

Abstract
We have developed an efficient chemoenzymatic synthesis of heparan sulfate oligosaccharides employing the para-nitrophenyl (p-NP) β-glucuronide as an acceptor compatible with enzymatic elongation and one that significantly simplifies oligosaccharide purification on C-18 resin. Employing ceric ammonium nitrate as oxidative reagent to remove the p-NP group unexpectedly also removed the glucuronic acid residue at the reducing-end, affording a smaller oligosaccharide. The application of ceric ammonium sulfate allowed the removal of the p-NP without concomitant loss of the adjacent glucuronic acid offering a route to longer heparin sulfate oligosaccharide products.
Keywords: Heparan sulfate oligosaccharides, chemoenzymatic synthesis, para-nitrophenyl glucuronic acid, ceric ammonium salt, deprotection
Introduction
Heparan sulfate (HS) and heparin (HP) which belong to the family of glycosaminoglycans (GAGs) are composed of (1→4)-linked alternating glucosamine and uronic acid residues containing diverse substitution with O-sulfo, N-acetyl and N-sulfo groups.1 HS, which has ubiquitous distribution on the cell surface and in the extracellular matrix, contains more varied structure but less sulfo groups than does heparin.2 HS/HP GAGs play important roles in several physiological processes at the cell–tissue–organ interface including cell adhesion, enzyme regulation, cytokine action, and the most convincing anticoagulation, etc.3 HP widely used as an clinical anticoagulant is mainly used in the forms of unfractionated heparin (UFH), low molecular weight heparins (LMWHs) and the synthetic ultralow molecular weight heparins (ULMWHs), such as the drug known as Arixtra.4
Chemical synthesis of oligosaccharides, which includes construction of building blocks, stereo- and regioselective glycosylation and deprotection steps, etc., has becoming more and more critical to obtain pure substances for developing structure-activity relationships.5 However the chemical synthesis requires a large number of complicated steps, many that are low yielding and challenging purification of both intermediates and products. Chemoenzymatic synthesis of oligosaccharides represents a relatively more efficient approach to obtain diverse targets.6 In our previous work, 10-step and 12-step chemoenzymatic syntheses afforded two structurally homogeneous ULMWHs (molecular mass = 1778.5 and 1816.5) in 45% and 37% overall yield, respectively.7
Although recent advances in chemoenzymatic synthesis have made the construction of complex heparin-like oligosaccharide increasingly feasible,8 speeding up the purification procedure after each enzymatic reaction was still necessary to obtain pure oligosaccharide targets in a shorter time and in higher overall yields. The p-NP group is an ultraviolet detectable, hydrophobic tag that can be reversibly bound to reversed phase chromatography resins.9 C-18 silica resin was useful for purifying p-NP glycosides of oligosaccharides of various sizes. After the final purification step the p-NP tag needed to be cleaved from the product to achieve our final desired oligosaccharide target. However, general and practical methods for deprotection of p-NP group are not well established, even though some successful examples have been reported on the protected sugar substrates10 or the similar substrates taged with p-nitrobenzyl (p-NPM), p-methoxybenzyl (p-MPM) and p-pivaloylaminobenzyl (p-PAB) groups.11 In this communication, we examined the oxidative deprotection of p-NP group with cerium reagents under mild oxidative conditions.
The chemoenzymatic synthesis of HS oligosaccharide backbones based on the p-NP β-glucuronide (p-NPG, 1) is presented in Scheme 1. The strong UV absorbance and highly C-18 binding affinity of the p-NP group greatly simplifies the process of oligosaccharide purification. To construct the heparin oligosaccharide backbone, p-NPG 1 acceptor was incubated with uridine diphosphate (UDP)-N-acetylglucosamine (GlcNAc) or UDP-N-trifluoroacetyl glucosamine (GlcNTFA) donor and KfiA (N-acetylglucosaminyl transferase), expressed in Escherichia coli K5, at room temperature overnight to furnish the disaccharide. Next, the disaccharide was treated with UDP-glucuronic acid (GlcA) and pmHS2 (heparosan synthase-2), from Pasteurella multocida, and incubated for 4–5 h, followed by a second addition of UDP-GlcA and pmHS2 to drive the enzymatic reaction to completion. During both enzymatic reactions, high performance liquid chromatography (HPLC) on C18 silica column was used to monitor the consumption of acceptor and smooth generation of products. Alternatively, while thin layer chromatography (TLC) could also be employed to monitor the enzymatic reaction it required ten-fold more sample for detection. Trisaccharides 2 and 3, with p-NP group at the reducing-ends, were obtained after a flash chromatograhy on a reverse phase C-18 column. Another cycle of enzymatic reaction was carried out to construct the tetrasaccharide and pentasaccharide (Scheme 1). Reversed phase C-18 flash column was again performed in preparative scale to obtain sufficient pentasaccharide 4 and 5 backbone structures for further modification.12
Scheme 1.

Backbone construction of HS oligosaccharides
With these substrates in hand, we next explored the oxidative deprotection of p-NP group using ceric ammonium nitrate (CAN). Conversion of nitro group, on the p-NP on trisaccharide 2, to an acetamido group on was first carried out by the hydrogenation with Pd(OH)2/C in THF-H2O-acetic anhydride (4 : 5 : 1) furnishing the corresponding acetamidophenyl glycoside. We found that the intermediate with a free aminophenyl group was extremely sensitive to polymerization, so the freshly prepared acetic anhydride solution was added to quickly and quantitatively convert the amino intermediate stable acetamidophenyl glycoside. Oxidative cleavage reaction using 5 eq. of CAN at 0 °C for 30 min in water solution was based on the reported cleavage of the similar p-methoxyphenyl group.11 However, a yield of less than 5%, detected by LC-MS, of desired trisaccharide was observed. Instead, an unexpected disaccharide was obtained in 80% yield and its structure was confirmed, using MS and NMR, to be missing the reducing-end GlcA residue. A similar reaction was observed when the tetrasaccharide or pentasaccharide was treated under the same conditions, affording the corresponding trisaccharide and tetrasaccharide with loss of the reducing-end GlcA (Scheme 2.) Thus, CAN-mediated oxidation removed the entire p-NPG not just the p-NP group.
Scheme 2.

Deprotection of p-NP group with ceric ammonium nitrate
The UDP-GlcNTFA donor was used to prepare a more structurally complex oligosaccharide that could be deprotected using triethylamine and N-sulfonated using recombinant N-sulfotransferase to furnish HS hexasaccharide 9 (Scheme 3).13 The CAN-mediated oxidative approach was applied on 9 and expected to afford a relatively pure pentasaccharide after size exclusive chromatography. Unfortunately, no pentasaccharide or hexasaccharide was obtained on oxidative reaction with CAN. Therefore, we decided to the investigate deprotection using the milder oxidative reagent, ceric ammonium sulfate (CAS), with a lower reduction potential of +1.44 V (versus the normal hydrogen electrode) than the +1.61 V for CAN.14 On treatment with 5 eq CAS at 0 °C, hexasaccharide 10 was oxidized to afford the product 11 in ~90% yield after column purification (Scheme 3).15 Thus, the milder oxidative reaction with CAS proceeded smoothly removing the p-NP group without the loss of the reducing-end GlcA, affording only the desired hexasaccharide backbone.
Scheme 3.

Deprotection of p-NP with ceric ammonium sulfate on HS oligosaccharide
1D and 2D NMR spectroscopy of substrate 9 and HS hexasaccharide 11 clearly support the mild nature of the oxidative deprotection of the p-NP group with CAS, (Figure 1).16 The aromatic protons of intermediate 10 appear as doublets at 7.27 and 7.05 ppm in 1H NMR and are distinguished from the signals 8.14 and 7.11 ppm of the substrate 9, confirming the successful hydrogenation of nitro group. The disappearance of signals in aromatic region of 1H NMR after CAS treatment clearly demonstrates the deprotection of the aryl group. The anomeric protons on each sugar unit were assigned based on the 2D 1H-1H correlation spectroscopy (COSY) and 1H-13C heteronuclear single quantum coherence (HMQC) spectroscopy and the stereochemistry on glycosidic bond was confirmed through the analysis of 1JCH on each anomeric carbon atom. TLC analysis (HCOOH/BuOH/H2O, 4/8/1) also confirmed the reduction and removal of the p-NP group.
Figure 1.

NMR characterization of heparan sulfate oligosaccharides. Panel I shows the 1D 1H NMR spectrum of compound 9 (R1 = p-NP, R2 = H). Panel II shows the 1D 1H NMR spectrum of compound 11 (R1 = H, R2 = Ac). Peaks corresponding to the anomeric protons of these compounds can be clearly identified. Panel III shows the selected anomeric carbon region (91 ppm to 103 ppm) in 2D 1H-13C HMQC spectrum of compound 11.
Further experiments were carried out to investigate whether the deprotection of p-NP group with CAS could smoothly occur without removal of the reducing-end GlcA on other oligosaccharides as well. Similar results were observed in the cases of oxidative p-NP deprotection of compounds 2 and 4 with CSA, affording the corresponding trisaccharide 12 and pentasaccharides 13 in relatively high yields (Scheme 4). Further studies will be required to determine how the oxidative reactivity of the p-NP group is impacted by oligosaccharide size and the number and position of N-acetyl or N-sulfo groups within p-NP tagged oligosaccharides.
Scheme 4.
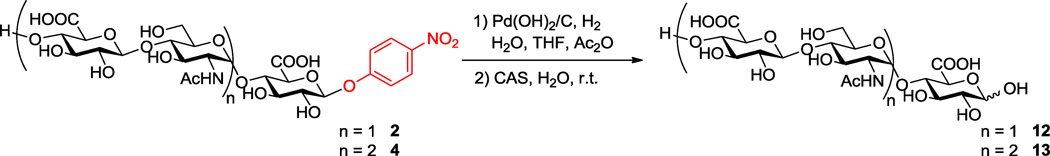
Deprotection of p-NP group with ceric ammonium sulfate
Conclusions
The HS hexasaccharide was successfully synthesized through a chemoenzymatic approach employing a p-NPG acceptor in which the p-NP group serves as a CAS removable tag. This protection/deprotection strategy should allow the construction of more structurally complex and highly sulfated HS oligosaccharides and ULMWHs. These targets are currently viewed as potential therapeutic agents for the modulation of anticoagulant and antithrombotic activities. The successful application of oxidative deprotection reaction using CAS supplies also provides a milder alternative for the deprotection of other sensitive substrates.
Acknowledgements
This work was supported by the National Institutes of Health in the form of grants HL62244, GM38060, and HL094463.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.a) Sasisekharan R, Venkataraman G. Curr. Opin. Chem. Biol. 2000;4:626–631. doi: 10.1016/s1367-5931(00)00145-9. [DOI] [PubMed] [Google Scholar]; b) Bishop JR, Schuksz M, Esko JD. Nature. 2007;446:1030–1037. doi: 10.1038/nature05817. [DOI] [PubMed] [Google Scholar]; c) Laremore TN, Zhang F, Dordick JS, Liu J, Linhardt RJ. Curr. Opin. Chem. Biol. 2009;13:633–640. doi: 10.1016/j.cbpa.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Salmivirta M, Lidholt K, Lindahl U. The FASEB Journal. 1996;10:1270–1279. doi: 10.1096/fasebj.10.11.8836040. [DOI] [PubMed] [Google Scholar]
- 3.Capila I, Linhardt RJ. Angew. Chem. Int. Ed. 2002;41:391–412. doi: 10.1002/1521-3773(20020201)41:3<390::aid-anie390>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 4.a) Linhardt RJ, Liu J. Current Opinion in Pharmacology. 2012;12:217–219. doi: 10.1016/j.coph.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sommers CD, Ye H, Kolinski RE, Nasr M, Buhse LF, Al-Hakim A, Keire DA. Anal. Bioanal. Chem. 2011;401:2445–2454. doi: 10.1007/s00216-011-5362-z. [DOI] [PubMed] [Google Scholar]
- 5.a) Petitou M, van Boeckel CAA. Angew. Chem. Int. Ed. 2004;43:3118–3133. doi: 10.1002/anie.200300640. [DOI] [PubMed] [Google Scholar]; b) Arungundram S, Boons GJ. J. Am. Chem. Soc. 2009;131:17394–17405. doi: 10.1021/ja907358k. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wang Z, Xu Y, Yang B, Tiruchinapally G, Sun B, Liu R, Dulaney S, Liu J, Huang X. Chem. Eur. J. 2010;16:8365–8375. doi: 10.1002/chem.201000987. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Hu Y–P, Lin S–Y, Huang C–Y, Zulueta MML, Liu J–Y, Chang W, Huang S–C. Nat. Chem. 2011;3:557–563. doi: 10.1038/nchem.1073. [DOI] [PubMed] [Google Scholar]; e) Cai C, Solakyildirim K, Yang B, Beaudet JM, Weyers A, Linhardt RJ, Zhang F. Carbohydrate Polymers. 2012;87:822–829. doi: 10.1016/j.carbpol.2011.08.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Kuberan B, Lech MZ, Beeler DL, Wu ZL, Rosenberg RD. Nat. Biotechnol. 2003;21:1343–1346. doi: 10.1038/nbt885. [DOI] [PubMed] [Google Scholar]; b) Lindahl U, Li JP, Kusche-Gullberg M, Salmivirta M, Alaranta S, Veromaa T, Emeis J, Roberts I, Taylor C, Oreste P, Zoppetti G, Naggi A, Torri G, Casu B. J. Med. Chem. 2005;48:349–352. doi: 10.1021/jm049812m. [DOI] [PubMed] [Google Scholar]
- 7.a) Xu Y, Masuko S, Takieddin M, Xu H, Liu R, Jing J, Mousa S, Linhardt RJ, Liu J. Science. 2011;334:498–501. doi: 10.1126/science.1207478. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Masuko S, Linhardt RJ. Future Med. Chem. 2012;4:289–296. doi: 10.4155/fmc.11.185. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Masuko S, Bera S, Green DE, Weïwer M, Liu J, DeAngelis PL, Linhardt RJ. J. Org. Chem. 2012;77:1449–1456. doi: 10.1021/jo202322k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeAngelis PL, Liu J, Linhardt RJ. Glycobiology. 2013 doi: 10.1093/glycob/cwt016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Minakuchi H, Nakanishi K, Soga N, Ishizuka N, Tanaka N. Anal. Chem. 1996;68:3498–3501. doi: 10.1021/ac960281m. [DOI] [PubMed] [Google Scholar]; b) Kerns RJ, Linhardt RJ. J. Chromatogr. A. 1995;705:369–373. doi: 10.1016/0021-9673(95)00293-v. [DOI] [PubMed] [Google Scholar]
- 10.a) Fukase K, Yasukochi T, Nakai Y, Kusumoto S. Tetrahedron Lett. 1996;37:3343–3344. [Google Scholar]; b) Fukase K, Yasukochi T, Suda Y, Yoshida M, Kusumoto S. Tetrahedron Lett. 1996;37:6763–6766. [Google Scholar]
- 11.a) Vaxelaire C, Souquet F, Lannou M, Ardisson J, Royer J. Eur. J. Org. Chem. 2009;2009:3138–3140. [Google Scholar]; b) Fukase K, Egusa K, Nakai Y, Kusumoto S. Mol. Divers. 1996;2:182–188. doi: 10.1007/BF01715633. [DOI] [PubMed] [Google Scholar]
- 12.Reverse phase C-18 flash column: 2% acetonitrile aqueous solution containing 0.1% TFA elute the enzyme, salt and UTP etc. 70% acetonitrile aqueous solution with 0.1% TFA elute the product with p-NP tag.
- 13.UDP-GlcNTFA was chemoenzymatically synthesized as described in Ref. 7c. Expression of enzyme including KfiA, pmHS2, NST and chemoenzymatic reactions were all handled as described in Ref. 7a.
- 14.a) Hajipour AR, Khazdooz L, Ruoho AE. J. Iran. Chem. Soc. 2011;8:382–387. [Google Scholar]; b) Maverick AW, Yao Q. Inorg. Chem. 1993;32:5626–5628. [Google Scholar]
- 15.General experimental procedure for the synthesis of compounds 10 and 11. To the freshly prepared solution of THF/H2O/acetic anhydride (4:5:1, 1 mL), 10% Pd(OH)2/C (w/w, 2 mg) and compound 9 (1.31 mg, 1.0 µmol) were added without special exclusion of air or moisture. The mixture was stirred under a hydrogen atmosphere at room temperature for 1.5 h, and progress of the reaction was monitored by TLC (HCOOH/BuOH/H2O, 4/8/1, Rf = 0.36). The mixture was filtered through a thin pad of Celite, which was subsequently washed with water (5 mL). The solvent was removed by rotary evaporation to yield 10 (1.28 mg, 94%). Compound 10 (1.28 mg, 0.94 µmol) was dissolved in 940 µL H2O which was subsequently added 5 mM CAS aqeous solution (940 µL) at 0 °C. The mixture was stirred for 0.5 h, and progress of the reaction was monitored by TLC (HCOOH/BuOH/H2O, 4:8:1, Rf = 0.31). The product was subjected on Biogel P-2 chromatography (1.0 × 60 cm) which was equilibrated with 0.1 M ammonium bicarbonate at a flow rate of 25 ml/h. The fraction containing the product was lyophilized to yield 11 (1.02 mg, 89%).
- 16.Spectroscopic data for compounds 9–11. Compound 9: 1H NMR (800 MHz, D2O): δ 8.14 (d, 1 H, J = 9.2 Hz), 7.11 (d, 1 H, J = 9.2 Hz), 5.63 (d, 1 H, J = 3.5 Hz, H-1A), 5.58 (d, 1 H, J = 3.4 Hz, H-1C), 5.55 (d, 1 H, J = 3.6 Hz, H-1E), 5.23 (d, 1 H, J = 7.8 Hz, H-1F), 4.46 (d, 1 H, J = 7.9 Hz, H-1B), 4.44 (d, 1 H, J= 7.8 Hz, H-1D), 4.36-4.26 (m, 3 H), 4.24-4.16 (m, 2 H), 4.14 (s, 2 H), 4.04-3.97 (m, 1 H), 3.95 (d, 1 H, J = 9.7 Hz), 3.91 (t, 1 H, J = 9.3 Hz), 3.89-3.84 (m, 2 H), 3.84-3.77 (m, 4 H), 3.78-3.73 (m, 4 H), 3.72-3.65 (m, 3H), 3.64-3.58 (m, 3 H), 3.52-3.47 (m, 1 H), 3.41 (t, 1 H, J = 9.7 Hz), 3.35 (t, 1 H, J = 9.2 Hz), 3.33 (t, 1 H, J =8.3 Hz), 3.24 (dd, 1 H, J= 3.6, 10.6 Hz), 3.22 (dd, 1 H, J= 3.9, 9.9 Hz), 3.19 (dd, 1 H, J= 4.0, 10.2 Hz); 13C NMR (200 MHz, D2O): δ 102.2 (C-1B, C-1D), 99.5 (C-1F), 96.9 (C-1C, C-1E), 95.1 (C-1A), 83.9, 83.1, 77.6, 76.6, 76.3, 75.8, 73.8, 73.1, 72.6, 72.2, 70.3, 70.2, 69.8, 69.4, 68.9, 65.1, 64.9, 64.4, 63.8, 63.4, 59.6, 59.3, 57.6, 54.5, 54.2. HRMS-FAB: [M−H]− m/z calcd. for C42H62N4O39S2, 1309.2354; found: 1309.2366. Compound 10: 1H NMR (800 MHz, D2O): δ 7.27 (d, 1 H, J = 9.0 Hz), 7.05 (d, 1 H, J = 9.0 Hz), 5.57 (d, 1 H, J = 3.7 Hz, H-1C), 5.54 (d, 1 H, J = 4.2 Hz, H-1E), 5.34 (d, 1 H, J = 3.6 Hz, H-1A), 5.05 (d, 1 H, J = 8.0 Hz, H-1F), 4.46 (d, 1 H, J = 7.8 Hz, H-1B), 4.43 (d, 1 H, J = 7.9 Hz, H-1D), 4.41 (d, 1 H, J = 7.6 Hz), 4.36-4.34 (m, 1 H), 4.32-4.30 (m, 1 H), 4.29-4.28 (m, 2 H), 4.22-4.13 (m, 4H), 4.00-3.96 (m, 1 H), 3.94-3.91 (m, 1 H), 3.87 (t, 2 H, J = 7.5 Hz), 3.82-3.78 (m, 3 H), 3.77-3.75 (m, 2 H), 3.75-3.71 (m, 3 H), 3.70-3.64 (m, 3 H), 3.63-3.58 (m, 4 H), 3.54 (t, 1 H, J = 8.48 Hz), 3.51 (t, 1 H, J = 7.2 Hz), 3.39 (t, 1 H, J = 9.6 Hz), 3.32 (t, 1 H, J = 8.3 Hz), 3.29 (t, 1 H, J = 8.9 Hz), 3.21-3.18 (m, 2 H). HRMS-FAB: [M−H]− m/z calcd. for C46H68N4O39S2, 1363.2824; found: 1363.2830. Compound 11: 1H NMR (800 MHz, D2O): δ 5.48 (s, 2 H, H-1C, H-1E) 5.28 (d, 1 H, J = 3.6 Hz, H-1A), 5.08 (d, 1 H, J = 3.4 Hz, H-1αF), 4.51 (d, 1 H, J = 8.0 Hz, H-1βF), 4.39 (d, 1 H, J = 7.9 Hz, H-1B), 4.35 (d, 1 H, J = 7.8 Hz, H-1D), 4.00 (d, 1 H, J = 9.9 Hz), 3.86 (t, 1 H, J = 9.5 Hz), 3.81 (m, 1 H), 3.73 (t, 5 H, J = 9.2 Hz), 3.71-3.62 (m, 10 H), 3.62-3.56 (m, 4 H), 3.57-3.51 (m, 6 H), 3.47 (dd, 1 H, J = 4.0, 10.2 Hz), 3.34 (t, 1 H, J = 9.6 Hz), 3.27-3.20 (m, 2 H), 3.16 (t, 1 H, J = 9.7 Hz), 3.14-3.10 (m, 2 H); 13C NMR (200 MHz, D2O): δ 102.0 (C-1B, C-1D), 96.8 (C-1C, C-1E), 96.7 (C-1A), 95.8 (C-1βF), 91.7 (C-1αF), 77.6, 76.6, 76.2, 75.7, 73.3, 72.6, 71.7, 70.5, 70.2, 70.1, 69.3, 69.2, 59.9, 59.8, 59.1, 57.2, 53.4. HRMS-FAB: [M−H]− m/z calcd. for C38H60N3O38S2, 1230.2296; found: 1230.2312.