively (Figure 1B) – consistent with the formation of 1:1 complexes. When both EgA1 and FabC225 are added, a larger species (6.6S) consistent with a trimolecular complex is seen. By contrast, binding of FabC225 and 7D12 are mutually exclusive (not shown). Similar experiments reveal that 7D12 and EgA1 can both bind to the same sEGFR molecule to form a ternary EgA1:7D12:sEGFR complex of 5.9S (Figure 1C). When EgA1 and 7D12 are linked with a 10 amino acid glycine/serine linker (7D12-EgA1) formation of a 1:2 7D12-EgA1:sEGFR complex of 8.3S is favored (Figure S1). This suggests that the increased potency of a 7D12-EgA1-like biparatopic inhibitor CONAN-1 (Roovers et al., 2011) is not due to the simultaneous binding of both VHH modules of the biparatopic inhibitor to a single EGFR molecule.
The VHH 7D12 epitope on domain III
To visualize the molecular details of 7D12 binding to EGFR, we determined the crystal structure of 7D12 bound to sEGFRd3. The 7D12:sEGFRd3 complex crystallized in two distinct conditions: crystals grown at pH 6.0 diffracted to 2.9Å resolution, and those grown at pH 3.5 diffracted to 2.7Å resolution (Table 2). Both structures were solved using molecular replacement (MR) in PHASER (McCoy et al., 2007), using sEGFRd3 (PDB ID 3B2U) and the framework region of VHH EgA1 (see below) as search models. Models were rebuilt in Coot (Emsley and Cowtan, 2004), and refined using REFMAC (CCP4, 1994), CNS (Brunger et al., 1998) and PHENIX (Adams et al., 2010)h. The structures from the two crystal forms superimpose with RMSD < 1.0Å, and the contacts stabilizing the interaction are identical in both structures.
Table 2.
Crystallographic Statistics
| Data Collection Statisticsa | EgA1 | 7D12:sEGFRd3 (pH 6.0) | 7D12:sEGFRd3 (pH 3.5) | EgA1:FabC225:sEGFR | 9G8:FabC225:sEGFR |
|---|---|---|---|---|---|
|
| |||||
| Space group | P43 | P6122 | P212121 | P21 | P21 |
|
| |||||
| Cell dimensions | a, b = 52.7 Å, c = 62.5 Å | a, b = 148.0 Å, c = 82.5 Å | a = 78.7 Å, b = 147.2 Å, c = 254.8 Å | a = 66.2 Å, b = 96.3 Å, c = 128.1 Å, β = 100.7° | a = 66.4 Å, b = 95.8 Å, c =129.5 Å, β = 99.9° |
|
| |||||
| X-ray source | APS 23-ID-B | APS 23-ID-B | APS 23-ID-B | CHESS F1 | APS 23-ID-B |
|
| |||||
| Wavelength (Å) | 0.980 | 1.033 | 0.980 | 0.918 | 0.980 |
|
| |||||
| Resolution limit (Å) | 1.55 | 2.85 | 2.65 | 3.05 | 2.80 |
|
| |||||
| Measured / Unique | 107,430 / 24,694 | 136,787 / 22,976 | 568,198 / 83,116 | 69,621 / 29,807 | 138,909 / 37,840 |
|
| |||||
| Fold Redundancy | 4.4 | 6.0 | 6.8 | 2.3 | 1.9 |
|
| |||||
| Completeness (%) | 99.2 (94.5) | 97.0 (73.4) | 96.6 (75.4) | 98.8 (99.6) | 99.0 (92.4) |
|
| |||||
| Rsymb | 0.037 (0.283) | 0.117 (0.574) | 0.108 (0.614) | 0.099 (0.463) | 0.096 (0.413) |
|
| |||||
| <I/σ> | 47.5 (4.4) | 21.9 (2.0) | 20.7 (2.0) | 10.7 (2.2) | 14.8 (2.3) |
|
| |||||
| Refinement Statistics | |||||
|
| |||||
| Resolution limits (Å) | 27 – 1.55 | 41.8 – 2.85 | 49.52 – 2.66 | 38.2-3.05 | 47.92 – 2.82 |
|
| |||||
| # reflections / # test set | 23,399/1,252 | 12,061/617 | 78,792/4,233 | 28,283/1,507 | 35,947/1,889 |
|
| |||||
| R factor (Rfree)c | 0.19 (0.21) | 0.22 (0.27) | 0.21 (0.24) | 0.22 (0.28) | 0.22 (0.26) |
|
| |||||
| Model VHH | 1-73,77-129 | 1-122 | 6 × 1-124d | 1-7, 20-127 | 2-11, 20-124 |
| sEGFR or sEGFRd3 | - | 307-511 | 6 × 307-503d | 4-100, 108-183, 208-604 | 4-132, 208-612 |
| FabC225 heavy chain | - | - | - | 1-135, 141-218 | 1-136, 141-218 |
| light chain | - | - | 1-211 | 1-211 | |
| Ions | 1 sulfate ion | 2 MES ions; 1 iodide ion, | - | - | - |
| Water molecules | 84 | 36 | 170 | 12 | 3 |
|
| |||||
| Total number of atoms | 1,074 | 2,527 | 14,923 | 7,952 | 7,866 |
|
| |||||
| Ramachandran: Favored | 96.9 | 96.3 | 96.4 | 91.4 | 92.1 |
|
| |||||
| Outliers (%) | 0 | 0.3 | 0.2 | 0.2 | 0.1 |
|
| |||||
| RMSD bond lengths (Å) | 0.007 | 0.005 | 0.005 | 0.006 | 0.006 |
|
| |||||
| RMSD bond angles (°) | 1.111 | 0.876 | 0.858 | 0.902 | 0.948 |
Numbers in parenthesis refer to last resolution shell.
Rsym = Σ|Ih-<Ih>|/ΣIh, where <Ih> = average intensity over symmetry equivalent measurements.
R factor = Σ|Fo-Fc|/ΣFo, where summation is over data used in the refinement; Rfree includes only 5% of the data excluded from the refinement.
Number of missing amino acids varies by chain.
The framework region of 7D12 has a typical VH Ig fold, aligning to the framework region of VHH cAb-Lys3 (Desmyter et al., 1996) with an RMSD of 0.52Å. The sEGFRd3 aligns with previously reported crystal structures of this domain (Ferguson et al., 2003; Garrett et al., 2002; Li et al., 2008; Li et al., 2005; Ogiso et al., 2002; Schmiedel et al., 2008) with RMSD < 1.0Å. 7D12 binds to a flat surface on domain III (Figure 2) that corresponds to the location of the epitope for cetuximab and of the domain III ligand binding site (Figure 3). As shown in Figure 2C, VHH CDR1 and CDR3 contact the first two turns of the domain III β-helix (amino acids 310–375); CDR2 makes no contacts with sEGFR. The complex buries about 700Å2 on each protein, 48% of which is hydrophobic in nature. The interface has a shape complementarity parameter (Lawrence and Colman, 1993) of 0.68, which is typical for antibody/antigen interfaces (Table S1).
Figure 2. The 7D12 binding site on domain III of sEGFR.
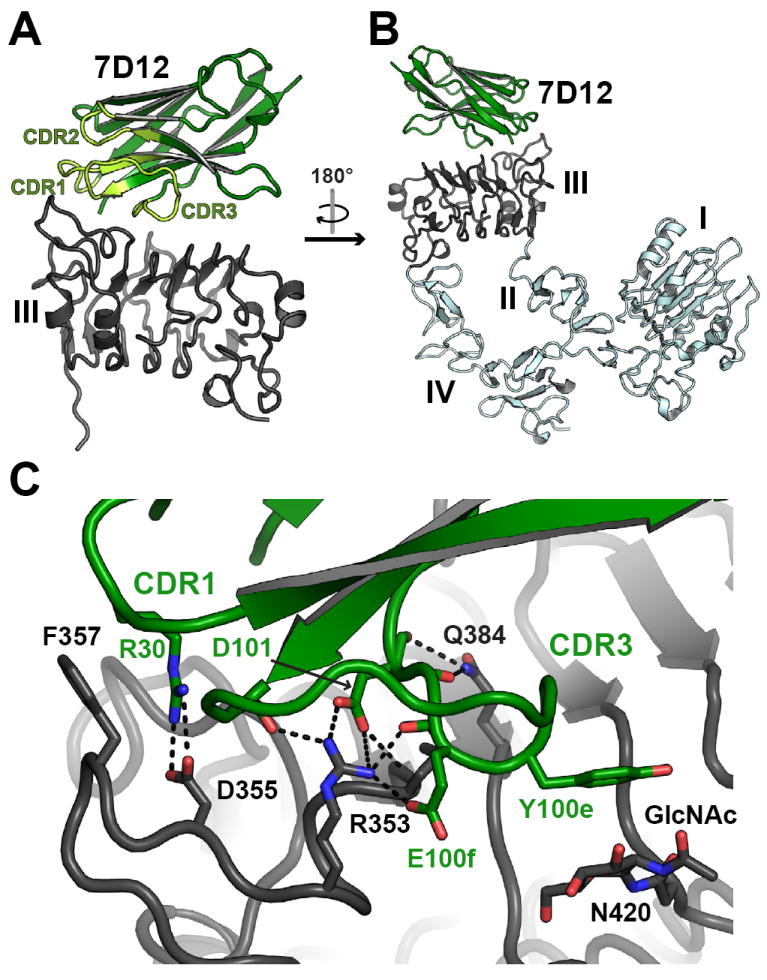
A. Cartoon is shown with 7D12 colored green and sEGFRd3 colored gray. CDRs are highlighted in light green and labeled. B. In this view the structure has been rotated approximately 180° about a vertical axis relative to A. The expected locations of domains I, II and IV of sEGFR are in light blue, based on the structure of tethered sEGFR in PDB ID 1NQL (Ferguson et al., 2003). C. View of the interface region between 7D12 and sEGFRd3 in a similar orientation to A. Side chains that participate in key interactions are shown as sticks, as is the sugar group on sEGFRd3. Predicted salt bridge (≤ 4.5 Å) or hydrogen bond (≤ 3.5 Å) interactions are indicated with dashed lines. Kabat numbering is used. See also Figure S2.
Figure 3. Comparison of the interactions of EGF, FabC225 and 7D12 with EGFR.
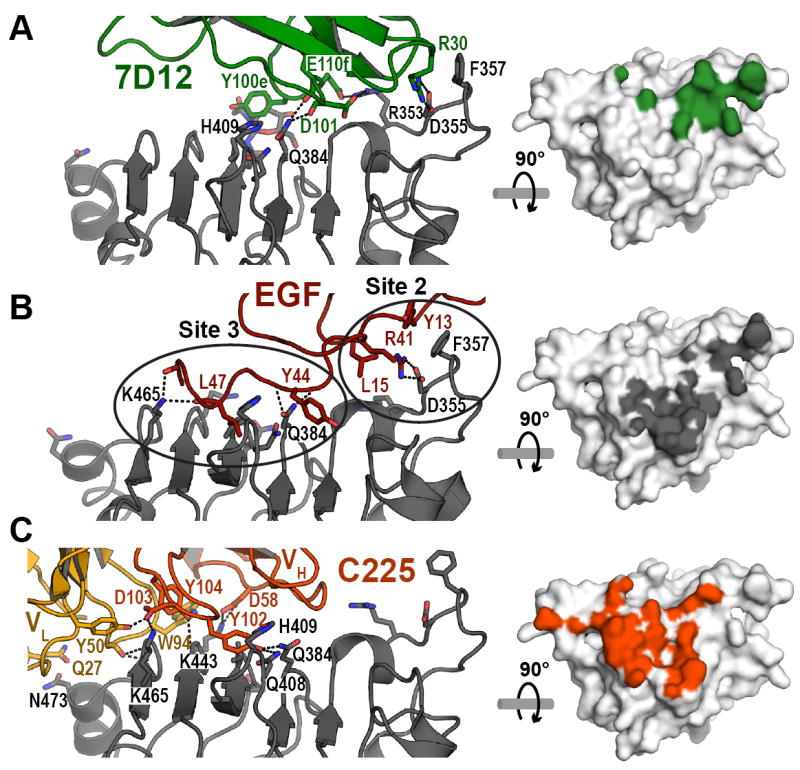
A. Cartoon of the 7D12:sEGFRd3 complex. Orientation is similar to Figure 2B. On the right hand panel the footprint (green) of 7D12 on the surface of domain III of EGFR highlights all atoms within 4Å of the bound 7D12. This view has been rotated by 90° about a horizontal axis so as to look down on to the flat domain III binding surface. B. The interaction of EGF with domain III. Domain III from PDB ID 1IVO was overlaid with domain III in the 7D12 complex. Orientations are as in A. The two groups of interactions between EGF and domain III (sites 2 and 3) as defined by Ogiso et al. (Ogiso et al., 2002) are ringed. The footprint for EGF is in grey. C. The interaction of FabC225 with domain III. Domain III from PDB ID 1YY9 was overlaid with domain III in the 7D12 complex. The footprint for FabC225 is in red. See also Table S1.
A cluster of polar and electrostatic interactions occurs in the center of the 7D12 epitope (Figure 2C). An arginine on CDR1 (R30) makes a salt bridge to EGFR D355 (a critical side-chain in EGF binding), while two acidic side chains (D101 and E100f) plus main chain carbonyls from CDR3 engage the side chains of R353 and Q384 on EGFR. These central polar interactions are flanked by apolar contacts. On one side the aliphatic portion of 7D12 R30 packs against EGFR F357, while on the other side Y100e from CDR3 packs against an N-acetylglucosamine (GlcNac) linked to EGFR N420 that is rotated ≈90° with respect to its typical position in EGFR domain III (Ferguson et al., 2003; Li et al., 2005; Ogiso et al., 2002) to contribute to this interaction.
In line with the observed importance of electrostatic interactions in the interaction of 7D12 with sEGFR, substitution of 7D12 R30 with alanine abolishes the interaction with sEGFR, and replacing D101 with alanine weakens binding to the receptor three-fold (Table 3).
Table 3.
Effects of epitope and paratope alterations on the equilibrium binding of sEGFR to immobilized VHHs.
| Immobilized VHH1 | KD value (nM) | |
|---|---|---|
| sEGFR | sEGFR (D355T/F357A) | |
| 7D12 | 219 ± 20 | > 5000 |
| 7D12 (R30A) | > 4,000 | ND |
| 7D12 (D101A) | 656 ± 22 | ND |
| EgA1 | 276 ± 7.0 | 342 ± 6.5 |
| EgA1 (R27A) | > 4,000 | ND |
| EgA1 (D101A) | > 3,500 | ND |
| 9G8 | 166 ± 1.2 | 303.5 ± 12 |
| 9G8 (E100iA) | > 2,500 | ND |
Kabat numbering is used for VHH residues.
Comparison of EGF, cetuximab, and 7D12 binding
The epitope for 7D12 partially overlaps both the ligand binding site and the cetuximab epitope on domain III (Figure 3). As previously described (Ogiso et al., 2002), EGF interactions with domain III fall in two groups (ringed in Figure 3B: sites 2 and 3 of Ogiso et al.(Ogiso et al., 2002)). Interactions of 7D12 with domain III are primarily centered on Site 2, mimicking (with R30 in CDR1) the salt bridge with EGFR D355 (and van der Waals contact with F357) made by R41 in EGF. 7D12 binding does not utilize the hydrophobic pocket on the domain III surface that lies at the center of Site 3 (Figure 3B) into which L47 of EGF projects. By contrast with the Site 2 focus of 7D12, C225 binding is largely centered on Site 3 (Figure 3C) – and C225 does not engage Site 2 on domain III (Li et al., 2005). The different foci of the interactions of 7D12, EGF, and C225 are most apparent when comparing the projection of the binding footprint of EGF with the 7D12 and cetuximab epitopes on domain III (Figure 3). The EGF-binding site encompasses Sites 2 and 3, whereas 7D12 engages Site 2, and cetuximab only Site 3. Consistent with the structural observations, substitution of F357 and D355 in EGFR (D355T/F357A) reduces 7D12 binding (Table 3) but not cetuximab binding (Li et al., 2005).
EgA1 and 9G8 bind to an epitope on domain III adjacent to domain II
We next sought to understand the structural basis of EGFR binding and inhibition by EgA1 and 9G8. Although EgA1 and sEGFR stably associate in solution (Figure 1B), we were unable to crystallize an EgA1:sEGFR complex. We were able to obtain crystals of the EgA1:FabC225:sEGFR ternary complex observed in our SV-AUC experiments in Figure 1B, which diffracted to 3.05Å resolution (Table 2) – using the Fab fragment to aid crystallization in common with many other studies (Koide, 2009). We exploited the same strategy to crystallize a 9G8:FabC225:sEGFR ternary complex. These crystals were of the same space group and diffracted to 2.8Å resolution. Both structures were solved by MR methods using the FabC225:sEGFR and free EgA1 structures as search models. The two VHH:FabC225:sEGFR structures align with an overall backbone RMSD of 0.54Å – revealing that EgA1 and 9G8 bind to the same region on sEGFR.
EgA1 and 9G8 (hereafter EgA1/9G8) bind to sEGFR in its tethered conformation, and the bound FabC225 makes identical contacts with EGFR domain III as seen in the previously reported FabC225:sEGFR complex structure (Li et al., 2005) (RMSD ≈ 0.5 Å). Crystal packing is mediated predominantly by contacts between the Fab and domain III of sEGFR from symmetry related molecules, suggesting a mechanism by which FabC225 promotes crystallization. FabC225 and sEGFR domain III are well ordered, as are portions of domains II, domain IV, and the VHH paratope region (Figure S3). However, EGFR domain I, the N-terminal portion of domain II, and the distal portion of the VHH are poorly resolved. Parts of these regions are absent or present as backbone atoms only in the refined model, and dihedral angles in domain I were restrained to those from PDB ID 1YY9.
EgA1/9G8 bind sEGFR in the cleft formed between domains II and III (Figure 4), consistent with their ability to bind sEGFRvIII but not sEGFRd3 in the binding studies described above. Interaction with sEGFR buries an average surface area on each partner of 701Å2 for EgA1 and 636Å2 for 9G8 (Table S1). In both cases approximately 100Å2 of surface area on the VHH is occluded from solvent by domain II – and the rest by domain III. As with 7D12, CDRs 1 and 3 of EgA1/9G8 contribute the key interactions, which are all made with domain III. CDR2 does not participate in the interaction. The CDR1 sequence is identical in EgA1 and 9G8 (Figure S2) and in each case R27 and Y32 participate in a cluster of interactions involving E431 and backbone moieties in EGFR. CDR3 of EgA1 and 9G8 share key sequence features, including Y96 (at the beginning), D101, and Y102 (at the end), which make similar interactions with EGFR domain III in each complex (Figure 4B, C). Y102 in each VHH domain engages E431 from EGFR – also engaged by CDR1. D101 and Y96 contribute to a second cluster of polar and electrostatic interactions with two arginines of EGFR (R403 and 405) and E400. The remainder of CDR3 is quite different for each VHH (Figure S2). In each case, a side chain from the VHH (D100 in EgA1 and E100i in 9G8) augments a network of salt bridges formed by charged side chains in EGFR – R403 to E376 to R310. This network is observed in all crystal structures of tethered sEGFR, but is disrupted in ligand bound structures due to reorientation of the domain II/III linker region that leads to displacement of the R310 side chain by over 7Å. The importance of R310 in sEGFR binding to EgA1/9G8 explains, in part, the lack of binding of these VHH domains to sEGFRd3. In this truncation variant the first 4 amino acids of mature sEGFR (LEEK) are fused to amino acid 311 of domain III. Not only does sEGFRd3 lack the R310 of domain III, but the two N-terminal glutamic acids that replace it may also disrupt the largely electrostatic interaction. In support of the importance of electrostatic contacts in the interaction of these VHH domains with sEGFR, we find that substitution with alanine of R27 or D101 in EgA1 or of E100i in 9G8 reduces binding by more than 20-fold (Table 3).
Figure 4. EgA1 and 9G8 have highly divergent CDR3s but bind to almost identical epitopes on sEGFR.
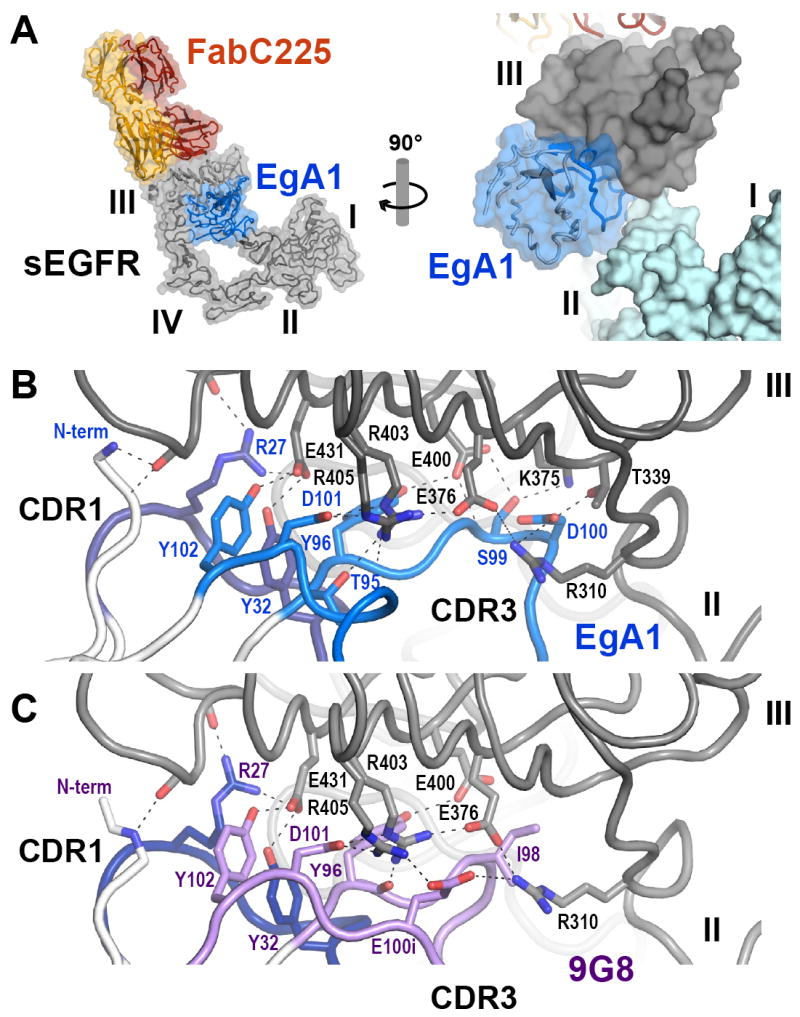
A. Overview of the EgA1:FabC225:sEGFR complex showing a cartoon plus transparent molecular surface. The sEGFR is in grey, FabC225 is colored orange (heavy chain) and yellow (light chain) and EgA1 is in blue. The right hand panel shows a close up view rotated approximately 90° about a vertical axis in which the location of EgA1 in a cleft between domains II and III can be appreciated. In this panel a solid surface has been rendered on sEGFR and domains I and II are colored light blue for contrast. B. Close up view of the interface between EgA1 and sEGFR in approximately the same orientation as in the right hand panel of part A. Only CDR1 (dark blue) and CDR3 (light blue) participate in the interaction. Key side chains are shown as sticks and predicted salt bridge (≤ 4.5 Å) or hydrogen bond (≤ 3.5 Å) interactions are indicated with dashed lines. C. The same view of the 9G8 complex is shown. CDR1 is identical and colored as in B. CDR3 is different (Figure S2) and colored light purple. See also Figure S3.
No well-ordered interactions between the VHH and sEGFR domain II proper are observed in either crystal structure. However, in each case the central region of CDR3 (near Y100d in EgA1 and N100b in 9G8) is close enough that direct or water mediated hydrogen bonds and van der Waals contacts may occur. Water mediated contacts are common in antibody:antigen interactions (Davies and Gerson, 1996) but not well resolved at the resolution of the VHH:FabC225:sEGFR structures.
The conformations of free and bound EgA1
We also determined the X-ray crystal structure of EgA1 in the absence of EGFR to a resolution of 1.55Å. The framework region of EgA1 is essentially identical in the bound and free structures, which superimpose with a backbone RMSD < 1.0 Å. However, differences are apparent in the paratope (Figure 5). In free EgA1, the N-terminal region projects between CDRs 1 and 3, such that the side chain of V2 lies in a hydrophobic crevice also occupied by Y32 (Figure 5B). In the bound structure, the N-terminal region is instead oriented away from the Ig core and makes polar contacts with domain III (Figure 4B). The side chain of Y32 also reorients in the bound structure to participate in interactions with sEGFR. The N-terminal part of CDR1, which does not participate in direct interactions with sEGFR, adopts a slightly different conformation in the bound structure (backbone RMSD with free VHH 1.98Å), and the orientation of the T28 and F29 side-chains switch direction such that in the bound structure F29 is flipped in towards the core of the protein, compensating in part for the reorientation of V2 and Y32 (Figure 5B). Less extensive rearrangements occur in CDRs 2 and 3. Of note however is a shift in the positions of two tyrosine side chains (Y100d and Y56; Figure 5C), which in the bound VHH structure adopt orientations that place them closer to domain II than they would be in their unbound orientations.
Figure 5. Conformational changes in EgA1 upon binding to sEGFR.
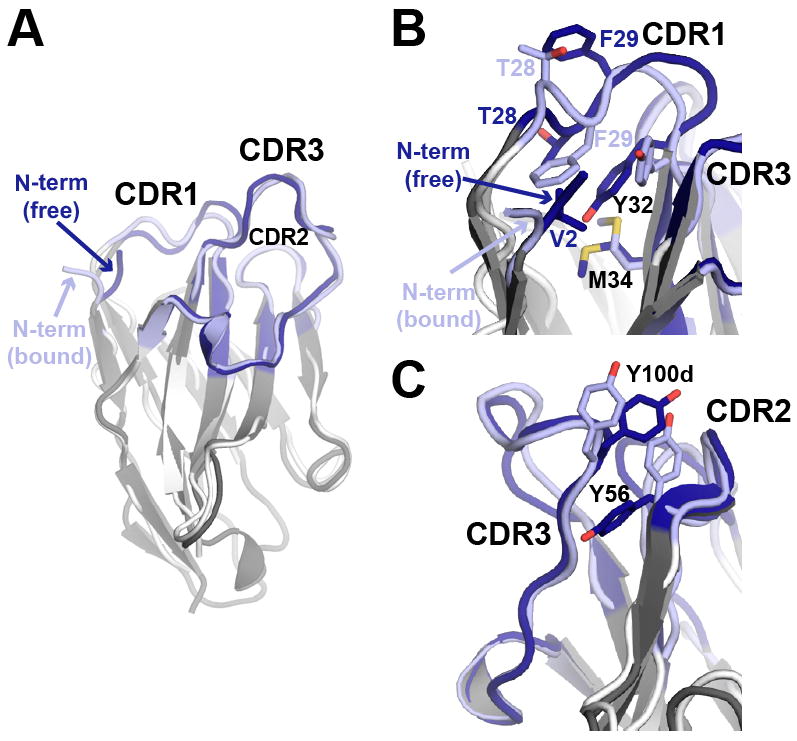
A. Superposition of free EgA1 (dark grey) and EgA1 bound to sEGFR (white), with CDRs in dark and light blue respectively. The N-terminus of free EgA1 packs against CDR1 and the hydrophobic core of the Ig fold, whereas in the bound structure this segment is oriented away from the VHH and makes (presumed) polar contacts with domain III. Detailed views of the differences in side chain orientations near the N-terminus (B) and in CDR2 and CDR3 (C). See also Figure S4
The conformation of sEGFR in the EgA1 and 9G8 complex
EGFR domains I and III in the EgA1/9G8 complex adopt identical conformations to those seen in all other crystal structures of sEGFR. The orientation and conformation of the first 4 disulfide-bonded modules of domain II of sEGFR in the VHH:FabC225:sEGFR ternary complex is also similar to that observed for all unliganded ErbB receptors (Alvarado et al., 2009; Bouyain et al., 2005; Liu et al., 2012) (Figure S4A). The same is true for the last disulfide-bonded module (m8) of domain II and the domain II/III linker region (Figure S4B). The intervening three disulfide bonded modules of domain II (m5-m7) are substantially altered in position, as a result of changes in the orientation of one module with respect to its neighboring module in the m5/m6, m6/m7 and m7/m8 connections. As shown in Figure S4C, this results in a bend in domain II that reorients domain I in the ternary VHH:FabC225:sEGFR with respect to domain III. This difference could be the result of steric effects of bound VHH, or may simply reflect inherent flexibility in sEGFR.
Although it is clear that domain II and IV are in close proximity in the EgA1 complex, the structure is too poorly ordered to determine whether specific domain II/IV contacts are maintained. The crystallographic data for the 9G8 complex yield interpretable electron density in the region of the EGFR domain II/IV tether interaction, and suggest that at least two of the four hydrogen bonds between domains II and IV are maintained in this complex (backbone carbonyl of Y251 to side chain of H566 sidechain, and between the side chains of Y246 and D563). Together, these observations argue that the binding of EgA1 and 9G8 does not substantially perturb the conformation of tethered sEGFR.
Comparison of EgA1, 9G8, and matuzumab binding to EGFR
The EgA1 (Figure 6A) and 9G8 (Figure 6B) epitopes are essentially identical. These VHH epitopes partly overlap that of the inhibitory antibody matuzumab (Figure 6C) (Schmiedel et al., 2008) and, like matuzumab, do not overlap with the EGF binding region of domain III (Figure 6D). Matuzumab binding is stabilized by extensive contacts to the domain III β-helix coil 449-463. This is on the periphery of the EgA1/9G8 epitope, with several main-chain hydrogen bonds predicted between the VHH N-terminal region and R27 (upper left hand corner of Figure 4B/C). Simultaneous binding of EgA1/9G8 and matuzumab would result in steric clash of the VHH and Fab domains, consistent with binding and competition experiments indicating that EgA1 and 9G8 compete with matuzumab for receptor binding (Figure 1 and (Roovers et al., 2011)). Aside from this small region of overlap, the matuzumab and EgA1/9G8 epitopes are quite distinct and share no specificity determining interactions. The VHH epitopes are located further towards the N-terminal end of domain III, and are further from the domain III ligand-binding site (Figure 6).
Figure 6. Comparison of the interactions of EgA1, 9G8, matuzumab and EGF with sEGFR.
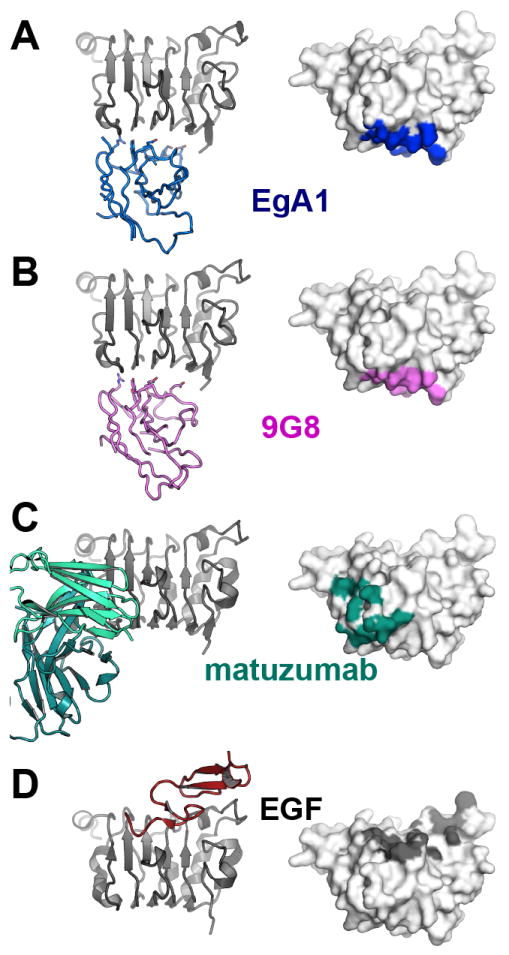
For each case the right hand panel shows a cartoon of just domain III of sEGFR (grey) with bound VHH, Fab or ligand. The left hand panel shows a surface representation of domain III in the same orientation with the footprint of VHH, Fab or ligand highlighted. A. EgA1 (blue); B. 9G8 (violet); C. matuzumab Fab (teal, PDB ID 3C09); D. EGF (dark red, PDB ID 1IVO). See also Figure S5.
DISCUSSION
Modes of inhibition of EGFR activation by VHH inhibitors
The VHH domains used in this study fall into two categories of inhibitory antibodies, one typified by cetuximab, the other by matuzumab. Like cetuximab, 7D12 is a ligand-competitive inhibitor. The 7D12 epitope overlaps the ligand-binding site on domain III of EGFR. In addition, as argued for cetuximab (Li et al., 2005), bound 7D12 would sterically prevent EGFR from adopting the extended conformation required for dimerization. By contrast, EgA1/9G8 do not directly occlude the ligand binding site on EGFR. Rather, as argued for matuzumab, inhibition is achieved by preventing the EGFR extracellular region from adopting the extended conformation that can dimerize. Like matuzumab, EgA1/9G8 impose a steric block on this conformational rearrangement (Schmiedel et al., 2008) (Figure S5). In addition, EgA1/9G8 appear to stabilize the tethered conformation of the receptor by binding at the junction between domains II and III, and interacting with key side chains in this region. The domain II/III junction acts as a hinge in the large scale domain rearrangement required to take the receptor from the tethered to the extended (dimerization competent) conformation, and is the site of the most dramatic local differences in backbone conformation between the unliganded and ligand bound structures of sEGFR. In tethered sEGFR, the side chain of R310, at the C-terminal end of domain II, projects towards domain III and makes an electrostatic interaction with the side chain of E376. This interaction is broken in ligand-bound structures. The altered main chain trajectory in the domain II/III interface region results in the R310 side chain projecting away from domain III. As shown in Figure 4, EgA1 and 9G8 interact with R310 and E376, effectively locking this region of EGFR into the conformation seen in tethered, inactive structures. This mode of inhibition may confer a therapeutic advantage to molecules such as EgA1/9G8 since binding, and hence inhibition, should not be impacted by the presence of excess ligand.
Comparison of VHH and Fab binding to EGFR
Single-domain antibody fragments have certain unique features that offer modes of interaction with antigens that are under exploited with conventional monoclonal antibodies (Muyldermans et al., 2001). Several such VHH specific binding features are observed in the interactions of EgA1/9G8 with sEGFR, and distinguish the modes of antigen binding by these VHH domains from those utilized by Fab fragments from cetuximab and matuzumab.
The single VHH domain of a heavy chain only camelid antibody functionally replaces the entire Fab fragment of a conventional antibody. To retain binding diversity, VHH domains exhibit greater variability in length and accessible conformations of their 3 CDRs compared to the 6 CDRs in an Fab fragment (Muyldermans et al., 2001). Crystal structures of VHH domains in complex with more than a dozen different protein antigens show a range of binding modes that utilize unique features of VHH domains, and explain how these smaller antigen recognition modules can maintain diverse specificity and high affinity. EgA1/9G8 use a binding mode – with a convex paratope – that has also been observed for several lysozyme specific VHH domains. Like EgA1/9G8 these lysozyme specific VHH domains have particularly long CDR3s that extend out from the VHH framework to recognize a concave epitope at the enzyme active site (De Genst et al., 2006; Desmyter et al., 1996). The VHH antigen recognition domain is uniquely suited to presentation of highly convex paratopes and this shape increases the surface available for interaction with antigen. The long CDR3 loops of EgA1 and 9G8 contribute to convex paratopes on these VHH domains that are ideally suited to bind at the EgA1/9G8 epitope, concave because it lies within a cleft at the domain II/III junction. The larger antigen-binding surface of an Fab is less able to access this epitope due in part to steric clash of the VL domain. The matuzumab epitope does partly overlap with that of EgA1/9G8, as expected since the mouse antecedent of matuzumab (mAb425) competes with EgA1/9G8 for binding to EGFR. However, the matuzumab epitope is displaced to the domain III side of the domain II/III junction. Bound matuzumab sterically prevents EGFR from adopting the extended conformation observed in the ligand bound structures, but likely permits EGFR to access a much greater range of conformations than are accessible to the EgA1/9G8:EGFR complex. EgA1/9G8 lock EGFR in the tethered, inactive conformation by binding at a key pivot point. It is also interesting to note that the interaction of matuzumab with EGFR utilizes a binding mode only accessible to a Fab/antigen interaction. A loop from domain III of sEGFR binds between the VH and VL domains of the Fab – a convex epitope interacting with a concave paratope. Other than a substantial difference in planarity of the interface for the VHH versus the Fab fragments, the general characteristics of the interfaces formed by the two classes of antibody are very similar (Table S1), as was reported in a study of VHH and Fab fragments binding to lysozyme (De Genst et al., 2006).
By contrast to EgA1/9G8, 7D12 interacts with a relatively flat epitope using only CDR1 and CDR3 in a manner reminiscent of the binding of several VHH domains to ribonuclease A (Koide et al., 2007). CDR3 folds back against the framework of the VHH domain and is stabilized by van der Waals contacts and hydrogen bonds (although not by a disulfide bond, as often occurs in camel HCAbs (Nguyen et al., 2001)). This creates a stable flat paratope that interacts with a region close to, although not entirely overlapping, the cetuximab epitope.
Implications for VHH domains as diagnostic and therapeutic agents
The structures presented here illustrate two important features of VHH domains that make them valuable additions to the drug discovery toolkit. For EgA1/9G8 the unique structure of the VHH domain allows recognition of an epitope that is inaccessible to a conventional mAb – generating an inhibitor with a new mode of EGFR inhibition. The structure of 7D12 bound to domain III of EGFR reveals how this smaller and readily engineered binding unit can mimic inhibitory features of the intact monoclonal antibody drug cetuximab. Multimerization of 7D12 with other VHH domains generates a potent EGFR inhibitor (Roovers et al., 2011). 7D12 is thus a cassette that can be used to combine cetuximab-like inhibition with modules of synergistic and/or complementary inhibitory properties. Availability of a repertoire of similarly well-characterized inhibitory VHH domains could facilitate the generation of multivalent/multispecific drugs that can ultimately be “personalized” for optimal effect against a patient’s tumor.
EXPERIMENTAL PROCEDURES
Protein production
sEGFR (amino acids [aa] 1-618 of mature EGFR), sEGFR501 (aa 1-501) and sEGFRd3 (aa 1-4 followed by 311-514) were produced and purified as described (Ferguson et al., 2000; Li et al., 2005). To generate sEGFRvIII codons 6–273 of wild-type sEGFR were replaced with a single glycine codon using standard molecular biology methods, sEGFRvIII expressed and purified as for sEGFR. DNA coding VHH fragments 7D12, EgA1, and 9G8 were cloned into pET-22b (EMD). For crystallization proteins included a C-terminal hexa-histidine (H6) tag (VHH-H6), whereas for binding experiments a C-terminal E. coli BirA biotinylation sequence (GLNDIFEAQKIEWH) (Beckett et al., 1999) followed by a H6 tag (VHH-AVI) was included. An upstream pelB leader sequence directed periplasmic expression. VHHs were overexpressed in LB media by lactose auto-induction (Studier, 2005). VHHs were extracted by freeze/thaw in PBS (25mM Na2HPO4/NaH2PO4, 150mM NaCl, 10% glycerol, pH 8.0). Lysates were applied to Ni-NTA resin (Qiagen) and eluted with an imidazole gradient. Fractions containing VHHs were concentrated and further purified by size exclusion chromatography (Superose 12, GE Healthcare) using a buffer of 25 mM HEPES, 150 mM NaCl, pH 8.0. sEGFR and VHH variants incorporating site directed alterations were generated by standard PCR methods. VHH:sEGFR complexes for crystallization were purified by size exclusion chromatography. mAb 425 was a generous gift from Prof. Ulrich Rodeck (Jefferson University). The Fab fragments were prepared by papain cleavage and protein A purification using the Pierce Fab Preparation Kit (Thermo Scientific) and used without further purification. EGF was purchased from Chemicon, Inc.
Crystallization and Data Collection
Crystals were grown using the hanging drop vapor diffusion method at 20°C. In each case protein was mixed with crystallization buffer and equilibrated against a reservoir of this same buffer. The following conditions were used: (i) EgA1 alone, 0.5μl of 18mg/ml EgA1 plus 1.0μl of 30% PEG3350, 0.2M (NH4)2SO4, 0.1M MES, pH 6.0; (ii) 7D12:sEGFRd3 at pH 6, 0.5μl 10mg/ml complex plus 0.5μl 22.5% PEG3350, 50mM KI, 0.1M MES, pH 6.0; (iii) 7D12:sEGFRd3 at pH 3, 1.0μl 10mg/ml complex plus 1.5μl 22.5% PEG3350, 0.1M sodium citrate, pH 3.5; (iv) EgA1:FabC225:sEGFR, 0.5μl of 11mg/ml complex plus 0.5μl 17.5% PEG3350, 1.5M NaCl, 5% glycerol, 0.1M MES, pH 6.5; (v) 9G8:FabC225:sEGFR, 0.5μl of 7mg/ml protein and 2.5μl 10% PEG3350, 0.1M HEPES, pH 7.0). All crystals were flash frozen in liquid nitrogen from cryostabilizers. EgA1 were flash frozen directly from the crystallization drop. Other crystals were transfer to crystallization buffer supplemented with (ii) 12.5% glycerol, (iii) 5% ethylene glycol (iv) 2.5% PEG3350 and 7.5% glycerol, (v) 5% PEG3350 and 15% ethylene glycol. X-ray diffraction data were collected at GM/CA @ APS beamline ID 23-B (EgA1, 7D12:sEGFRd3, and 9G8:FabC225:sEGFR) and at CHESS beamline F1 (EgA1:FabC225:sEGFR).
Structure determination and refinement
Data were processed with HKL-2000 (Otwinowski and Minor, 1997). All structures were solved by molecular replacement (MR) in the program PHASER (McCoy et al., 2007). The framework region of a hapten binding VHH (PDB ID 1I3V) was used as the search model for the structure of EgA1 alone. EGFR domain III from PDB ID 3B2U (chain A) and the framework region of EgA1 were used as independent search models to solve 7D12:sEGFRd3 at pH 6.0; the resulting complex served as the search model to identify the six copies of 7D12:sEGFRd3 in the pH 3.5 data. For the EgA1:FabC225:sEGFR complex, EgA1, the C225 Fv region, the C225 CH2/CL region, EGFR domains I-II, and EGFR domains III-IV (PDB ID 1YY9) were used as independent search models. The EgA1:FabC225:sEGFR structure was used as the search model to solve the 9G8:FabC225:sEGFR complex. Protein models were built in COOT (Emsley and Cowtan, 2004), and refined using REFMAC (CCP4, 1994), CNS (Brunger et al., 1998) and PHENIX (Adams et al., 2010). The presence of an iodide ion in the pH 6.0 7D12:sEGFRd3 model was confirmed by inspection of an anomalous difference map. In the EgA1/9G8 complexes, FabC225 and EGFR amino acids 208-605 are well ordered, as is most of the VHH. EGFR domain I, the N-terminal portion of domain II, and the distal portion of the VHH are poorly ordered. The dihedral angles in domain I were restrained to those in PDB ID 1YY9. Data collection and refinement statistics are summarized in Table 2.
Structure analysis
Shape complementarity values were determined by the sc module in CCP4 (CCP4, 1994), excluding solvent molecules. Interactions between chains were identified by NCONT in CCP4, using a distance cutoff of 3.5 Å for hydrogen bonds and van der Waals contacts, and 4.5 Å for electrostatic interactions. Average excluded surface areas were calculated by CONTACT in CCP4. The fraction of buried hydrophobic surface area was determined by dividing the surface area of buried carbon atoms by the total buried surface area. Structural alignments and RMSD values were generated within the programs PYMOL or COOT, and reflect main chain atoms only.
Analytical ultracentrifugation
Sedimentation velocity analytical ultracentrifugation (SV AUC) experiments were conducted in a Beckman Optima XL-A instrument in an An-Ti 60 rotor at 20°C, at 35,000-50,000 rpm in 10mM HEPES, 150mM NaCl, pH 8.0. Sample absorbance was monitored at 280nm. Buffer density, buffer viscosity, and protein partial specific volume were estimated in the program SEDNTERP (Philo et al., 1996). Size distribution c(S) analysis was performed with the program SEDFIT (Schuck, 2000). Only c(S) fits producing low residuals with no systematic error are reported.
Surface plasmon resonance studies
SPR binding experiments were performed with a Biacore 3000 instrument at 25°C. Immobilized species were diluted to 50μg/ml (Fabs), 100μg/ml (VHHs), or 200μg/ml (EGF) in acidic buffer (10mM sodium acetate at pH 4.0 for Fab425 and EGF; pH 5.0 for VHHs; pH 5.5 for FabC225) and were amine coupled to activated CM5 sensor chips at 5 μl/min for 5–10 minutes. Binding of sEGFR to these surfaces was determined as described (Ferguson et al., 2000; Li et al., 2005). VHH-AVI constructs were exogenously biotinylated by BirA as described (Abbott and Beckett, 1993; Beckett et al., 1999) and immobilized to streptavidin coated SA sensor chips. Surfaces were regenerated between data points with 5 μl of 1M NaCl at low pH (2.5–5.5) to remove residual sEGFR. Multiple cycles of regeneration did not impair sEGFR binding. Data were analyzed using Prism 4 (GraphPad Software, Inc.).
Supplementary Material
Highlights.
Structures of three nanobodies/VHH domains bound to EGFR extracellular region.
Two distinct modes of VHH binding indicate two different mechanism of inhibition.
One mimics Erbitux/cetuximab mechanism with this smaller antigen-binding module.
Second exploits the convex VHH paratope to reach a key conformational epitope.
Acknowledgments
We thank Mark Lemmon and members of the Ferguson and Lemmon laboratories for critical comments on the manuscript. The work was supported by the NIH grant R01 CA 112552 to K.M.F. K.R.S. was supported in part by T32 GM08275 and by a U.S. Army Breast Cancer Research Program Predoctoral Fellowship (BC051591). R.C.R was supported by the Dutch Technology Foundation (STW, grant 10074). This work is based in part upon research conducted (i) at GM/CA @ APS, which has been funded by NCI (Y1-CO-1020) and NIGMS (Y1-GM-1104); APS was supported by U.S. DoE (contract DE-AC02-06CH11357), and (ii) at CHESS, which is supported by the NSF & NIH via NSF award DMR-0936384, and MacCHESS that is supported by NIH award GM103485.
Footnotes
ACCESSION NUMBERS
The coordinates and structure factors have been deposited in the Protein Data Bank with accession number as follows: (i) EgA1 – 4KRN, (ii) 7D12:sEGFRd3 at pH 6 – 4KRL, (iii) 7D12:sEGFRd3 at pH 3 – 4KRM, (iv) EgA1:FabC225:sEGFR – 4KRO, (v) 9G8:FabC225:sEGFR – 4KRP.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbott J, Beckett D. Cooperative binding of the Escherichia coli repressor of biotin biosynthesis to the biotin operator sequence. Biochemistry. 1993;32:9649–9656. doi: 10.1021/bi00088a017. [DOI] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. 2010;D66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarado D, Klein DE, Lemmon MA. ErbB2 resembles an autoinhibited invertebrate epidermal growth factor receptor. Nature. 2009;461:287–291. doi: 10.1038/nature08297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baselga J, Arteaga CL. Critical update and emerging trends in epidermal growth factor receptor targeting in cancer. J Clin Oncol. 2005;23:2445–2459. doi: 10.1200/JCO.2005.11.890. [DOI] [PubMed] [Google Scholar]
- Beck A, Wurch T, Bailly C, Corvaia N. Strategies and challenges for the next generation of therapeutic antibodies. Nat Rev Immunol. 2010;10:345–352. doi: 10.1038/nri2747. [DOI] [PubMed] [Google Scholar]
- Beckett D, Kovaleva E, Schatz PJ. A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Prot Sci. 1999;8:921–929. doi: 10.1110/ps.8.4.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouyain S, Longo PA, Li S, Ferguson KM, Leahy DJ. The extracellular region of ErbB4 adopts a tethered conformation in the absence of ligand. Proc Natl Acad Sci U S A. 2005;102:15024–15029. doi: 10.1073/pnas.0507591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Burgess AW, Cho HS, Eigenbrot C, Ferguson KM, Garrett TP, Leahy DJ, Lemmon MA, Sliwkowski MX, Ward CW, Yokoyama S. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol Cell. 2003;12:541–552. doi: 10.1016/s1097-2765(03)00350-2. [DOI] [PubMed] [Google Scholar]
- CCP4. The CCP4 Suite: Programs for Protein Crystallography. Acta Crystallogr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Davies DR, Gerson HC. Interactions of Protein Antigens with Antibodies. Proc Natl Acad Sci U S A. 1996;93:7–12. doi: 10.1073/pnas.93.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Genst E, Silence K, Decanniere K, Conrath K, Loris R, Kinne J, Muyldermans S, Wyns L. Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc Natl Acad Sci U S A. 2006;103:4586–4591. doi: 10.1073/pnas.0505379103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmyter A, Transue TR, Ghahroudi MA, Thi MH, Poortmans F, Hamers R, Muyldermans S, Wyns L. Crystal structure of a camel single-domain VH antibody fragment in complex with lysozyme. Nat Struct Biol. 1996;3:803–811. doi: 10.1038/nsb0996-803. [DOI] [PubMed] [Google Scholar]
- Emmerson CD, van der Vlist EJ, Braam MR, Vanlandschoot P, Merchiers P, de Haard HJ, Verrips CT, van Bergen En Henegouwen PM, Dolk E. Enhancement of Polymeric Immunoglobulin Receptor Transcytosis by Biparatopic VHH. PLoS One. 2011;6:e26299. doi: 10.1371/journal.pone.0026299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Ferguson KM, Berger MB, Mendrola JM, Cho HS, Leahy DJ, Lemmon MA. EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol Cell. 2003;11:507–517. doi: 10.1016/s1097-2765(03)00047-9. [DOI] [PubMed] [Google Scholar]
- Ferguson KM, Darling PJ, Mohan MJ, Macatee TL, Lemmon MA. Extracellular domains drive homo- but not hetero-dimerization of erbB receptors. EMBO J. 2000;19:4632–4643. doi: 10.1093/emboj/19.17.4632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainkam LO, Huang L, Caveliers V, Keyaerts M, Hernot S, Vaneycken I, Vanhove C, Revets H, De Baetselier P, Lahoutte T. Comparison of the biodistribution and tumor targeting of two 99mTc-labeled anti-EGFR nanobodies in mice, using pinhole SPECT/micro-CT. J Nucl Med. 2008;49:788–795. doi: 10.2967/jnumed.107.048538. [DOI] [PubMed] [Google Scholar]
- Garrett TP, McKern NM, Lou M, Elleman TC, Adams TE, Lovrecz GO, Zhu HJ, Walker F, Frenkel MJ, Hoyne PA, et al. Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha. Cell. 2002;110:763–773. doi: 10.1016/s0092-8674(02)00940-6. [DOI] [PubMed] [Google Scholar]
- Gill GN, Kawamoto T, Cochet C, Le A, Sato JD, Masui H, McLeod C, Mendelsohn J. Monoclonal anti-epidermal growth factor receptor antibodies which are inhibitors of epidermal growth factor binding and antagonists of epidermal growth factor binding and antagonists of epidermal growth factor-stimulated tyrosine protein kinase activity. J Biol Chem. 1984;259:7755–7760. [PubMed] [Google Scholar]
- Gullick WJ. Prevalence of aberrant expression of the epidermal growth factor receptor in human cancers. Br Med Bull. 1991;47:87–98. doi: 10.1093/oxfordjournals.bmb.a072464. [DOI] [PubMed] [Google Scholar]
- Hamers-Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hamers C, Songa EB, Bendahman N, Hamers R. Naturally occurring antibodies devoid of light chains. Nature. 1993;363:446–448. doi: 10.1038/363446a0. [DOI] [PubMed] [Google Scholar]
- Harmsen MM, De Haard HJ. Properties, production, and applications of camelid single-domain antibody fragments. Appl Microbiol Biotechnol. 2007;77:13–22. doi: 10.1007/s00253-007-1142-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofman EG, Ruonala MO, Bader AN, van den Heuvel D, Voortman J, Roovers RC, Verkleij AJ, Gerritsen HC, van Bergen En Henegouwen PM. EGF induces coalescence of different lipid rafts. J Cell Sci. 2008;121:2519–2528. doi: 10.1242/jcs.028753. [DOI] [PubMed] [Google Scholar]
- Huang PH, Xu AM, White FM. Oncogenic EGFR signaling networks in glioma. Sci Signal. 2009;2:re6. doi: 10.1126/scisignal.287re6. [DOI] [PubMed] [Google Scholar]
- Jahnichen S, Blanchetot C, Maussang D, Gonzalez-Pajuelo M, Chow KY, Bosch L, De Vrieze S, Serruys B, Ulrichts H, Vandevelde W, et al. CXCR4 nanobodies (VHH-based single variable domains) potently inhibit chemotaxis and HIV-1 replication and mobilize stem cells. Proc Natl Acad Sci U S A. 2010;107:20565–20570. doi: 10.1073/pnas.1012865107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koide A, Tereshko V, Uysal S, Margalef K, Kossiakoff AA, Koide S. Exploring the capacity of minimalist protein interfaces: interface energetics and affinity maturation to picomolar KD of a single-domain antibody with a flat paratope. J Mol Biol. 2007;373:941–953. doi: 10.1016/j.jmb.2007.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koide S. Engineering of recombinant crystallization chaperones. Curr Opin Struct Biol. 2009;19:449–457. doi: 10.1016/j.sbi.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MC, Colman PM. Shape complementarity at protein-/protein interfaces. J Mol Biol. 1993;234:946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- Li S, Kussie P, Ferguson KM. Structural basis for EGF receptor inhibition by the therapeutic antibody IMC-11F8. Structure. 2008;16:216–227. doi: 10.1016/j.str.2007.11.009. [DOI] [PubMed] [Google Scholar]
- Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Can Cell. 2005;7:301–311. doi: 10.1016/j.ccr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Liu P, Bouyain S, Eigenbrot C, Leahy DJ. The ErbB4 extracellular region retains a tethered-like conformation in the absence of the tether. Prot Sci. 2012;21:152–155. doi: 10.1002/pro.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muyldermans S, Atarhouch T, Saldanha J, Barbosa JA, Hamers R. Sequence and structure of VH domain from naturally occurring camel heavy chain immunoglobulins lacking light chains. Protein Eng. 1994;7:1129–1135. doi: 10.1093/protein/7.9.1129. [DOI] [PubMed] [Google Scholar]
- Muyldermans S, Cambillau C, Wyns L. Recognition of antigens by single-domain antibody fragments: the superfluous luxury of paired domains. Trends Biochem Sci. 2001;26:230–235. doi: 10.1016/s0968-0004(01)01790-x. [DOI] [PubMed] [Google Scholar]
- Nguyen VK, Desmyter A, Muyldermans S. Functional heavy-chain antibodies in Camelidae. Adv Immunol. 2001;79:261–296. doi: 10.1016/s0065-2776(01)79006-2. [DOI] [PubMed] [Google Scholar]
- Ogiso H, Ishitani R, Nureki O, Fukai S, Yamanaka M, Kim JH, Saito K, Sakamoto A, Inoue M, Shirouzu M, Yokoyama S. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell. 2002;110:775–787. doi: 10.1016/s0092-8674(02)00963-7. [DOI] [PubMed] [Google Scholar]
- Oliveira S, van Dongen GA, Stigter-van Walsum M, Roovers RC, Stam JC, Mali W, van Diest PJ, van Bergen en Henegouwen PM. Rapid visualization of human tumor xenografts through optical imaging with a near-infrared fluorescent anti-epidermal growth factor receptor nanobody. Mol Imaging. 2012;11:33–46. [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Carter CW Jr, Sweet RM, editors. Meth Enzymol. New York: Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Philo JS, Aoki KH, Arakawa T, Narhi LO, Wen J. Dimerization of the extracellular domain of the erythropoietin (EPO) receptor by EPO: one high-affinity and one low-affinity interaction. Biochemistry. 1996;35:1681–1691. doi: 10.1021/bi9524272. [DOI] [PubMed] [Google Scholar]
- Roovers RC, Laeremans T, Huang L, De Taeye S, Verkleij AJ, Revets H, de Haard HJ, van Bergen en Henegouwen PM. Efficient inhibition of EGFR signaling and of tumour growth by antagonistic anti-EFGR Nanobodies. Cancer Immunol Immunother. 2007;56:303–317. doi: 10.1007/s00262-006-0180-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roovers RC, Vosjan MJ, Laeremans T, El Khoulati R, de Bruin RC, Ferguson KM, Verkleij AJ, van Dongen GA, van Bergen En Henegouwen PM. A biparatopic anti-EGFR nanobody efficiently inhibits solid tumour growth. Int J Cancer. 2011;129:2013–2024. doi: 10.1002/ijc.26145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saerens D, Ghassabeh GH, Muyldermans S. Single-domain antibodies as building blocks for novel therapeutics. Curr Opin Pharmacol. 2008;8:600–608. doi: 10.1016/j.coph.2008.07.006. [DOI] [PubMed] [Google Scholar]
- Sato JD, Kawamoto T, Le AD, Mendelsohn J, Polikoff J, Sato GH. Biological effects in vitro of monoclonal antibodies to human epidermal growth factor receptors. Mol Biol Med. 1983;1:511–529. [PubMed] [Google Scholar]
- Schmiedel J, Blaukat A, Li S, Knochel T, Ferguson KM. Matuzumab binding to EGFR prevents the conformational rearrangement required for dimerization. Can Cell. 2008;13:365–373. doi: 10.1016/j.ccr.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz KR, Ferguson KM. Interaction of antibodies with ErbB receptor extracellular regions. Exp Cell Res. 2009;315:659–670. doi: 10.1016/j.yexcr.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steyaert J, Kobilka BK. Nanobody stabilization of G protein-coupled receptor conformational states. Curr Opin Struct Biol. 2011;21:567–572. doi: 10.1016/j.sbi.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Sugawa N, Ekstrand AJ, James CD, Collins VP. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc Natl Acad Sci U S A. 1990;87:8602–8606. doi: 10.1073/pnas.87.21.8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tijink BM, Laeremans T, Budde M, Stigter-van Walsum M, Dreier T, de Haard HJ, Leemans CR, van Dongen GA. Improved tumor targeting of anti-epidermal growth factor receptor Nanobodies through albumin binding: taking advantage of modular Nanobody technology. Mol Cancer Ther. 2008;7:2288–2297. doi: 10.1158/1535-7163.MCT-07-2384. [DOI] [PubMed] [Google Scholar]
- Vaneycken I, D’Huyvetter M, Hernot S, De Vos J, Xavier C, Devoogdt N, Caveliers V, Lahoutte T. Immuno-imaging using nanobodies. Curr Opin Biotechnol. 2011 doi: 10.1016/j.copbio.2011.06.009. [DOI] [PubMed] [Google Scholar]
- Wong AJ, Ruppert JM, Bigner SH, Grzeschik CH, Humphrey PA, Bigner DS, Vogelstein B. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc Natl Acad Sci U S A. 1992;89:2965–2969. doi: 10.1073/pnas.89.7.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You B, Chen EX. Anti-EGFR Monoclonal Antibodies for Treatment of Colorectal Cancers: Development of Cetuximab and Panitumumab. J Clin Pharmacol. 2011 doi: 10.1177/0091270010395940. [DOI] [PubMed] [Google Scholar]
- Zhang H, Berezov A, Wang Q, Zhang G, Drebin J, Murali R, Greene MI. ErbB receptors: from oncogenes to targeted cancer therapies. J Clin Invest. 2007;117:2051–2058. doi: 10.1172/JCI32278. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.