

Abstract
C-reactive protein (CRP) largely has been studied in white non-Hispanic cohorts. There is limited information on CRP’s range of values, heritability and relation to cardiovascular disease (CVD) risk factors in African Americans. We sought to evaluate the distribution, clinical correlates, heritability and genetic linkage of log-transformed CRP in participants of the middle-aged to elderly African American community-based Jackson Heart Study. The distribution and correlates of CRP were analyzed for the entire study cohort who underwent the first examination (2001–2004). Heritability was estimated for the family cohort nested within the larger Jackson Heart Study (246 families, n=1,317). The relation between CRP and CVD risk factors were tested with multivariable stepwise regression analyses. Heritability was estimated using a variance components method. Linkage analysis was performed using the multipoint variance components approach. The study sample consisted of 4,919 participants (mean age 55±13 years, 63% women); median CRP concentration was 2.7 mg/L. In stepwise models traditional risk factors explained 23.8% of CRP’s variability, with body mass index (BMI, partial R2=13.6%) explaining 57.1% of the variability of CRP due to traditional risk factors. The heritability of CRP (adjusted for age, sex and BMI) was 0.45. The strongest linkage evidence for CRP was observed on chromosome 11 (11p13–11p11.2) with a logarithm of odds score of 2.72. In conclusion, in this large population-based cohort of African Americans, circulating CRP concentration was heritable and associated with several traditional cardiovascular risk factors, particularly BMI.
Keywords: C-reactive protein, risk factors, genetics, heritability, blood pressure, cholesterol, body mass index, African Americans
C-reactive protein (CRP) represents the inflammatory marker whose relation with cardiovascular disease (CVD) risk factors and CVD has been most intensively studied. CRP concentrations have been correlated with female sex, advancing age,1 diabetes,2 and higher glucose, 2 cholesterol,2 and triglyceride concentrations, lower HDL,3 increasing blood pressure,2 smoking,2 and body mass index (BMI). Beyond CRP’s relation to CVD risk factors, there are numerous investigations supporting an association between CRP and peripheral vascular disease,4 ischemic stroke,5 and myocardial infarction.6 A recent meta-analysis by Danesh et al. examined 7 prospective studies of CRP and long term coronary heart disease risk that had a total of 1053 events and found an adjusted relative risk of 1.7 for coronary heart disease comparing the top to the bottom tertile of CRP.7 Given the higher rates of CVD and CVD mortality in African Americans and the relation of CRP to CVD events, the environmental and genetic determinants of CRP in African Americans is of interest. We hypothesized that CRP concentrations are related to CVD risk factors, and that there is a significant heritable component after accounting for environmental risk factors. In this report, we have evaluated the relation of CRP concentrations to age, sex, and CVD risk factors in the middle-aged to elderly African American cohort of the Jackson Heart Study. We subsequently investigated the heritability of CRP and examined genetic linkage in the family cohort.
Methods
The Jackson Heart Study is a longitudinal population-based observational cohort that was initiated in 2000 to prospectively investigate the epidemiology and determinants of CVD in African Americans.8 Thirty percent of study participants were former members of the Atherosclerosis Risk in Communities study, and had been recruited by random selection from the driver’s license registry.9 Among the remaining participants, 23% were recruited by random selection from the “Accudata” list, a commercial listing that represents the overall tri-county population. An additional 23% were members of a constrained volunteer sample, in which recruitment was distributed among defined demographic cells in proportions designed to mirror those in the overall population, and 24% were recruited through the Jackson Heart Study Family Study, as described.10 Among the 5,302 participants recruited for Examination 1, a total of 4919 were used in the analysis performed in this study. The difference of 383 participants was due to: lack of consent for the use of their lab data for analysis (n=23); no CRP values (n=82); and missing data on covariates used in the various analyses (n=278). The Jackson Heart Study was approved by the University of Mississippi Medical Center Institutional Review Board and the participants gave written informed consent.
All clinical covariates were classified at the first examination. BMI was determined as fasting weight divided by height squared (kg/m2); obesity, as BMI ≥30 kg/m2. Systolic and diastolic blood pressure were taken in the sitting position by trained technicians using a random-zero sphygmomanometer after 5-minute rest; an average of the second and third readings was used. Hypertension was defined as systolic blood pressure ≥140 mmHg, or diastolic ≥90 mmHg, or reported use of antihypertensive medications within 2 weeks prior to the visit.11 Diabetes mellitus was defined as fasting serum glucose of ≥126 mg/dL or use of diabetic medications within 2 weeks of the clinic visit, or history of physician-diagnosed diabetes.12 Insulin resistance was estimated by the homeostasis model assessment (HOMA-IR)13 using the following formula: HOMA-IR=(fasting plasma insulin[microU/ml])x(fasting plasma glucose[mmol/l])/22.5. Insulin resistance was defined as a HOMA-IR score greater than 4.6 for non-diabetics. Fasting serum total cholesterol and high density lipoprotein cholesterol and triglyceride concentrations were assessed with Roche enzymatic methods using a Cobras centrifuge analyzer (Hoffman-La Roche), with the laboratory certified by the CDC-NHLBI Lipid Standardization Program. Prevalent CVD was defined as a stated history of physician-diagnosed myocardial infarction or stroke, electrocardiographic evidence of myocardial infarction, or history of a revascularization procedure (percutaneous transluminal coronary angioplasty, coronary bypass surgery).
CRP was measured in duplicate by the immunoturbidimetric CRP-Latex assay from Kamiya Biomedical Company using a Hitachi 91l analyzer, according to the manufacturer’s high-sensitivity protocol. The inter-assay coefficients of variation on control samples repeated in each assay were 4.5% at a CRP concentration of 0.45 mg/L and 4.4% at 1.56 mg/L. Any of the duplicates that were not within 3 assay standard deviations from one another were rerun. For external quality control (to assess performance at the clinical center and in the laboratory), approximately 6% of samples were measured as masked replicates on different dates to assess repeatability of measurements of CRP. The reliability coefficient for masked quality control replicates was 0.95 for the CRP assay.
Genotyping was performed by the Mammalian Genotyping Service as described at http://research.marshfieldclinic.org/genetics/lab_methods/methods/html. A total of 1501 individuals were genotyped for 374 polymorphic markers (Marshfield marker set 16). Average heterozygosity was 0.76 and the sex-averaged mean inter-marker spacing was 9.3 cM. After extensive quality checks to verify consistency of marker genotyping and stated pedigree relationships, a total of 246 families (1,317 individuals) were included in the final genetic analysis dataset.10
We performed descriptive statistics to determine the mean, standard deviation and/or percentages for study participants’ characteristics. Frequency distributions were used for categorical data. Descriptive statistics were also used to determine the mean, median, SD, 25th percentile and 75th percentile of CRP, by age and sex in the study population. Similar descriptive analysis was performed for the family sample. To establish reference ranges for CRP concentrations in African Americans, a reference sample was selected from the study population. The reference sample excluded participants with CRP concentrations >10 mg/L, hypertension, diabetes, obesity, chronic kidney disease (defined as estimated glomerular filtration rate <60 mL/min/1.73m2), or prevalent CVD and excluded participants taking lipid lowering and hormone replacement therapy. The reference sample was compared both with the broad sample, consisting of all other participants and with the family sample.
Since its distribution was skewed, CRP concentration was log-transformed for clinical correlates and genetic analyses.14 Age- and sex-adjusted linear regression was used to assess the relation of traditional cardiovascular risk factors to CRP concentrations (dependent variable). We performed stepwise linear regression (p<0.05 to remain in the model; see legend Table 4 for candidate covariates) with age and sex forced in, to determine which correlates were significantly associated with CRP; the partial R-square was computed to assess the contribution of each significant correlate to CRP variability. We also assessed the fold change of CRP for each predictor. Analyses were conducted with SAS version 9.2 (SAS Institute, Cary, NC). A 2-sided p-value <0.05 was considered statistically significant.
Table 4.
Clinical correlates of C-reactive protein concentrations: Stepwise linear regression model, R2 = 0.238
| Fold Change in CRP (95% CI) | Partial R2* | p Value | |
|---|---|---|---|
| Age, 10 years | 1.05 (1.02,1.08) | 0.0027 | 0.0018 |
| Sex, women vs. men | 0.91 (0.80, 1.04) | 0.0543 | 0.1833† |
| Body mass index, 5 kg/m2 | 1.27 (1.22, 1.32) | 0.1360 | <0.0001 |
| Current hormone replacement, yes vs. no | 1.61 (1.47, 1.77) | 0.0153 | <0.0001 |
| Current cigarette smoking, yes vs. no | 1.43 (1.31, 1.57) | 0.0107 | <0.0001 |
| Waist circumference, 15 cm | 1.17 (1.12, 1.23) | 0.0071 | <0.0001 |
| Triglycerides, 100 mg/dL | 1.10 (1.05, 1.15) | 0.0038 | <0.0001 |
| Lipid lowering therapy, yes vs. no | 0.78 (0.71,0.86) | 0.0034 | <0.0001 |
| Fasting glucose, 40 mg/dL | 1.06 (1.02, 1.10) | 0.0021 | 0.0010 |
| Diastolic blood pressure, 10 mm Hg | 0.96 (0.94, 0.99) | 0.0010 | 0.0113 |
| Pulse pressure, 10 mm Hg | 1.03 (1.01, 1.05) | 0.0012 | 0.0061 |
Partial R2 indicates the increment to R2 as each variable was added in the order listed, with age and sex forced into the model.
Candidate covariates for selection included body mass index, waist circumference, current cigarette smoking, total/HDL cholesterol, triglycerides, blood glucose, diabetes, systolic, diastolic and pulse pressure, medications (anti-hypertensive, lipid lowering, aspirin therapy, hormone replacement) and prevalent CVD
Sex not significant in the stepwise model though accounting for 5.5% of total R2 because sex was forced into the model and correlates with other risk factors in the final model.
In a subgroup analysis of 4748 participants who had fasting insulin and fasting glucose information available, we analyzed the relation of obesity, insulin resistance and diabetes to age-and sex-adjusted CRP concentration using general linear models (PROC GLM).
To analyze heritability, both “ASPEX” and “RELTEST” were used to check the consistency of the pedigrees. After resolution of all instances of non-paternity and other errors in the family structures, the maximum likelihood heritability estimate for serum CRP concentration was obtained using a variance components method.15
To analyze genetic linkage, the potential confounding influences of age, sex and BMI on the distribution of CRP were removed by regression. Quantitative trait locus linkage analysis was performed using the multipoint variance components approach as implemented in MERLIN.16 A logarithm of odds score ≥3.3 was taken as evidence of significant linkage, and a logarithm of odds score ≥1.9 but <3.3 as evidence of suggestive linkage.17 The Marshfield age-and sex-averaged maps were used in the linkage analyses.
To estimate the probability of obtaining false positive evidence of linkage, we conducted gene-dropping simulations using MERLIN. Marker data were simulated under the null hypothesis of no linkage or association to observed phenotypes while retaining the same pedigree structures, maps, marker allele frequencies and missing data patterns. We simulated 2,000 replicates and conducted the same linkage analyses as described earlier. The probability of obtaining a false positive result was defined as the proportion of replicates for which we obtained a specified logarithm of odds score or higher.
Results
In our large community-based cohort of African Americans, the mean CRP concentration was 5.1±9.0 for the entire cohort and 1.8±1.9 for the reference subsample. The characteristics of the study sample of 4919 participants (mean age 55±13 years, 63% female) are displayed in Table 1. The age of the entire cohort of 5302 was the same as the study sample (mean ± SD= 55±13 years). There was only a percentage point difference in the proportion of females in the entire cohort (64%) compared to the study sample. Subsequent results presented in this study will be limited to the study sample of 4919. A large percentage of the participants were obese (40.9% men; 60.0% women). Approximately 18% had diabetes. Also shown in Table 1 are the characteristics of participants in the reference subsample and the family cohort. As anticipated given the exclusion of individuals with major CVD risk factors and CVD, the reference sample participants were younger than the broad sample.
Table 1.
Jackson Heart Study participant characteristics by study sample
| Sample | ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Variable | Broad | Reference | Family | |||
| Men (N=1810) | Women (N=3109) | Men (N=408) | Women (N=393) | Men (N=457) | Women (N=860) | |
| Age (years) | 54 ± 13 | 55 ± 13 | 47± 13 | 48 ± 12 | 49 ± 15 | 50 ± 14 |
| Current cigarette smoker | 18.5% | 10.1% | * | * | 20.1% | 10.7% |
| Systolic blood pressure (mm Hg) | 128 ± 18 | 127 ± 19 | 116 ± 10 | 112 ± 12 | 126 ± 17 | 124 ± 19 |
| Diastolic blood pressure (mm Hg) | 81 ± 11 | 77 ± 10 | 77 ± 8 | 73 ± 8 | 81 ± 10 | 78 ± 10 |
| Pulse pressure (mm Hg) | 46 ± 15 | 49 ± 16 | 39 ± 10 | 39 ± 10 | 45 ± 15 | 46 ± 16 |
| Hypertensive medication | 45.5% | 57.1% | * | * | 36.7% | 49.0% |
| Hypertension | 59.8% | 64.3% | * | * | 50.6% | 57.3% |
| Body mass index (kg/m2) | 30 ± 6 | 33 ± 7 | 26 ± 3 | 26 ± 3 | 30 ± 7 | 33 ± 8 |
| Obese, Body mass index ≥30kg/m2 | 40.9% | 60.0% | * | * | 37.9% | 63.8% |
| Waist (centimeters) | 101 ± 15 | 100 ± 17 | 90 ± 9 | 84 ± 9 | 100 ± 16 | 100 ± 17 |
| Fasting glucose (mg/dL) | 102 ± 38 | 102 ± 38 | 89 ± 7 | 86 ± 8 | 100 ± 40 | 98 ± 36 |
| Diabetes mellitus | 16.6% | 19.2% | * | * | 12.5 | 15.7% |
| Total/High density lipoprotein cholesterol | 4.6 ± 1.5 | 3.9 ± 1.2 | 4.3 ± 1.4 | 3.6 ± 1.1 | 4.6 ± 1.5 | 3.9 ± 1.2 |
| Lipid lowering medication | 12.2% | 12.6% | * | * | 8.8% | 10.4% |
| Hormone replacement therapy | -- | 21.6% | -- | * | -- | 20.1% |
| Prevalent cardiovascular disease | 12.6% | 8.9% | * | * | 8.4% | 7.1% |
Characteristics are mean ± SD or %; -- Signifies not applicable;
Signifies exclusion criteria for reference sample, which consisted of participants with CRP concentration <10mg/L and without the following: hypertension, obesity, diabetes, chronic kidney disease, current smoking, prevalent cardiovascular disease, lipid lowering therapy or hormone replacement therapy.
Table 2 displays the distributions of CRP concentrations by sex and age in the broad study cohort (n=4,919), and the reference sample (n=801). For both the broad sample and the reference sample, CRP concentrations were higher in women than in men.
Table 2.
Distribution of C-reactive protein (mg/L) in Jackson Heart Study by age, sex and sample
| Variable | n | mean | S.D. | 25th percentile | Median | 75th percentile |
|---|---|---|---|---|---|---|
|
Broad Sample
| ||||||
| Total | 4919 | 5.1 | 9.0 | 1.1 | 2.7 | 5.6 |
| Men | 1810 | 3.6 | 9.8 | 0.8 | 1.7 | 3.7 |
| Women | 3109 | 6.0 | 8.4 | 1.5 | 3.5 | 6.9 |
|
| ||||||
| Age (years) | ||||||
| 21 – 34 | 229 | 3.7 | 5.5 | 0.6 | 1.6 | 4.1 |
| 35 – 44 | 943 | 4.8 | 7.7 | 0.9 | 2.4 | 5.3 |
| 45 – 54 | 1206 | 5.1 | 7.0 | 1.1 | 2.8 | 6.0 |
| 55 – 64 | 1327 | 5.4 | 11.9 | 1.2 | 3.0 | 5.9 |
| 65 – 74 | 927 | 5.3 | 9.0 | 1.3 | 2.8 | 5.7 |
| ≥75 | 287 | 4.8 | 7.7 | 1.1 | 2.3 | 5.1 |
|
| ||||||
|
Reference Sample
| ||||||
| Total | 801 | 1.8 | 1.9 | 0.5 | 1.0 | 2.5 |
|
| ||||||
| Men | 408 | 1.5 | 1.6 | 0.4 | 0.9 | 2.0 |
| Women | 393 | 2.1 | 2.1 | 0.5 | 1.2 | 2.8 |
|
| ||||||
| Age (years) | ||||||
| 21 – 34 | 93 | 1.2 | 1.4 | 0.3 | 0.7 | 1.7 |
| 35 – 44 | 279 | 1.7 | 1.7 | 0.4 | 1.0 | 2.3 |
| 45 – 54 | 208 | 2.0 | 2.2 | 0.5 | 1.0 | 2.8 |
| 55 – 64 | 139 | 2.1 | 2.0 | 0.6 | 1.4 | 2.7 |
| 65 – 74 | 60 | 1.9 | 1.8 | 0.8 | 1.3 | 2.6 |
| ≥75 | 22 | 2.3 | 2.2 | 0.7 | 1.4 | 3.6 |
In age- and sex-adjusted regressions CRP concentration was significantly related to advancing age, female sex, higher blood pressure (systolic and pulse pressure), larger body size (BMI, waist circumference), glucose-measures (diabetes, fasting glucose concentration), current cigarette smoking, higher lipid concentrations (total cholesterol/high density lipoprotein ratio, triglycerides), medications (higher with hypertensive, and hormone replacement, lower with lipid lowering medication) and prevalent CVD (Table 3).
Table 3.
Age- and Sex-Adjusted Correlates of log transformed C-Reactive Protein
| Variable | Fold Change in CRP (95% CI) | p Value |
|---|---|---|
| Age per 10 years* | 1.05 (1.03, 1.08) | 0.0002 |
| Women vs. men† | 1.81 (1.69, 1.94) | <0.0001 |
| Systolic blood pressure per 20 mm Hg | 1.07 (1.03, 1.11) | 0.0004 |
| Diastolic blood pressure per 10 mm Hg | 1.00 (0.97, 1.03) | 0.95 |
| Pulse pressure, 10 mm Hg | 1.06 (1.03, 1.08) | <0.0001 |
| Waist circumference, 15 cm | 1.50 (1.46, 1.54) | <0.0001 |
| Body mass index, 5 kg/m2 | 1.40 (1.37, 1.43) | <0.0001 |
| Current cigarette smoking, yes vs. no | 1.30 (1.18, 1.44) | <0.0001 |
| Fasting glucose, 40 mg/dL | 1.16 (1.12, 1.20) | <0.0001 |
| Diabetes status, yes vs. no | 1.30 (1.19, 1.42) | <0.0001 |
| Total/HDL cholesterol, ratio | 1.11 (1.08, 1.14) | <0.0001 |
| Triglyceride, 100 mg/dL | 1.22 (1.17, 1.28) | <0.0001 |
| Hypertension treatment, yes vs. no | 1.42 (1.32, 1.53) | <0.0001 |
| Lipid lowering therapy, yes vs. no | 0.78 (0.71, 0.86) | 0.009 |
| Hormone replacement therapy, yes vs. no | 1.50 (1.35, 1.66) | <0.0001 |
| Prevalent cardiovascular disease, yes vs. no | 1.13 (1.01, 1.27) | 0.03 |
Sex-adjusted;
age-adjusted
In step-wise regression, the amount of variability in CRP explained by clinical covariates was 23.8% (Table 4). After forcing in age and sex, we found increasing BMI, hormone replacement therapy, current smoking status, waist circumference, triglycerides, fasting glucose, and pulse pressure were positively related, and lipid lowering therapy and diastolic blood pressure were inversely related to CRP variability. BMI contributed 57% of the variability in CRP explained by standard clinical covariates (partial R2=0.1360, p<0.0001).
Figure 1 shows the relation of age- and sex-adjusted CRP concentrations by obesity, diabetes, and insulin resistance (in non diabetic participants) status. Mean CRP concentrations were higher for participants with, compared to those without obesity, regardless of insulin resistance or diabetes status. For both obese and non-obese participants, the age- and sex-adjusted linear models suggested that mean CRP concentrations were significantly higher in individuals without diabetes but with insulin resistance compared to those without insulin resistance. There was no significant difference between individuals without diabetes with insulin resistance and participants with diabetes for obese or non-obese participants. There was no interaction between insulin resistance status, diabetes status and obesity (p=0.46).
Figure 1. The Age- and Sex-Adjusted Relation of Obesity, Insulin Resistance (In Participants without Diabetes) and Diabetes to C-Reactive Protein Concentration.
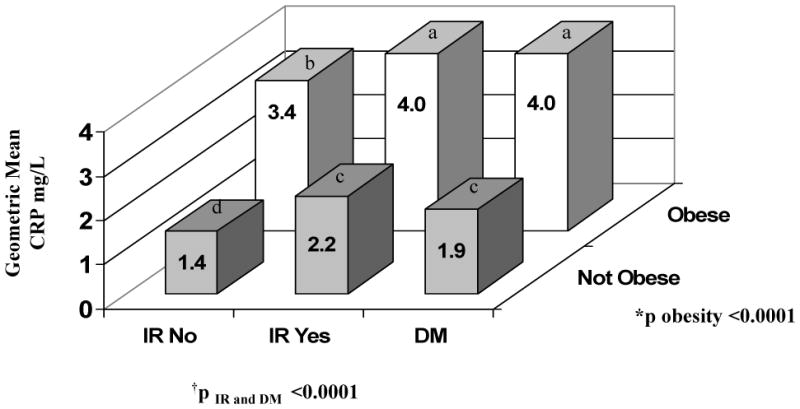
Log CRP concentrations were age- and sex-adjusted: back transformed geometric means are displayed. SAS omits records with a single missing data. Note that missing data may occur for different measures and not the same measure.
IR, insulin resistance in those without diabetes; DM, diabetes mellitus
*p obesity assessed for a significant difference in CRP concentration between individuals with versus without obesity after adjusting for diabetes and insulin resistance.
†p IR and diabetes assessed for a significant difference in CRP concentrations adjusting for obesity between participants without diabetes who do not have insulin resistance (referent group), versus participants without diabetes who have insulin resistance and participants with diabetes.
There was no significant interaction between obesity, insulin resistance (in participants without diabetes) and diabetes.
Note: There are 171 missing data on IR status (IR no and IR yes) this was due to missing data on Fasting Insulin and/or fasting glucose. There was no significant difference between DM and IR Yes when persons are not obese. Similarly results are true for obese persons. Groups of similar alphabet indicate no difference and group with different alphabet are different.
The heritability for the age-, sex- and BMI-adjusted log-CRP was 0.45 with a standard error of 0.06 and kurtosis of −0.0172. Plots of the logarithm of odds scores on all 22 autosomes obtained from the initial multipoint variance component linkage analyses for log-CRP are presented in Figure 2. The peak logarithm of odds scores in this genome scan are shown in Table 5. The strongest evidence for linkage to CRP was observed on chromosome 11 (maximum logarithm of odds score 2.72, nominal p=0.0002, empirical genome wide p=0.065) near marker D11S1993; in the 11p13 – 11p11.2 region, 54 cM from p-ter.
Figure 2. Multipoint LOD scores by chromosome for adjusted log transformed CRP.
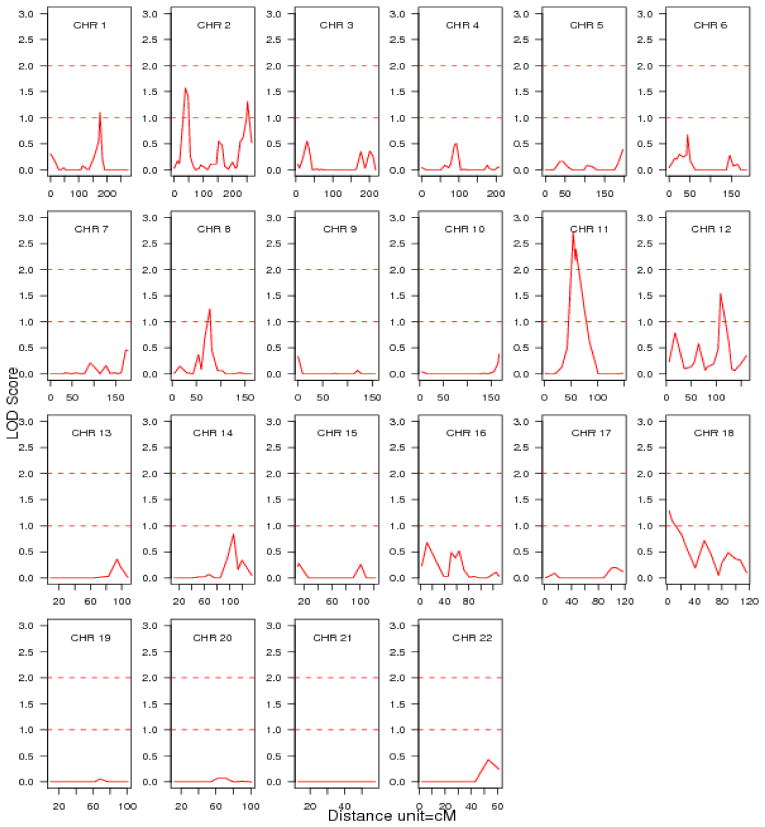
LOD, logarithm of odds;
CRP, C-reactive protein;
CRP adjusted for age, sex and body mass index.
Table 5.
Adjusted LOD scores > 1.0 for log transformed CRP residual values obtained from multiple regression analysis.
| Chromosome | Nearest marker | Max location | LOD score | Nominal p Value | Empiric p Value |
|---|---|---|---|---|---|
| 1 | D1S1677 | 175.6 | 1.10 | 0.01 | 0.97 |
| 2 | D2S1360 | 38.3 | 1.57 | 0.004 | 0.60 |
| 2 | D2S2968 | 251.9 | 1.31 | 0.007 | 0.89 |
| 8 | D8S1113 | 78 | 1.25 | 0.008 | 0.89 |
| 11 | D11S1993 | 54 | 2.72 | 0.0002 | 0.065 |
| 12 | PAH | 109 | 1.54 | 0.004 | 0.60 |
Discussion
We report the distribution, reference ranges, correlates, heritability and genetic linkage for CRP concentrations in the cohort, a large population-based study of African Americans with a high risk of CVD. Traditional CVD risk factors explained 24% of the variability in CRP concentrations, with BMI explaining 14% of the variability. Further, we observed that CRP concentrations were moderately heritable (0.45), and have evidence for suggestive linkage to chromosome 11 (logarithm of odds score of 2.72).
Similar to most cohorts, CRP concentrations were higher in women relative to men.18 Recent data suggest that CRP concentrations differ by race. In the racially-mixed Dallas Heart Study cohort, CRP concentrations were higher in African Americans compared to whites (median 3.0 mg/l vs. 2.3 mg/l, p<0.001). After adjustment for traditional cardiovascular risk factors, estrogen and statin use, and body mass index, a CRP concentration >3 mg/l remained more common in white women (odds ratio [OR] 1.6; 95% confidence interval [CI] 1.1 to 2.5) and black women (OR 1.7; 95% CI 1.2 to 2.6) but not in black men (OR, 1.3; 95% CI, 0.8 to 1.9) compared with white men.19 Ethnic/racial variation in CRP concentrations was noted in the Third National Health and Nutrition Examination Survey with the highest concentrations seen in non-Hispanic black men and Mexican-American women.20 In the Women’s Health Study, CRP concentrations were notably higher in African American women.21
In the stepwise linear regression model, BMI, waist circumference, diastolic blood pressure and fasting glucose significantly contributed to the variability of CRP, in addition to hormonal and lipid lowering therapy. The relation of CRP to traditional cardiovascular risk factors observed in the cohort is similar to findings in other cohorts. In the Third National Health and Nutrition Examination Survey cigarette smoking, increased age, BMI and systolic blood pressure in men, and BMI and diabetes in women were strongly associated with CRP concentrations.18 In the Coronary Artery Risk Development in Young Adults Study, CRP was associated with hypertension in white and African American young adults, but in contrast to the finding in older cohorts such as ours, the association was no longer present after adjusting for BMI.22
In our cohort, we found that individuals with diabetes and those without diabetes but with insulin resistance had higher CRP concentrations compared to those without insulin resistance or diabetes. There was no significant difference between CRP concentrations in individuals with diabetes and individuals without diabetes but with IR. Additionally, obesity was not an effect modifier on the relation between insulin resistance, diabetes and CRP. Our data are consistent with several other studies.23 For example, In the Risk Factors in Impaired Glucose Tolerance for Atherosclerosis and Diabetes study, those who were at risk for type 2 diabetes had higher CRP concentrations and were likely to be insulin resistant.24
Several recent human studies have indicated significant genetic effects on variation in inflammatory markers.25 Until now, however, there have been limited data on the heritability of CRP concentrations in African Americans. In the predominantly white NHLBI Family Heart Study, investigators studied the familial aggregation of 3 systemic markers of inflammation, CRP and found evidence of substantial heritability (35–40%) for CRP concentrations.26 In Framingham, investigators conducted variance-component linkage analyses of blood concentrations of 4 biomarkers of vascular inflammation including ; the heritability of CRP was estimated to be 28.2%, and the only adjusted logarithm of odds score >1.5 for CRP was located at Chromosome 14q31.3.27
The results of this study need to be confirmed by replication in multiple studies and ethnic cohorts. Our study was cross-sectional and observational. Hence, we cannot establish causal relations or the temporality of the observed association; we acknowledge that increased CRP concentration contributes to the development of risk factors, risk factors may precede and predispose to inflammation, the relations may be bidirectional, or the associations may be secondary to other factors that we did not model. In addition, the generalizability to other ethnicities/races, or individuals of African ancestry in other states or countries is uncertain. Balanced against these caveats is the routine ascertainment of CRP concentrations and covariates in a large community-based cohort of African Americans.
Our study suggests that both genetic and environmental risk factors, particularly obesity, contribute substantially to concentrations of CRP in African Americans. The high prevalence of obesity in our cohort, and the important contribution of obesity to variability in CRP concentrations suggest that weight loss may be a critical strategy to attenuate the contribution of systemic inflammation in the development of cardiovascular risk factors and outcomes in this group. Given the disproportionate burden of CVD in the African American community, future investigations should focus on the relation of CRP to incident CVD to establish optimal threshold values for estimating risk in this group. In addition, the substantial heritability of CRP, and recent data suggesting that CRP polymorphisms contribute to CVD risk underscore the importance of further research into genetic factors contributing to CRP variation in African Americans.28
Acknowledgments
The Jackson Heart Study is a collaborative study supported by the National Institutes of Health and the National Center on Minority Health and Health Disparities (study ID numbers: 5001; N01 HC95170; N01 HC95171; N01 HC95172) in partnership with 3 local institutions (University of Mississippi Medical Center, Jackson State University and Tougaloo College). Dr. Fox’s work on this project is supported by American Heart Association (0555209B). Dr. Benjamin’s work on this project is supported by HL076784 and AG028321.
The authors thank the staff and participants in the Jackson Heart Study for their important contributions.
Abbreviations
- CRP
- BMI
- CVD
- HOMA-IR
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kathiresan S, Larson MG, Vasan RS, Guo CY, Gona P, Keaney JF, Jr, Wilson PW, Newton-Cheh C, Musone SL, Camargo AL, Drake JA, Levy D, O’Donnell CJ, Hirschhorn JN, Benjamin EJ. Contribution of clinical correlates and 13 C-reactive protein gene polymorphisms to interindividual variability in serum C-reactive protein level. Circulation. 2006;113(11):1415–1423. doi: 10.1161/CIRCULATIONAHA.105.591271. [DOI] [PubMed] [Google Scholar]
- 2.Koenig W, Sund M, Frohlich M, Fischer HG, Lowel H, Doring A, Hutchinson WL, Pepys MB. C-Reactive protein, a sensitive marker of inflammation, predicts future risk of coronary heart disease in initially healthy middle-aged men: results from the MONICA (Monitoring Trends and Determinants in Cardiovascular Disease) Augsburg Cohort Study, 1984 to 1992. Circulation. 1999;99(2):237–242. doi: 10.1161/01.cir.99.2.237. [DOI] [PubMed] [Google Scholar]
- 3.Tracy RP, Psaty BM, Macy E, Bovill EG, Cushman M, Cornell ES, Kuller LH. Lifetime smoking exposure affects the association of C-reactive protein with cardiovascular disease risk factors and subclinical disease in healthy elderly subjects. Arterioscler Thromb Vasc Biol. 1997;17(10):2167–2176. doi: 10.1161/01.atv.17.10.2167. [DOI] [PubMed] [Google Scholar]
- 4.Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Plasma concentration of C-reactive protein and risk of developing peripheral vascular disease. Circulation. 1998;97(5):425–428. doi: 10.1161/01.cir.97.5.425. [DOI] [PubMed] [Google Scholar]
- 5.Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med. 1997;336(14):973–979. doi: 10.1056/NEJM199704033361401. [DOI] [PubMed] [Google Scholar]
- 6.Ridker PM, Glynn RJ, Hennekens CH. C-reactive protein adds to the predictive value of total and HDL cholesterol in determining risk of first myocardial infarction. Circulation. 1998;97(20):2007–2011. doi: 10.1161/01.cir.97.20.2007. [DOI] [PubMed] [Google Scholar]
- 7.Danesh J, Collins R, Appleby P, Peto R. Association of fibrinogen, C-reactive protein, albumin, or leukocyte count with coronary heart disease: meta-analyses of prospective studies. JAMA. 1998;279(18):1477–1482. doi: 10.1001/jama.279.18.1477. [DOI] [PubMed] [Google Scholar]
- 8.Taylor HA, Jr, Wilson JG, Jones DW, Sarpong DF, Srinivasan A, Garrison RJ, Nelson C, Wyatt SB. Toward resolution of cardiovascular health disparities in African Americans: design and methods of the Jackson Heart Study. Ethn Dis. 2005;15(4 Suppl 6):S6–17. [PubMed] [Google Scholar]
- 9.Fuqua SR, Wyatt SB, Andrew ME, Sarpong DF, Henderson FR, Cunningham MF, Taylor HA., Jr Recruiting African-American research participation in the Jackson Heart Study: methods, response rates, and sample description. Ethn Dis. 2005;15(4 Suppl 6):S6–29. [PubMed] [Google Scholar]
- 10.Wilson JG, Rotimi CN, Ekunwe L, Royal CD, Crump ME, Wyatt SB, Steffes MW, Adeyemo A, Zhou J, Taylor HA, Jr, Jaquish C. Study design for genetic analysis in the Jackson Heart Study. Ethn Dis. 2005;15(4 Suppl 6):S6–37. [PubMed] [Google Scholar]
- 11.Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, Izzo JL, Jr, Jones DW, Materson BJ, Oparil S, Wright JT, Jr, Roccella EJ. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003;289(19):2560–2572. doi: 10.1001/jama.289.19.2560. [DOI] [PubMed] [Google Scholar]
- 12.Genuth S, Alberti KG, Bennett P, Buse J, Defronzo R, Kahn R, Kitzmiller J, Knowler WC, Lebovitz H, Lernmark A, Nathan D, Palmer J, Rizza R, Saudek C, Shaw J, Steffes M, Stern M, Tuomilehto J, Zimmet P. Follow-up report on the diagnosis of diabetes mellitus. Diabetes Care. 2003;26(11):3160–3167. doi: 10.2337/diacare.26.11.3160. [DOI] [PubMed] [Google Scholar]
- 13.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28(7):412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 14.Pearson TA, Mensah GA, Alexander RW, Anderson JL, Cannon RO, III, Criqui M, Fadl YY, Fortmann SP, Hong Y, Myers GL, Rifai N, Smith SC, Jr, Taubert K, Tracy RP, Vinicor F. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: A statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation. 2003;107(3):499–511. doi: 10.1161/01.cir.0000052939.59093.45. [DOI] [PubMed] [Google Scholar]
- 15.Almasy L, Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet. 1998;62(5):1198–1211. doi: 10.1086/301844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30(1):97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 17.Lander E, Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet. 1995;11(3):241–247. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
- 18.Wong ND, Pio J, Valencia R, Thakal G. Distribution of C-reactive protein and its relation to risk factors and coronary heart disease risk estimation in the National Health and Nutrition Examination Survey (NHANES) III. Prev Cardiol. 2001;4(3):109–114. doi: 10.1111/j.1520-037x.2001.00570.x. [DOI] [PubMed] [Google Scholar]
- 19.Khera A, McGuire DK, Murphy SA, Stanek HG, Das SR, Vongpatanasin W, Wians FH, Jr, Grundy SM, de Lemos JA. Race and gender differences in C-reactive protein levels. J Am Coll Cardiol. 2005;46(3):464–469. doi: 10.1016/j.jacc.2005.04.051. [DOI] [PubMed] [Google Scholar]
- 20.Wener MH, Daum PR, McQuillan GM. The influence of age, sex, and race on the upper reference limit of serum C-reactive protein concentration. J Rheumatol. 2000;27(10):2351–2359. [PubMed] [Google Scholar]
- 21.Albert MA, Glynn RJ, Buring J, Ridker PM. C-reactive protein levels among women of various ethnic groups living in the United States (from the Women’s Health Study) Am J Cardiol. 2004;93(10):1238–1242. doi: 10.1016/j.amjcard.2004.01.067. [DOI] [PubMed] [Google Scholar]
- 22.Lakoski SG, Herrington DM, Siscovick DM, Hulley SB. C-Reactive Protein Concentration and Incident Hypertension in Young Adults: The CARDIA Study. Arch Intern Med. 2006;166(3):345–349. doi: 10.1001/archinte.166.3.345. [DOI] [PubMed] [Google Scholar]
- 23.Festa A, Hanley AJ, Tracy RP, D’Agostino R, Jr, Haffner SM. Inflammation in the prediabetic state is related to increased insulin resistance rather than decreased insulin secretion. Circulation. 2003;108(15):1822–1830. doi: 10.1161/01.CIR.0000091339.70120.53. [DOI] [PubMed] [Google Scholar]
- 24.Temelkova-Kurktschiev T, Siegert G, Bergmann S, Henkel E, Koehler C, Jaross W, Hanefeld M. Subclinical inflammation is strongly related to insulin resistance but not to impaired insulin secretion in a high risk population for diabetes. Metabolism. 2002;51(6):743–749. doi: 10.1053/meta.2002.32804. [DOI] [PubMed] [Google Scholar]
- 25.Reiner AP, Carlson CS, Ziv E, Iribarren C, Jaquish CE, Nickerson DA. Genetic ancestry, population sub-structure, and cardiovascular disease-related traits among African-American participants in the CARDIA Study. Hum Genet. 2007;121(5):565–575. doi: 10.1007/s00439-007-0350-2. [DOI] [PubMed] [Google Scholar]
- 26.Pankow JS, Folsom AR, Cushman M, Borecki IB, Hopkins PN, Eckfeldt JH, Tracy RP. Familial and genetic determinants of systemic markers of inflammation: the NHLBI family heart study. Atherosclerosis. 2001;154(3):681–689. doi: 10.1016/s0021-9150(00)00586-4. [DOI] [PubMed] [Google Scholar]
- 27.Dupuis J, Larson MG, Vasan RS, Massaro JM, Wilson PW, Lipinska I, Corey D, Vita JA, Keaney JF, Jr, Benjamin EJ. Genome scan of systemic biomarkers of vascular inflammation in the Framingham Heart Study: evidence for susceptibility loci on 1q. Atherosclerosis. 2005;182(2):307–314. doi: 10.1016/j.atherosclerosis.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 28.Lange LA, Carlson CS, Hindorff LA, Lange EM, Walston J, Durda JP, Cushman M, Bis JC, Zeng D, Lin D, Kuller LH, Nickerson DA, Psaty BM, Tracy RP, Reiner AP. Association of polymorphisms in the CRP gene with circulating C-reactive protein levels and cardiovascular events. JAMA. 2006;296(22):2703–2711. doi: 10.1001/jama.296.22.2703. [DOI] [PubMed] [Google Scholar]