

Abstract
A series of N-(2-methoxyphenyl)homopiperazine analogs was prepared and their affinities for dopamine D2, D3, and D4 receptors were measured using competitive radioligand binding assays. Several ligands exhibited high binding affinity and selectivity for the D3 dopamine receptor compared to the D2 receptor subtype. Compounds 11a, 11b, 11c, 11f, 11j and 11k had Ki values ranging from 0.7–3.9 nM for the D3 receptor with 30- to 170-fold selectivity for the D3 vs. D2 receptor. Calculated log P values (log P = 2.6–3.6) are within the desired range for passive transport across the blood brain barrier. When the binding and the intrinsic efficacy of these phenylhomopiperazines was compared to those of previously published phenylpiperazine analogues, it was found that a) affinity at D2 and D3 dopamine receptors generally decreased, b) the D3 receptor binding selectivity (D2:D3 Ki value ratio) decreased and, c) the intrinsic efficacy, measured using a forskolin-dependent adenylyl cyclase inhibition assay, generally increased.
Keywords: Dopamine D2-like receptors, D3 dopamine receptors, receptor subtype selective ligands, homopiperazine analogs
1. Introduction
The neurotransmitter dopamine has been implicated in a variety of physiological and pathophysiological processes. Dopamine’s effects are mediated by dopamine receptors, which belong to the family of G protein coupled receptors (GPCR) and share the characteristic structural architecture of seven transmembrane spanning regions. There are five different dopamine receptor subtypes that are classified into two protein families, the D1-like (D1 and D5) and the D2-like (D2, D3, and D4) receptors, based upon related pharmacological and structural properties.1, 2 Abnormalities within the dopaminergic system are thought to play a role in psychiatric and neurological disorders, including Parkinson’s disease, substance abuse and schizophrenia.
The D2 and D3 receptor subtypes are highly homologous, especially within the helical transmembrane segments, which serve as the orthosteric binding site for dopamine. Therefore, while there has been interest in developing agonists, partial agonists and antagonists that are selective for the dopamine D2 and D3 receptor subtypes, this task has been difficult due to the high sequence homology for these two receptors.3–5
Receptor autoradiography studies have shown that D2 and D3 receptors are widely distributed in striatal regions of human6 and nonhuman primate7 brain. The expression of D3 receptors in limbic regions suggests that this receptor subtype may play an important role in the pathological abnormalities associated with many neuropsychiatric disorders. Autoradiography studies have revealed decreased D3 receptor expression in the frontal cortex and increased expression in the ventral striatum of schizophrenics compared to normal individuals.8, 9 A selective dopamine D3 receptor antagonist may provide antipsychotic properties in the absence of extrapyramidal side effects.10
Recent findings suggest that dopamine D3 receptor agonists may also have beneficial effects for Parkinson’s patients.11, 12 Dopamine D3 receptor partial agonists have also been reported to attenuate L-DOPA induced dyskinetic-like movements in animal models of Parkinson’s disease. D3 receptor partial agonists normalize involuntary movement in MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine)-treated monkeys, a primate model for Parkinson’s disease.13, 14
Finally, the activation of dopamine receptors in the nucleus accumbens is involved in the rewarding properties and/or the development of motivation for drug seeking behaviors for psychostimulants, such as cocaine. Therefore, partial agonists or antagonists that can reduce the interaction of psychostimulant-induced increases in synaptic dopamine levels with the D3 receptor may be useful as pharmacotherapeutics for the treatment of cocaine abuse.4, 15–17
A number of conformationally-flexible benzamide analogs displaying high affinity and binding selectivity for D3 versus D2 dopamine receptors have been reported in recent years. Examples include BP 897, NGB 2904, and the structural congeners 1–5 (Fig. 1).18–20 A common structural feature of these conformationally-flexible benzamides is the N-arylpiperazine ring and the 4-carbon spacer group separating the benzamide and the piperazine moieties.18, 21–25 The lipophilic residue on the arylamide moiety permits diverse modifications including aryl, biphenyl, heteroaryl, or cycloalkyl substituents. However, the relatively high lipophilicity of these analogs is above the optimal range for passive transport across the blood brain barrier. For example, the calculated log P values of benzamide analogs BP 897, 1 and 2 (Fig. 1) are 4.7, 5.9 and 7.1, respectively, which are not within the range of log P values for compounds that can readily cross the blood brain barrier.26, 27
Figure 1.

Structure and binding properties of D3 receptor selective substituted N-phenylpiperazines as reported in the literature.
Previously, our group synthesized a series of N-phenylpiperazine analogs22 and evaluated their affinities and intrinsic activities at dopamine D2 and D3 receptors.3 These compounds share structural elements with the classic D2-like dopamine receptor antagonists, including the 4-(2,3-dichlorophenyl)piperazino moiety. We have expanded upon those studies by synthesizing a new series of structurally related compounds and evaluating their binding affinities at D2-like receptors. These modifications include: (a) replacement of the 2,3-dimethoxy-5-bromobenzene ring of 5 with other aromatic or heteroaromatic moieties to explore the structure activity relationship of the benzamide group, (b) replacement of the piperazine ring with homopiperazine and (c) comparison of the N-(2,3-dichlorophenyl)piperazine of NGB 2904 and compounds 1–5 with the N-(2-methoxyphenyl)piperazine analog of BP 897 to determine the effect of this substitution on dopamine receptor affinity and calculated log P values (Fig. 1).
The results of this study has led to the identification of a number of compounds possessing a high affinity (nM) and moderate selectivity (10 to 100-fold) for dopamine D3 versus D2 receptors with a log P value within the range desired for crossing the blood brain barrier through passive diffusion.
2. Chemistry
The syntheses of all target compounds (Fig. 2) are outlined in Scheme 1. The homopiperazine was protected to afford its N-Boc derivative 6. Compound 6 was reacted with 2-bromoanisole to obtain the N-phenyl homopiperazine, 7. Deprotection of 7 using trifluoroacetic acid yielded the deprotected N-phenylhomopiperazine 8. Finally, 8 was alkylated with N-(4-bromobutyl)phthalimide to give the corresponding phthalimido derivative 9, which was hydrolyzed with hydrazine hydrate to afford the amine 10. Condensation of this amine with the appropriate carboxylic acids in the presence of N,N′-dicyclohexylcarbodiimide (DCC) and 1-hydroxybenzotriazole (HOBt) gave the expected final compounds 11a–11t. Synthesis of 4-(2-hydroxyethyl)benzoic acid, 14 and 4-(2-fluoroethyl)benzoic acid, 16, was accomplished using the reaction sequence outlined in Scheme 2.
Figure 2.
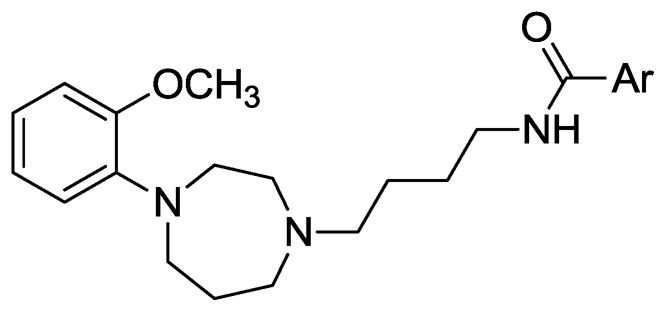
General structure of target compounds 11a–11t.
Scheme 1.

Reagents: (a) di-tert-butyl dicarbonate, MeOH; (b) 2-bromoanisole, BINAP, Pd2(dba)3, KOtBu/Et3N, toluene, reflux; (c) trifluroacetic acid; (d) N-(4-bromobutyl)phthalimide, K2CO3, acetonitrile; (e) NH2NH2, EtOH; (f) ArCOOH, HOBt, DCC, dichloromethane.
Scheme 2.
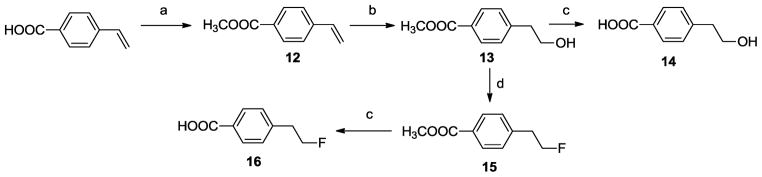
Reagents: (a) (CH3)3OBF4, CH2Cl2, N(C2H5)3; (b) (1) BH3/THF, (2) NaOH, 35% H2O2; (c) (1) NaOH, CH3OH/H2O, (2) HCl; (d) (C2H5)2NSF3, CH2Cl2;
3. Radioligand binding studies
Competitive radioligand binding studies were performed as previously described to determine the equilibrium dissociation constants of each compound at human D2, D3, and D4 dopamine receptors (Table 1). Briefly, D2-like receptors expressed in stably transfected HEK 293 cells were used in conjunction with the radioligand 125I-IABN. We previously reported that the benzamide 125I-IABN binds with high affinity and selectively to D2-like dopamine receptors, but it binds non-selectively to the D2 and D3 dopamine receptor subtypes.24 Measures were made of the affinity at D2 and D3 dopamine receptors of the new homopiperazine analogs, which have structural variations within the alkylamine moiety. When a comparison was made of the affinity of N-phenylhomopiperazine and the previously described corresponding N-phenylpiperazine analogues,20 several differences between the two series of compounds became evident. First, the affinity of the homopiperazine analogs for D2, there is a 2- to 6-fold lower, for D3, 3.8- to 8-fold lower compared to the piperazine congener. Second, the D2:D3 affinity ratio is generally lower for the homopiperazine analogs compared to the corresponding piperazine compounds (e.g., reduced D3 receptor selectivity).20
Table 1.
Binding affinities for dopamine D2-like receptors
| Compound |
Ki (nM)a
|
D2/D3e | Log Pf | ||
|---|---|---|---|---|---|
| D2b | D3c | D4d | |||
| 11a | 128±8.8 | 0.7±0.10 | 187±21 | 183 | 3.50 |
| 11b | 119±3.5 | 3.9±0.3 | 608±50 | 30 | 2.33 |
| 11c | 101±15.5 | 2.9±0.5 | 372±7.1 | 35 | 3.62 |
| 11d | 125±6.5 | 5.5±1.3 | 429 ±19 | 23 | 2.77 |
| 11e | 108±7.5 | 9.0±0.7 | 522±51 | 12 | 2.19 |
| 11f | 128±9.7 | 1.8±0.01 | 728±100 | 71 | 2.41 |
| 11g | 111±9.2 | 3.7±0.3 | 350±47 | 30 | 2.64 |
| 11h | 95.2±3.8 | 8.8±0.9 | 159±25 | 11 | 2.60 |
| 11i | 56.5±5.9 | 7.5±0.7 | 188±43 | 8 | 2.16 |
| 11j | 138±18.4 | 2.5±0.4 | 161±9.1 | 56 | 2.86 |
| 11k | 125±7.0 | 1.5±0.2 | 297±35 | 82 | 4.01 |
| 11l | 107±16.3 | 9.0±0.5 | 342±37 | 12 | 3.59 |
| 11m | 151±11.2 | 13.2±1.7 | 748±36 | 11 | 1.01 |
| 11n | 100±10.0 | 11.6±2.2 | 657±10 | 9 | 3.52 |
| 11o | 43.7±7.0 | 4.8±1.0 | 177±19 | 9 | 2.81 |
| 11p | 280±13.2 | 8.0±1.5 | 403±33 | 35 | 3.60 |
| 11q | 119±7.5 | 15.0±0.7 | 641±57 | 8 | 1.89 |
| 11r | 145±8.2 | 17.9±1.5 | 830±57 | 8 | 2.02 |
| 11s | 152±25.6 | 44.2±3.7 | 441±98 | 3 | 1.23 |
| 11t | 169±10.9 | 7.2±0.2 | 706±67 | 23 | 1.66 |
| NGB-2904 | 112±22 | 2.0±0.4 | NDg | 56 | 6.94h |
Mean ± SEM, Ki values were determined by at least three experiments.
Ki values for D2 receptors were measured using human D2 (long) expressed in HEK cells with [125I]ABN as the radioligand.
Ki values for D3 receptors were measured using human D3 expressed in HEK cells with [125I]ABN as the radioligand.
Ki values for D4 receptors were measured using human D4.4 expressed in HEK cells with [125I]ABN as the radioligand.
Ki for D3 receptors/ Ki for D2 receptors.
Calculated C log P values using the program C log P by Advanced Chemistry Development, Inc. Toronto, Canada (ACD/Labs).
Not determined.
Published data, Leopoldo et al, 2002. 24
The substitution of the 4-position of the benzamide group with a 3-thiophene ring resulted in compound 11a. This analogue displayed both the highest D3 binding affinity (0.7 nM) and greatest D3 vs. D2 receptor selectivity (187-fold) of the panel of compounds reported in this communication. Other potent and selective compounds included 11b, 11c, 11f, 11g, 11j and 11k (Table 1).
The phenylhomopiperazine compounds had uniformly low affinity at the D4 dopamine receptor subtype (Table 1), with Ki values of >100 nM. The log P value for the homopiperazine analogs ranged from 1.0 to 4.0 (Table 1).
4. Adenylyl cyclase inhibition studies
D2 and D3 dopamine receptors are negatively coupled to adenylyl cyclase. Therefore, a forskolin-dependent adenylyl cyclase inhibition assay was used to determine the intrinsic efficacies of the new panel of homopiperazine compounds; these results were compared with the previously published values for the piperazine analogs (Table 2).22 The intrinsic efficacy of the homopiperazine compounds was generally found to be higher at D2 dopamine receptors. The effect of this structural modification on efficacy appears to vary at D3 receptors. The efficacy was comparable for some analogs (i.e., WC-26 vs. 11c, WC-28 vs. 11k and WC-34 vs. 11j) while the efficacy of the homopiperazine was higher for others (i.e., WC-10 vs. 11b, WC-21 vs. 11d and WC-23 vs. 11q) at D3 dopamine receptors (Table 2). WC-44 was previously reported to be a full agonist at D3 receptors but the homopiperazine analog, 11e, was found to be a strong partial agonist.
Table 2.
Comparison of the efficacy D3 dopamine receptor for selective phenylhomopiperazine and phenylpiperazine (WC) analogues.
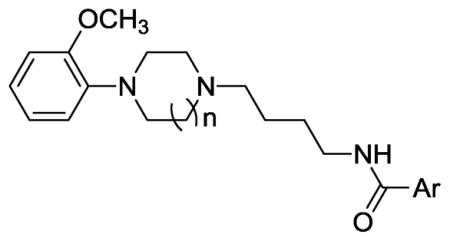
| ||||
|---|---|---|---|---|
| Compound | Ar | n | Percent Intrinsic Efficacy | |
| %IA D2 | %IAD3 | |||
| WC-10 |
|
n = 1 | 33.5±3.1 | 18.7±2.2 |
| 11b | n = 2 | 61.2±7.3 | 80.3±9.2 | |
|
| ||||
| WC-26 |
|
n = 1 | 29.88±4.8 | 68.7±4.1 |
| 11c | n = 2 | 65.2±7.3 | 67.9±6.7 | |
|
| ||||
| WC-21 |
|
n = 1 | 21.8±2.1 | 47.8±2.2 |
| 11d | n = 2 | 68.2±3.9 | 63.7±9.0 | |
|
| ||||
| WC-44 |

|
n = 1 | 35.3±1.0 | 96.2±4.2 |
| 11e | n = 2 | 57.1±3.4 | 77.8±7.8 | |
|
| ||||
| WC-28 |
|
n = 1 | 26.0±3.0 | 57.2±5.5 |
| 11k | n = 2 | 48.8±11.1 | 64.9±7.2 | |
|
| ||||
| WC-34 |
|
n = 1 | 20.8±4.4 | 62.4±3.3 |
| 11j | n = 2 | 50.8±3.5 | 59.5±12.2 | |
|
| ||||
| WC-23 |
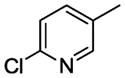
|
n = 1 | 33.4±4.9 | 59.9±0.8 |
| 11q | n = 2 | 84.2±5.5 | 74.5±10.5 | |
The intrinsic efficacy of the test compounds was evaluated by determining the percent inhibition of a forskolin-dependent whole cell adenylyl cyclase assay. The results were normalized to the percent inhibition obtained using the full agonist quinpirole at human D2 (1 μM) and D3 (100 nM) receptors expressed in stably transfected HEK 293 cells. The test drug was used at a concentration equal to approximately 10 × the Ki value that was determined from the radioligand binding analysis. The mean ± the S.E.M. values are reported for n ≥ 3.
Figure 3A shows a graph displaying the Ki values of the homopiperazine analogs at D3 receptors versus their corresponding piperazine congeners. Figure 3B shows a similar representation between the homopiperazine/piperazine congeners with respect to intrinsic activity at the D3 receptor. There was a linear correlation between the Ki values of the homopiperazine/piperazine congeners for binding to the D3 receptor, but no such correlation was observed with respect to intrinsic activity (IA) at the D3 receptor. These data suggest that although the homopiperazines and piperazines bind in a similar manner to the D3 receptor, there is a fundamental difference in the ability of the structural congeners to activate D3 receptor coupling to G proteins. This low correlation in IA is caused by the uniformly high intrinsic activity of the homopiperazine analogs at the D3 receptor (ranging from 60–60%), whereas there was a large range in IA of the piperazine analogs at the D3 receptor (ranging from 20–96%).
Figure 3.
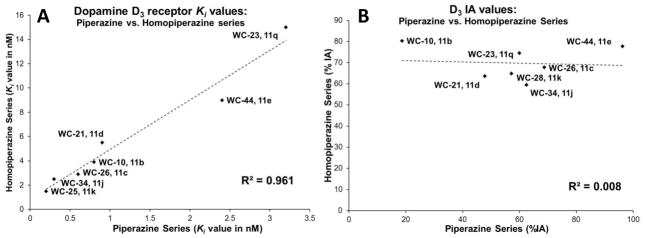
(A) Comparison of the Ki values of the homopiperazine and piperazine analogs at D3 receptors. (B) Similar representation for the Intrinsic Activity at D3 receptors.
5. Modeling studies
In an attempt to better understand the structure-activity relationship of the homopiperazine analogs, we utilized the 3D-QSAR models previously built to predict the binding activities for this series of compounds. The ligand alignments were obtained following essentially the protocol previously described by our group.3 Specifically, a conformer library for each ligand was generated using the MCMM method available in MacroModel. ROCS (version 2.3.1, OpenEye Scientific Software, Santa Fe NM)28 was used subsequently to retrieve the conformer from each library with the maximum shape alignment against a reference structure, the antagonist haloperidol which is bound to the orthosteric site of the refined homology models of D2 and D3.3 This procedure was applied to obtain two separate sets of ligand alignments for both the D2 and D3 binding sites. A salt bridge constraint between the highly conserved Asp carbonyl group in the third helical transmembrane spanning region and the protonated ligand amine was specified for both the piperazine and homopiperazine moieties.
Table 3 shows the data from both the a) experimental radioligand binding studies and b) the predicted binding affinities from the modeling studies. The residuals range from −0.41 to 1.24, indicating that our modeling studies are in good agreement with our experimental values. Figure 4 shows the phenylpiperazine WC-10 and the phenylhomopiperazine compound 11b as they occupy the human dopamine D2 and D3 receptor binding sites. We previously found that the orthosteric binding site, which is occupied by the substituted phenylpiperazine or phenylhomopiperazine moiety, is situated deeper in the D3 receptor molecular than in the D2 receptor. Therefore, the ligands assume a more linear conformation in the D3 receptor binding site (Figure 4B). When situated in the D2 receptor binding site, the 4-carbon chain assumes a bent conformation (Figure 4A). It is apparent from the modeling studies that the orientation of the piperazine andhomopiperazine moieties within the D3 receptor binding site are very similar. However, the orientation of both the arylamide moiety and the substituted phenyl is slightly different between the piperazine and homopiperazine analogues when bound by the D2 receptor. Our binding data indicates while the affinity of the homopiperazine analogs is lower for both the D2 and the D3 receptors, the difference in affinity is generally greater for the D3 receptor than for D2 receptor binding. Hence D3 receptor selectivity decreases.
Table 3.
Comparison of predicted and experimental affinities values for the homopiperazine analogs.
| Compound | D2 (log Ki) (nM) | D3 (log Ki) (nM) | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| Experiment | Predicted | Residual | Experiment | Predicted | Residual | |
| 11a | 2.107 | 1.385 | 0.722 | −0.155 | 0.256 | −0.411 |
| 11b | 2.074 | 1.524 | 0.550 | 0.591 | 0.204 | 0.387 |
| 11c | 2.003 | 1.380 | 0.623 | 0.462 | 0.212 | 0.250 |
| 11d | 2.097 | 1.839 | 0.258 | 0.740 | 0.145 | 0.595 |
| 11e | 2.032 | 1.547 | 0.485 | 0.954 | 0.354 | 0.600 |
| 11f | 2.106 | 1.533 | 0.573 | 0.255 | 0.359 | −0.104 |
| 11g | 2.043 | 1.249 | 0.794 | 0.568 | 0.403 | 0.165 |
| 11h | 1.979 | 1.176 | 0.803 | 0.944 | 0.298 | 0.646 |
| 11i | 1.752 | 1.607 | 0.145 | 0.875 | 0.422 | 0.453 |
| 11j | 2.138 | 1.433 | 0.705 | 0.398 | 0.079 | 0.319 |
| 11k | 2.096 | 1.927 | 0.169 | 0.176 | 0.186 | −0.010 |
| 11l | 2.029 | 1.965 | 0.064 | 0.954 | 0.216 | 0.738 |
| 11m | 2.179 | 1.511 | 0.668 | 1.121 | 0.123 | 0.998 |
| 11n | 1.998 | 1.256 | 0.742 | 1.064 | −0.163 | 1.227 |
| 11o | 1.640 | 1.391 | 0.249 | 0.681 | 0.438 | 0.243 |
| 11p | 2.446 | 1.988 | 0.458 | 0.903 | 0.283 | 0.620 |
| 11q | 2.073 | 1.573 | 0.500 | 1.176 | 0.279 | 0.897 |
| 11r | 2.162 | 1.636 | 0.526 | 1.253 | 0.340 | 0.913 |
| 11s | 2.180 | 1.455 | 0.725 | 1.645 | 0.401 | 1.244 |
| 11t | 2.227 | 1.522 | 0.705 | 0.857 | 0.170 | 0.687 |
Figure 4. Structural alignment of 11b and WC-10 as they occupy the D2 and D3 dopamine receptor binding sites.
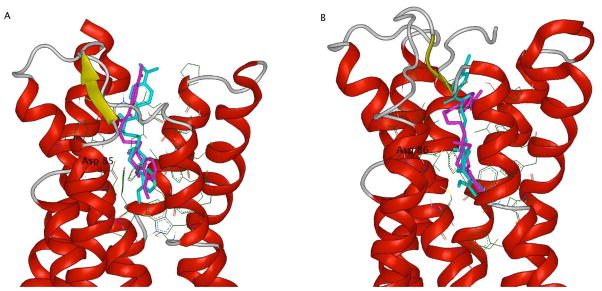
Compounds 11b (shown in cyan) and the piperazine analogue WC-10 (shown in magenta) as they occupy the binding sites of A) the human dopamine D2 receptor and B) the human D3 dopamine receptor. The compounds sit much deeper in D3 than in D2 and overall adopt a more linear orientation.
In an attempt to understand the interactions between the ligands and respective receptors, the “hybrid” docking program implemented in the OEdocking toolkit (version 3.0.0, OpenEye Scientific Software, Santa Fe NM)29 was used to dock the conformer libraries of each homopiperazine compound into the orthosteric site into both D2 and D3. This process finds and scores the best fit of each conformer to the bound haloperidol in the orthosteric site. A custom constraint was imposed ensuring a conserved salt bridge between the protonated amine and conserved Asp (comparable with what was done for the QSAR alignment). The docked molecules were further optimized using the program “szybki” (version 1.7.0, OpenEye Scientific Software, Santa Fe NM) which performs a molecular mechanics optimization of the ligand within the binding site. This process produces protein ligand binding energies for each ligand. The average binding energies of the homopiperazines were found to be essentially equivalent between the two receptors; −12.81 kcal/mol with D2, and −10.95 kcal/mol with D3. Gratifyingly, the interaction of compound 11a was 18-fold better for D3 than for D2 (1.35 kcal/mol vs −24.67 kcal/mol) consistent with what was experimentally observed.
6. Discussion
The goal of the current study was to identify ligands having a high affinity and selectivity for D3 versus D2 dopamine receptors, with calculated log P values within the desired range for passive transport across the blood brain barrier. Although many potent and selective D3 ligands have been reported in the literature, the majority of these ligands have calculated log P values > 5.0, which should limit their ability to cross the blood brain barrier. Previous studies with 11C-labeled aliphatic alcohols have indicated that the optimal log P values for a compound to have a high brain uptake (i.e., % brain extraction > 85% at a cerebral blood flow of 100 mL/min) range from −0.32 to 3.2.24 Given the structural requirements of the lead compounds for the current study (Figure 1), which consisted of a benzamide ring and an N-phenylhomopiperazine moiety separated by a 4-carbon spacer unit, it was unlikely that we would prepare an analog near the low end of the log P range described above. However, it should be possible to prepare analogs of the lead compounds that would fall within the upper limit of this log P range. Based on the data shown in Table 1, this goal was accomplished by a) replacing the N-(2,3-dichlorophenyl)piperazine group with an N-(2-methoxyphenyl) homopiperazine moiety, and b) attaching a heteroatom into the benzamide aromatic ring. The most promising analogs were achieved by substituting the para position of the benzamide ring with a 3-thiophene ring (i.e., 11a). This compound had a D2:D3 selectivity ratio >100 and a log P value of 3.5, which, based on the data presented by Dischino et al.,24 should result in high brain uptake, barring other factors such as being substrates for P glycoprotein. This compound may be useful a probe in the study of the behavioral pharmacology of dopamine D3 receptors. In addition, the presence of the 2-methoxy group of compound 11a indicates that the corresponding 11C-labeled versions of the compound can be prepared via alkylation of the corresponding de-methyl precursor with [11C]iodomethane. Therefore, [11C]11a may be a useful radiotracer for studying the regulation of dopamine D3 receptors in a variety of CNS disorders using Positron Emission Tomography (PET).
In an attempt to better understand the structure-activity relationship of the homopiperazine analogs, we utilized the 3D-QSAR models previously built for the piperazine based ligands to predict the binding activities of these homopiperazine based compounds. The predictions for the binding affinities of these homopiperazine analogs are in a reasonable range in comparison with the measured values by minimal residuals. It also further indicated they have moderate D3 selectivity.
In addition to 3D-QSAR, docking studies of these compounds in both the D2 and D3 receptors were consistent with what was found in previous QSAR studies. The D2 orthosteric site is shallower and closer to the solvent and favors a bent conformation in the ligands. The D3 site is deeper and the ligands further from exposure to solvent.
This simplistic docking evaluation highlights one limitation of modeling studies. The protein models described above have been optimized using an antagonist (haloperidol) in the ligand binding site. Therefore, differences in the increased intrinsic efficacy of the homopiperazines compared to the piperazines were not captured by the current docking studies.
Over the last decade, it has become clear that ligand-dependent GPCR activation is likely a multi-state process. Therefore, each of these states result in a different orientation of the helices and loops which make up the binding site of the D2 and D3 receptors. Consequently, the use of a static structure is unable to assess this dynamic process. Differences in intrinsic activity between the piperazine and homopiperazine analogs result from unique interactions with each of the different conformations, and are not likely to be predicted in a model based on the binding of a D2 and D3 antagonist. This is illustrated in Figure 4 where the top graph shows an excellent correlation in Ki values for the homopiperazine and piperazine based compounds found in Table 2, while the lower graph shows no correlation in terms of the observed IA. Therefore, more advanced models are needed in order to better understand the interaction of ligands with the D3 receptor leading to increased activation of second messenger systems.
In conclusion, we have completed a structure activity relationship study on a series of conformationally-flexible benzamides with the goal of identifying potential probes for studying the behavioral pharmacology and developing radiotracers for imaging dopamine D3 receptors with PET. We also have compared the affinity and intrinsic efficacy D3 dopamine receptor selective phenylpiperazine and phenylhomopiperazine analogues in Table 3. The conclusion is that D2 and D3 affinity decreases compare to piperazine analogs. The results of this study identified a number of compounds having a high affinity and selectivity for D3 versus D2 receptor with a log P value that may result in a high uptake in brain in vivo.
7. Experimental
7.1. Chemical analysis
Nuclear magnetic resonance (NMR) spectra were recorded on a Varian 300 MHz NMR spectrometer. Chemical shifts were reported in δ values (parts per million, ppm) relative to an internal standard of tetramethylsilane (TMS). The following abbreviations are used for multiplicity of NMR signals: br s = broad singlet, d = doublet, dd = doublet of doublets, dt = doublet of triplets, m = multiplet, s = singlet, t = triplet. Melting points were determined using MEL-TEMP 3.0 apparatus and uncorrected. Elemental analyses (C, H, N) were determined by Atlantic Microlab, Inc. and the analytical results were within ±0.4% of the theoretical values. HR-MS was performed on a Waters ZQ 4000 single quadrupole mass spectrometer equipped with an electrospray ionization (ESI) LC-MS interface. All reactions were carried out under an inert atmosphere of nitrogen. Lipophilicity measurements of the compounds were estimated using the computational program, Clog P (Advanced Chemistry Development, Inc., Toronto, Canada).
7.2. tert-Butyl 1,4-diazepane-1-carboxylate (6)
A solution of di-tert-butyl dicarbonate (2.18 g, 10 mmol) in MeOH (25 mL) was slowly added to a stirring solution of homopiperazine (2.0 g, 20 mmol) in MeOH (50 mL) at 0 °C. The mixture was then stirred for 2 days at room temperature, and the solvent removed under reduced pressure. The crude solid was redissolved in diethyl ether (100 mL) with warming, and the white precipitate was removed by filtration. The product was extracted from the mother liquor with 1 M citric acid solution (3×50 mL), and the aqueous layer was washed with EtOAc (3×50 mL), basified with Na2CO3 (pH 11), and extracted with EtOAc (3×50 mL). The organic layer was dried over Na2SO4 and evaporated in vacuum to give tert-butyl 1-homopiperazinecarboxylate 6 as a waxy white liquid (crude, 1.61 g, 80%); 1H NMR (300 MHz, CDCl3) δ 3.44 (m, 4H), 2.87 (m, 4H), 2.04 (s, 1H), 1.77 (m, 2H), 1.46 (s, 9H).
7.3. tert-Butyl 4-(2-methoxyphenyl)-1,4-diazepane-1-carboxylate (7)
In a flask was added to degassed toluene (5 ml) in the following order 2-bromoanisole (929 mg, 5 mmol, 1.0 equiv), tert-butyl 1-homopiperazinecarboxylate (6) (1.10 g, 5.5 mmol, 1.1 equiv of the amine), Pd2(dba)3 (50 mg, 0.054 mmol, 1–2.5 mol% of the amine), (±)-BINAP (100 mg, 0.16 mmol, 1.5 equiv/Pd), Et3N (0.30 mL, ~0.5 equiv), and KOtBu (1.68 g, 15 mmol, 3.0 equiv). The resulting dark red mixture was heated to 110 °C (bath temperature) and stirred at this temperature for 21 hours. Once TLC indicated complete consumption of the aryl bromide, the brownish mixture was cooled to room temperature, then filtered over a thin layer of Celite, and the filter cake was thoroughly washed with EtOAc. The filtrate was concentrated in vacuo and the residue purified by flash chromatography using mixtures of hexanes and EtOAc (4/1) to give an oil (928 mg, 61%). TLC Rf 0.28 (hexanes/ethyl acetate 4/1); 1H NMR (300 MHz, CDCl3) δ 6.95-6.83 (m, 4H), 3.84 (s, 3H), 3.51 (m, 4H), 3.24 (m, 4H), 2.04 (s, 1H), 1.93 (m, 2H), 1.46 (s, 9H).
7.4. 1-(2-Methoxyphenyl)-1,4-diazepane (8)
tert-Butyl 4-(2-methoxyphenyl)homopiperazine-1-carboxylate (7) (570 mg, 1.86 mmol) was dissolved in a mixture of trifluoroacetic acid (10 mL) and water (2.5 mL). The solution was stirred at room temperature overnight, and then was evaporated to dryness in vacuo. The residue was dissolved in water (10 mL) and then basified with Na2CO3 (pH = 11), and extracted with EtOAc (3×50 mL). The organic layer was washed with brine (2×50 mL) and dried over Na2SO4 overnight. Evaporation under reduced pressure gave N-4-(2-methoxyphenyl)homopiperazine (8) as a yellow liquid (252 mg, 66%), which was used without further purification.
7.5. 2-(4-(4-(2-Methoxyphenyl)-1,4-diazepan-1-yl)butyl)isoindoline-1,3-dione (9)
To a solution of N-4-(2-methoxyphenyl)homopiperazine (8) (283 mg, 1.37 mmol) and N-(4-bromobutyl)phthalimide (387 mg, 1.37 mmol) in acetonitrile (20 mL) was added K2CO3 (567 mg, 4.11 mmol). The mixture was refluxed overnight under N2, then filtered to remove the excess K2CO3, the filtrate was concentrated in vacuo and the residue purified with a mixture of hexanes and EtOAc (1:2) to give a yellow liquid (150 mg, 99%). TLC Rf 0.3 (hexanes/ethyl acetate 1:2); 1H NMR (300 MHz, CDCl3) δ 7.84 (d, J = 8.4 Hz, 2H), 7.72 (d, J = 8.4 Hz, 2H), 6.85–6.92 (m, 4H), 3.83 (s, 3H), 3.71 (m, 2H), 3.32 (m, 4H), 2.81 (m, 2H), 2.71 (m, 2H), 2.53 (m, 2H), 2.01 (m, 2H), 1.68 (m, 2H), 1.54 (m, 2H). 13C NMR (CDCl3, 300 MHz): 167.2, 162.4, 141.3, 132.4, 123.6, 122.2, 121.6, 113.8, 59.6, 58.3, 56.8, 53.9, 52.2, 39.5, 26.9, 25.7. HRMS (ESI) Calcd for C24H29N3O3 (M+H)+: 408.2287. Found: 408.2275.
7.6. 4-(4-(2-Methoxyphenyl)-1,4-diazepan-1-yl)butan-1-amine (10)
Hydrazine hydrate (2 mL) was added into the solution of N-[4-[4-(2-methoxyphenyl)-1-homopiperazinyl)butyl]-phthalimide (9) ( 328 g, 0.80 mmol) in ethanol (10 mL). The reaction mixture was heated at reflux for 2 h. The reaction was cooled, filtered and concentrated in vacuo. The residue was dissolved in CH2Cl2 (20 mL) and washed with 1N NaOH (20 mL × 3) and brine solution. The organic phase was dried over Na2SO4 and concentrated in vacuo to afford target product as a light yellow oil (223 mg, 100%); 1H NMR (300 MHz, CDCl3) δ 6.92-6.85 (m, 4H), 3.83 (s, 3H), 3.33 (m, 4H), 2.83 (m, 2H), 2.71 (m, 4H), 2.51 (m, 2H), 1.96 (m, 2H), 1.54-1.44 (m, 4H). 13C NMR (CDCl3, 300 MHz): 162.3, 141.3, 123.6, 121.9, 113.4, 59.6, 58.3, 56.8, 52.2, 41.5, 26.9, 26.2, 25.7. HRMS (ESI) Calcd for C16H27N3O (M+H)+: 278.2232. Found: 278.2243.
7.7. Methyl 4-vinylbenzoate (12)
A solution of 4-vinylbenzoic acid (2.08 g, 14.0 mmol) and trimethyloxonium tetrafluoroborate (2.60 g,17.5 mmol) in CH2Cl2 (200 mL) was added triethylamine (1.56 g, 15.4 mmol) at ambient temperature. The reaction mixture was stirred for 20 hours at ambient temperature, then washed with saturated Na2CO3 (50 mL), saturated NaCl (50 mL) and dried over Na2SO4. After evaporation of the solvent in vacuo, the crude product was purified by silica gel column chromatography with hexane and ether (10/1) to afford 1.72 g (76%) of 12 as a white solid which was used directly in the next step. TLC Rf 0.25 (hexane and ether 10/1); 1H NMR (300 MHz, CDCl3) δ 8.01 (d, J = 8.4 Hz, 2H), 7.48 (d, J = 8.4 Hz, 2H), 6.76 (dd, J = 17.5 Hz, J = 10.8 Hz, 1H), 5.88 (d, J = 17.5 Hz, 1H), 5.40 (d, J = 10.8 Hz), 3.93 (s, 3H).
7.8. Methyl 4-(2-hydroxyethyl)benzoate (13)
Compound 12 (1.95 g, 12.0 mmol) in 1M BH3 in THF (24 mL) was stirred 1h at 0 °C, then 1h at ambient temperature. A solution of 1N NaOH (36 mL) was added to the reaction mixture at 0 °C, then a solution of 35% H2O2 (20 mL) was added. The mixture was stirred 30 min at 0 °C, then 30 min at ambient temperature. Ethyl acetate (100 mL) was added, the organic layer was separated, washed with water (3×50 mL), saturated Na2CO3 (3×50 mL), saturated NaCl (3×50 mL) and dried over Na2SO4. After evaporation of the solvent in vacuo, the crude product was purified by chromatography with hexane ether (1:1) to afford 1.16 g (54%) of 13 as a colorless oil. TLC Rf 0.31 (hexane and ether 1:1); 1H NMR (300 MHz, CDCl3) δ 8.00 (d, J = 8.4 Hz, 2H), 7.32 (d, J = 8.4 Hz, 2H), 3.92 (s, 3H), 3.91 (t, J = 6.6 Hz, 2H), 2.95 (t, J = 6.6 Hz, 2H).
7.9. 4-(2-Hydroxyethyl)benzoic acid (14)
A solution of 13 (174 mg, 0.96 mmol) in methanol (3 mL) and water (1 mL) was added NaOH (58 mg, 1.43 mmol) at ambient temperature. The mixture was stirred for 2 days at ambient temperature, water (5 mL) was then added, and extracted with ether (3×20 mL). The aqueous layer was acidified with HCl (1:1) to pH = 1, the white solid was filtered out to afford 158 mg (100%) of 14 which was used directly in the next step. 1H NMR (300 MHz, CDCl3) δ 7.97 (d, J = 8.4 Hz, 2H), 7.27 (d, J = 8.4 Hz, 2H), 3.83 (t, J = 6.3 Hz, 2H), 2.88 (t, J = 6.3 Hz, 2H).
7.10. Methyl 4-(2-fluoroethyl)benzoate (15)
A solution of 13 (1.16 g, 6.44 mmol) in CH2Cl2 (20 mL) was added DAST (1.56 g, 9.66 mmol) at 0 °C. The mixture was warmed to ambient temperature and stirred overnight, then ethyl acetate (100 mL) was added, washed with water (50 mL), saturated Na2CO3 (50 mL), saturated NaCl (50 mL), and dried over Na2SO4. After evaporation of the solvent in vacuo, the crude product was purified by chromatography with hexane ether (10:1) to afford 1.06 g (91%) of 15 as a colorless oil. TLC Rf 0.25 (hexane ether 10:1); 1H NMR (300 MHz, CDCl3) δ 8.01 (d, J = 8.4 Hz, 2H), 7.33 (d, J = 8.1 Hz, 2H), 4.67 (dt, J = 47.1 Hz, J = 6.3 Hz, 2H), 3.93 (s, 3H), 3.08 (dt, J = 24.6 Hz, J = 6.3 Hz, 2H).
7.11. 4-(2-Fluoroethyl)benzoic acid (16)
Solid NaOH (58 mg, 1.43 mmol) was added to a solution of 15 (174 mg, 0.96 mmol) in methanol (3 mL) and water (1 mL) at ambient temperature. The mixture was stirred for 2 days at ambient temperature, water (5 mL) was added, extracted with ether (20 mL). The aqueous layer was acidified with HCl (1:1) to pH = 1, the white solid was filtered out to afford 160 mg (100%) of 16 which was used directly in the next step. 1H NMR (300 MHz, CDCl3) δ 8.07 (d, J = 8.4 Hz, 2H), 7.36 (d, J = 8.4 Hz, 2H), 4.68 (dt, J = 46.8 Hz, J = 6.3 Hz, 2H), 3.10 (dt, J = 24.6 Hz, J = 6.3 Hz, 2H).
7.12. General Procedure for Preparing the Substituted Benzamide Analogues N-(4-(4-(2-Methoxyphenyl)-1,4-diazepan-1-yl)butyl)-4-(thiophen-3-yl)benzamide (11a)
A mixture of compound 10 (135 mg, 0.49 mmol) and 4-(thiophen-3-yl)benzoic acid (99.4 mg, 0.49 mmol) in dichloromethane (10 mL) was stirred at 0 °C (ice-water bath). Dicyclohexylcarbodiimide (DCC) (121 mg, 0.59 mmol) and hydroxybenzotriazole (HOBt) (80 mg, 0.59 mmol) were added to the above solution. The ice bath was removed, and the reaction mixture was stirred at ambient temperature for 15 hours. Dichloromethane (20 mL) was added into the reaction mixture, and the solution was washed with saturated aqueous NaHCO3 solution (3×10 mL). The organic layer was dried over Na2SO4, concentrated under reduced pressure, and the crude product was purified by silica gel column chromatography using dichloromethane methanol (20:1) as the mobile phase to give 11a as a white solid (194 mg, 86%). TLC Rf 0.20 (dichloromethane methanol 20:1); mp 125.5–126.6 °C. 1H NMR (300 MHz, CDCl3) δ 7.75 (d, J = 8.7 Hz, 2H), 7.54 (d, J = 8.7 Hz, 2H), 7.43 (s, 1H), 7.33 (s, 2H), 7.19 (s, 1H), 6.81–6.94 (m, 4H), 3.85 (s, 3H), 3.44 (q, J = 5.1 Hz, 2H), 3.25-3.20 (m, 4H), 2.83-2.76 (m, 4H), 2.57 (t, J = 5.2 Hz, 2H), 1.93 (t, J = 4.2 Hz, 2H), 1.61–1.69 (m, 4H). 13C NMR (CDCl3, 300 MHz): 167.4, 151.4, 142.1, 141.2, 138.8, 133.4, 127.5, 126.6, 126.3, 126.1, 121.3, 121.0, 120.7, 117.9, 111.6, 57.1, 56.1, 55.3, 54.3, 52.5, 51.8, 39.4, 27.7, 27.3, 25.5. HRMS (ESI) Calcd for C27H33N3O2S (M+H)+: 464.2372. Found: 464.2379. Anal. (C27H33N3O2S·1.5H2C2O4) C, H, N.
7.13. 4-(Dimethylamino)-N-(4-(4-(2-methoxyphenyl)-1,4-diazepan-1-yl)butyl)benzamide(11b)
Compound 11b was prepared according to the procedure for compound 11a except using 4-(dimethylamino)benzoic acid, which afford 102 mg (80%) of 11b as a white solid. TLC Rf 0.15 (dichloromethane methanol 20:1); mp 55–56 °C. 1H NMR (300 MHz, CDCl3) δ 7.89 (d, J = 9.0 Hz, 2H), 7.27 (m, 1H), 6.97 (m, 2H), 6.89 (m, 2H), 6.68 (d, J = 9.0 Hz, 2H), 3.82 (s, 3H), 3.54-3.48 (m, 8H), 3.28 (m, 2H), 3.12 (m, 2H), 3.07 (s, 6H), 2.46 (m, 2H), 2.04 (m, 2H), 1.74 (m, 2H). 13C NMR (CDCl3, 300 MHz): 167.5, 162.4, 154.3, 141.1, 130.4, 123.5, 122.9, 121.4, 113.5, 111.7, 57.1, 56.4, 55.2, 54.3, 52.5, 51.8, 39.3, 27.7, 26.7, 25.4. HRMS (ESI) Calcd for C25H36N4O2 (M+H)+: 425.2917. Found: 425.2936. Anal. (C25H36N4O2·H2C2O4·0.5H2O) C, H, N.
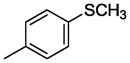
7.14. N-(4-(4-(2-Methoxyphenyl)-1,4-diazepan-1-yl)butyl)-4-(methylthio)benzamide(11c)
Compound 11c was prepared according to the procedure for compound 11a except using 4-(methylthio)benzoic acid, which afford 71 mg (79%) of 11c as a white solid. TLC Rf 0.21 (dichloromethane methanol 20:1); mp 92.2–93 °C. 1H NMR (300 MHz, CDCl3) δ 7.63 (d, J = 7.5 Hz, 2H), 7.14 (d, J = 7.5 Hz, 2H), 6.85-6.77 (m, 5H), 3.87 (s, 3H), 3.53 (d, J = 4.2 Hz, 2H), 3.22 (m, 4H), 2.81-2.73 (m, 4H), 2.53 (t, J = 6.9 Hz, 2H), 2.41 (s, 3H), 1.91 (m, 2H), 1.57 (m, 4H). 13C NMR (CDCl3, 300 MHz): 167.3, 162.2, 142.8, 141.1, 130.5, 127.8, 126.9, 123.0, 122.2, 113.5, 57.2, 56.2, 55.4, 54.4, 52.7, 51.8, 39.4, 27.7, 26.6, 25.4. 14.7. HRMS (ESI) Calcd for C24H33N3O2S (M+H)+: 428.2372. Found: 428.2369. Anal. (C24H33N3O2S·H2C2O4) C, H, N.
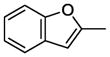
7.15. N-(4-(4-(2-Methoxyphenyl)-1,4-diazepan-1-yl)butyl)benzofuran-2-carboxamide(11d)
Compound 11d was prepared according to the procedure for compound 11a except using 2-benzofurancarboxylic acid, which afford 65 mg (66%) of 11d as a white solid. TLC Rf 0.30 (Ethyl acetate methanol 10:1); mp 57.5–59 °C. 1H NMR (300 MHz, CDCl3) δ 7.67 (m, 1H), 7.48 (m, 1H), 7.40 (m, 1H), 7.23 (m, 1H), 7.19 (m, 1H), 6.85-6.77 (m, 5H), 3.87 (s, 3H), 3.47 (q, J = 6.0 Hz, 2H), 3.28 (m, 4H), 2.83-2.73 (m, 4H), 2.52 (t, J = 6.6 Hz, 2H), 1.93 (m, 2H), 1.63 (m, 4H). 13C NMR (CDCl3, 300 MHz): 162.3, 157.2, 156.3, 150.1, 141.1, 128.8, 123.3, 123.0, 122.0, 120.9, 113.4, 111.5, 108.9, 57.2, 56.2, 55.4, 54.4, 52.7, 51.8, 39.3, 27.7, 26.7, 25.4. HRMS (ESI) Calcd for C25H31N3O3 (M+H)+: 422.2444. Found: 422.2427. Anal. (C25H31N3O3·H2C2O4·0.25H2O) C, H, N.
7.16. 4-(2-Fluoroethyl)-N-(4-(4-(2-methoxyphenyl)-1,4-diazepan-1-yl)butyl)benzamide(11e)
Compound 11e was prepared according to the procedure for compound 11a except using 16, purified with ether methanol (10:1) to afford 112 mg (84%) of 11e as a white solid. TLC Rf 0.29 (dichloromethane methanol 15:1); mp 64.2–65.3 °C. 1H NMR (300 MHz, CDCl3) δ 7.67 (d, J = 8.4 Hz, 2H), 7.18 (d, J = 8.4 Hz, 2H), 6.85-6.77 (m, 5H), 4.62 (dt, J = 46.8 Hz, J = 6.3 Hz, 2H), 4.47 (dt, J = 46.8 Hz, J = 6.3 Hz, 2H), 3.86 (s, 3H), 3.49 (q, J = 6.0 Hz, 2H), 3.23 (m, 4H), 3.01-2.98 (m, 4H), 2.80-2.73 (m, 4H), 2.52 (m, 2H), 1.91 (m, 2H), 1.59 (m, 4H). 13C NMR (CDCl3, 300 MHz): 167.4, 162.2, 142.8, 141.2, 131.4, 127.4, 123.0, 122.0, 113.5, 86.2, 57.5, 56.6, 55.4, 54.4, 52.6, 51.8, 39.3, 36.4, 27.7, 26.7, 25.4. HRMS (ESI) Calcd for C25H34FN3O2 (M+H)+: 428.2713. Found: 428.2730. Anal. (C25H34FN3O2· H2C2O4) C, H, N.
7.17. N-(4-(4-(2-Methoxyphenyl)-1,4-diazepan-1-yl)butyl)-4-vinylbenzamide (11f)
Compound 11f was prepared according to the procedure for compound 11a except using 4-vinylbenzoic acid, purified with ether methanol (10:1) to afford 92 mg (81%) of 11f as a white solid. TLC Rf 0.31 (dichloromethane methanol 15:1); mp 72.5–74 °C. 1H NMR (300 MHz, CDCl3) δ 7.75 (d, J = 8.4 Hz, 2H), 7.43 (d, J = 8.4 Hz, 2H), 6.89-6.85 (m, 5H), 6.63 (dd, J = 17.7 Hz, J = 10.8 Hz, 1H), 5.74 (d, J = 17.7 Hz, 1H), 5.26 (d, J = 10.8 Hz, 1H), 3.86 (s, 3H), 3.49 (m, 2H), 3.31 (m, 4H), 2.88-2.73 (m, 4H), 2.59 (m, 2H), 1.99 (m, 2H), 1.67 (m, 4H). 13C NMR (CDCl3, 300 MHz): 167.6, 162.2, 141.3, 136.1, 133.4, 129.2, 127.5, 123.0, 122.0, 114.4, 113.4, 57.2, 56.2, 55.4, 54.4, 52.7, 51.8, 39.3, 27.8, 26.6, 25.4. HRMS (ESI) Calcd for C25H33N3O2 (M+H)+: 408.2651. Found: 408.2638. Anal. (C25H33N3O2· H2C2O4) C, H, N.
7.18. 4-Chloro-N-(4-(4-(2-methoxyphenyl)-1,4-diazepan-1-yl)butyl)benzamide(11g)
Compound 11g was prepared according to the procedure for compound 11a except using 4-chlorobenzoic acid, purified with ether methanol (10:1) to afford 83 mg (80%) of 11g as a white solid. TLC Rf 0.25 (dichloromethane methanol 20:1); mp 62–63.2 °C. 1H NMR (300 MHz, CDCl3) δ 7.88 (d, J = 8.4 Hz, 2H), 7.78 (s, 1H), 7.39 (d, J = 8.4 Hz, 2H), 6.95-6.85 (m, 4H), 3.82 (s, 3H), 3.53 (d, J = 4.2 Hz, 2H), 3.36-3.24 (m, 6H), 3.07 (m, 2H), 2.30 (m, 2H), 1.91 (m, 2H), 1.64 (m, 4H). 13C NMR (CDCl3, 300 MHz): 167.5, 162.3, 141.1, 137.4, 132.4, 130.3, 128.8, 123.0, 122.0, 113.5, 57.2, 56.4, 55.4, 54.3, 52.7, 51.8, 39.5, 27.7, 26.6, 25.4. HRMS (ESI) Calcd for C23H30ClN3O2 (M+H)+: 416.2105. Found: 416.2129. Anal. (C23H30ClN3O2·H2C2O4·H2O) C, H, N.
7.19. 4-Fluoro-N-(4-(4-(2-methoxyphenyl)-1,4-diazepan-1-yl)butyl)benzamide(11h)
Compound 11h was prepared according to the procedure for compound 11a except using 4-fluorobenzoic acid, purified with ether methanol (10:1) to afford 75 mg (79%) of 11h as a white solid. TLC Rf 0.24 (dichloromethane methanol 20:1); mp 52.8–54 °C. 1H NMR (300 MHz, CDCl3) δ 7.95 (d, J = 8.1 Hz, 2H), 7.72 (s, 1H), 7.05 (d, J = 8.4 Hz, 2H), 6.97-6.88 (m, 4H), 3.82 (s, 3H), 3.50 (d, J = 4.2 Hz, 2H), 3.48-3.24 (m, 6H), 3.07 (m, 2H), 2.30 (m, 2H), 1.89 (m, 2H), 1.69 (m, 4H). 13C NMR (CDCl3, 300 MHz): 167.7, 166.4, 162.5, 141.3, 130.0, 129.3, 123.1, 122.0, 115.7, 113.8, 57.2, 56.2, 55.4, 54.4, 52.7, 51.8, 39.4, 27.7, 26.7, 25.4. HRMS (ESI) Calcd for C23H30FN3O2 (M+H)+: 400.2400. Found: 400.2415. Anal. (C23H30FN3O2·H2C2O4·0.5H2O) C, H, N.
7.20. N-(4-(4-(2-Methoxyphenyl)-1,4-diazepan-1-yl)butyl)-5-methylthiophene-2-carboxamide(11i)
Compound 11i was prepared according to the procedure for compound 11a except using 5-methylthiophene-2-carboxylic acid, which afforded 100 mg (83%) of 11i as a white solid. TLC Rf 0.21 (dichloromethane methanol 20:1); mp 58.7–59.2 °C. 1H NMR (300 MHz, CDCl3) δ 7.40 (d, J = 3.6 Hz, 1H), 6.91-6.84 (m, 5H), 6.65 (d, J = 3.6 Hz, 1H), 3.85 (s, 3H), 3.45 (m, 2H), 3.35-3.26 (m, 4H), 3.06-2.98 (m, 4H), 2.77 (m, 2H), 2.46 (s, 3H), 2.14 (m, 2H), 1.74-1.66 (m, 4H). 13C NMR (CDCl3, 300 MHz): 162.0, 151.7, 144.8, 142.7, 136.6, 128.5, 125.9, 120.9, 120.7, 117.9, 111.5, 57.6, 56.2, 55.6, 54.4, 52.7, 51.9, 39.5, 28.1, 27.6, 25.5. 15.5. HRMS (ESI) Calcd for C22H31N3O2S (M+H)+: 402.2215. Found: 402.2229. Anal. (C22H31N3O2S·H2C2O4·0.5H2O) C, H, N
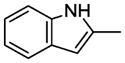
7.21. N-(4-(4-(2-Methoxyphenyl)-1,4-diazepan-1-yl)butyl)-1H-indole-2-carboxamide(11j)
Compound 11j was prepared according to the procedure for compound 11a except using indole-2-carboxylic acid, which afford 87 mg (75%) of 11j as a white solid. TLC Rf 0.25 (ethyl acetate methanol 15:1); mp 72.9–74 °C. 1H NMR (300 MHz, CDCl3) δ 9.83 (br, 1H), 7.66 (d, J = 8.4 Hz, 1H), 7.46 (d, J = 8.4 Hz, 1H), 7.30 (t, J = 7.5 Hz, 1H), 7.16 (t, J = 7.5 Hz, 1H), 7.02 (m, 1H), 6.9-6.82 (m, 5H), 3.88 (s, 3H), 3.55 (m, 2H), 3.32 (m, 4H), 2.72 (m, 4H), 2.62 (t, J = 6.9 Hz, 2H), 2.02 (m, 2H), 1.73 (m, 4H). 13C NMR (CDCl3, 300 MHz): 162.2, 160.7, 141.3, 139.4, 138.6, 131.1, 123.1, 122.0, 120.7, 119.8, 114.9, 113.5, 111.2, 57.9, 56.2, 55.5, 54.4, 52.7, 51.9, 39.3, 27.7, 26.7, 24.6. HRMS (ESI) Calcd for C25H32N4O2 (M+H)+: 421.2604. Found: 421.2621. Anal. (C25H32N4O2·H2C2O4·0.25H2O) C, H, N
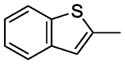
7.22. N-(4-(4-(2-Methoxyphenyl)-1,4-diazepan-1-yl)butyl)benzo[b]thiophene-2-carboxamide(11k)
Compound 11k was prepared according to the procedure for compound 11a except using thianaphthene-2-carboxylic acid, which afford 81 mg (78%) of 11k as a white solid. TLC Rf 0.32 (ethyl acetate methanol 10:1); mp 66–67 °C. 1H NMR (300 MHz, CDCl3) δ 7.86-7.78 (m, 3H), 7.44-7.36 (m, 2H), 7.02-6.84 (m, 5H), 3.85 (s, 3H), 3.51 (q, J = 6.3 Hz, 2H), 3.1 (m, 4H), 2.86 (m, 4H), 2.58 (t, J = 6.9 Hz, 2H), 1.98 (m, 2H), 1.67 (m, 4H). 13C NMR (CDCl3, 300 MHz): 162.6, 161.4, 146.6, 145.7, 141.1, 139.8, 124.3, 123.1, 122.8, 113.7, 57.6, 56.4, 55.3, 54.4, 52.7, 51.9, 39.6, 27.6, 26.8, 25.4. HRMS (ESI) Calcd for C25H31N3O2S (M+H)+: 438.2215. Found: 438.2236. Anal. (C25H31N3O2S·H2C2O4·0.5H2O) C, H, N.
7.23. N-(4-(4-(2-Methoxyphenyl)-1,4-diazepan-1-yl)butyl)-5-phenylthiophene-2-carboxamide(11l)
Compound 11l was prepared according to the procedure for compound 11a except using 4-phenyl-thiophene-2-carboxylic acid, which afford 80 mg (81%) of 11l as a white solid. TLC Rf 0.22 (dichloromethane methanol 15:1); mp 77–78 °C. 1H NMR (300 MHz, CDCl3) δ 7.79 (m, 1H), 7.57-7.53 (m, 3H), 7.40-7.25 (m, 3H), 6.87-6.75 (m, 5H), 3.80 (s, 3H), 3.47 (q, J = 6.3 Hz, 2H), 3.29 (m, 4H), 2.86 (m, 4H), 2.58 (m, 2H), 1.96 (m, 2H), 1.66 (m, 4H). 13C NMR (CDCl3, 300 MHz): 162.4, 151.3, 142.5, 141.1, 140.2, 135.0, 128.9, 127.5, 127.3, 126.2, 124.4, 122.3, 120.9, 117.9, 111.5, 110.0, 57.2, 56.9, 55.3, 54.4, 52.7, 51.9, 38.7, 27.5, 26.4, 25.3. HRMS (ESI) Calcd for C27H33N3O2S (M+H)+: 464.2372. Found: 464.2358. Anal. (C27H33N3O2S·H2C2O4·0.25H2O) C, H, N.
7.24. 4-(2-Hydroxyethyl)-N-(4-(4-(2-methoxyphenyl)-1,4-diazepan-1-yl)butyl)benzamide(11m)
Compound 11m was prepared according to the procedure for compound 11a except using 14, purified with ether methanol (10:1) to afford 102 mg (84%) of 11m as a white solid. TLC Rf 0.27 (dichloromethane methanol 12:1); mp 52–53 °C. 1H NMR (300 MHz, CDCl3) δ 7.77 (d, J = 6.9 Hz, 2H), 7.26 (m, 3H), 6.86 (m, 4H), 3.89-3.82 (m, 5H), 3.48 (q, J = 5.4 Hz, 2H), 3.26 (m, 4H), 3.06-2.98 (m, 4H), 2.89 (m, 2H), 2.76 (m, 2H), 2.09 (m, 2H), 1.77-1.69 (m, 4H). 13C NMR (CDCl3, 300 MHz): 167.5, 152.4, 141.8, 141.2, 131.6, 127.8, 127.4, 123.0, 122.1, 121.9, 113.7, 61.3, 57.5, 56.2, 55.6, 54.4, 52.7, 51.7, 39.4, 38.9, 27.7, 26.5, 25.0. HRMS (ESI) Calcd for C25H35N3O3 (M+H)+: 426.2757. Found: 426.2742.Anal. (C25H35N3O3·H2C2O4) C, H, N.
7.25. N-(4-(4-(2-Methoxyphenyl)-1,4-diazepan-1-yl)butyl)-9-oxo-9H-fluorene-4-carboxamide(11n)
Compound 11n was prepared according to the procedure for compound 11a except using 9-oxofluorene-4-carboxylic acid, which afford 80 mg (81%) of 11n as a white solid. TLC Rf 0.30 (ethyl acetate methanol 10:1); mp 61–62 °C. 1H NMR (300 MHz, CDCl3) δ 7.98 (m, 1H), 7.77 (d, J = 7.2 Hz, 1H), 7.69 (d, J = 7.2 Hz, 2H), 7.57 (m, 3H), 7.35-7.25 (m, 3H), 6.87-6.75 (m, 5H), 3.80 (s, 3H), 3.47 (m, 2H), 3.14-3.04 (m, 4H), 2.72 (m, 2H), 2.63 (m, 2H), 2.55 (m, 2H), 1.77-1.69 (m, 6H). 13C NMR (CDCl3, 300 MHz): 193.8, 162.3, 144.6, 141.5, 139.5, 137.0, 134.2, 133.0, 131.8, 130.2, 127.4, 126.8, 123.1, 122.1, 121.9, 113.8, 57.6, 56.5, 55.6, 54.4, 52.7, 51.8, 39.4, 27.5, 25.4, 24.4. HRMS (ESI) Calcd for C30H33N3O3 (M+H)+: 484.2600. Found: 484.2612. Anal. (C30H33N3O3·H2C2O4·0.25H2O) C, H, N.
7.26. 5-Bromo-N-(4-(4-(2-methoxyphenyl)-1,4-diazepan-1-yl)butyl)thiophene-2-carboxamide(11o)
Compound 11o was prepared according to the procedure for compound 11a except using 5-methyl-thiophene-2-carboxylic acid, which afford 103 mg (81%) of 11o as a white solid. TLC Rf 0.21 (dichloromethane methanol 15:1); mp 69–70 °C. 1H NMR (300 MHz, CDCl3) δ 7.35 (d, J = 4.2 Hz, 1H), 7.01-6.87 (m, 6H), 3.85 (s, 3H), 3.44 (m, 2H), 3.22 (m, 4H), 2.85 (m, 2H), 2.77 (m, 2H), 2.60 (m, 2H), 2.00 (m, 2H), 1.69 (m, 4H). 13C NMR (CDCl3, 300 MHz): 162.7, 161.3, 141.4, 140.1, 138.8, 132.5, 124.6, 123.4, 122.1, 121.8, 113.7, 57.6, 56.2, 55.2, 54.4, 52.7, 51.7, 38.6, 26.9, 25.5, 24.5. HRMS (ESI) Calcd for C21H28BrN3O2S (M+H)+: 466.1164. Found: 466.1182. Anal. (C21H28BrN3O2S·H2C2O4·0.5H2O) C, H, N.
7.27. 5-(4-Fluorophenyl)-N-(4-(4-(2-methoxyphenyl)-1,4-diazepan-1-yl)butyl)thiophene-2-carboxamide(11p)
Compound 11p was prepared according to the procedure for compound 11a except using 5-(4-fluorophenyl)thiophene-2-carboxylic acid, which afford 93 mg (75%) of 11p as a white solid. TLC Rf 0.23 (dichloromethane methanol 15:1); mp 130–131 °C. 1H NMR (300 MHz, CDCl3) δ 7.50 (m, 3H), 7.08-6.94 (m, 4H), 6.85-6.76 (m, 4H), 3.85 (s, 3H), 3.34 (m, 2H), 3.20 (m, 4H), 2.99 (m, 2H), 2.93 (m, 2H), 2.67 (m, 2H), 2.03 (m, 2H), 1.67 (m, 4H). 13C NMR (CDCl3, 300 MHz): 163.0, 162.2, 161.3, 148.1, 141.1, 138.2, 137.3, 129.4, 129.1, 123.1, 121.9, 121.1, 116.0, 113.5, 57.6, 56.2, 55.6, 54.4, 52.7, 51.9, 49.1, 39.9, 33.9, 27.5, 26.4, 25.3. HRMS (ESI) Calcd for C27H32FN3O2S (M+H)+: 482.2278. Found: 482.2259. Anal. (C27H32FN3O2S·H2C2O4·0.5H2O) C, H, N.
7.28. 6-Chloro-N-[4-[4-(2-methoxyphenyl)-1,4-diazepan-1-yl)butyl)nicotinamide (11q)
Compound 11q was prepared according to the procedure for compound 11a except using 6-chloronicotinic acid, which afford 95 mg (82%) of 11q as a white solid. TLC Rf 0.21 (dichloromethane methanol 15:1); mp 43–44 °C. 1H NMR (300 MHz, CDCl3) δ 9.00 (d, J = 2.4 Hz, 1H), 8.45 (dd, J = 8.6 Hz, J = 2.4 Hz, 1H), 8.32 (d, J = 8.4 Hz, 1H), 7.35 (d, J = 8.4 Hz, 1H), 7.03-6.84 (m, 4H), 3.85 (s, 3H), 3.49 (m, 2H), 3.39-3.26 (m, 4H), 3.07 (m, 2H), 2.34 (m, 2H), 2.00 (m, 2H), 1.71 (m, 4H). 13C NMR (CDCl3, 300 MHz): 165.7, 162.4, 154.7, 150.4, 141.1, 140.4, 128.4, 126.1, 123.0, 122.1, 121.9, 113.5, 57.6, 56.2, 55.6, 54.4, 52.7, 51.9, 39.9, 27.6, 25.6, 24.4. HRMS (ESI) Calcd for C22H29ClN4O2 (M+H)+: 417.2057. Found: 417.2049. Anal. (C22H29ClN4O2·H2C2O4) C, H, N.
7.29. 6-Bromo-N-[4-[4-(2-methoxyphenyl)-1,4-diazepan-1-yl)butyl)nicotinamide (11r)
Compound 11r was prepared according to the procedure for compound 11a except using 6-bromonicotinic acid, which afford 91 mg (83%) of 11r as a white solid. TLC Rf 0.21 (dichloromethane methanol 15:1); mp 50–51 °C. 1H NMR (300 MHz, CDCl3) δ 8.95 (d, J = 2.4 Hz, 1H), 8.36 (dd, J = 8.6 Hz, J = 2.4 Hz, 1H), 8.20 (d, J = 8.4 Hz, 1H), 7.53 (d, J = 8.4 Hz, 1H), 7.03-6.86 (m, 4H), 3.85 (s, 3H), 3.53 (m, 2H), 3.38-3.31 (m, 4H), 3.04 (m, 2H), 2.33 (m, 2H), 1.97 (m, 2H), 1.72 (m, 4H). 13C NMR (CDCl3, 300 MHz): 167.5, 162.3, 141.1, 137.4, 132.4, 130.3, 128.8, 123.0, 122.0, 113.5, 57.6, 56.2, 55.6, 54.4, 52.7, 51.9, 39.5, 27.7, 26.6, 25.4. HRMS (ESI) Calcd for C23H30ClN3O2 (M+H)+: 416.2105. Found: 416.2129. Anal. (C22H29BrN4O2·H2C2O4·0.5H2O) C, H, N.
7.30. 2,3-Dimethoxy-N-[4-[4-(2-Methoxyphenyl)-1,4-diazepan-1-yl)butyl)benzamide (11s)
Compound 11s was prepared according to the procedure for compound 11a except using 2,3-dimethoxybenzoic acid, which afford 70 mg (79%) of 11s as a white solid. TLC Rf 0.26 (dichloromethane methanol 15:1); mp 43–44 °C. 1H NMR (300 MHz, CDCl3) δ 8.05 (m, 1H), 7.67 (d, J = 7.8 Hz, 1H), 7.14 (t, J = 8.1 Hz, 1H), 7.02 (d, J = 7.8 Hz, 1H), 6.91-6.83 (m, 4H), 3.90 (s, 3H), 3.83 (s, 3H), 3.51 (d, J = 4.2 Hz, 2H), 3.33 (m, 4H), 2.99-2.91 (m, 4H), 2.71 (t, J = 6.9 Hz, 2H), 2.01 (m, 2H), 1.67 (m, 4H). 13C NMR (CDCl3, 300 MHz): 169.3, 162.7, 154.0, 149.8, 141.1, 128.5, 123.0, 122.1, 120.8, 118.7, 113.8, 61.0, 57.6, 56.2, 55.6, 54.4, 52.7, 51.9, 39.6, 27.7, 26.8, 24.4. HRMS (ESI) Calcd for C25H35N3O4 (M+H)+: 442.2706. Found: 442.2718. Anal. (C25H35N3O4·H2C2O4·0.25H2O) C, H, N.
7.31. N-[4-[4-(2-Methoxyphenyl)-1,4-diazepan-1-yl)butyl)-4-(methylamino)benzamide (11t)
Compound 11t was prepared according to the procedure for compound 11a except using 4-(methylamino)benzoic acid, which afford 102 mg (81%) of 11t as a white solid. TLC Rf 0.19 (dichloromethane methanol 15:1); mp 58–59 °C. 1H NMR (300 MHz, CDCl3) δ 7.72 (d, J = 6.9 Hz, 2H), 6.94-6.81 (m, 5H), 6.55 (d, J = 6.9 Hz, 2H), 3.82 (s, 3H), 3.45 (m, 2H), 3.34 (m, 2H), 3.28 (m, 2H), 3.07-3.00 (m, 4H), 2.85 (s, 3H), 2.78 (m, 2H), 2.16 (m, 2H), 1.79-1.65 (m, 4H). 13C NMR (CDCl3, 300 MHz): 167.9, 162.1, 154.0, 141.1, 130.4, 123.2, 122.6, 121.4, 113.9, 112.7, 57.6, 56.2, 55.6, 54.4, 52.7, 51.9, 41.4, 39.5, 29.8, 26.7, 25.6, 24.9. HRMS (ESI) Calcd for C24H34N4O2 (M+H)+: 411.2760. Found: 411.2749. Anal. (C24H34N4O2·H2C2O4·0.5H2O) C, H, N.
7.32. Dopamine receptor binding assays
A filtration binding assay was used to characterize the binding properties of membrane-associated receptors.26 For human D2 long, D3, and D4 dopamine receptors expressed in HEK 293 cells, membrane homogenates (50 μL) were suspended in 50 mM Tris HCl/150 mM NaCl/10 mM EDTA buffer, pH 7.5 and incubated with 50 μL of 125I-IABN at 37 °C for 60 min. Non-specific binding was defined using 20 μM (+)-butaclamol. For competition experiments the radioligand concentration is generally equal to 0.5 times the Kd value and the concentration of the competitive inhibitor ranges over 5 orders of magnitude. Each competition curve was performed using two concentrations of inhibitor per decade and all assays are performed in triplicate. Binding was terminated by the addition of cold wash buffer (10 mM Tris HCl/150 mM NaCl, pH 7.5) and filtration over a glass-fiber filter (Schleicher and Schuell No. 32). Filters were washed with 10 mL of cold buffer and the radioactivity was measured using a Packard Cobra gamma counter. Estimates of the equilibrium dissociation constant and maximum number of binding sites were obtained using unweighted nonlinear regression analysis of data modeled according to the equation describing mass action binding.30 Data from competitive inhibition experiments were modeled using nonlinear regression analysis to determine the concentration of inhibitor that inhibits 50% of the specific binding of the radioligand. Competition curves were modeled for a single site and the IC50 values will be converted to equilibrium dissociation constants (Ki values) using the Cheng and Prusoff equation.31 Mean Ki values ± S.E.M are reported for at least three independent experiments.
Acknowledgments
This research was funded by DA023957, MH081281 and DA29840 awarded by the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Jackson DM, Westlind-Danielsson A. Pharmacology & Therapeutics. 1994;64:291. doi: 10.1016/0163-7258(94)90041-8. [DOI] [PubMed] [Google Scholar]
- 2.Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Physiol Rev. 1998;78:189. doi: 10.1152/physrev.1998.78.1.189. [DOI] [PubMed] [Google Scholar]
- 3.Wang Q, Mach RH, Luedtke RR, Reichert DE. J Chem Inf Model. 2010;50:1970. doi: 10.1021/ci1002747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luedtke RR, Mach RH. Curr Pharm Des. 2003;9:643. doi: 10.2174/1381612033391199. [DOI] [PubMed] [Google Scholar]
- 5.Hackling AE, Stark H. Chembiochem. 2002;3:946. doi: 10.1002/1439-7633(20021004)3:10<946::AID-CBIC946>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 6.Morissette M, Goulet M, Grondin R, Blanchet P, Bedard PJ, Di Paolo T, Levesque D. Eur J Neurosci. 1998;10:2565. doi: 10.1046/j.1460-9568.1998.00264.x. [DOI] [PubMed] [Google Scholar]
- 7.Ryoo HL, Pierrotti D, Joyce JN. Mov Disord. 1998;13:788. doi: 10.1002/mds.870130506. [DOI] [PubMed] [Google Scholar]
- 8.Gurevich EV, Joyce JN. Neuropsychopharmacology. 1999;20:60. doi: 10.1016/S0893-133X(98)00066-9. [DOI] [PubMed] [Google Scholar]
- 9.Gurevich EV, Bordelon Y, Shapiro RM, Arnold SE, Gur RE, Joyce JN. Arch Gen Psychiatry. 1997;54:225. doi: 10.1001/archpsyc.1997.01830150047009. [DOI] [PubMed] [Google Scholar]
- 10.Schwartz JC, Diaz J, Pilon C, Sokoloff P. Brain Research Reviews. 2000;31:277. doi: 10.1016/s0165-0173(99)00043-0. [DOI] [PubMed] [Google Scholar]
- 11.Pinter MM, Rutgers AWF, Hebenstreit E. Journal of Neural Transmission. 2000;107:1307. doi: 10.1007/s007020070020. [DOI] [PubMed] [Google Scholar]
- 12.Möller JC, Oertel WH. European Journal of Neurology. 2000;7:21. doi: 10.1046/j.1468-1331.2000.0070s1021.x. [DOI] [PubMed] [Google Scholar]
- 13.Bézard E, Ferry S, Mach U, Stark H, Leriche L, Boraud T, Gross C, Sokoloff P. Nature Medicine. 2003;9:762. doi: 10.1038/nm875. [DOI] [PubMed] [Google Scholar]
- 14.Guillin O, Griffon N, Diaz J, Foll BL, Bezard E, Gross C, Lammers C, Stark H, Carroll P, Schwartz JC, Sokoloff P. Int Rev Neurobiol. 2004;59:425. doi: 10.1016/S0074-7742(04)59016-5. [DOI] [PubMed] [Google Scholar]
- 15.Grundt P, Carlson EE, Cao J, Bennett CJ, McElveen E, Taylor M, Luedtke RR, Newman AH. J Med Chem. 2005;48:839. doi: 10.1021/jm049465g. [DOI] [PubMed] [Google Scholar]
- 16.Newman AH, Blaylock BL, Nader MA, Bergman J, Sibley DR, Skolnick P. Biochemical Pharmacology. 2012;84:882. doi: 10.1016/j.bcp.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Newman AH, Grundt P, Nader MA. J Med Chem. 2005;48:3663. doi: 10.1021/jm040190e. [DOI] [PubMed] [Google Scholar]
- 18.Bettinetti L, Schlotter K, Hubner H, Gmeiner P. J Med Chem. 2002;45:4594. doi: 10.1021/jm025558r. [DOI] [PubMed] [Google Scholar]
- 19.Newman AH, Grundt P, Cyriac G, Deschamps JR, Taylor M, Kumar R, Ho D, Luedtke RR. J Med Chem. 2009;52:2559. doi: 10.1021/jm900095y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chu W, Tu Z, McElveen E, Xu J, Taylor M, Luedtke RR, Mach RH. Bioorg Med Chem. 2005;13:77. doi: 10.1016/j.bmc.2004.09.054. [DOI] [PubMed] [Google Scholar]
- 21.Glase SA, Akunne HC, Heffner TG, Johnson SJ, Kesten SR, MacKenzie RG, Manley PJ, Pugsley TA, Wright JL, Wise LD. Bioorg Med Chem Lett. 1996;6:1361. [Google Scholar]
- 22.Robarge MJ, Husbands SM, Kieltyka A, Brodbeck R, Thurkauf A, Newman AH. J Med Chem. 2001;44:3175. doi: 10.1021/jm010146o. [DOI] [PubMed] [Google Scholar]
- 23.Newman AH, Cao J, Bennett CJ, Robarge MJ, Freeman RA, Luedtke RR. Bioorg Med Chem Lett. 2003;13:2179. doi: 10.1016/s0960-894x(03)00389-5. [DOI] [PubMed] [Google Scholar]
- 24.Leopoldo M, Berardi F, Colabufo NA, De Giorgio P, Lacivita E, Perrone R, Tortorella V. J Med Chem. 2002;45:5727. doi: 10.1021/jm020952a. [DOI] [PubMed] [Google Scholar]
- 25.Yuan J, Chen X, Brodbeck R, Primus R, Braun J, Wasley JWF, Thurkauf A. Bioorganic & Medicinal Chemistry Letters. 1998;8:2715. doi: 10.1016/s0960-894x(98)00469-7. [DOI] [PubMed] [Google Scholar]
- 26.Dischino DD, Welch MJ, Kilbourn MR, Raichle ME. Journal of Nuclear Medicine. 1983;24:1030. [PubMed] [Google Scholar]
- 27.Waterhouse RN. Mol Imaging Biol. 2003;5:376. doi: 10.1016/j.mibio.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 28.Hawkins PCD, Skillman AG, Nicholls A. J Med Chem. 2007;50:74. doi: 10.1021/jm0603365. [DOI] [PubMed] [Google Scholar]
- 29.McGann M. J Chem Inf Model. 2011;51:578. doi: 10.1021/ci100436p. [DOI] [PubMed] [Google Scholar]
- 30.McGonigle P, Molinoff PB. In: Basic Neurochemistry: Molecular, Cellular, and Medical Aspects. Siegel GJ, Agranoff BW, Albers W, Molinoff PB, editors. Raven Press; New York, New York: 1989. p. 183. [Google Scholar]
- 31.Cheng Y, Prusoff WH. Biochem Pharmacol. 1973;22:3099. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]