

Abstract
Objective
Prolonged human immunodeficiency virus-1 (HIV-1) infection leads to neurological debilitation, including motor dysfunction and frank dementia. Although pharmacological control of HIV infection is now possible, HIV-associated neurocognitive disorders (HAND) remain intractable. Here, we report that chronic treatment with erythropoietin (EPO) and insulin-like growth factor-I (IGF-I) protects against HIV/gp120-mediated neuronal damage in culture and in vivo.
Methods
Initially, we tested the neuroprotective effects of various concentrations of EPO, IGF-I, or EPO+IGF-I from gp120-induced damage in vitro. To assess the chronic effects of EPO+IGF-I administration in vivo, we treated HIV/gp120-transgenic or wild-type mice transnasally once a week for 4 months and subsequently conducted immunohistochemical analyses.
Results
Low concentrations of EPO+IGF-I provided neuroprotection from gp120 in vitro in a synergistic fashion. In vivo, EPO+IGF-I treatment prevented gp120-mediated neuronal loss, but did not alter microgliosis or astrocytosis. Strikingly, in the brains of both humans with HAND and gp120-transgenic mice, we found evidence for hyperphosphorylated tau protein (paired helical filament-I tau), which has been associated with neuronal damage and loss. In the mouse brain following transnasal treatment with EPO+IGF-I, in addition to neuroprotection we observed increased phosphorylation/activation of Akt (protein kinase B) and increased phosphorylation/inhibition of glycogen synthase kinase (GSK)-3β, dramatically decreasing downstream hyperphosphorylation of tau. These results indicate that the peptides affected their cognate signaling pathways within the brain parenchyma.
Interpretation
Our findings suggest that chronic combination therapy with EPO+IGF-I provides neuroprotection in a mouse model of HAND, in part, through cooperative activation of phosphatidylinositol 3-kinase/Akt/GSK-3β signaling. This combination peptide therapy should therefore be tested in humans with HAND.
Human immunodeficiency virus-1 (HIV-1) infection can lead to progressive motor and cognitive dysfunction.1-3 Although survival and morbidity from acquired immunodeficiency syndrome (AIDS) have improved through the use of highly active antiretroviral therapy (HAART), the prevalence of HIV-associated neurologic complications is increasing.2 The increasing prevalence of HIV-associated neurocognitive disorders (HAND) indicates that HAART does not provide sufficient protection against neurological complications and suggests a need for neuroprotective therapies to ameliorate this condition. Although there are several rodent models of HAND, including HIV-1–infected macrophages in subacute combined immunodeficiency mice,4,5 HIV/gp120 envelope protein-expressing transgenic mice have been shown to develop several neuropathologic features associated with HAND, such as dendritic damage, synaptic degeneration, and frank neuronal loss in numerous reports.6,7 It has proven worthwhile to follow these features in gp120-transgenic mice, because they can be predictive of effects in subsequent human clinical trials.8
Concerning potential neuroprotective treatments, erythropoietin (EPO), which is classically known as a kidney-generated hematopoietic growth factor, is also expressed in the central nervous system.9 Insulin-like growth factor-I (IGF-I), which affects differentiation and survival in a variety of cells and tissues, is also expressed in the brain.10,11 Acting via the EPO receptor (EPO-R) and IGF-I receptor, EPO and IGF-I are neuroprotective in a variety of in vitro and in vivo animal models, including damage from excitotoxins, ischemia, and gp120/HIV envelope protein.12-14 Previously, we demonstrated that Janus tyrosine kinase-2 (Jak2) is phosphorylated/activated after EPO binds to the EPO-R, initiating a neuroprotective signaling pathway (Fig 1).15 Phosphorylated Jak2 activates a nuclear factor kappa B (NF-κB) signaling cascade that counteracts caspase activity by upregulating both XIAP and Bcl-2 family members.15,16 Additionally, activation of the EPO and IGF-I receptors leads to activation of multiple biochemical cascades, including the PI3K/Akt signaling pathway, which phosphorylates glycogen synthase kinase (GSK)-3β and several other targets critical to cell survival (see Fig 1).17 Phosphorylation inactivates GSK-3β and thus decreases tau phosphorylation, which in the hyperphosphorylated state is causally associated with neuronal cell injury and death.18,19
FIGURE 1.
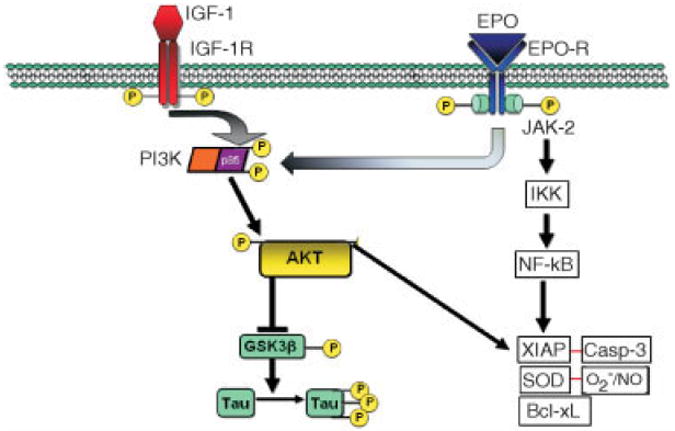
Schematic model of erythropoietin (EPO) and insulin-like growth factor-I (IGF-I) signaling pathways, showing synergistic effects of EPO and IGF-I against gp120-induced neurodegeneration. The signaling mechanism includes phosphorylation and activation of the receptors (R) on EPO or IGF-I binding, activation of PI3K/Akt signaling pathways, hyperphosphorylation of tau, and activation of nuclear factor kappa B (NF-κB). JAK-2 = Janus tyrosine kinase-2; IKK = IκB kinase; AKT = protein kinase B; GSK = glycogen synthase kinase; XIAP = X-linked inhibitor of apoptosis; SOD = superoxide dismutase; Casp-3 = caspase-3.
Recently, we demonstrated that relatively low concentrations of EPO+IGF-I exert a synergistic effect on activation of the PI3K/Akt pathway in rat cerebrocortical cultures, resulting in neuroprotection from excitotoxic insults and thus potentially avoiding side effects that occur with administration of high doses of either EPO or IGF-I alone.20 Considering this potent neuroprotective action of EPO+IGF-I in culture, the present study was undertaken to investigate if application of these cytokines could not only ameliorate the harmful effects of HIV/gp120 in vitro but also repair neuronal dendritic damage that is known to occur in the gp120-transgenic murine model of HAND in vivo. Here, we show that EPO+IGF-I exerts such effects, at least in part, by decreasing tau hyperphosphorylation via PI3K/Akt-mediated inhibition of GSK-3β activity.
Materials and Methods
Cerebrocortical Cultures, Apoptosis Analysis, and PI3K Inhibitors
Mixed neuronal/glial cerebrocortical cells were derived from embryonic (E17) Sprague-Dawley rats and plated at a density of 4.5 × 105 per 35 mm dish containing poly-l-lysine–coated glass coverslips in Dulbecco modified Eagle medium with Ham F12 and heat-inactivated iron-supplemented calf serum (Hyclone, Logan, UT) plus 2mM glutamine, 24mM HEPES, and penicillin-streptomycin. The culture medium was changed every other day. After 17 to 21 days in culture, the cells were exposed to HIV/gp120 (200pM) for 24 hours in the presence or absence of various concentrations of EPO, IGF-I, or a combination of EPO+IGF-I. Neuronal viability and apoptosis were analyzed in these cultures as previously described.21 In brief, apoptosis was scored in a blinded fashion as the percentage of NeuN-positive neurons that stained for TUNEL and exhibited a condensed nuclear morphology under epifluorescence microscopy. For the experiments with inhibitors to PI3K, cortical neurons were preincubated in the presence or absence of 50μM of LY294002 for 1 hour prior to EPO+IGF-1 treatment, then exposed to gp120 for 24 hours. Afterwards, cells were homogenized in RIPA buffer and subject to Western blot.
HIV/gp120-Transgenic Mice
Transgenic mice expressing HIV/gp120 under the control of a modified murine glial fibrillary acidic protein (GFAP) promoter were obtained from Dr Lennart Mucke (Gladstone Institute, University of California, San Francisco, San Francisco, CA). Characteristics of these mice have been previously presented.7,8
Human Brain Samples
HIV−/age-matched controls (35–45 years old) were obtained from the University of California, San Diego Medical Center Autopsy Service. HIV+ cases were from the California NeuroAIDS Tissue Network cohort. The brain regions examined included frontal cortex (gray and white matter), hippocampus, caudate, and basal ganglia (putamen and globus pallidus). All HIV+ patients died of respiratory failure due to bronchopneumonia, and the general autopsy findings were consistent with AIDS. The associated pathology was most frequently due to systemic cytomegalovirus, Kaposi sarcoma, and liver disease. Five-micron sections were mounted on glass slides for immunohistochemistry. After deparaffinization in Histoclear, rehydration through graded alcohol solutions, and blockade of endogenous peroxidases in 3% H2O2 for 30 minutes, tissue sections were processed for antigen retrieval using the pressure cooker method. This consisted of a 20-minute incubation at the high setting in Coplin jars filled with Dako Target Retrieval Solution (Dako-Cytomation, Carpinteria, CA). After cooling for 20 minutes, the tissue sections were incubated for an additional 30 minutes in normal goat serum to decrease nonspecific staining. Primary monoclonal antibodies were incubated for 2 hours at room temperature (RT). Following a 30-minute incubation in secondary antibody (biotinylated goat-antimouse) and 30 minutes in Avidin: Biotinylated enzyme Complex (ABC), color reactions were developed with NovaRed (Vector Labs, Burlingame, CA). Sections were counterstained with Meyer hematoxylin.
Transnasal Delivery of EPO/IGF-I
Mice were anesthetized with isoflurane and maintained under anesthesia for the duration of the treatment. Mice were placed in a supine position, and EPO and/or IGF-I were administered into the nares by pipette in a dropwise fashion in 2μl aliquots, alternating between each nostril every 2 minutes, over a total of 12 minutes. As drug was administered to one naris, the other side was plugged with soft putty. After an initial dose-ranging set of experiments to find the maximally effective concentration of this combination of cytokines, 12μl of a mixture of EPO (50U; Ortho Biotech Products, Raritan, NJ) and IGF-I (2,000ng; Invitrogen, Carlsbad, CA) was administered transnasally.23,24 Prior studies using I125-radiolabeled cytokines had shown that these approximate doses of EPO and IGF-I passed the blood-brain barrier (BBB) and acted synergistically in vivo in the brain during acute neuroprotective experiments.25
Western Blot Analysis
For determination of phospho(p)Akt and pGSK-3β activity, olfactory bulb or forebrain was homogenized in ice-cold lysis buffer (Cell Lysis Buffer, Cell Signaling Technology, Beverly, MA) containing protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN). Tissue lysates were centrifuged (15 minutes at 14,000g, 4°C), and supernatants were collected. Protein concentration was determined using a BCA Protein Assay (Pierce Biotechnology, Rockford, IL). Proteins were separated on 4 to 12% NU-PAGE gels (Invitrogen) and transferred onto polyvinylidene difluoride membrane (Immobilon-P, Millipore, Billerica, MA). After blocking with 5% nonfat milk in Tris-buffered saline containing 0.05% Tween 20, the membranes were incubated with primary antibodies overnight at 4°C followed by horseradish peroxidase-conjugated secondary antibody for 1 hour at RT. Protein bands were visualized with enhanced chemoluminescent detection reagents (ECL, Amersham Biosciences, Fairfield, CT; GE Healthcare, Pittsburgh, PA) and exposed to X-ray film (Hyperfilm ECL, Amersham Bioscience). Autoradiographic films were scanned, and densitometric analysis was performed. The following primary antibodies were used: rabbit polyclonal antibody against Akt, pAkt (Ser 473), GSK-3β, pGSK-3β (Cell Signaling Technology), mouse monoclonal antiactin antibody (clone AC-40, Sigma, St. Louis, MO), and phospho-tau (PHF-1) (pSer404) antibody (Sigma). Horseradish peroxidase-conjugated goat antirabbit or antimouse immunoglobulin G (Pierce Biotechnology) was used as a secondary antibody.
Quantitative Neuropathological Analysis
For each immunostain, 3 serial sections of corresponding mouse brain regions were analyzed. For assessment of neuronal changes, sections were immunolabeled with antibodies against microtubule-associated protein-2 (MAP-2) (Chemicon, Temecula, CA) to label neuronal cell bodies and dendrites, NeuN (Chemicon) to label neuronal nuclei and cell bodies, GFAP (Chemicon) to label astrocytes in this context, Iba1 (Wako Chemicals, Richmond, VA) to label microglia, and paired helical filament-1 (PHF-1, a gift from P. Davies) to label hyperphosphorylated tau in abnormal filaments. Primary antibody staining was identified with fluorescently tagged secondary antibodies or immunoperoxidase. Sections were examined using a Bio-Rad MRC-1000 laser scanning confocal microscope. Digitized images of 3 optical sections (40μm in thickness) were transferred to a Macintosh computer, running a public domain program (Wayne Rasband), and analyzed as previously described.25 For each case, the frontal cortex (layers 2, 3, and 5) and the hippocampus (molecular layer of dentate gyrus and pyramidal layer of CA1 region) were analyzed quantitatively for each antibody label.
Enzyme-Linked Immunosorbent Assays for IGF-I and EPO
Whole brains from littermate, sex matched, wild-type (WT), and gp120 transgenic 6-week-old mice were homogenized in 2.4ml ice-cold lysis buffer (Cell Lysis Buffer, Cell Signaling Technology) containing protease inhibitor cocktail (Roche Applied Science). Tissue lysates were sonicated and centrifuged for 10 minutes at 14,000g, 4°C, and supernatants were collected and stored at −80°C until used. Protein concentration was determined using a BCA Protein Assay (Pierce Biotechnology) with bovine serum albumin as standard. Quantitative determination of EPO was performed using Quantikine mouse EPO Immunoassay (R&D Systems, Minneapolis, MN). Immunoreactive IGF-I levels were analyzed by a mouse-specific sandwich enzyme-linked immunosorbent assay, developed with a commercially available kit (DuoSet ELISA Development System, R&D Systems). Optical densities were read at 450nm (correction wavelength set at 540nm) by using an automated plate reader, and cytokine levels were calculated by interpolation from the standard curve. Values were corrected for protein concentration. Data were reported as mean ± standard error of the mean and statistically evaluated for difference by Student t test.
Results
EPO+IGF-I Prevents HIV/gp120-Induced Neuronal Apoptosis in a Synergistic Fashion In Vitro
We found that 200pM HIV/gp120 increased neuronal apoptosis in mixed neuronal/glial cerebrocortical cultures by ~200% over control (Fig 2). High concentrations of EPO (100U/ml) nearly completely prevented gp120-induced apoptosis, whereas lower concentrations of EPO (10U/ml) produced a substantially smaller reduction in gp120 neurotoxicity, in agreement with prior studies.20 IGF-I (100ng/ml) also offered modest neuroprotection from gp120. Interestingly, our previous studies had demonstrated that low concentrations of EPO+IGF-I act synergistically in cerebrocortical cultures to exert neuroprotection from excitotoxic (N-methyl-d-aspartate [NMDA]) insults.20 Additionally, we had shown that HIV/gp120-induced damage is mediated at least in part via excessive NMDA receptor activation.2,27 Therefore, in the present study, we investigated whether the combination of low concentrations of EPO+IGF-I could protect cultured cerebrocortical neurons from exposure to gp120. We found that the combination of 10U/ml EPO plus 100ng/ml IGF-I completely abrogated gp120-induced neuronal apoptosis, an effect that was more than additive. Thus, the effect of low-dose EPO+IGF-I was at least as protective from gp120-induced toxicity as high-dose EPO alone. Importantly, when administered individually, high doses of EPO or IGF-I exert potentially harmful side effects,28,29 so the attainment of neuroprotection at low doses is of potential clinical utility.
FIGURE 2.
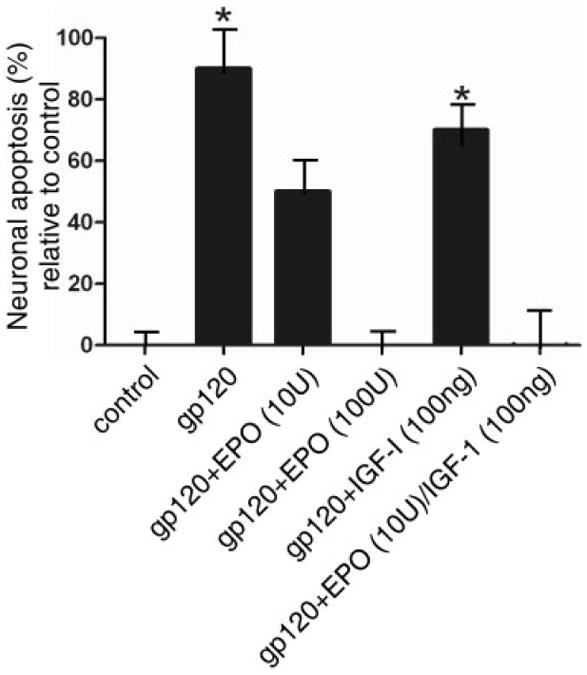
Neuroprotection of rat cerebrocortical neurons from gp120-induced apoptosis, showing protection by erythropoietin (EPO), insulin-like growth factor-I (IGF-I), or a combination of both. Mixed neuronal/glial cerebrocortical cell cultures were exposed for 24 hours to gp120 (200pM) in the presence or absence of the indicated concentrations of EPO and IGF-I. Each experiment was performed in triplicate and repeated at least 3 ×. Approximately 1,000 neurons were scored for each value. The y (ordinate) axis represents the percentage of neurons undergoing apoptosis above control levels. Approximately 20% of the neurons not exposed to gp120 died by apoptosis in the control cultures, as expected among developing neurons. *p < 0.01 by ANOVA with post hoc Scheffé test.
Mechanism of Neuroprotection by EPO+IGF-I
We had previously demonstrated that the PI3K/Akt signaling pathway mediates the synergistic effect of EPO+IGF-I and that inhibiting phosphorylation of Akt, either by dominant negative molecular interference or by pharmacological antagonism, abrogated the neuroprotective effect of these cytokines in cultured cerebrocortical neurons.20 Here, to further dissect the mechanism whereby EPO+IGF-I prevents HIV/gp120-induced neuronal cell death in these cultures, we examined the phosphorylation state of GSK-3β after exposure to gp120 in the presence and absence of EPO+IGF-I. We found that EPO+IGF-I increased both Akt phosphorylation and GSK-3β phosphorylation (Fig 3A), consistent with the notion that EPO+IGF-I exerts its neuroprotective effect at least in part by phosphorylating and thus inactivating GSK-3β, thereby inhibiting tau hyperphosphorylation. Furthermore, inhibiting Akt activation using the PI3K inhibitor LY294002 completely prevented EPO+IGFI-I–induced Akt phosphorylation and GSK-3β phosphorylation compared to control (see Fig 3A), confirming the involvement of the PI3K/Akt/GSK-3β pathway.
FIGURE 3.
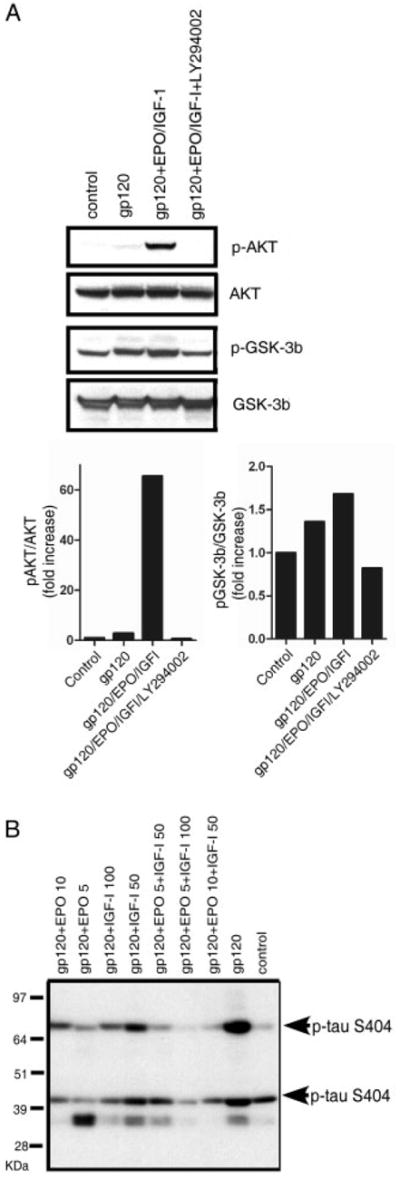
Erythropoietin (EPO)+insulin-like growth factor-I (IGF-I) activates the PI3K/Akt/glycogen synthase kinase (GSK)-3β pathway to counteract gp120-induced tau hyperphosphorylation. Western blots of cortical culture lysates following a 1-day exposure to 200pM gp120 are shown. (A) EPO+IGF-I treatment produced Akt phosphorylation and GSK-3β phosphorylation, whereas total Akt and GSK-3β remained constant. Pretreatment with 50μM of the PI3K inhibitor LY294002 completely abrogated Akt phosphorylation and largely blocked the phosphorylation of GSK-3β engendered by EPO+IGF-I. (B) Hyperphosphorylation of tau following exposure of cerebrocortical cultures to gp120. Antitau clone 5E2 (Upstate Biotechnology, Lake Placid, NY) was used to immunoprecipitate (IP) total tau protein from cortical cell culture lysates (this antibody brings down tau isoforms in the molecular weight range of ~45–70kDa). The IPs were then run on a 10% NuPage gel and probed with phospho-tau antibody (directed against ser404, which is known to represent tau hyperphosphorylation). In the blot, 2 isoforms of tau are seen to be phosphorylated at serine 404 (bands at ~45 and 70kDa). Tau phosphorylation increased after exposure to 200pM gp120, but was prevented by treatment with EPO+IGF-I.
Next, we examined the phosphorylation state of tau in these cultures. Tau hyperphosphorylation increased in response to gp120, whereas EPO and/or IGF-I decreased tau hyperphosphorylation, as revealed by immunoblotting (see Fig 3B). Prior work has suggested that hyperphosphorylated tau may contribute to neuronal injury in a number of neurodegenerative disorders, including HAND,30,31 so our finding that EPO+IGF-I completely abrogated PHF-1 formation suggests a possible mechanism for the observed neuroprotection.
To extend our findings on tau hyperphosphorylation to humans, brain samples from patients with HAND were analyzed by Western blot and immunostaining with PHF-1 antibody (Fig 4). The non-HAND cases did not manifest significant neurological impairment. The HAND cases were characterized by the presence of HIV p24 by Western blot and immunohistochemistry, astrogliosis, microglial nodules, and multinucleated giant cells on neuropathological examination, coupled with significant deficits on neuropsychological testing.8 Similar to findings of hyperphosphorylated tau in Alzheimer brains, we found increased staining in the neuropil of the hippocampus and to a lesser extent in the frontal cortex in HAND. Unlike Alzheimer disease, however, frank tangles of hyperphosphorylated tau proteins were not present in the HAND brains.
FIGURE 4.
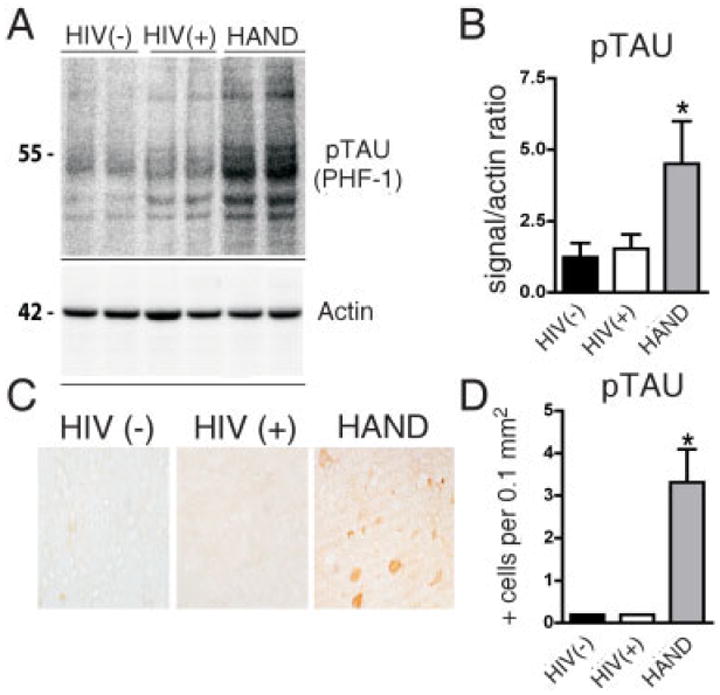
Alterations in phospho-tau levels in basal ganglia from brains of patients with human immunodeficiency virus (HIV)-associated neurocognitive disorders (HAND). For each group, 5 cases are included. Significantly increased hyperphosphorylated tau (pTAU) was observed in autopsied brain tissue from the basal ganglia (putamen) in all patients with HAND. (A) Western blot analysis of tissue homogenates from the putamen probed with an antibody against pTAU (PHF-1); pTAU is identified as a complex of several bands at an estimated molecular weight of 50–60kDa. (B) Analysis of pixel intensity of the bands using the ImageQuant system. Results are expressed as a ratio of pTAU over actin. (C) Immunohistochemical analysis with antibodies against PHF-1 in the putamen. In HAND brains, occasional positive midsize spine neurons were identified. (D) Image analysis of the numbers of pTAU-positive cells.
Akt/GSK-3β Phosphorylation in the Olfactory Bulb and Forebrain after Transnasal Administration of EPO+IGF-I In Vivo
To begin to apply our findings with EPO+IGF-I in vivo, we measured endogenous IGF-I levels in the gp120-transgenic mouse brain and found that they were significantly lower compared to WT littermates (Supplementary Fig). In the same experimental groups, EPO was not detectable in either WT or gp120-transgenic mouse brain. These findings added further impetus for treating gp120-transgenic mice with EPO+IGF-I.
Next, we investigated if transnasal application of the cytokines could exert an effect across the BBB. Initially, we studied if the Akt/GSK-3β pathway was activated in the olfactory bulb and forebrain following transnasal administration of these peptides. It is well known that transnasal delivery of peptides can facilitate their transport across the BBB, for example, via receptor-mediated endocytosis of endothelial cells,23,32-34 and these cells are known to possess receptors for EPO and IGF-I.32 Transnasal delivery of EPO+IGF-I can limit systemic administration25 and thus the myriad of consequent side effects seen with high-dose parenteral administration of EPO and IGF-I in prior clinical trials.35-38 Based on our in vitro work, we made estimates of the dosage of the cyto-kines that had to be administered in vivo, and then bracketed these amounts for further studies. We thus delivered EPO (50 or 100U) and IGF-I (1,000 or 2,000ng) by transnasal application to 6-month-old HIV/gp120-transgenic or WT mice, and 30 minutes later dissected out the olfactory bulbs/forebrains and performed Western blots. Immunoblotting revealed that Akt was phosphorylated following EPO and/or IGF-I administration (Fig 5A). In particular, the combination of 50U EPO plus 2,000ng IGF-I dramatically increased Akt phosphorylation. Next, we examined whether phosphorylation of GSK-3β was affected in response to transnasal delivery of EPO+IGF-I. As expected, GSK-3β phosphorylation increased after a single administration of EPO+IGF-I and remained elevated in the forebrain for over 24 hours (see Fig 5B, C). These data demonstrate that transnasal application of EPO+IGF-I can activate signaling pathways in the brain that could be potentially neuroprotective in vivo.
FIGURE 5.
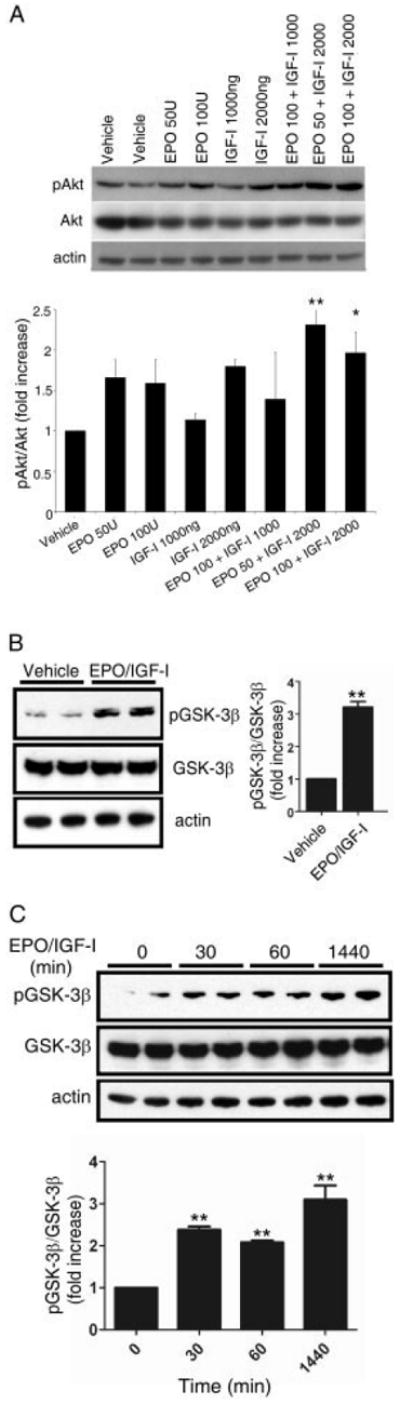
Effect of transnasal delivery of erythropoietin (EPO)/insulin-like growth factor-I (IGF-I) on Akt/glycogen synthase kinase (GSK)-3β phosphorylation in mouse forebrain/olfactory bulb. Six-month-old wild-type (A) or gp120-transgenic (B and C) mice received the indicated dose of EPO (in U/ml) and/or IGF-I (in ng/ml) dissolved in vehicle (10mM sodium succinate buffer, pH 6.2) via transnasal delivery. Mice were sacrificed 30 minutes (A), 10 minutes (B), or 30 minutes, 60 minutes, and 24 hours (C) after the last application of peptide. The forebrain (C) or olfactory bulb (A and B) was then dissected to prepare lysates. Proteins from individual samples were subjected to immunoblot analysis with anti–phospho-Akt antibody (pAkt) (A) or anti–phospho-GSK-3β antibody (pGSK-3β) (B and C). Blots were then stripped and reprobed with anti-Akt antibody (Akt) (A), anti-GSK-3β (B and C), or antiactin antibody for total protein as a loading control. By densitometric analysis, combined treatment with EPO+IGF-I resulted in an increase in Akt phosphorylation compared to EPO or IGF-I alone. EPO (50U) plus IGF-I (2,000ng) also increased GSK-3β phosphorylation in gp120-transgenic mouse brain (B and C). For densitometric quantification, the ratio of pAkt to total Akt (A) or the ratio of pGSK-3β to total GSK-3β (B and C) for each lane was normalized to vehicle-treated samples. Data are expressed as mean + standard error of the mean of 3 to 6 independent experiments. *p < 0.05, **p < 0.01 versus vehicle (analysis of variance followed by Tukey-Kramer test for multiple comparisons).
Importantly, transnasal administration of EPO did not increase the hematocrit (Hct) after chronic treatment (Hct of vehicle-treated animals, 36%; Hct of EPO+IGF–treated animals, 32%). This result is consistent with the notion that very little systemic absorption occurred with this route of delivery, as we have also recently documented with I125-radiolabeld cytokines; this study also demonstrated that the vast majority of transnasally administered EPO+IGF-I entered the brain.25 Taken together, these findings suggest that fewer systemic side effects will occur following transnasal administration than with standard parenteral administration, and that these cytokines can bypass the BBB and enter the brain from the nasal mucosa.
Neuroprotective Effect of EPO+IGF-I in HIV/gp120-Transgenic Mice
To study the neuroprotective effect of EPO+IGF-I in an animal model of HAND, we treated gp120-transgenic mice at a time point when damage would have otherwise progressed to cause severe dendritic and neuronal loss.7,8 Starting at 6 months of age, we administered 50U of EPO plus 2,000ng of IGF-I for a period of 4 months to both WT and gp120-transgenic mice. We then analyzed the effects of this treatment on brain sections under laser scanning confocal microscopy. The extent of neuronal damage in the brains of these 10-month-old gp120-transgenic mice was examined by immunostaining for NeuN, MAP-2, GFAP, Iba1, and PHF-1(Fig 6). Significant dendritic damage and frank neuronal loss were detected throughout the cortex and hippocampus of the gp120-transgenic mice when compared with WT mice of the same age, as shown by MAP-2 and NeuN staining. The neuronal damage in the cortex and hippocampus was largely reversed by chronic EPO+IGF-I treatment (see Fig 6D, H and 7A–D). Quantitative analysis of confocal fluorescent images revealed statistically significant increases in the percentage area of neuropil after EPO+IGF-I treatment compared with vehicle-treated gp120 mice, indicating significant protection of the dendritic field. Brain sections of gp120-transgenic mice revealed an increase in neocortical and hippocampal astrogliosis (GFAP staining) and microgliosis (Iba1 staining) compared to WT mice (see Figs 6K, O and 7E–H). The degree of astrocytosis and microglial reactivity was similar in EPO+IGF-I–treated and vehicle-treated gp120 mice (see Figs 6L, P and Fig 7E-H). In line with our evidence for hyperphosphorylation of cortical neurons in vitro after gp120 exposure and with our PHF-1 staining in human brain sections with HAND, both cortical and hippocampal brain sections of gp120-transgenic mice showed statistically significant increases in PHF-1 tau compared to WT mice (p < 0.0001). These data suggest that HIV/gp120 induces hyperphosphorylated tau in a pathophysiologically relevant manner. Moreover, treatment with EPO+IGF-I greatly reduced PHF-1 tau (p < 0.003 and p < 0.01 in cortex and hippocampus, respectively) compared to vehicle-treated gp120-transgenic mice. Taken together, these findings demonstrate that chronic treatment with transnasal EPO+IGF-I produced significant improvements in dendritic complexity and prevention of neuronal degeneration compared to untreated gp120-transgenic mice. On the other hand, EPO+IGF-I manifested no significant effect on astrocytosis or microglial responses.
FIGURE 6.
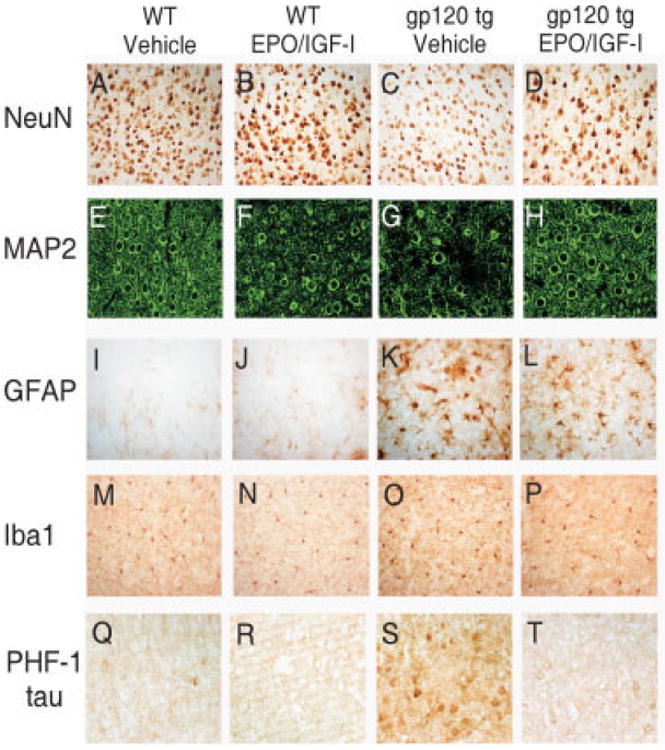
Neuropathological characterization of the protective effect of erythropoietin (EPO)+insulin-like growth factor-I (IGF-I) against gp120 toxicity in the mouse cortex. Brain sections of 10-month-old wild-type (WT) and gp120-transgenic mice treated chronically with EPO+IGF-I versus control were analyzed for degree of cortical neuropathology. Serial 40μm brain sections were immunostained for NeuN (A–D), microtubule-associated protein-2 (MAP-2) (E–H), glial fibrillary acidic protein (GFAP) (I–L), Iba1 (M-P), or phospho-tau (PHF-1) (Q-T). Note the loss of NeuN and MAP-2 staining in gp120-transgenic mice (C and G), but NeuN and MAP-2 staining approached WT levels after chronic treatment with EPO+IGF-I (D and H). GFAP and Iba1 staining in gp120-transgenic mice did not change after EPO+IGF-I treatment. Human immunodeficiency virus/gp120-transgenic mice manifested increased PHF-1 tau staining (S) compared to WT (Q) or WT treated with EPO+IGF-I (R). PHF-1 tau was dramatically reduced in gp120-transgenic mice after chronic treatment of EPO+IGF-I (T).
FIGURE 7.
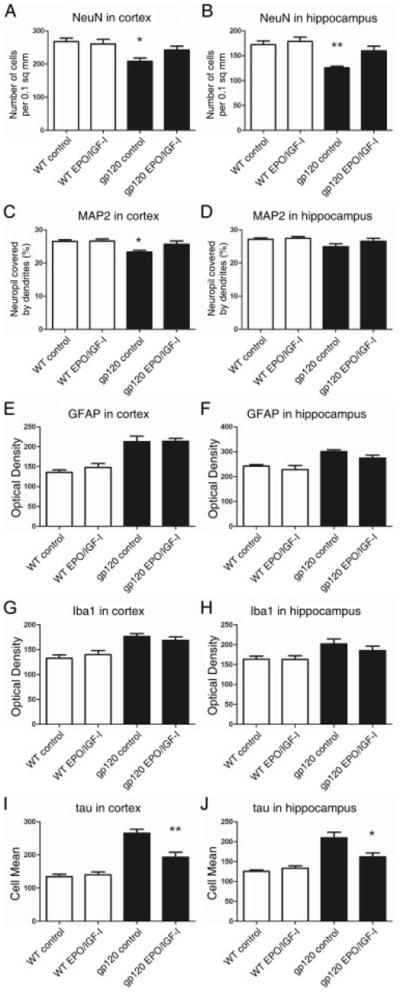
Computer-aided image analysis of neurodegeneration in the cortex and hippocampus of wild-type (WT) and human immunodeficiency virus/gp120-transgenic mice treated with erythropoietin (EPO)+insulin-like growth factor-I (IGF-I). Laser scanning confocal images of immunolabeled neocortical (A, C, E, G, and I) and hippocampal sections (B, D, F, H, and J; 5–7 mice per group) were analyzed quantitatively using NIH Image. Representative laser scanning confocal images revealed significant increases in NeuN (A and B) and microtubule-associated protein-2 (MAP-2) (C), but a decrease in phospho-tau (I and J) in gp120-transgenic mouse brains treated with EPO+IGF-I compared to vehicle. Values represent mean + standard error of the mean. *p < 0.05, **p < 0.005, by 1-tailed Student t-test comparing vehicle versus EPO+IGF-I–treated gp120-transgenic mice. GFAP = glial fibrillary acidic protein.
Discussion
In the present study, we demonstrate that the HIV envelope glycoprotein gp120 induces neuronal damage and tau hyperphosphorylation in vitro and in vivo. We show that chronic EPO+IGF-I treatment in vitro or in vivo in HIV/gp120-transgenic mice ameliorates this neuronal damage by histological criteria and decreases tau hyperphosphorylation. Prominent HAND neuropathologic features include astrogliosis, activation of microglia, decreased synaptic and dendritic density, and neuronal apoptosis.2,42 Previously, several groups, including our own, had shown that EPO or IGF-I could exert a neuroprotective effect against various forms of neuronal insults.13-15,43 However, the high doses of EPO or IGF-I used in these studies are known to cause systemic side effects. Recently, we found a synergistic effect of EPO+IGF-I at much lower doses,20 so in the present study we took advantage of this synergistic effect for chronic treatment with these cytokines. Additionally, we found that the endogenous IGF-I levels in the gp120-transgenic mouse brain were significantly lower compared to WT littermates, and EPO was not detectable in either WT or gp120 mouse brain. Therefore, together with our finding that EPO+IGF-I synergistically activated the PI3K/Akt neuroprotective pathway, these findings provided a strong rationale for treating gp120-transgenic mice with EPO+IGF-I.
Additional recent studies have shown that acute transnasal treatment of IGF-I and/or EPO can offer protection from experimental brain injury in rat stroke models.25,44,45 It has been suggested that transnasal administration of IGF-I can cross the BBB by an extracellular route along the olfactory bulb or trigeminal neural pathways, as observed by tracing studies with [125I]-IGF-I. Additional reports have suggested that IGF-I and EPO can cross the BBB by endocytosis after binding to IGF-I or EPO receptors that are expressed on the endothelium of brain capillaries.32,34 Thus, transnasal delivery of EPO+IGF-I to the brain may occur via endocytosis as a result of binding of these cytokines to their cognate receptors, or possibly via an extracellular route.
We found several lines of evidence for neuroprotection by the chronic administration of EPO+IGF-I in gp120-transgenic mice, and to our knowledge this represents the first report of successful, low-dose treatment with these cytokines for a prolonged period. From our quantitative confocal microscopy assessments, dendritic area and neuronal survival were improved by this treatment. We have previously demonstrated that 1 mechanism whereby EPO+IGF-I elicits neuroprotection involves activation of the antiapoptotic PI3K/Akt signaling pathway. As shown here, treatment with EPO+IGF-I results in phosphorylation/activation of Akt. Activated Akt then phosphorylates GSK-3β, thus inactivating it and, in turn, preventing phosphorylation of tau protein.
Additionally, we demonstrate here for the first time that hyperphosphorylated tau is more abundant in the brains of human AIDS patients with HAND than in controls, as well as in the brains of HIV/gp120-transgenic mice. It has been surmised by several groups that hyperphosphorylated tau is involved in the pathogenesis of HAND,30,31,46 and several lines of evidence suggest that hyperphosphorylated tau may contribute to neurodegeneration.39,47 Tau can be phosphorylated by various kinases, including protein kinase A, protein kinase C, Jun kinase, p38 mitogen-activated protein kinase, cyclin-dependent kinase 5, and GSK-3β.48-50 Although the mechanism whereby HIV-1 causes tau hyperphosphorylation in HAND brains has yet to be fully elucidated, based on our data we suggest that HIV envelope protein gp120 activates GSK-3β by dephosphorylation, leading to tau hyperphosphorylation, which in turn may potentially contribute to the pathology of HAND. Additionally, in the present study, we found that brains from gp120-transgenic mice expressed abundant levels of PHF-1 tau staining, representing Ser-396/404 PHF-1 sites, which are known to be phosphorylated by GSK-3β. Importantly, we observed a significant decrease in this staining after chronic transnasal treatment with EPO+IGF-I. Taken together, our results suggest that EPO+IGF-I improved the pathological state of gp120 mouse brains, and this neuroprotective effect may possibly have been mediated in part by dephosphorylation of tau, although additional mechanisms of protective action are also possible.
In summary, in the present study we show that chronic transnasal application of EPO+IGF-I induces neuroprotection from gp120 both in vitro and in vivo. We also show that the transnasal route effectively delivers EPO+IGF-I to the brain, and this method can avoid systemic side effects such as a rise in Hct (with consequent thrombosis) or induction of severe cachexia. Moreover, we provide evidence that HAND is associated with hyperphosphorylated tau in the human brain, and that HIV/gp120 can induce hyperphosphorylation of tau. Because reduction in tau phosphorylation after treatment with EPO+IGF-I was accompanied by neuroprotection in the gp120-transgenic mice, we speculate that PHF-1 may serve as a biomarker for the disease process in HAND. Because both EPO and IGF-I are approved for clinical use by the US Food and Drug Administration for other indications, we suggest based on these results that transnasal delivery of EPO+IGF-I should be considered for expedited human therapeutic trials for HAND.
Supplementary Material
Acknowledgments
This work was supported in part by NIH grants R01 NS047973, R01 NS046994, R01 NS43242, and R01 EY09024 to S.A.L, MH076681 and MH072529 to C.L.A., and R01 NS050621 to M.K. Additional support was provided by the NIH Blueprint Grant for La Jolla Interdisciplinary Neuroscience Center Cores P30 NS057096 to S.A.L.
We thank T. Fang for preparation of the cerebrocortical cultures, A. Adame and R. Dowen for technical help, K. Walsh for providing adenoviral constructs, and L. Mucke for HIV/gp120-transgenic mice.
Footnotes
Potential Conflicts of Interest
Dr Lipton has served as a consultant to Ortho Biotech, Inc. (J&J, Inc.). Drs Lipton and Digicaylioglu are the named inventors on patent applications filed by their institution for the use of erythropoietin plus insulin like growth factor-I as a disease-modifying treatment for neurodegenerative disorders, including HIV-related conditions.
Additional supporting information can be found in the online version of this article.
References
- 1.Kolson DL. Neuropathogenesis of central nervous system HIV-1 infection. Clin Lab Med. 2002;22:703–717. doi: 10.1016/s0272-2712(02)00009-4. [DOI] [PubMed] [Google Scholar]
- 2.McArthur JC. HIV dementia: an evolving disease. J Neuroimmunol. 2004;157:3–10. doi: 10.1016/j.jneuroim.2004.08.042. [DOI] [PubMed] [Google Scholar]
- 3.Mocchetti I, Bachis A, Masliah E. Chemokine receptors and neurotrophic factors: potential therapy against aids dementia? J Neurosci Res. 2008;86:243–255. doi: 10.1002/jnr.21492. [DOI] [PubMed] [Google Scholar]
- 4.Gorantla S, Liu J, Sneller H, et al. Copolymer-1 induces adaptive immune anti-inflammatory glial and neuroprotective responses in a murine model of HIV-1 encephalitis. J Immunol. 2007;179:4345–4356. doi: 10.4049/jimmunol.179.7.4345. [DOI] [PubMed] [Google Scholar]
- 5.Potula R, Poluektova L, Knipe B, et al. Inhibition of indoleamine 2,3-dioxygenase (IDO) enhances elimination of virus-infected macrophages in an animal model of HIV-1 encephalitis. Blood. 2005;106:2382–2390. doi: 10.1182/blood-2005-04-1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toggas SM, Masliah E, Rockenstein EM, et al. Central nervous system damage produced by expression of the HIV-1 coat protein gp120 in transgenic mice. Nature. 1994;367:188–193. doi: 10.1038/367188a0. [DOI] [PubMed] [Google Scholar]
- 7.Garden GA, Budd SL, Tsai E, et al. Caspase cascades in human immunodeficiency virus-associated neurodegeneration. J Neurosci. 2002;22:4015–4024. doi: 10.1523/JNEUROSCI.22-10-04015.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schifitto G, Navia BA, Yiannoutsos CT, et al. Memantine and HIV-associated cognitive impairment: a neuropsychological and proton magnetic resonance spectroscopy study. AIDS. 2007;21:1877–1886. doi: 10.1097/QAD.0b013e32813384e8. [DOI] [PubMed] [Google Scholar]
- 9.Morishita E, Masuda S, Nagao M, et al. Erythropoietin receptor is expressed in rat hippocampal and cerebral cortical neurons, and erythropoietin prevents in vitro glutamate-induced neuronal death. Neuroscience. 1997;76:105–116. doi: 10.1016/s0306-4522(96)00306-5. [DOI] [PubMed] [Google Scholar]
- 10.Dore S, Kar S, Quirion R. Rediscovering an old friend, IGF-I: potential use in the treatment of neurodegenerative diseases. Trends Neurosci. 1997;20:326–331. doi: 10.1016/s0166-2236(96)01036-3. [DOI] [PubMed] [Google Scholar]
- 11.Brooker GJ, Kalloniatis M, Russo VC, et al. Endogenous IGF-1 regulates the neuronal differentiation of adult stem cells. J Neurosci Res. 2000;59:332–341. doi: 10.1002/(sici)1097-4547(20000201)59:3<332::aid-jnr6>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 12.Nitta A, Zheng WH, Quirion R. Insulin-like growth factor 1 prevents neuronal cell death induced by corticosterone through activation of the PI3k/Akt pathway. J Neurosci Res. 2004;76:98–103. doi: 10.1002/jnr.20057. [DOI] [PubMed] [Google Scholar]
- 13.Brines M, Cerami A. Emerging biological roles for erythropoietin in the nervous system. Nat Rev Neurosci. 2005;6:484–494. doi: 10.1038/nrn1687. [DOI] [PubMed] [Google Scholar]
- 14.Digicaylioglu M, Kaul M, Fletcher L, et al. Erythropoietin protects cerebrocortical neurons from HIV-1/gp120-induced damage. Neuroreport. 2004;15:761–763. doi: 10.1097/00001756-200404090-00004. [DOI] [PubMed] [Google Scholar]
- 15.Digicaylioglu M, Lipton SA. Erythropoietin-mediated neuroprotection involves cross-talk between Jak2 and NF-kappaB signalling cascades. Nature. 2001;412:641–647. doi: 10.1038/35088074. [DOI] [PubMed] [Google Scholar]
- 16.Ito T, Deng X, Carr B, May WS. Bcl-2 phosphorylation required for anti-apoptosis function. J Biol Chem. 1997;272:11671–11673. doi: 10.1074/jbc.272.18.11671. [DOI] [PubMed] [Google Scholar]
- 17.Kermer P, Klocker N, Labes M, Bahr M. Insulin-like growth factor-I protects axotomized rat retinal ganglion cells from secondary death via PI3-K-dependent Akt phosphorylation and inhibition of caspase-3 In vivo. J Neurosci. 2000;20:2–8. [PubMed] [Google Scholar]
- 18.Johnson GV, Stoothoff WH. Tau phosphorylation in neuronal cell function and dysfunction. J Cell Sci. 2004;117:5721–5729. doi: 10.1242/jcs.01558. [DOI] [PubMed] [Google Scholar]
- 19.Ishihara T, Hong M, Zhang B, et al. Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron. 1999;24:751–762. doi: 10.1016/s0896-6273(00)81127-7. [DOI] [PubMed] [Google Scholar]
- 20.Digicaylioglu M, Garden G, Timberlake S, et al. Acute neuroprotective synergy of erythropoietin and insulin-like growth factor I. Proc Natl Acad Sci U S A. 2004;101:9855–9860. doi: 10.1073/pnas.0403172101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaul M, Lipton SA. Chemokines and activated macrophages in HIV gp120-induced neuronal apoptosis. Proc Natl Acad Sci U S A. 1999;96:8212–8216. doi: 10.1073/pnas.96.14.8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Green DA, Masliah E, Vinters HV, et al. Brain deposition of beta-amyloid is a common pathologic feature in HIV positive patients. Aids. 2005;19:407–411. doi: 10.1097/01.aids.0000161770.06158.5c. [DOI] [PubMed] [Google Scholar]
- 23.Born J, Lange T, Kern W, et al. Sniffing neuropeptides: a transnasal approach to the human brain. Nat Neurosci. 2002;5:514–516. doi: 10.1038/nn849. [DOI] [PubMed] [Google Scholar]
- 24.Thorne RG, Pronk GJ, Padmanabhan V, Frey WH., Jr Delivery of insulin-like growth factor-I to the rat brain and spinal cord along olfactory and trigeminal pathways following intranasal administration. Neuroscience. 2004;127:481–496. doi: 10.1016/j.neuroscience.2004.05.029. [DOI] [PubMed] [Google Scholar]
- 25.Fletcher L, Kohli S, Sprague SM, et al. Intranasal delivery of erythropoietin plus insulin-like growth factor-I for acute neuroprotection in stroke. J Neurosurg. 2009;111:164–170. doi: 10.3171/2009.2.JNS081199. [DOI] [PubMed] [Google Scholar]
- 26.Rockenstein E, Torrance M, Adame A, et al. Neuroprotective effects of regulators of the glycogen synthase kinase-3beta signaling pathway in a transgenic model of Alzheimer’s disease are associated with reduced amyloid precursor protein phosphorylation. J Neurosci. 2007;27:1981–1991. doi: 10.1523/JNEUROSCI.4321-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410:988–994. doi: 10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- 28.Rosenzweig MQ, Bender CM, Lucke JP, et al. The decision to prematurely terminate a trial of R-HuEPO due to thrombotic events. J Pain Symptom Manage. 2004;27:185–190. doi: 10.1016/j.jpainsymman.2003.06.010. [DOI] [PubMed] [Google Scholar]
- 29.Miller SB, Moulton M, O’Shea M, Hammerman MR. Effects of IGF-I on renal function in end-stage chronic renal failure. Kidney Int. 1994;46:201–207. doi: 10.1038/ki.1994.260. [DOI] [PubMed] [Google Scholar]
- 30.Stanley LC, Mrak RE, Woody RC, et al. Glial cytokines as neuropathogenic factors in HIV infection: pathogenic similarities to Alzheimer’s disease. J Neuropathol Exp Neurol. 1994;53:231–238. doi: 10.1097/00005072-199405000-00003. [DOI] [PubMed] [Google Scholar]
- 31.Dou H, Ellison B, Bradley J, et al. Neuroprotective mechanisms of lithium in murine human immunodeficiency virus-1 encephalitis. J Neurosci. 2005;25:8375–8385. doi: 10.1523/JNEUROSCI.2164-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reinhardt RR, Bondy CA. Insulin-like growth factors cross the blood-brain barrier. Endocrinology. 1994;135:1753–1761. doi: 10.1210/endo.135.5.7525251. [DOI] [PubMed] [Google Scholar]
- 33.Pardridge WM. Brain drug development and brain drug targeting. Pharm Res. 2007;24:1729–1732. doi: 10.1007/s11095-007-9387-0. [DOI] [PubMed] [Google Scholar]
- 34.Brines ML, Ghezzi P, Keenan S, et al. Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc Natl Acad Sci U S A. 2000;97:10526–10531. doi: 10.1073/pnas.97.19.10526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jabri N, Schalch DS, Schwartz SL, et al. Adverse effects of recombinant human insulin-like growth factor I in obese insulinresistant type II diabetic patients. Diabetes. 1994;43:369–374. doi: 10.2337/diab.43.3.369. [DOI] [PubMed] [Google Scholar]
- 36.Holloway L, Butterfield G, Hintz RL, et al. Effects of recombinant human growth hormone on metabolic indices, body composition, and bone turnover in healthy elderly women. J Clin Endocrinol Metab. 1994;79:470–479. doi: 10.1210/jcem.79.2.7519191. [DOI] [PubMed] [Google Scholar]
- 37.Raine AE. Hypertension, blood viscosity, and cardiovascular morbidity in renal failure: implications of erythropoietin therapy. Lancet. 1988;1:97–100. doi: 10.1016/s0140-6736(88)90293-0. [DOI] [PubMed] [Google Scholar]
- 38.Singbartl G. Adverse events of erythropoietin in long-term and in acute/short-term treatment. Clin Investig. 1994;72:S36–S43. [PubMed] [Google Scholar]
- 39.Fulga TA, Elson-Schwab I, Khurana V, et al. Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat Cell Biol. 2007;9:139–148. doi: 10.1038/ncb1528. [DOI] [PubMed] [Google Scholar]
- 40.Cruz JC, Tseng HC, Goldman JA, et al. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron. 2003;40:471–483. doi: 10.1016/s0896-6273(03)00627-5. [DOI] [PubMed] [Google Scholar]
- 41.Wittman CW, Wszolek MF, Shulman JM, et al. Taupathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science. 2001;293:711–714. doi: 10.1126/science.1062382. [DOI] [PubMed] [Google Scholar]
- 42.Petito CK, Roberts B. Evidence of apoptotic cell death in HIV encephalitis. Am J Pathol. 1995;146:1121–1130. [PMC free article] [PubMed] [Google Scholar]
- 43.Ehrenreich H, Aust C, Krampe H, et al. Erythropoietin: novel approaches to neuroprotection in human brain disease. Metab Brain Dis. 2004;19:195–206. doi: 10.1023/b:mebr.0000043969.96895.3c. [DOI] [PubMed] [Google Scholar]
- 44.Liu XF, Fawcett JR, Thorne RG, et al. Intranasal administration of insulin-like growth factor-I bypasses the blood-brain barrier and protects against focal cerebral ischemic damage. J Neurol Sci. 2001;187:91–97. doi: 10.1016/s0022-510x(01)00532-9. [DOI] [PubMed] [Google Scholar]
- 45.Yu YP, Xu QQ, Zhang Q, et al. Intranasal recombinant human erythropoietin protects rats against focal cerebral ischemia. Neurosci Lett. 2005;387:5–10. doi: 10.1016/j.neulet.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 46.Wyss-Coray T, Masliah E, Toggas SM, et al. Dysregulation of signal transduction pathways as a potential mechanism of nervous system alterations in HIV-1 gp120 transgenic mice and humans with HIV-1 encephalitis. J Clin Invest. 1996;97:789–798. doi: 10.1172/JCI118478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kins S, Beyreuther K. Teasing out the tangles. Nat Med. 2006;12:764–765. doi: 10.1038/nm0706-764. discussion 765. [DOI] [PubMed] [Google Scholar]
- 48.Billingsley ML, Kincaid RL. Regulated phosphorylation and dephosphorylation of tau protein: effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochem J. 1997;323(pt 3):577–591. doi: 10.1042/bj3230577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Han P, Dou F, Li F, et al. Suppression of cyclin-dependent kinase 5 activation by amyloid precursor protein: a novel excitoprotective mechanism involving modulation of tau phosphorylation. J Neurosci. 2005;25:11542–11552. doi: 10.1523/JNEUROSCI.3831-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pei JJ, Braak E, Braak H, et al. Distribution of active glycogen synthase kinase 3beta (GSK-3beta) in brains staged for Alzheimer disease neurofibrillary changes. J Neuropathol Exp Neurol. 1999;58:1010–1019. doi: 10.1097/00005072-199909000-00011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.