

Abstract
Purpose of review
Recent progress in understanding the production, processing, and function of the cystic fibrosis gene product, the cystic fibrosis transmembrane conductance regulator (CFTR), has revealed new therapeutic targets to repair the mutant protein. Classification of CFTR mutations and new treatment strategies to address each will be described here.
Recent findings
High-throughput screening and other drug discovery efforts have identified small molecules that restore activity to mutant CFTR. Compounds such as VX-770 that potentiate CFTR have demonstrated exciting results in recent clinical trials and demonstrate robust effects across several CFTR mutation classes in the laboratory. A number of novel F508del CFTR processing correctors restore protein to the cell surface and improve ion channel function in vitro and are augmented by coadministration of CFTR potentiators. Ongoing discovery efforts that target protein folding, CFTR trafficking, and cell stress have also indicated promising results. Aminoglycosides and the novel small molecule ataluren induce translational readthrough of nonsense mutations in CFTR and other genetic diseases in vitro and in vivo and have shown activity in proof of concept trials, and ataluren is now being studied in confirmatory trials.
Summary
An improved understanding of the molecular mechanisms underlying the basic genetic defect in cystic fibrosis have led to new treatment strategies to repair the mutant protein.
Keywords: cystic fibrosis, cystic fibrosis transmembrane conductance regulator, new therapies
Introduction
Remarkable progress in the understanding of cystic fibrosis (CF) pathogenesis has led to a number of therapeutic opportunities in the disease [1]. Several therapies including mucolytics, inhaled antimicrobials, systemic anti-inflammatories, and nutritional support are the mainstays of CF treatment, and these supportive therapies are largely responsible for the marked improvement in life expectancy over time, resulting in a median survival of 37 years [2]. Based on the discovery of the CF gene by Collins, Riordan, and Tsui [3–5], coupled with our emerging understanding of the function of its gene product, the cystic fibrosis transmembrane conductance regulator (CFTR), new treatments that target the basic ion transport defect in the disease are now in various stages of development, and promise to complement supportive therapies already available to patients. Recent progress in the treatment strategies that target CFTR through protein repair will be described here. Other promising treatment strategies that intend to circumvent CFTR by restoring depleted airway surface liquid and augmenting mucus clearance through osmotic forces in the airway, activate alternative chloride transporters or inhibit hyperactive sodium absorption through the epithelial sodium channel, and replacement of the CFTR gene itself through gene therapy are reviewed elsewhere [6,7].
Cystic fibrosis transmembrane conductance regulator mutations and molecular approaches to protein repair
CFTR is an epithelial ion channel expressed in exocrine glands that conducts chloride and bicarbonate across the plasma membrane and regulates transepithelial transport of sodium. Absence of functional CFTR in CF engenders gross dysfunction of multiple organs, causing excess morbidity and early mortality owing to mucus obstruction and leading to profound pulmonary failure [7].
Over 1500 putative mutations have been described in CFTR, which can be divided into classes according to the molecular mechanism of the genetic defect [1]. An understanding of the biology of each of these mutations has led to therapeutic strategies based on the particular mutation type (Fig. 1). Class I mutations include premature termination codons (PTCs, e.g. nonsense mutations) within the coding region of CFTR, which cause premature truncation of normal protein translation. These mutations are found in ~10% of CF patients, but are particularly common in Ashkenazi Jews (~75% of mutant CFTR alleles) [8]. Class II CFTR mutations include F508del CFTR, the most common mutation in humans (accounting for ~75% of alleles and found in approximately 90% of CF patients). The deletion of phenylalanine at the 508 positions causes CFTR to exhibit abnormal folding characterized by deficient stabilization by domain–domain interactions between the nucleotide-binding domain 1 (NBD1) and the transmembrane domains [9,10]. The misfolded protein is recognized by cellular chaperones within the endoplasmic reticulum (ER), directed to the proteasome, and rapidly degraded prior to reaching its active site at the cell surface. Because the cellular machinery responsible for the recognition and degradation of the misfolded protein is not 100% efficient, particular individuals exhibit low levels of surface expression of F508del CFTR, which may account for partial CFTR activity (and a more mild CF phenotype) observed in individuals homozygous for F508del CFTR, and could represent a population more amenable to protein repair [11]. Even when at the cell surface, F508del CFTR exhibits reduced gating, suggesting that misfolded CFTR also exhibits reduced CFTR ion channel activity.
Figure 1. Therapeutic approaches to address various cystic fibrosis transmembrane conductance regulator mutation classes.
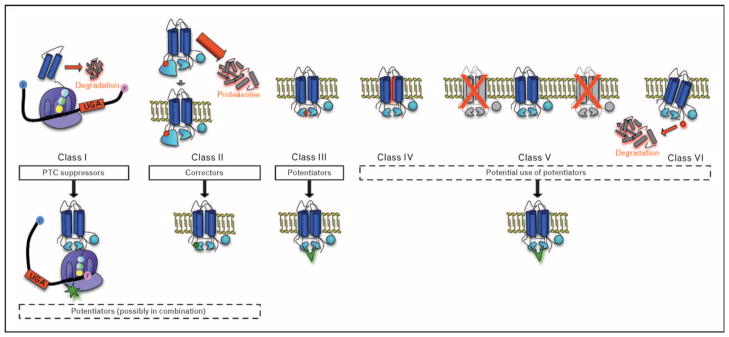
Classes of defects in the cystic fibrosis transmembrane conductance regulator (CFTR) gene include PTCs causing truncated protein translation (class I); misfolded CFTR, including deletion of phenylalanine at position 508 (class II); full-length CFTR that reaches the cell surface, but exhibits abnormal channel gating (class III) or reduced pore conductivity (class IV); CFTR with splicing errors that reduce surface expression (class V); and C-terminus mutations (class VI) that reduce membrane residence time. PTC suppressors (e.g. aminoglycosides and ataluren) bind to ribosomal subunits (green star) to allow suppression of PTCs and expression of full-length protein. Class II mutations like F508del can respond to small-molecule corrector compounds to restore folding defects and/or enhance expression of the channel at the cell membrane. Without correction, almost all F508del CFTR is shunted to the proteosome, leaving detectable surface protein in only select individuals. CFTR potentiators are currently in human clinical trials (VX-770, green chevron) for patients with G551D and F508del CFTR. Future directions include exploring the use of CFTR potentiators for other CFTR mutations known to reside at the cell surface. Combination therapy with potentiators has also been proposed for classes I and II CFTR mutations. PTC, premature termination codon.
Class III and IV CFTR mutations are characterized by full-length CFTR that reaches the cell surface but exhibit reduced ion transport activity owing to abnormal channel gating (Class III, e.g. G551D) or reduced conductivity of the ion channel pore (Class IV, e.g. R117H). Similarly, splicing mutants (Class V) and mutations within the C-terminus (Class VI) are also full length, but exhibit reduced activity owing to reduced numbers of active channels within the plasma membrane [12,13].
Although the molecular basis of CFTR mutants is complex and as yet incomplete, the classification of CFTR mutants can be simplified into the therapeutically relevant groups based on the activity of agents in development. Both traditional and high-throughput drug discovery programs have resulted in discovery of novel compounds that address specific mutant CFTR alleles.
These ‘CFTR modulators’ are pharmacological agents intended to repair the CFTR protein and are described in each section that follows. Although the molecular defect of many CFTR mutants have not been fully characterized, ongoing efforts within the international ‘CFTR2’ project are intended to systematically catalog the phenotype associated with reported CFTR mutations, and will be complemented with functional and biochemical measures of CFTR activity and expression in vitro. These data should provide a path for systematic categorization of many CFTR mutations commonly present in the CF population, and provide guidance as to the most applicable therapy for each specific mutation.
Potentiators of cell-surface cystic fibrosis transmembrane conductance regulator
CFTR mutation classes that result in dysfunctional CFTR that resides at the plasma membrane include Class III, IV, V, and VI mutations and represent potential targets for CFTR activators. G551D CFTR represents an archetype CFTR allele for this category of agents, as it exhibits normal surface expression and half-life, but confers a severe defect in channel gating owing to an amino acid substitution in the adenosine triphosphate (ATP) binding pocket within the nucleotide binding domains [14]. Flavonoids are well known activators of mutant CFTR and were among the first to be studied for beneficial effects in human individuals (including topical administration) [15]. Although agents such as genistein were affected by lack of efficacy in the nasal airway, more recent efforts have demonstrated activity of the flavonoid quercetin in the nose [16]. However, flavonoid agents are challenged by poor solubility and systemic absorption, and are poor development candidates for inhaled therapeutics.
More recent discovery strategies have focused on identification of compounds that ‘potentiate’ CFTR activity, restoring endogenous regulation (e.g. cyclic adenosine monosphosphate (cAMP) dependent regulation) and ion transport without excessive, constitutive activation that may potentially be detrimental (such as excessive CFTR activation seen with certain diarrheal illnesses) [17]. Identification of agents of this type is amenable to high-throughput screening-based strategies to discover agents that activate mutant CFTR by measuring the effects on anion conductance in cell-based screening assays. A number of specific strategies have been used for screens of this sort, including chloride sensitive dyes [18,19], fluorescence resonance energy transfer-based analysis of membrane potential [17], and cell conductance of airway monolayers [20]. Identification and characterization of small molecule potentiators of mutant CFTR have led to the development of agents with pronounced activity in vitro and in the clinic. Secondary testing of a lead compound developed by Vertex Pharmaceuticals (Cambridge, Massachusetts, USA), VX-770, indicated robust activation of mutant CFTR, including G551D expressed in heterologous cell culture systems and primary airway epithelial cell monolayers. The underlying mechanism of this agent is through augmentation of the open probability (Po), as judged by single-channel analysis of excised membrane patches [21••]. The agent is highly specific to human (but not murine) CFTR, a characteristic shared by CFTR potentiators identified by other developmental strategies, and likely indicative of a protein conformation in human CFTR that is dependent on the human amino acid sequence [22]. Although the binding site of this molecule has not been reported, other agents (e.g. VRT-532) that exhibit potentiator activity have been shown to directly bind F508del CFTR as indicated by protease stability experiments [22], and have also been shown to increase the ATPase activity (in addition to chloride conductance) of G551D CFTR [23•].
Testing of VX-770 has been conducted in CF individuals harboring at least one G551D CFTR mutation. Preliminary findings from a two-part phase 2 randomized, double-blind, placebo-controlled, dose-ranging clinical trial to evaluate safety, efficacy, and pharmacokinetics of VX-770 were recently reported [24]. Results of this two-part study indicated significant and dose-dependent improvements of sweat chloride (remarkably, mean sweat chloride achieved the diagnostic threshold at the most efficacious doses tested), improvement of CFTR-dependent chloride conductance in the airways as measured by nasal potential difference (NPD), and a significant increase in lung function over 2 weeks. The second part of this study confirmed initial findings, established that beneficial effects on ion transport and lung function were sustained at 4 weeks, and indicated a positive trend in measures of respiratory quality of life (e.g. the Cystic Fibrosis Questionaire-Revised (CFQ-R), a validated patient outcome measure [25,26]. These results represent the first evidence that any drug treatment improves CFTR activity in the airways (thus addressing the basic CF defect), corrects the sweat chloride abnormality, and ameliorates lung function in CF individuals. The results firmly establish CFTR as a viable therapeutic target in CF and support confirmatory studies of VX-770 in large randomized placebo-controlled studies. VX-770 has now progressed to phase 3 testing in CF individuals harboring G551D (Clinical Trials.gov reference NCT00909532 and NTC00909727) and phase 2 testing in individuals homozygous for F508del (Clinical Trials.gov reference number NCT00953706). An important question to be addressed in these long-term studies is the relationship between measures of CFTR activity and degree of lung function improvement, as a correlation between these parameters was not observed in the preliminary analysis of the phase 2 study [26].
Correction of F508del cystic fibrosis transmembrane conductance regulator misprocessing
Significant effort has been directed toward the goal of correcting the folding of F508del CFTR, thus restoring ion channel activity to the misfolded protein. A diverse array of cellular targets have been explored, commensurate with the large number of proteins now known to interact with CFTR biogenesis. Agents such as 4-phenyl butyrate downregulate Hsc70 (or other cell chaperones) central to the folding process, and represent an early example of compounds tested in the clinic [27,28]. Other more recent efforts have resulted from high-throughput library screens for chloride channel function following incubation of test compounds with F508del expressing cells [17,29,30•]. A number of these strategies have identified F508del correctors that may address cell biogenesis through chaperone pathways. Pharmacologic activity of such agents has also been reported to augment F508del CFTR half-life in the plasma membrane through altered surface recycling attributed to features of the cellular processing machinery [31] or reduced endocytic trafficking [32••]. This class of agents may be potential drug development candidates if their safety in vivo is confirmed. Other compounds have been shown to directly interact with CFTR [33•,34•] and may offer greater specificity than agents that alter general aspects of cell folding or cellular quality control.
Initial results of a phase 2 trial of VX-809, a lead F508del CFTR corrector in the Vertex program, established that systemic administration of the compound for 4 weeks modestly improved sweat chloride at the highest dose tested compared with placebo [35]. These remarkable results further demonstrate that rescue of F508del CFTR in human individuals is achievable by a systemically delivered small molecule; although no effect on NPD or lung function was observed in the first clinical trial with this agent, ongoing efforts to enhance the degree of CFTR rescue to levels that also clearly confer clinically meaningful improvements in lung function are in progress. One straightforward approach is to coadminister a potentiator of CFTR channel gating. Because F508del CFTR exhibits other defects in addition to misfolding, including abnormal channel gating and membrane residence time, this strategy provides a high likelihood of increasing F508del CFTR following treatment with VX-809, as indicated by in-vitro results with combination therapy [36].
Other discovery platforms seek to identify agents that augment F508del CFTR folding, independent of ion channel function. Using a trafficking assay based on epitope tagged CFTR, phosphodiesterase inhibitors including sildenafil and other active analogues have been shown to improve surface localization of the mutant protein. The same agents augment short-circuit current in F508del CFTR expressing cell lines [37] and enhance NPD in CF mice [38,39•]. More recently, the orally bioavailable compound glafanine was identified using the same assay [40]. Other approaches to discover F508del CFTR correctors utilize fluorescently tagged CFTR labels [41]. Cell-free systems to examine the stability of purified NBD1, CFTR folding (as judged by proteolytic degradation patterns), or other methods may complement available cell-based screens.
The global cellular response to misfolded protein may also represent a target. Histone deacetylases (HDAC) have far-ranging effects on gene expression, and specific members of the HDAC family are involved in the ER-associated degradation pathway promoting degradation of F508del CFTR [42]. Treatment of CF cells with HDAC inhibitors can modulate ER stress, and HDACs such as suberoylanilide hydroxamic acid, as well as siRNA-silencing of HDACs, increase levels of F508del CFTR in the cell membrane [43•]. The combination of approaches such as these reveal a number of potential pharmacologic agents for F508del correction. Additive or synergistic rescue of F508del CFTR using more than one such strategy may offer hope of achieving ion transport activity sufficient to confer a normal phenotype in CF respiratory epithelia [30•,44].
Translational readthrough of premature termination codons
Readthrough of PTCs represents another exciting approach to address the underlying cause of CF, and many other genetic diseases caused by PTCs. Certain aminoglycosides and other agents have the capacity to interact with the eukaryotic rRNA within the ribosomal subunits [45]. Although this interaction is much weaker than that seen in prokaryotes and is distinct from the primary cause of aminoglycoside toxicity in human individuals, it can modestly reduce the fidelity of eukaryotic translation by interrupting the normal proofreading function of the ribosome [46–52]. Insertion of a near cognate amino acid at a premature stop codon allows protein translation to continue until one of several stop codons normally present at the end of the mRNA transcript is reached and properly utilized [48]. The specificity of the strategy has been attributed to greater stop codon fidelity at the authentic (3′) end of mRNA and has been established in vitro by demonstrating no detectable elongation beyond native stop codons [53–56] together with an acceptable safety profile in both preclinical and phase I and II clinical studies.
Proof of concept experiments with aminoglycosides established that premature stop mutations within CFTR in human individuals can be suppressed, resulting in the synthesis of full-length, functional CFTR protein [53–56,57••]. As expected, the approach is not specific to CF, as in-vitro experiments have demonstrated efficacy of the approach in other diseases caused by PTCs, including Duchenne’s muscular dystrophy, Hurler’s syndrome, ceroid lipofuscinosis, nephropathic cystinosis, and expression of mutated p53 [46,51,58–61], and has also demonstrated success in mouse models of CF [62,63]. In a double-blind, placebo-controlled trial from Israel using gentamicin, bioelectric correction of nasal ion transport was seen specifically in individuals with nonsense mutations, and, as expected, not observed in CF controls homozygous for F508del [55]. This followed two small pilot trials also indicating restoration of chloride secretion in CF individuals harboring stop codons [54,56]. A trial examining systemic gentamicin in seven French individuals with Y122X CFTR, a mutation highly susceptible to readthrough, also indicated rescue of CFTR activity in the airway and sweat duct [53]. Not all aminoglycoside trials in CF have demonstrated success, suggesting low levels of protein correction, or the possibility of genetic founder effects. Regardless due to the known toxicity and poor bioavailability of aminoglycosides, more efficacious agents that avoid undesirable properties of aminoglycosides will be required in CF, where ~10% of CFTR function is likely required to confer clinical improvement based on genotype–phenotype correlations in the disease [64]. One such approach includes medicinal chemistry approaches to isolate the antimicrobial, toxic, and readthrough effects of the base scaffolds, a strategy demonstrating initial success using in-vitro reporters of efficacy and toxicity [65•], and cell-based and animal-based models of CFTR rescue [66]. Another is to identify entirely novel compounds that confer advantages over the aminoglycoside class.
PTC Therapeutics, Inc. (South Plainfield, New Jersey, USA), screened and evaluated over 800 000 compounds to identify new agents more suitable than gentamicin for inducing translational readthrough [67]. The effort resulted in the identification of ataluren (formerly PTC124), a novel, orally bioavailable agent that exhibits PTC suppression at concentrations readily achievable in serum [62,68]. The agent is also efficacious in vivo in an animal model of nonsense mediated CF [69]. Surface localized full-length CFTR was substantially improved in cross-sections of intestinal tissues following administration to CF mice carrying the G542X mutation and restored CFTR function by Ussing chamber analysis in intestinal samples of mice after 2 weeks of treatment with ataluren. The drug is well tolerated in normal and CF individuals [68], leading to a series of clinical trials examining ataluren in CF individuals harboring nonsense alleles.
Two recent studies utilizing orally administered ataluren in Israel [70] and France/Belgium [57••] detected rescue of CFTR activity (as assayed by the NPD) in open label, two-dose crossover phase II trials in CF individuals possessing PTCs. Similarly to the results of clinical studies with gentamicin, a complementary trial conducted at United States centers did not demonstrate improvement in CFTR function [71], raising questions as to the lack of efficacy. The reasons for failure are not completely clear, but include the challenge of NPD studies in multicenter trials (a critique that has been subsequently addressed by improvement in the testing method [72]), relative susceptibility of the W1282X mutation found commonly in Israel [73], and genetic founder effects, including the degree of CFTR mRNA expression at baseline [70,74]. Alternatively, ataluren has recently been reported to induce stabilization of the firefly luciferase [75•], which induces a paradoxical increase when used as a reporter of PTC suppression [76]. Although the concentration that is observed is greater than the dose required to induce readthrough, it has raised the question as to whether chemical optimization using an alternate readthrough assay might yield even more efficacious compounds.
A follow-up study examining the effect of prolonged treatment with ataluren (3 months) in 19 individuals previously studied for 2 weeks indicated significant improvement in CFTR activity following a 3-month treatment period, and was accompanied by a trend toward improved pulmonary function and a significant reduction in quantitative cough [77]. Importantly, all individuals who failed to respond to treatment in the previous 2-week cycle exhibited an NPD response after 3 months of therapy, suggesting time-dependent effects in individuals resistant to initial treatment [77]. Based on these results, a phase 3 randomized, blinded, placebo-controlled study is currently ongoing and is of sufficient size and duration to discern time-dependent effects on a CF population harboring a diverse array of CFTR PTCs (Clinical Trials.gov reference number NCT00803205).
Conclusion
Discovery of the CFTR gene and an improved understanding of the role of ion transport in disease pathogenesis have led to the development of new therapies that target the underlying defect in cystic fibrosis. Several high-throughput-based screening efforts have come to fruition, demonstrating rescue of the CFTR protein in the clinic in phase 2 trials. A new generation of strategies that address F508del CFTR misfolding promise new opportunities to address the most common cause of CF. Combined with steady improvements in the supportive care of CF, these advances promise an optimistic future for CF patients and their families.
Acknowledgments
The authors are grateful for the thoughtful advice provided by Eric J. Sorscher, MD. Omnigraffle Professional v5.2 (Seattle, Washington, USA) was used to compose Fig. 1. Financial support was provided by the National Institutes of Health (1K23 DK075788-01 and 1R03DK084110-01, each to S.M.R.).
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest
Additional references related to this topic can also be found in the Current World Literature section in this issue (pp. 000–000).
- 1.Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005;352:1992–2001. doi: 10.1056/NEJMra043184. [DOI] [PubMed] [Google Scholar]
- 2.Cystic Fibrosis Foundation. National Patient Registry Annual Data Report 2009. Bethesda (MD): Cystic Fibrosis Foundation; 2009. [Google Scholar]
- 3.Kerem B, Rommens JM, Buchanan JA, et al. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245:1073–1080. doi: 10.1126/science.2570460. [DOI] [PubMed] [Google Scholar]
- 4.Riordan JR, Rommens JM, Kerem B, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 5.Rommens JM, Iannuzzi MC, Kerem B, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245:1059–1065. doi: 10.1126/science.2772657. [DOI] [PubMed] [Google Scholar]
- 6.Mogayzel PJ, Jr, Flume PA. Update in cystic fibrosis. Am J Respir Crit Care Med. 2010;181:539–544. doi: 10.1164/rccm.200912-1943UP. [DOI] [PubMed] [Google Scholar]
- 7.O’Sullivan BP, Freedman SD. Cystic fibrosis. Lancet. 2009;373:1891–1904. doi: 10.1016/S0140-6736(09)60327-5. [DOI] [PubMed] [Google Scholar]
- 8.Kerem E, Kerem B. Genotype-phenotype correlations in cystic fibrosis. Pediatr Pulmonol. 1996;22:387–395. doi: 10.1002/(SICI)1099-0496(199612)22:6<387::AID-PPUL7>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 9.Loo TW, Bartlett MC, Clarke DM. Correctors enhance maturation of DeltaF508 CFTR by promoting interactions between the two halves of the molecule. Biochemistry. 2009;48:9882–9890. doi: 10.1021/bi9004842. [DOI] [PubMed] [Google Scholar]
- 10.Serohijos AW, Hegedus T, Aleksandrov AA, et al. Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc Natl Acad Sci U S A. 2008;105:3256–3261. doi: 10.1073/pnas.0800254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bronsveld I, Mekus F, Bijman J, et al. Chloride conductance and genetic background modulate the cystic fibrosis phenotype of Delta F508 homozygous twins and siblings. J Clin Invest. 2001;108:1705–1715. doi: 10.1172/JCI12108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zielenski J, Tsui LC. Cystic fibrosis: genotypic and phenotypic variations. Annu Rev Genet. 1995;29:777–807. doi: 10.1146/annurev.ge.29.120195.004021. [DOI] [PubMed] [Google Scholar]
- 13.Haardt M, Benharouga M, Lechardeur D, et al. C-terminal truncations destabilize the cystic fibrosis transmembrane conductance regulator without impairing its biogenesis: a novel class of mutation. J Biol Chem. 1999;274:21873–21877. doi: 10.1074/jbc.274.31.21873. [DOI] [PubMed] [Google Scholar]
- 14.Welsh MJ, Smith AE. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 1993;73:1251–1254. doi: 10.1016/0092-8674(93)90353-r. [DOI] [PubMed] [Google Scholar]
- 15.Weinreich F, Wood PG, Riordan JR, Nagel G. Direct action of genistein on CFTR. Pflugers Arch. 1997;434:484–491. doi: 10.1007/s004240050424. [DOI] [PubMed] [Google Scholar]
- 16.Pyle LC, Fulton JC, Sloane PA, et al. Activation of CFTR by the flavonoid quercetin: potential use as a biomarker of ΔF508 CFTR rescue. Am J Respir Cell Mol Biol. 2009 doi: 10.1165/rcmb.2009-0281OC. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van Goor F, Straley KS, Cao D, et al. Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1117–L1130. doi: 10.1152/ajplung.00169.2005. [DOI] [PubMed] [Google Scholar]
- 18.Ma T, Vetrivel L, Yang H, et al. High-affinity activators of cystic fibrosis transmembrane conductance regulator (CFTR) chloride conductance identified by high-throughput screening. J Biol Chem. 2002;277:37235–37241. doi: 10.1074/jbc.M205932200. [DOI] [PubMed] [Google Scholar]
- 19.Yang H, Shelat AA, Guy RK, et al. Nanomolar affinity small molecule correctors of defective Delta F508-CFTR chloride channel gating. J Biol Chem. 2003;278:35079–35085. doi: 10.1074/jbc.M303098200. [DOI] [PubMed] [Google Scholar]
- 20.Thakerar A, Van Driessch A, Bridges RJ. The conductance assay: a simple assay to measure ΔF508-CFTR channel activity. Ped Pulmonl Suppl. 2009;32:Abstract 267. [Google Scholar]
- 21••.Van Goor F, Hadida S, Grootenhuis PD, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A. 2009;106:18825–18830. doi: 10.1073/pnas.0904709106. This article describes a new potentiator ameliorating the conductance of both F508del-CFTR and G551D-CFTR in recombinant cells and greatly increased chloride secretion in human airway cells with both mutant alleles. Future studies will employ VX-770 on other mutations yielding surface CFTR in combination with processing correctors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galietta LJ. Problems and opportunities in mutant CFTR rescue. Pediatric Pulmonol Suppl. 2009;32 Symposium 9.1. [Google Scholar]
- 23•.Pasyk S, Li C, Ramjeesingh M, Bear CE. Direct interaction of a small-molecule modulator with G551D-CFTR, a cystic fibrosis-causing mutation associated with severe disease. Biochem J. 2009;418:185–190. doi: 10.1042/BJ20081424. These studies provide evidence supporting the proposed role of the small molecule potentiator VRT-532 binding to G551D-CFTR to rescue impaired ATP binding and gating. [DOI] [PubMed] [Google Scholar]
- 24.Accurso F, Rowe SM, Durie P, et al. Interim results of a phase IIa study of VX-770 to evalaute safety, pharmacokinetics and biomarkers of CFTR activity in cystic fibrosis subjects with G551D. Pediatr Pulmonol. 2008;43:Abstract 295. [Google Scholar]
- 25.Clancy JP, Rowe SM, Durie PR, et al. NPD evaluation of ion transport in G551D CF patients treated with a CFTR potentiator. Ped Pulmonol Suppl. 2009;32:Abstract 222. [Google Scholar]
- 26.Boyle M, Clancy JP, Rowe SM, et al. Effect of VX-770, a CFTR potentiator, on spirometry and QOL assessment in subjects with CF and the G551D-CFTR mutation. Ped Pulmonol Supp. 2009;32:Abstract 217. [Google Scholar]
- 27.Rubenstein RC, Egan ME, Zeitlin PL. In vitro pharmacologic restoration of CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis epithelial cells containing delta F508-CFTR. J Clin Invest. 1997;100:2457–2465. doi: 10.1172/JCI119788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rubenstein RC, Zeitlin PL. A pilot clinical trial of oral sodium 4-phenylbutyrate (Buphenyl) in deltaF508-homozygous cystic fibrosis patients: partial restoration of nasal epithelial CFTR function. Am J Respir Crit Care Med. 1998;157:484–490. doi: 10.1164/ajrccm.157.2.9706088. [DOI] [PubMed] [Google Scholar]
- 29.Pedemonte N, Lukacs GL, Du K, et al. Small-molecule correctors of defective DeltaF508-CFTR cellular processing identified by high-throughput screening. J Clin Invest. 2005;115:2564–2571. doi: 10.1172/JCI24898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30•.Pedemonte N, Tomati V, Sondo E, Galietta LJ. Influence of cell background on pharmacological rescue of mutant CFTR. Am J Physiol Cell Physiol. 2010;298:C866–C874. doi: 10.1152/ajpcell.00404.2009. A recent evaluation of CFTR modulators demonstrated that potentiators more consistently rescue CFTR function across cell types than corrector compounds do, suggesting the intracellular environment may alter susceptibility to particular F508del corrector strategies. [DOI] [PubMed] [Google Scholar]
- 31.Varga K, Goldstein RF, Jurkuvenaite A, et al. Enhanced cell-surface stability of rescued DeltaF508 cystic fibrosis transmembrane conductance regulator (CFTR) by pharmacological chaperones. Biochem J. 2008;410:555–564. doi: 10.1042/BJ20071420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32••.Young A, Gentzsch M, Abban CY, et al. Dynasore inhibits removal of wild-type and DeltaF508 cystic fibrosis transmembrane conductance regulator (CFTR) from the plasma membrane. Biochem J. 2009;421:377–385. doi: 10.1042/BJ20090389. This article describes the inhibition of F508del-CFTR endocytosis combined with a known CFTR biosynthesis corrector as a novel synergistic avenue of therapy. [DOI] [PubMed] [Google Scholar]
- 33•.Kim Chiaw P, Wellhauser L, Huan LJ, et al. A chemical corrector modifies the channel function of F508del-CFTR. Mol Pharmacol. 2010;78:411–418. doi: 10.1124/mol.110.065862. The success here of direct interaction with a small-molecule corrector with mutant CFTR demonstrates the possibilities for wider application in other protein folding disorders. [DOI] [PubMed] [Google Scholar]
- 34•.Wellhauser L, Kim Chiaw P, Pasyk S, et al. A small-molecule modulator interacts directly with deltaPhe508-CFTR to modify its ATPase activity and conformational stability. Mol Pharmacol. 2009;75:1430–1438. doi: 10.1124/mol.109.055608. The findings described here suggest that the CFTR modulator VRT-532 directly interacts with F508del CFTR to confer improvements in channel biogenesis and activation, as indicated by stabilization of proteolytic fragmentation patterns of F508del CFTR upon incubation of the compound. [DOI] [PubMed] [Google Scholar]
- 35.Press release. Vertex Pharmaceuticals, Inc; http://investors.vrtx.com/releasedetail.cfm?releaseid=442429. [Google Scholar]
- 36.Van Goor F, Haditha S, Grootenhuis PD, et al. VX-809, a CFTR corrector, increases the cell surface density of functional F508del-CFTR in preclinical models of cystic fibrosis. Ped Pulmonol Supp. 2009;32:Abstract 154. [Google Scholar]
- 37.Robert R, Carlile GW, Pavel C, et al. Structural analog of sildenafil identified as a novel corrector of the F508del-CFTR trafficking defect. Mol Pharmacol. 2008;73:478–489. doi: 10.1124/mol.107.040725. [DOI] [PubMed] [Google Scholar]
- 38.Lubamba B, Lecourt H, Lebacq J, et al. Preclinical evidence that sildenafil and vardenafil activate chloride transport in cystic fibrosis. Am J Respir Crit Care Med. 2008;177:506–515. doi: 10.1164/rccm.200703-344OC. [DOI] [PubMed] [Google Scholar]
- 39•.Lubamba B, Lebacq J, Reychler G, et al. Inhaled PDE5 inhibitors restore chloride transport in cystic fibrosis mice. Eur Respir J. 2010 doi: 10.1183/09031936.00013510. [Epub ahead of print]. This article offers preclinical evidence that the phosphodiesterase inhibitors sildenafil and verdenafil administered intravenously increase chloride conductance (without affecting sodium conductance) in F508del CFTR mice. [DOI] [PubMed] [Google Scholar]
- 40.Robert R, Carlile GW, Liao J, et al. Correction of the Delta phe508 cystic fibrosis transmembrane conductance regulator trafficking defect by the bioavailable compound glafenine. Mol Pharmacol. 2010;77:922–930. doi: 10.1124/mol.109.062679. [DOI] [PubMed] [Google Scholar]
- 41.Amaral MD. High-content fluorescence microscopy siRNA screens to track function/traffic of ENaC & CFTR. European CF Society: new frontiers in basic science of cystic fibrosis. 2010 symposium 9.2. [Google Scholar]
- 42.Wiech NL, Fisher JF, Helquist P, Wiest O. Inhibition of histone deacetylases: a pharmacological approach to the treatment of noncancer disorders. Curr Top Med Chem. 2009;9:257–271. doi: 10.2174/156802609788085241. [DOI] [PubMed] [Google Scholar]
- 43•.Hutt DM, Herman D, Rodrigues AP, et al. Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nat Chem Biol. 2010;6:25–33. doi: 10.1038/nchembio.275. This manuscript describes how HDAC7 inhibitors can be integral in restoring F508del CFTR function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mu TW, Ong DS, Wang YJ, et al. Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell. 2008;134:769–781. doi: 10.1016/j.cell.2008.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hermann T. Aminoglycoside antibiotics: old drugs and new therapeutic approaches. Cell Mol Life Sci. 2007;64:1841–1852. doi: 10.1007/s00018-007-7034-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keeling KM, Brooks DA, Hopwood JJ, et al. Gentamicin-mediated suppression of Hurler syndrome stop mutations restores a low level of alpha-L-iduronidase activity and reduces lysosomal glycosaminoglycan accumulation. Hum Mol Genet. 2001;10:291–299. doi: 10.1093/hmg/10.3.291. [DOI] [PubMed] [Google Scholar]
- 47.Howard M, Frizzell RA, Bedwell DM. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat Med. 1996;2:467–469. doi: 10.1038/nm0496-467. [DOI] [PubMed] [Google Scholar]
- 48.Bedwell DM, Kaenjak A, Benos DJ, et al. Suppression of a CFTR premature stop mutation in a bronchial epithelial cell line. Nat Med. 1997;3:1280–1284. doi: 10.1038/nm1197-1280. [DOI] [PubMed] [Google Scholar]
- 49.Tok JB, Bi L. Aminoglycoside and its derivatives as ligands to target the ribosome. Curr Top Med Chem. 2003;3:1001–1019. doi: 10.2174/1568026033452131. [DOI] [PubMed] [Google Scholar]
- 50.Bonetti B, Fu L, Moon J, Bedwell DM. The efficiency of translation termination is determined by a synergistic interplay between upstream and downstream sequences in Saccharomyces cerevisiae. J Mol Biol. 1995;251:334–345. doi: 10.1006/jmbi.1995.0438. [DOI] [PubMed] [Google Scholar]
- 51.Barton-Davis ER, Cordier L, Shoturma DI, et al. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J Clin Invest. 1999;104:375–381. doi: 10.1172/JCI7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Manuvakhova M, Keeling K, Bedwell DM. Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. RNA. 2000;6:1044–1055. doi: 10.1017/s1355838200000716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sermet-Gaudelus I, Renouil M, Fajac A, et al. In vitro prediction of stop-codon suppression by intravenous gentamicin in patients with cystic fibrosis: a pilot study. BMC Med. 2007;5:5. doi: 10.1186/1741-7015-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Clancy JP, Bebok Z, Ruiz F, et al. Evidence that systemic gentamicin suppresses premature stop mutations in patients with cystic fibrosis. Am J Respir Crit Care Med. 2001;163:1683–1692. doi: 10.1164/ajrccm.163.7.2004001. [DOI] [PubMed] [Google Scholar]
- 55.Wilschanski M, Yahav Y, Yaacov Y, et al. Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N Engl J Med. 2003;349:1433–1441. doi: 10.1056/NEJMoa022170. [DOI] [PubMed] [Google Scholar]
- 56.Wilschanski M, Famini C, Blau H, et al. A pilot study of the effect of gentamicin on nasal potential difference measurements in cystic fibrosis patients carrying stop mutations. Am J Respir Crit Care Med. 2000;161 (3 Pt 1):860–865. doi: 10.1164/ajrccm.161.3.9904116. [DOI] [PubMed] [Google Scholar]
- 57••.Sermet-Gaudelus I, De Boeck K, Casimir GJ, et al. Ataluren (PTC124) Induces CFTR Protein Expression and Activity in Children with Nonsense Mutation Cystic Fibrosis. Am J Respir Crit Care Med. 2010 doi: 10.1164/rccm.201001-0137OC. [Epub ahead of print] This is the first study describing the use of ataluren in pediatric CF patients, indicating improvement of chloride transport in CF human airways and augmented protein expression in the nasal airway. [DOI] [PubMed] [Google Scholar]
- 58.Helip-Wooley A, Park MA, Lemons RM, Thoene JG. Expression of CTNS alleles: subcellular localization and aminoglycoside correction in vitro. Mol Genet Metab. 2002;75:128–133. doi: 10.1006/mgme.2001.3272. [DOI] [PubMed] [Google Scholar]
- 59.Sleat DE, Sohar I, Gin RM, Lobel P. Aminoglycoside-mediated suppression of nonsense mutations in late infantile neuronal ceroid lipofuscinosis. Eur J Paediatr Neurol. 2001;5 (Suppl A):57–62. doi: 10.1053/ejpn.2000.0436. [DOI] [PubMed] [Google Scholar]
- 60.Keeling KM, Bedwell DM. Clinically relevant aminoglycosides can suppress disease-associated premature stop mutations in the IDUA and P53 cDNAs in a mammalian translation system. J Mol Med. 2002;80:367–376. doi: 10.1007/s00109-001-0317-z. [DOI] [PubMed] [Google Scholar]
- 61.Wagner KR, Hamed S, Hadley DW, et al. Gentamicin treatment of Duchenne and Becker muscular dystrophy due to nonsense mutations. Ann Neurol. 2001;49:706–711. [PubMed] [Google Scholar]
- 62.Du M, Jones JR, Lanier J, et al. Aminoglycoside suppression of a premature stop mutation in a Cftr−/− mouse carrying a human CFTR-G542X transgene. J Mol Med. 2002;80:595–604. doi: 10.1007/s00109-002-0363-1. [DOI] [PubMed] [Google Scholar]
- 63.Du M, Keeling KM, Fan L, et al. Clinical doses of amikacin provide more effective suppression of the human CFTR-G542X stop mutation than gentamicin in a transgenic CF mouse model. J Mol Med. 2006;84:573–582. doi: 10.1007/s00109-006-0045-5. [DOI] [PubMed] [Google Scholar]
- 64.Wilschanski M, Dupuis A, Ellis L, et al. Mutations in the cystic fibrosis transmembrane regulator gene and in vivo transepithelial potentials. Am J Respir Crit Care Med. 2006;174:787–794. doi: 10.1164/rccm.200509-1377OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65•.Nudelman I, Rebibo-Sabbah A, Cherniavsky M, et al. Development of novel aminoglycoside (NB54) with reduced toxicity and enhanced suppression of disease-causing premature stop mutations. J Med Chem. 2009;52:2836–2845. doi: 10.1021/jm801640k. Rational chemical optimization of aminoglycosides is shown here to enhance stop codon suppression in vitro and reduce cellular toxicity, suggesting designer compounds could be developed that are optimized for translational readthrough. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rowe SM, Backer K, Tang L, et al. Enhanced activity for translational read-through and reduced toxicity demonstrated by the synthetic aminoglycoside NB54. Ped Pulmonol Supp. 2009;32:Abstract 218. [Google Scholar]
- 67.Welch EM, Barton ER, Zhuo J, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447:87–91. doi: 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]
- 68.Hirawat S, Welch EM, Elfring GL, et al. Safety, tolerability, and pharmacokinetics of PTC124, a nonaminoglycoside nonsense mutation suppressor, following single- and multiple-dose administration to healthy male and female adult volunteers. J Clin Pharmacol. 2007;47:430–444. doi: 10.1177/0091270006297140. [DOI] [PubMed] [Google Scholar]
- 69.Du M, Liu X, Welch EM, et al. PTC124 is an orally bioavailable compound that promotes suppression of the human CFTR-G542X nonsense allele in a CF mouse model. Proc Natl Acad Sci U S A. 2008;105:2064–2069. doi: 10.1073/pnas.0711795105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kerem E, Hirawat S, Armoni S, et al. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trial. Lancet. 2008;372:719–727. doi: 10.1016/S0140-6736(08)61168-X. [DOI] [PubMed] [Google Scholar]
- 71.Clancy JP, Konstan MW, Rowe SM, et al. A phase II study of PTC124 in CF patients harboring premature stop mutations. Ped Pulmonol Suppl. 2006;41:Abstract 269. [Google Scholar]
- 72.Solomon GM, Konstan MW, Wilschanski M, et al. An international randomized multicenter comparison of nasal potential difference techniques. Chest. 2010 doi: 10.1378/chest.10-0179. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rowe SM, Varga K, Rab A, et al. Restoration of W1282X CFTR activity by enhanced expression. Am J Respir Cell Mol Biol. 2007;37:347–356. doi: 10.1165/rcmb.2006-0176OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Linde L, Boelz S, Nissim-Rafinia M, et al. Nonsense-mediated mRNA decay affects nonsense transcript levels and governs response of cystic fibrosis patients to gentamicin. J Clin Invest. 2007;117:683–692. doi: 10.1172/JCI28523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75•.Auld DS, Thorne N, Maguire WF, Inglese J. Mechanism of PTC124 activity in cell-based luciferase assays of nonsense codon suppression. Proc Natl Acad Sci U S A. 2009;106:3585–3590. doi: 10.1073/pnas.0813345106. PTC124 (ataluren) induces a paradoxical increase in luciferase bioluminescence, which may have biased the measurement of its readthrough activity in a luciferase-based high-throughput screen. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Auld DS, Lovell S, Thorne N, et al. Molecular basis for the high-affinity binding and stabilization of firefly luciferase by PTC124. Proc Natl Acad Sci U S A. 2010;107:4878–4883. doi: 10.1073/pnas.0909141107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wilschanski M, Armoni S, Yaacov Y, et al. PTC124 treatment over 3 months improves pharmacodynamic and clinical parameters in patients with nonsense-mutation-mediated CF. J Cyst Fibros. 2008;7 Symposium 22. [Google Scholar]