

Abstract

It is established that drugs targeting viral proteins are at risk of generating resistant strains. However, drugs targeting host factors can potentially avoid this problem. Herein, we report structure–activity relationship studies leading to the discovery of a very potent lead compound 6-fluoro-2-(5-isopropyl-2-methyl-4-phenoxyphenyl)quinoline-4-carboxylic acid (C44) that inhibits human dihydroorotate dehydrogenase (DHODH) with an IC50 of 1 nM and viral replication of VSV and WSN-Influenza with an EC50 of 2 nM and 41 nM. We also solved the X-ray structure of human DHODH bound to C44, providing structural insight into the potent inhibition of biaryl ether analogues of brequinar.
Keywords: High-throughput screening, structure−activity relationship, antiviral, human DHODH inhibitor
In the last 50 years, several antiviral drugs with high therapeutic value for the treatment of several life threatening diseases such as AIDS, Influenza, Herpes, Hepatitis C, and/or Hepatitis B infections have emerged.1 Most of these drugs inhibit specific virus-encoded enzymes essential for viral replication. However, this type of antiviral drug therapy has several drawbacks. First, because these drugs are designed to target and/or inhibit specific viral enzymes, their efficacy can be quite narrow, sometimes targeting only a subtype of a specific virus. Although this strategy reduces the risks associated with the toxicity caused by targeting viral enzymes nonspecifically, it restricts the usage of these agents.2 Furthermore, most viruses mutate rapidly, leading to drug resistance and thereby limiting the usage of viral specific inhibitors. Resistance to viral inhibitors is a major problem especially for important small RNA viruses such as HIV, HCV, and influenza virus. For viruses with genomes encoding only 10–12 genes, of which only a few function as enzymes, it is difficult to find drugable targets for small molecules despite substantial medicinal chemistry efforts.3
Virus physiology and often their pathogenicity are highly dependent upon host cell factors. Viral proteins and genomes often use host cell machinery to generate highly infectious viral progeny and, more often than not, evade and antagonize host antiviral immune response.4 This raises the possibility that host genes, proteins, and/or pathways that are needed for viral replication and pathogenesis, can be used as targets for antiviral drug discovery. Some examples of potential host cell targets include protein kinases, cell surface receptors, the proteasome, and nuclear receptors.5−8 One inherent problem might be the toxicity associated with inhibiting host cell pathways and/or enzymes. Nonetheless, complete dependence of the viruses on the cellular functions of the host cell, availability of only a handful of broad spectrum antiviral drugs,9−11 and the presence of several unexplored host cell targets interested us in this approach.
Among potential viral drug targets, dihydroorotate dehydrogenase (DHODH) is unique. It is the fourth enzyme of pyrimidine de novo synthesis that catalyzes the conversion of dihydroorotate to orotate.12 Pyrimidines are essential for the biosynthesis of DNA, RNA, and phospholipids.13 Inhibition of DHODH causes reduced levels of pyrimidine nucleotides that in turn trigger various activities such as anticancer,14 immunosuppressive,15 antimalarial,16 and antifungal.17 Additionally, inhibition of DHODH can be beneficial for spinal cord injury and rheumatoid arthritis.18 Various DHODH inhibitors have evolved over the years out of which brequinar15 and leflunomide18 have gained a lot of attention due to their high potency. Recently, we described the discovery of 4-quinoline carboxylic acids with antiviral (IC50 = 110 nM for VSV) activity via a high-throughput screen and brief SAR study19 and disclosed the target enzyme for the antiviral activity to be human DHODH.19 Other inhibitors of pyrimidine biosynthesis were also reported to have antiviral activity.20,21 Herein, we report an extensive structure–activity relationship study leading to the identification of compound C44 with excellent inhibitory activity for viruses (EC50 = 1.9 nM for VSV and 41 nM for WSN) and human DHODH (IC50 = 1.0 nM).
Given the structural relationship of HTS hit C1 to brequinar, we recognized that there are several key pharmacophoric regions where appropriate substituents are necessary (Figure 1).15,22−24 For example, a free carboxylic acid is crucial, whereas a hydrophobic moiety is necessary at C(2). Additionally, electron withdrawing groups such as fluorine, chlorine, and trifluoromethyl have a beneficial effect at C(7). Lastly, it was reported that although the methyl group at C(3) of brequinar did not influence DHODH activity, it did significantly improve cellular activity in a mixed lymphocyte reaction (MLR) assay.15
Figure 1.
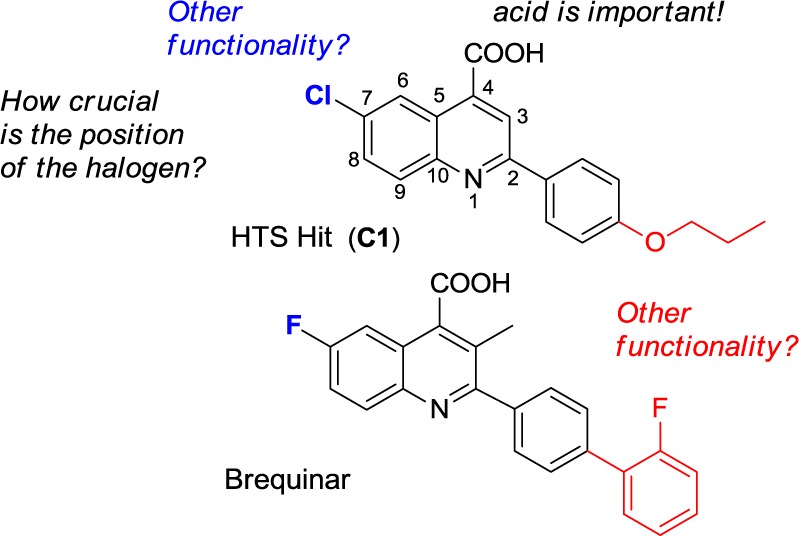
SAR starting point.
Guided by the structure activity relationship insights of brequinar, we started our structure optimization study of C1. Initially, we kept the chloroquinoline part of the molecule constant and synthesized compounds C2 to C11.25 As shown in Table 1, replacing the propyl ether of C1 by methyl, ethyl, or butyl (C2-C4) ethers did not result in improved activity in the vesicular stomatitis virus (VSV) viral replication assay.26 However, replacing the propoxy group by fluorine (C6), bromine (C7), or trifluoromethoxy (C5) resulted in significant higher activity. In order to elucidate the importance of the ether linkage, we prepared analogues C8–C10 by replacing the alkoxy groups of C2, C3, and C5 with their respective alkyl chain counterparts. In general, the alkyl-substituted analogues were equipotent or less active than their corresponding alkoxy counterparts. Given the beneficial effect of an additional aromatic ring (2-F-Ph) connected to the C(2)-phenyl ring in brequinar, we decided to explore the diaryl ether analogue C11. Although less active than the trifluoromethoxy-substituted analogue C5 (0.479 μM), diaryl ether analogue C11 demonstrated improved activity versus the other analogues in Table 1 (1.293 μM versus 1.770–35.32 μM). This observation opens the possibility to significantly expand SAR studies via exploration of a large set of diverse diaryl ethers (vide infra).
Table 1. SAR of the Right-Hand Aryl Ring.
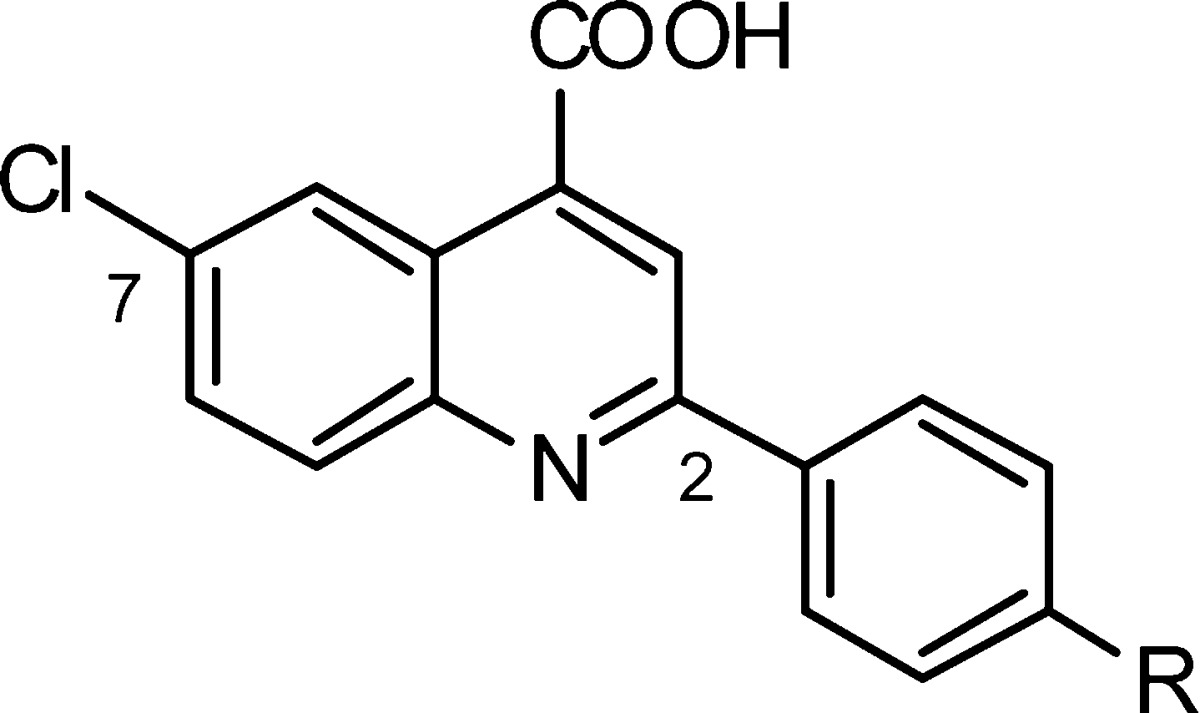
| compd | R | EC50 (μM)a |
|---|---|---|
| brequinar | see Figure 1 | 0.3 |
| C1 | O(CH2)2CH3 | 4.7 |
| C2 | OCH3 | 7.1 |
| C3 | OCH2CH3 | 5.7 |
| C4 | O(CH2)3CH3 | 6.3 |
| C5 | OCF3 | 0.5 |
| C6 | F | 1.8 |
| C7 | Br | 2.2 |
| C8 | CH3 | 8.7 |
| C9 | CH2CH3 | 5.7 |
| C10 | CF3 | 35.3 |
| C11 | OPh | 1.3 |
Inhibition of VSV replication in MDCK epithelial cells.
Our approach thus focused on the optimization of C11. With a series of analogues displayed in Table 2, we explored functional group tolerance in the 4-quinoline carboxylic acid ring system. Replacement of the C(7)-chlorine with fluorine (C12) showed a significant 10-fold boost in activity (0.110 μM), whereas bromine (C13) and nitro (C16) functionality at this position was only moderately tolerated (EC50 = 3.10 and 1.98 μM, respectively). However, trifluoromethoxy (C14), trifluoromethyl (C17), and methoxy (C15) functionality is detrimental for activity (EC50 = 95.64, 16.58, and 2611 μM, respectively). Interestingly, the unsubstituted analogue C18 exhibited increased activity (EC50 = 0.96 μM), indicating that the presence of small groups like fluorine and hydrogen are better tolerated. To investigate how crucial the position of the fluorine atom is, additional compounds were synthesized and screened against the in vitro assay. The C(9)-fluoro and the C(7),C(8)-difluoro analogues C19 and C21 were about 10-fold less potent than the C(7)-fluoro analogue C 12, whereas introduction of fluorine at the C(8)-position completely abolished activity (C20).
Table 2. SAR of the Left-Hand Ring.
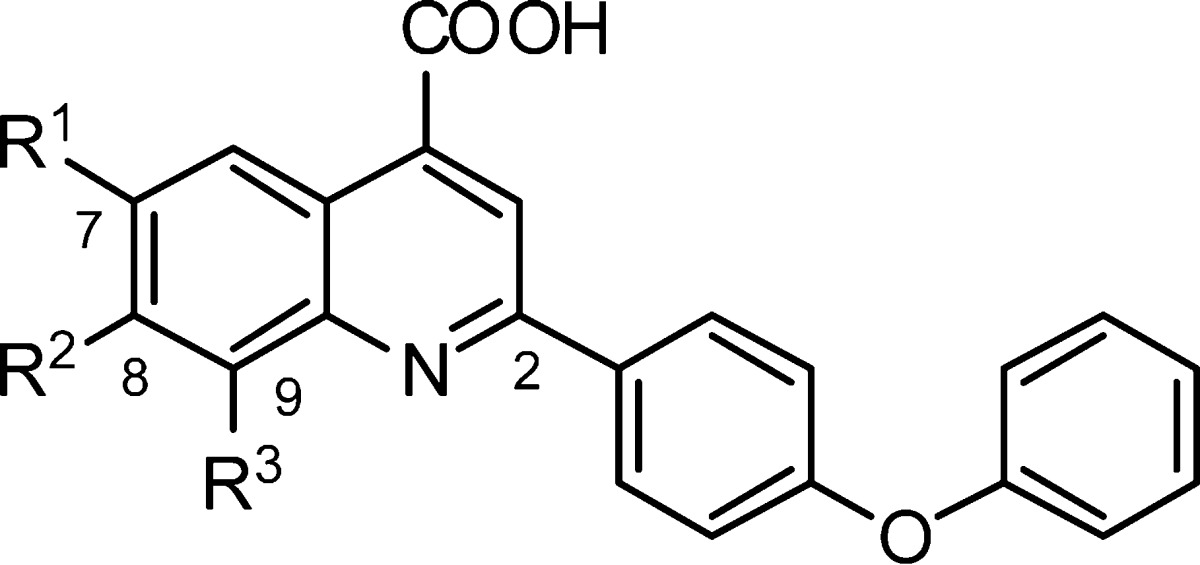
| compd | R1 | R2 | R3 | EC50 (μM)a |
|---|---|---|---|---|
| C11 | Cl | H | H | 1.3 |
| C12 | F | H | H | 0.1 |
| C13 | Br | H | H | 3.1 |
| C14 | OCF3 | H | H | 95.6 |
| C15 | OCH3 | H | H | 2611 |
| C16 | NO2 | H | H | 2.0 |
| C17 | CF3 | H | H | 16.6 |
| C18 | H | H | H | 1.0 |
| C19 | H | H | F | 1.2 |
| C20 | H | F | H | 158.8 |
| C21 | F | F | H | 1.6 |
Inhibition of VSV replication in MDCK epithelial cells.
At this point, it is useful to make some comparisons to analogous SAR studies reported for brequinar.15 In a cellular assay (mixed lymphocyte reaction), which presumably captures similar issues of cell uptake as in our cellular assay (VSV-replication), the potency of the C(7)-chloro-analogue was reduced only 2-fold (compared to C(7)-fluoro) versus >10-fold in our series. In addition, the cellular activity of the nonsubstituted (C(7)-H) analogue was reduced >100-fold (compared to C(7)-F) versus a 9-fold difference reported here. More significantly, where we observe a significantly decreased activity (∼150-fold) when substituting a trifluoromethyl for the C(7)-fluorine (C17 vs C12), an increased 2-fold cellular activity was reported for a similar replacement in the brequinar series of compounds.15 Finally, as reported for the brequinar series,15 methyl ester formation (C12-methyl ester) virtually abolished activity in the VSV assay (EC50 = 86.46 μM).
With C12 as our best analogue thus far, we decided to return to the C(2) diaryl ether moiety of the molecule and synthesized various analogues of C12 (C22–C36, Table 3).25 Following the same trend as observed for the C(7)-chloro series (Table 1), the diaryl ether C12 was more potent than the corresponding aryl-alkyl ethers C22–C27. The 3,4-(methylenedioxy)phenoxy (C30), 4-chlorophenoxy (C28), and 4-nitrophenoxy (C29) analogues were approximately 9- to 45-fold less active than the nonsubstituted analogue C12. Of the three fluorophenyl analogues, C31 and C33 performed slightly less, whereas the 3-F-Ph analogue C32 displayed an activity similar to lead C12. Finally, heteroaromatic substitution was less fruitful as indicated by the weak activity exhibited by the pyridyl and thiazolyl analogues C34–C36.
Table 3. SAR of the Right-Hand Phenol.

| compd | R | EC50 (μM)a |
|---|---|---|
| C12 | Ph | 0.1 |
| C22 | CH3 | 6.4 |
| C23 | CH2CH3 | 0.6 |
| C24 | (CH2)2CH3 | 19.0 |
| C25 | (CH2)3CH3 | 2.1 |
| C26 | CH2(C3H5) | 6.8 |
| C27 | CF3 | 1.0 |
| C28 | 4-Cl–Ph | 2.0 |
| C29 | 4-NO2–Ph | 4.9 |
| C30 | 3,4-(OCH2O)–Ph | 0.9 |
| C31 | 2-F–Ph | 0.3 |
| C32 | 3-F–Ph | 0.1 |
| C33 | 4-F–Ph | 1.0 |
| C34 | 2-pyridyl | 22.9 |
| C35 | 3-pyridyl | 2.5 |
| C36 | 2-thiazolyl | 14.6 |
Inhibition of VSV replication in MDCK epithelial cells.
Because SAR studies leading to brequinar indicated that a C(3)-methyl substituent increased cellular activity 10-fold (DHOD enzyme activity did not change however),15 we synthesized C(3)-methyl substituted analogue C37 (Chart 1). Surprisingly, this compound was significantly less potent (∼70-fold) compared to the nonsubstituted congener C12. Restricting conformational freedom with a planarizing fusion of the quinoline and C(2)-aromatic ring proved also unbeneficial (compound C38 in Chart 1).22
Chart 1. Conformationally Restricted Analogues.

With C12 as our most potent analogue, we made a final push to improve activity by exploring a set of diaryl ether analogues (Chart 2).25 The activities of C39–C41 were found to be only slightly less than our most potent compound C12. Interestingly, the t-butyl substituted analogue C42 was more potent, and sterically hindered diaryl ether C43 showed much improved potency. On the basis of this result, we envisioned that restriction of rotation across the diaryl ether linkage coupled with the increase in hydrophobicity might be crucial for activity. This hypothesis led to our final analogue C44 that showed a significant 50-fold increase in potency versus C12 (2 nM) in the VSV in vitro assay.
Chart 2. VSV Inhibition with Diaryl Ether Analoguesa.

a Inhibition of VSV replication in MDCK epithelial cells.
In Table 4, we compare several analogues in assays measuring inhibition of human DHODH enzyme activity and in vitro influenza-WSN viral replication. The data for inhibition of VSV viral replication are included for comparison. Thus, starting from compound C1 with modest activity, a submicromolar analogue C12 was developed and further optimized to the low nanomolar biaryl ether analogue C44. In the VSV assay, compound C44 was 2 orders of magnitude more potent than brequinar at nontoxic concentrations (cytotoxicity in human bronchial epithelial and Caco-2 cells: IC50 ≥ 10 μM; see Figures S2 and S3 in the Supporting Information).
Table 4. DHODH Enzyme, VSV, and WSN Viral Growth Inhibition for Selected Analoguesa.
| compd | DHODH IC50 (nM) | VSV EC50 (nM) | WSN EC50 (nM) |
|---|---|---|---|
| C1 | 260 | 4650 | 7600 |
| C5 | 310 | 479 | NAb |
| C6 | 1600 | 1770 | NA |
| C8 | 5900 | 8650 | NA |
| C12 | 36 | 110 | 1380 |
| C11 | 95 | 1330 | 1884 |
| C42 | 19 | 65 | 55 |
| C43 | 16 | 23 | 71 |
| C44 | 1 | 2 | 41 |
| brequinar | 12 | 304 | 460 |
Assay conditions are described in the Supporting Information.
NA: not assayed.
The X-ray structure of C44 in complex with human DHODH was solved to provide insight into the structural basis for the potent binding.27 Previous X-ray structures of human DHODH utilized a truncated soluble enzyme containing amino acids 30–396, and the highest resolution was obtained for the complex with brequinar,12 solved to 1.6 Å. In order to improve crystallization, we generated a new expression construct (pET28b-hDHODH33–396) containing amino acids 33–396. Using this construct, we were able to obtain crystals of human DHODH bound to C44 that diffracted to 1.22 Å resolutions. The crystals formed in a space group of P3221 with the cell dimension a = b = 90.87 and c = 122.19, and the structure was refined by molecular replacement to an Rfree/Rcrysal of 16.1 and 14%, respectively. The refined structure contained residues 34–396, with the exception that density was not observed for a loop formed by residues 71–73 and 222–225. Cofactor FMN, substrate l-DHO, inhibitor C44, 300 water molecules, and a bound Zwittergent 3–10 detergent molecule were also present in the structure.
The structure of the bound C44 shows that the inhibitor binds in the same site as was observed for brequinar (Figure 2), overlaying with an rmsd of 0.7 Ǻ when the structures were superimposed with DaliLite.28 The binding site is predominantly hydrophobic, and the only H-bond interaction is formed between the inhibitor carboxylate and R136. Several amino acid residues (L46, F62, L67, and Y38) in the binding site have rotated to accommodate the isopropyl group on the diaryl ether moiety. Thus, as has been observed previously,29,30 structural plasticity in the DHODH inhibitor binding site allows it to accommodate a more diverse range of ligands than would be expected of a rigid structure. The phenyl ring of the F62 appears to interact with the terminal phenyl ring of the inhibitor via a T-shaped π-stacking fashion. Additionally, the isopropyl moiety appears to add additional hydrophobic interactions within the pocket that provide additional binding energy, thus explaining the potent inhibition of this compound.
Figure 2.
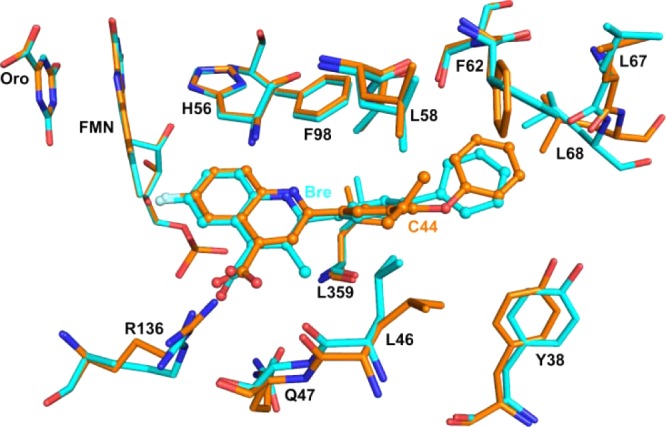
X-ray structure of human DHODH bound to C44 solved to 1.2 Å resolutions. The 4 Å shell around C44 is displayed, and the structure is aligned to the previously reported human DHODH structure complexed with brequinar (1D3G). The protein chain for the C44 structure is shown in orange, and the brequinar structure is shown in turquoise. The bound C44 and brequinar are shown as ball and stick. The bound FMN cofactor and the product orotate are also shown.
In conclusion, we have discovered a new class of diaryl ether-substituted 4-quinolinecarboxylic acid analogues of brequinar with potent human DHODH inhibitory and antiviral activity. Analogue C44 was identified as a promising candidate that showed broad spectrum antiviral activity (2 orders of magnitude more potent than brequinar) via the inhibition of human DHODH enzyme. A cocrystal structure of analogue C44 bound to human DHODH was solved and provides structural insight into the potent inhibition of this particular biaryl ether analogue.
Acknowledgments
Results shown in this report are partially derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357. We thank Dr. Soumya Krishnamurthy, Department of Biochemistry, for generating the data presented in Supplemental Figure S3. J.K.D.B. holds the Julie and Louis Beecherl, Jr., Chair in Medical Science, M.A.P. the Carolyn R. Bacon Professorship in Medical Science and Education and the Beatrice and Miguel Elias Distinguished Chair in Biomedical Science, and M.G.R. the Diane and Hal Brierely Distinguished Chair in Biomedical Research.
Glossary
Abbreviations
- DHODH
dihydroorotate dehydrogenase
- FMN
flavin mononucleotide
- l-DHO
l-dihydroorotate
- MDCK
Madin–Darby canine kidney
- rmsd
root-mean-square deviation
- SAR
structure–activity relationship
- VSV
vesicular stomatitis virus
- WSN virus
influenza A/WSN/33 (H0N1) virus
Supporting Information Available
Synthetic procedures and characterization data, assay conditions, and details regarding the crystal structure solution of human DHODH bound to analogue C44. This material is available free of charge via the Internet at http://pubs.acs.org.
Financial support was provided by the National Institutes of Health through grants GM07159 (to B.M.A.F.), AI079110 (to B.M.A.F. and M.G.R.), AI089539 (to B.M.A.F. and M.G.R.), and AI53680 (to M.A.P.). J.K.D.B and M.A.P thank the Robert A. Welch Foundation for support (grants I-1422 and I-1257).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- De Clercq E., Ed. Antiviral Drug Strategies, 1st ed.; Wiley-VCH Verlag GMBH & Co KGaA: Berlin, Germany, 2011; Chapter 1. [Google Scholar]
- Richman D. D. The implications of drug resistance for strategies of combination antiviral chemotherapy. Antiviral Res. 1996, 29, 31–33. [DOI] [PubMed] [Google Scholar]
- Magden J.; Kääriäinen L.; Ahola T. Inhibitors of virus replication: recent developments and prospects. Appl. Microbiol. Biotechnol. 2005, 66, 612–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katze M. G.; He Y.; Michael G. Jr. Viruses and interferon: a fight for supremacy. Nat. Rev. Immunol. 2002, 2, 675–687. [DOI] [PubMed] [Google Scholar]
- DeFilippis V.; Raggo C.; Moses A.; Früh K. Functional genomics in virology and antiviral drug discovery. Trends Biotechnol. 2003, 21, 452–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Früh K.; Simmen K.; Luukkonen B. G. M.; Bell Y. C.; Ghazal P. Virogenomics: a novel approach to antiviral drug discovery. Drug Discovery Today 2001, 6, 621–627. [DOI] [PubMed] [Google Scholar]
- Tan S. L.; Gangi G.; Paeper B.; Proll S.; Katze M. G. Systems biology and the host response to viral infection. Nat. Biotechnol. 2007, 25, 1383–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFilippis V.; Früh K. Host cell targets for antiviral therapy: an update. Future Virol. 2006, 1, 509–518. [Google Scholar]
- Rider T. H.; Zook C. E.; Boettcher T. L.; Wick S. T.; Pancoast J. S.; Zushman B. D. Broad-spectrum antiviral therapeutics. PLoS One 2011, 6, e22572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf M. C.; Freiberg A. N.; Zhang T.; Akyol-Ataman Z.; Grock A.; Hong P. W.; Li J.; Watson N. F.; Fang A. Q.; Aguilar H. C.; Porotto M.; Honko A. N.; Damoiseaux R.; Miller J. P.; Woodson S. E.; Chantasirivisal S.; Fontanes V.; Negrete O. A.; Krogstad P.; Dasgupta A.; Moscona A.; Hensley L. E.; Whelan S. P.; Faull K. F.; Holbroock M. R.; Jung M. E.; Lee B. A broad-spectrum antiviral targeting entry of enveloped viruses. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 3157–3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R. A.; Kirkpatrick W.; Eds. Ribavarin: A Broad Spectrum Antiviral Agent; Academic Press: New York, 1980; pp 1–21. [Google Scholar]
- Liu S.; Neidhardt E. A.; Grossman T. H.; Ocain T.; Clardy J. Structures of human dihydroorotate dehydrogenase in complex with antiproliferative agents. Structure 2000, 8, 25–33. [DOI] [PubMed] [Google Scholar]
- Löeffler M.; Zameitat E. Pyrimidine biosynthesis. Encycl. Biol. Chem. 2004, 3, 600–605. [Google Scholar]
- Shawver L. K.; Schwartz D. P.; Mann E.; Chen H.; Tsai J.; Chu L.; Taylorson L.; Longhi M.; Meredith S.; Germain L.; Jacobs J. S.; Tang C.; Ullrich A.; Berens M. E.; Hersh E.; McMahon G.; Hirth K. P.; Powell T. J. Inhibition of platlet-derived growth factor-mediated signal transduction and tumor growth by N-[4-(trifluoromethyl)-phenyl]-5-methylisooxazole-4-carboxamide. Clin. Cancer Res. 1997, 3, 1167–1177. [PubMed] [Google Scholar]
- Batt D. G.; Copeland R. A.; Dowling R. L.; Gardner T. L.; Jones E. A.; Orwat M. J.; Pinto D. J.; Pitts W. J.; Magolda R. L.; Jaffee B. D. Immunosuppressive structure–activity relationships of brequinar and related chinchoninic acid derivatives. Bioorg. Med. Chem. Lett. 1995, 5, 1549–1554. [DOI] [PubMed] [Google Scholar]
- Phillips M. A.; Rathod P. K. Dihydroorotate dehydrogenase is a promising target for the identification of novel anti-malarial chemotherapy. Infect. Disord.: Drug Targets 2010, 10, 226–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson G.; Davis G.; Waldron C.; Smith A.; Henry M. Identification of a new antifungal target site through a dual biochemical and molecular-genetics approach. Curr. Genet. 1996, 30, 159–165. [DOI] [PubMed] [Google Scholar]
- Rosse G. Novel use of leflunomide and malononitrilamides. ACS Med. Chem. Lett. 2012, 3, 785–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Das P.; Schmolke M.; Manicassamy B.; Wang Y.; Deng X.; Cai L.; Tu B. P.; Forst V. F.; Roth M. G.; Levy D. E.; Garcia-Sastre A.; De Brabander J. K.; Phillips M. A.; Fontoura B. M. A. Inhibition of pyrimidine synthesis reverses viral virulence factor-mediated block of mRNA export. J. Cell. Biol. 2012, 196, 315–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman H. H.; Kunz A.; Simon V. A.; Palese P.; Shaw M. L. Broadspectrum antiviral that interferes with de novo pyrimidine biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 5777–5782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhoff E.; Tylden G. D.; Kjerpeseth L. J.; Gutteberg T. J.; Hirsch H. H.; Rinaldo C. H. Leflunomide inhibition of BK virus replication in renal tubular epithelial cells. J. Virol. 2010, 84, 2150–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitts W. J.; Jetter J. W.; Pinto D. J.; Orwat M. J.; Batt D. G.; Sherk S. R.; Petraitis J. J.; Jacobson I. C.; Copeland R. A.; Dowling R. L.; Jaffee B. D.; Gardner T. L.; Jones E. A.; Magolda R. L. Structure–activity relationships (SAR) of some tetracyclic heterocycles related to the immunosuppressive agent brequinar sodium. Bioorg. Med. Chem. Lett. 1998, 8, 307–312. [DOI] [PubMed] [Google Scholar]
- Batt D. G.; Petraitis J. J.; Sherk S. R.; Copeland R. A.; Dowling R. L.; Taylor T. L.; Jones E. A.; Magolda R. L.; Jaffee B. D. Heteroatom- and carbon-linked biphenyl analogs of brequinar as immunosuppressive agents. Bioorg. Med. Chem. Lett. 1998, 8, 1745–1750. [DOI] [PubMed] [Google Scholar]
- Chen S.-F.; Papp L. M.; Ardecky R. J.; Rao G. V.; Hesson D. P.; Forbes M.; Dexter D. L. Structure–activity relationship of quinoline carboxylic acids. A new class of inhibitors of dihydroorotate dehydrogenase. Biochem. Pharmacol. 1990, 40, 709–714. [DOI] [PubMed] [Google Scholar]
- See the Supporting Information for the synthesis of all new compounds.
- For assay conditions, see the Supporting Information.
- The coordinates have been deposited in the Protein Data Bank, with ID 4IGH.
- Holm L.; Park J. DaliLite workbench for protein structure comparison. Bioinformatics 2000, 16, 566–567. [DOI] [PubMed] [Google Scholar]
- Deng X.; Gujjar R.; El Mazouni F.; Goldsmith E. J.; Rathod P. K.; Phillips M. A. Structural plasticity of malaria dihydroorotate dehydrogenase allows selective binding of diverse chemical scaffolds. J. Biol. Chem. 2009, 284, 26999–27009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walse B.; Dufe V. T.; Svensson B.; Fritzson I.; Dahlberg L.; Khairoullina A.; Wellmar U.; Al-Karadaghi S. The structure of human dehydroorotate dehydrogenase with and without inhibitor reveal conformational flexibility in the inhibitor and substrate binding sites. Biochemistry 2008, 47, 8929–8936. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.