

Abstract
The inherited marrow failure syndromes are a diverse set of genetic disorders characterized by hematopoietic aplasia and cancer predisposition. The clinical phenotypes are highly variable and much broader than previously recognized. The medical management of the inherited marrow failure syndromes differs from that of acquired aplastic anemia or malignancies arising in the general population. Diagnostic workup, molecular pathogenesis, and clinical treatment are reviewed.
Keywords: aplastic anemia, Fanconi, Dyskeratosis congenita, Diamond-Blackfan anemia, Shwachman-Diamond syndrome, inherited bone marrow failure syndromes
Introduction
The inherited bone marrow failure syndromes (IBMFS) are undoubtedly underdiagnosed, in both pediatric and adult hematology/oncology practices. While the topic has been the subject of many earlier reviews, this report is current through 2009.1–3 The differential diagnosis that must be considered when a patient presents with pancytopenia due to apparently acquired aplastic anemia is summarized in the diagram in Figure 1. After examination of a bone marrow to confirm aplastic anemia and rule out acute leukemia or myelodysplastic syndrome (MDS), the pediatric approach usually begins with consideration of Fanconi anemia (FA) by physical examination and by testing for chromosome breakage, while the adult approach might be to rule out paroxysmal nocturnal hemoglobinuria (PNH) by flow cytometry for CD55 or CD59 negative clones. Although we estimate that about 30% of childhood aplasia is due to FA or the other syndromes to be discussed here, the proportion among adults is unknown. Appropriate classification of patients is imperative, since it impacts on medical and transplant management, choice of stem cell donors, estimated risks for complications including future neoplasms, and genetic and medical counseling and surveillance of the probands and their family members. Furthermore, patients with an IBMFS may not present with aplastic anemia, but may have characteristic physical anomalies, MDS, acute myeloid leukemia (AML), or a solid tumor, or even pulmonary fibrosis or liver disease as their first sign of an IBMFS. The major syndromes and their hematologic and neoplastic consequences are listed in Table 1.
Figure 1.

Overlapping Syndromes. The differential diagnosis for apparently acquired aplastic anemia includes paroxysmal nocturnal hemoglobinuria (PNH), myelodysplastic syndrome (MDS), acute myeloid leukemia (AML), and inherited bone marrow failure syndromes (IBMFS).
Table 1.
Inherited Bone Marrow Failure Syndromes
| Syndrome | Hematology* | Leukemia | Solid Tumors |
|---|---|---|---|
| Fanconi Anemia | Aplastic anemia | AML** | Squamous cell carcinomas |
| Dyskeratosis congenita | Aplastic anemia | AML | Squamous cell carcinomas |
| Diamond-Blackfan anemia | Anemia | AML | Sarcomas |
| Shwachman-Diamond syndrome | Neutropenia | AML | - |
| Severe congenital neutropenia | Neutropenia | AML | - |
| Amegakaryocytic thrombocytopenia | Thrombocytopenia | AML | - |
| Thrombocytopenia absent radii | Thrombocytopenia | AML | - |
Hematology means the usual initial presentation
AML, acute myelogenous leukemia
In this article we will discuss the presentation, physical and laboratory findings, pathophysiology, and management of the major syndromes within the classification of IBMFS. The tables provide a comprehensive review from the literature of the distinctive physical features for the major syndromes and the frequency of these findings, the types and frequencies of cancers specific to each syndrome, and the known mutated genes and their frequencies. The figures demonstrate ages at diagnosis, cumulative survivals, cancer risks, physical features, and pathophysiologic pathways. While the literature may have publication bias, and does not provide good quantitative epidemiologic data, case reports do offer some initial insights into frequencies and complications. Using that metric, the most frequently reported syndrome was FA (2002 cases), followed by Diamond-Blackfan anemia (DBA, 970 cases), Shwachman-Diamond syndrome (SDS, 560 cases), and Dyskeratosis congenita (DC, 550 cases). Large series of cases (without individual-level data) have reported on FA, DBA, SDS and DC.1–4 One large series included 374 patients with severe congenital neutropenia (SCN)1, and smaller series discussed amegakaryocytic thrombocytopenia (Amega) and thrombocytopenia absent radii.4,5
Fanconi Anemia: Clinical Features
Fanconi Anemia (MIM 607139) was first described in 1927 by Dr Fanconi, a Swiss pediatrician who noted a family with 3 brothers who had “perniziosiforme anemia”, i.e. macrocytic red cells and pancytopenia, along with several physical anomalies. Since then, more than 2000 cases have been reported with case descriptions, as well as several different cohorts of patients in recent years.6–9 Initially, cases were recognized only when they had the combination of aplastic anemia and birth defects, while current diagnostic criteria are more extensive, and rely on demonstration of chromosomal aberrations in cells cultured with DNA crosslinking agents (see below). Table 2A lists the physical findings that have been reported in FA, in approximate order of the frequency of the reports, and a “typical” patient is shown in Figure 2. Despite the lack of description in many reports, the relative frequencies provide important clues to diagnosis. The male:female ratio was 1.2:1, an unexplained significant increase in males compared with the expected 50% (p<0.001). Approximately 60% were reported with at least one physical finding. The most common were short stature, as well as café au lait and hyper- and hypo-pigmented areas (Figure 2). Abnormalities of the radial ray were described in one-third of the cases, all involving the thumb, with 7% absent or hypoplastic radii (along with absent thumbs). The next most common anomalies, in 20–25% of the cases, involved microcephaly, microphthalmia, structural renal anomalies, and hypogonadism. Other less common features are listed in Table 1. In addition, our own studies have indicated a high frequency (about 75%) of endocrine abnormalities in FA patients, including short stature and/or growth hormone deficiency, hypothyroidism, midline brain abnormalities, abnormal glucose/insulin metabolism, obesity, dyslipidemia, and metabolic syndrome.10 In our personal experience, all of the abnormalities occur more frequently than shown in Table 2A, when all patients are examined carefully and all findings reported. For example, 75% of our patients had hearing loss or structural otologic anomalies (unpublished), and 90% had small eyes.2 However, there are clearly patients who have no physical findings whatsoever, and are identified as affected family members of probands, or sporadic cases with aplastic anemia, leukemia, or solid tumors, in whom FA is diagnosed only because it is sought.
Table 2.
Physical Findings Reported in the Literature
A) Fanconi Anemia*
| Any Physical Abnormality 60%. Male:female 1.2:1 (p <0.001 vs expected 1.00) |
|---|
| Microsomia (40%): Short stature |
| Skin (40%): Generalized hyperpigmentation; cafe au lait spots, hypopigmented areas |
| Upper Limbs, unilateral or bilateral (35%): |
| Thumbs (35%): Absent or hypoplastic, bifid, duplicated, rudimentary, attached by a thread, triphalangeal, long, low set |
| Radii (7%): Absent or hypoplastic (only with abnormal thumbs), absent or weak pulse |
| Hands (5%): Flat thenar eminence, absent first metacarpal, clinodactyly, polydactyly |
| Ulnae (1%): Dysplastic, short |
| Skeletal: |
| Head (20%): Microcephaly, hydrocephaly, |
| Face (2%): Triangular, birdlike, dysmorphic, micrognathia, mid-face hypoplasia |
| Neck (1%): Sprengel, Klippel-Fiel, short, low hairline, web |
| Spine (2%): Spina bifida, scoliosis, hemivertebrae, abnormal ribs, coccygeal aplasia |
| Eyes (20%): Small, strabismus, epicanthal folds, hypotelorism, hypertelorism, cataracts, astigmatism, ptosis |
| Renal (20%): Horseshoe, ectopic or pelvic, abnormal, hypoplastic or dysplastic, absent, hydronephrosis or hydroureter |
| Gonads: |
| Males (25%): Hypogenitalia, undescended testes, hypospadias, micropenis, absent testes |
| Females (2%): Hypogenitalia, bicornuate uterus, malposition, small ovaries |
| Developmental Delay (10%): Mental retardation, developmental delay |
| Ears (10%): Deaf (usually conductive), abnormal shape, dysplastic, atretic, narrow ear canal, abnormal middle ear |
| Cardiopulmonary (6%): Congenital heart disease, patent ductus arteriosus, atrial septal defect, ventricular septal defect, coarctation, situs inversus, truncus arteriosus |
| Low Birth Weight (5%) |
| Lower Limbs (5%): |
| Feet: Toe syndactyly, abnormal toes, club feet |
| Legs: Congenital hip dislocation |
| Gastrointestinal (5%): Atresia (esophagus, duodenum, jejunum), imperforate anus, tracheoesophageal fistula, annular pancreas, malrotation |
| Central Nervous System (3%): Small pituitary, pituitary stalk interruption syndrome, absent corpus callosum, cerebellar hypoplasia, hydrocephalus, dilated ventricles |
Listed in approximate order of frequency. Percent is from 2000 cases reported in the literature from 1927 to 2009. Frequencies are very approximate, since many reports did not mention physical descriptions.
Figure 2.
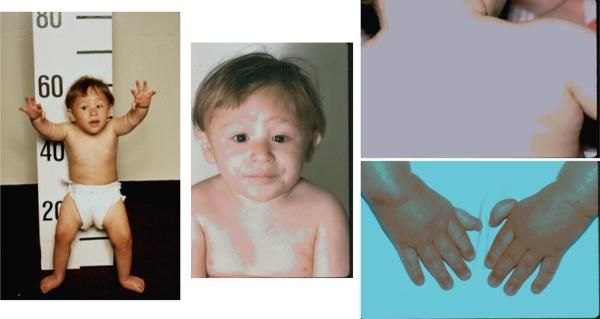
Patient with Fanconi Anemia. Features include short stature, microcephaly, dangling thumbs, epicanthal folds, micropthalmia, triangular face, café au lait and hypopigmented areas, dislocated hips which prevent him from standing straight, and rockerbottom feet. He also had an imperforate anus and ureter reimplantation. Consent for publication obtained.153
The median age at diagnosis was 6.5 years, ranging from birth to adults (Figure 3); we recently diagnosed FA in an asymptomatic physically and hematologically normal 55 year old who was identified only as a tissue match for a sibling with aplastic anemia.3 The diagnostic age in the reported cases was similar in both sexes. Blood pancytopenia was the most common presentation, particularly when the red cell mean cell volume (MCV) and fetal hemoglobin (Hb F) were elevated for age. Bone marrow biopsy examination most often showed hypocellularity for age, due to decreased numbers of hematopoietic precursors with normal morphology (on the aspirate).
Figure 3.

Age at diagnosis of cases reported in the literature in the major IBMFS. FA, Fanconi Anemia. DC, Dyskeratosis Congenita. DBA, Diamond-Blackfan Anemia. SDS, Shwachman-Diamond Syndrome. FA, age available in 1497/2002 case reports; median age 6.5 years, range 0–49. DC, age available for 467/550 case reports; median age 14 years, range 0–75. DBA, age available for 722/980 case reports; median 3 months, range birth-64 years. SDS, age available for 318/563 case reports; median 2 weeks, range birth-11 years. Note the different X-axes. Insets in DBA and SDS extend to older patients, representing <3% of the total number for each syndrome.
The suspected diagnosis is usually confirmed by demonstration of chromosomal aberrations in blood lymphocytes cultured with a DNA-crosslinking agent such as diepoxybutane (DEB) or mitomycin C (MMC) (Figure 4).4 The next step is determination of the complementation group by correction of the FA cellular phenotype by retroviral transfection of lymphoblasts or fibroblasts with one of the known FA genes; gene sequencing can then be performed to determine the relevant mutations.5
Figure 4.

Patterns of chromosomes in blood treated with DNA crosslinking agents. Left, MMC, mitomycin C, arrows show radial figures. Right, DEB, diepoxybutane, arrows show breaks, gaps, and rearrangement figures. Photograph courtesy of Lisa Moreau.
A significant albeit currently unknown proportion of patients with FA have hematopoietic somatic mosaicism. These cases are a diagnostic challenge, since they have a molecular event which has corrected one mutated allele in a bone marrow stem cell, leading to an acquired heterozygosity in the blood cells. For these cases, skin fibroblast cultures are required to demonstrate sensitivity to DNA-damaging agents. One example is a patient whose diagnosis of FA was made only after development of a head and neck cancer of the type seen in FA; genetic mosaicism was proven by the identification of two mutations in FANCA in fibroblasts, and only one in blood, due to gene conversion.6
The first adverse event in patients with FA is usually aplastic anemia, often sufficiently severe to lead to death, or to a hematopoietic stem cell transplant (SCT). We found that the annual hazard for severe bone marrow failure reached 4% per year by age 7, but was less than 1% per year in adults, while leukemia reached a hazard rate of 1% per year in teens and young adults, and the hazard of solid tumors rose steadily to more than 10% per year by age 45; the cumulative incidences of these respective complications were around 50, 25, and 10%.7,9 We also found that those patients who had many birth defects, defined as a high CABS (congenital abnormality) score, including anomalies of the radius, plus cardiopulmonary, kidney, hearing, head size, and developmental delay, were more likely to have early onset bone marrow failure, while the patients with the most normal physical appearance had the converse, with the highest risks of leukemia and solid tumors as young adults.7
In the literature cases, the median age for surviving free of any malignancy (leukemia or solid tumors) was 29 years; this is substantially younger than expected in the general population (Figure 5). The types of cancers in patients with FA who have not and have received an SCT are summarized in Table 3A. More than 300 patients were reported to have at least one cancer; 25 patients had between 2 to 4 malignancies; 14 had solid tumors plus leukemia and 6 plus liver tumors, and 6 with leukemia also had liver tumors. The most common malignancies were AML, head and neck squamous cell carcinoma (SCC), liver tumors, vaginal SCC, and brain tumors. We found that the relative risk of observed cancers in FA compared with the number expected according to the Surveillance Epidemiology and End Results (SEER) program was around 50-fold, and that the risk for AML was more than 600-fold, for HNSCC it was ~500-fold, and for vaginal SCC it was about 3000-fold.7,9 In addition, the risk for tongue cancer increased more than 4-fold above the already high baseline in FA patients following bone marrow transplantation, and occurred 16 years earlier; most of these cases had chronic graft-vs-host disease (Table 3B).8 One specific genotype (FANCD1/BRCA2, see below) had the highest risk for cancer, with a cumulative probability of 97% by age 6 years, and inordinately high risks for midline brain tumors, Wilms tumor, and AML, as well as a high frequency of birth defects in the VACTERL-H association category (vertebral, anal atresia, cardiac, trachea-esophageal fistula, renal, limb, +/− hydrocephalus).9 Additional genotype/phenotype/cancer associations are emerging from ongoing analyses.
Figure 5.

Probability of survival free of first cancer (solid tumors or leukemia) in cases reported in the literature.
Table 2.
Physical Findings Reported in the Literature
B) Dyskeratosis Congenita*
| Any Physical Abnormality 75%. Male:female 3.2:1. |
|---|
| Diagnostic Triad: All 3 components 46%, 2 of 3 features 22%, 1 of 3 features 9%; any or all 75%; nail dystrophy 70%, skin lacey reticular pigmentation 67%, oral leukoplakia 47% |
| Eyes (29%): Lacrimal duct stenosis, epiphora, blepharitis, exudative retinopathy, vascular retinopathy, strabismus, cataracts, absent eyelashes, ulcers |
| Hair (19%): Sparse, thin, alopecia, early loss, early grey, sparse eyebrows |
| Teeth (13%): Caries, missing, abnormal shape, periodontitis, decreased crown/root ratio, taurodontism |
| Development (13%): Developmental delay, retardation |
| Gastrointestinal (12%): 8% Esophageal stenosis, stricture, web; 2% liver cirrhosis, fibrosis, dysfunction |
| Short (12%) |
| Skeletal (10%): |
| Osteopenia (6%): osteoporosis |
| Hip (3%): avascular necrosis, aseptic necrosis |
| Head (9%): 9% Microcephaly, 5% cerebellar hypoplasia, 3% intracranial calcification, Dandy-Walker, tonsillar herniation |
| Low Birth Weight (9%) |
| Cardiopulmonary (7%): |
| Pulmonary (7%): pulmonary fibrosis, decreased perfusion, restrictive lung disease, arterio-venous fustulas |
| Cardiac (1%): atrial septal defect, ventricular septal defect, dilated cardiomyopathy |
| Genitourinary (7%): phimosis, meatal stenosis, urethral stricture, hypospadias, penile leukoplakia |
| Gonads (3%): |
| Male: Small testes, undescended testes |
| Female: atrophic, constriction, vaginal leukoplakia |
| Hyperhidrosis (6%) |
| Neurologic Signs (4%): Ataxia, spasticity, hypotonia |
| Ears (2%): deaf, rotated, low set |
| Hoyeraal-Hreidarsson 5% |
| Revesz 4% |
Listed in approximate order of frequency. Percent is from 550 cases reported in the literature from 1910 to 2009. Frequencies are very approximate, since many reports did not mention physical descriptions; in addition, many findings are age-dependent.
Atkinson
Recent analyses of MDS in the FA cohorts for which we have data indicate that the relative risk is more than 5000-fold compared with the general population (Alter et al, submitted). Many patients with FA have cytogenetic clones in their bone marrow, some of which may fluctuate in their frequency; the prognostic implications of specific types, such as monosomy 7 or 3q+ are not entirely clear, and our own data suggest that marrow morphology and significant cytopenias have more clinical significance than a clone alone.18,19
The major causes of death in FA include complications from aplastic anemia (sepsis, bleeding), SCT, and cancer. Data on survival in FA indicate a trend toward improvement in the most recent decade (Figure 6). Prior to 2000, the median survival in case reports was 21 years, while in more recent reports the median was 29 years of age. Factors responsible for this trend include better management with medical and transplantation regimens. However, the trend may also reflect better diagnosis of mild or asymptomatic patients, or those whose first presentation is as an adult with AML or a solid tumor. Whatever the explanation, patients with FA clearly frequently reach adulthood, with more than 80% achieving age 18 or more. It is important to note that FA females can have pregnancies, although they may require transfusions for worsening cytopenias and Caesarean section for failure of labor to advance; fertility in males with FA is very low due to hypogonadism and azospermia.10
Figure 6.

Overall survival of literature cases according to era of publication. Red, bottom line in each represents publications prior to 2000, while the green, upper line is for cases from 2000–2009. FA, median age for 1927–1999 was 21 years; 2000–2009 29 years, p <0.001. DC, median age for 1910–1999 was 34 years, 2000–2009 49 years, p = 0.009. DBA, median age for 1936–1999 was 38 years, 2000–2009 45 years, p = not significant. SDS, median age for 1949 was 35 years, 2000–2009 36 years, p = 0.01.
Fanconi Anemia: Molecular Features
FA is a multigenic disorder with 13 genes currently identified (Table 4A). With the exception of the X-linked FANCB gene, the remaining 12 FA genes are autosomal recessive. (Table 4). The encoded FA proteins function coordinately in the repair of DNA crosslinks. (Figure 7). Current evidence also points to additional functions of the FA proteins in stress signaling and apoptosis in response to oxidative damage and inflammatory cytokines.
Table 2.
Physical Findings Reported in the Literature
C) Diamond-Blackfan Anemia*
| Any Physical Abnormality 25%. Male:female 1.1:1. |
|---|
| Abnormal thumbs (8%): triphalangeal, bifid, duplicated, hypoplastic, subluxed, small, extra; flat thenar muscles |
| Low Birth Weight (5%) |
| Eyes (5%): small, epicanthal folds, hypertelorism, hypotelorism, strabismus, cataract, glaucoma |
| Short (13%) |
| Cleft lip/palate (4%): cleft palate (4%), cleft lip (0.2%) |
| Cardiac (3%): ventricular septal defect, atrial septal defect, tetralogy of Fallot, pulmonary stenosis |
| Genitourinary (3%): horseshoe, duplicated ureters, ectopic, absent |
| Abnormal Facies (3%): Cathie (tow-headed, snub nose, intelligent look), dysmorphic, mongoloid |
| Gonads (3%): undescended testes, hypospadias, inguinal hernia |
| Neck (2%): web, Sprengel, Klippel-Feil, short |
| Head (2%): microcephaly, hydrocephalus, wide fontanelle |
| Delayed Development (2%): Developmental delay, retardation |
| Ears (1%): deaf, low set, small |
| Central Nervous System (1%): hypopituitary, Chiari, myelomeningocele |
Listed in approximate order of frequency. Percent is from 970 cases reported in the literature from 1936 to 2009. Frequencies are very approximate, since many reports did not mention physical descriptions.
Figure 7.

FA/BRCA DNA damage response pathway. Following DNA damage, the proteins represented by A, B, C, E, F, G, L, and M form the core complex, which is required for ubiquitination of the I and D2 proteins, which are in turn required for the downstream complex of D2-ubi, I-ubi, and D1/BRCA2, N/PALB2, BRCA1, and J/BACH1/BRIP1 to form foci for DNA repair. Only BRCA1 is not yet known to be a Fanconi gene.
While many FA proteins lack homology to know protein functional domains, FANCL has an E3 ubiquitin ligase domain and FANCM contains a DNA helicase domain.
Several FA genes had been previously identified as cancer susceptibility genes involved in DNA repair.11,12FANCD1 was identified as BRCA2, the cancer susceptibility gene that functions in homologous recombination repair. FANCJ is BRIP1/BACH1, which encodes a 5' to 3' DNA helicase that binds to the BRCT domain of BRCA1. FANCN was identified as PALB2, a partner of BRCA2, important for BRCA2 stabilization and localization.
Eight of the FA proteins, FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, and FANCM, form a core complex that coordinately functions to monoubiquitinate FANCD2 and FANCI via the FANCL E3 ubiquitin ligase and the E2 conjugating enzyme UBE2T.11–13 FANCM recognizes replication forks stalled at sites of DNA damage such as interstrand crosslinks and is believed to recruit the FA core complex to chromatin. The ubiquitinated FANCD2/FANCI complex binds to discrete chromatin foci at presumed sites of DNA damage where it co-localizes with other DNA repair proteins including BRCA1, FANCD1/BRCA2, NBS1, RAD51, H2AX, and PCNA. Loss of any component of the FA core complex results in failure to monoubiquitinate FANCD2 and FANCI. FANCI is also required for FANCD2 monoubiquitination. FANCD2 mutations that prevent monoubiquitination render cells sensitive to DNA crosslinking agents, thus confirming the essential role of FANCD2 monoubiquitination in the FA pathway. Deubiquitination of FANCD2 and FANCI by USP1 in complex with UAF1 is also important for FA pathway function. FANCD1/BRCA2, FANCJ/BRIP1, and FANCN/PALB2 are not required for FANCD2 monoubiquitination and thus function downstream. Although the precise functions of the FA proteins in DNA repair are still under active investigation, interstrand crosslink repair is thought to involve several DNA repair pathways including nucleotide excision repair, translesion synthesis, and homologous recombination (reviewed in14).
Regulation of the FA pathway involves a complex network interacting with other DNA repair pathways.13 FANCD2, FANCA, and FANCJ/BRIP1/BACH1 associate with BRCA1, the familial breast cancer protein. ATR, which is deficient in Seckel syndrome, is required for efficient monoubiquitination of FANCD2. 15Cells from Seckel syndrome patients exhibit chromosomal instability. ATR together with CHK1 kinase functions in the phosphorylation of FA proteins including FANCA, FANCE, FANCD2, and FANCI, and affects FA pathway activation in response to DNA damage (reviewed in13). FANCD2 phosphorylation by the ataxia-telangiectasia protein ATM is important for its function in the S phase checkpoint but is not required for FANC2 monoubiquitination.16 ATM activation involves the MRE11/RAD50/NBS (MRN) complex, which senses double strand breaks. The FA pathway also intersects with NBS, which is mutated in Nijmegen breakage syndrome, a chromosomal instability syndrome sharing many clinical features with Fanconi anemia.17 The FA complex associates with the BLM helicase, whose loss results in Bloom's syndrome which is characterized by increased sister chromatid exchange.18 BLM also associates with FANCD2 within nuclear foci but BLM is not required for FANCD2 monoubiquitination.19
It is currently unclear why such a large multiprotein FA core complex is required for DNA repair. Although patients with other genomic instability syndromes, such as ataxia-telangiectasia, exhibit genotoxin sensitivity and cancer predisposition, marrow failure is generally not a typical clinical feature. Additional functions for specific FA proteins are emerging.20 FA cells are hypersensitive to cellular stress signals that activate apoptosis (reviewed in13,21).
Fanconi Anemia: Management
Currently the only cure for the hematological complications of FA remains hematopoietic stem cell transplant. The optimal timing of transplant is challenging since outcomes are best prior to the development of complications such as infections from chronic severe neutropenia, high transfusion burden to treat anemia/thrombocytopenia, and the development of MDS or AML. The definition of MDS can be challenging in patients with inherited marrow failure syndromes since the diagnostic findings of MDS in the general population, such as hypoproductive cytopenias, marrow dysplasias, and clonal cytogenetic abnormalities, are frequently present at baseline in FA patients. The progression and severity of the marrow dysplasia, rising blast counts, and possible high risk cytogenetic clones such as monosomy 7 or possibly amplification of chromosome 3q26q2922 are helpful markers of MDS in patients with Fanconi anemia. Marrow cellularity is patchy and subject to sampling bias; thus, marrow cellularity must be considered within the context of the peripheral blood counts. Many patients maintain stable mild cytopenias despite seemingly minimal marrow cellularity and do not warrant immediate transplant. Only a subset of patients with Fanconi anemia progress to severe marrow failure or leukemia, hence prediction of which patients would benefit from early preemptive transplant is currently difficult. The recommendation of a recent clinical consensus conference is to monitor the blood counts at least every 3–4 months and evaluate the bone marrow at least once a year to detect evolving complications early.23 As with any rare disorder, consultation with a hematologist experienced with the care of FA patients is recommended.
Patients with FA are exquisitely sensitive to genotoxic agents such as cyclophosphamide, busulfan and ionizing radiation. FA patients are also susceptible to the damaging inflammatory side effects of graft versus host disease. For these reasons, efforts have focused on reducing the doses required for transplant preparative regimens, choosing nongenotoxic regimens to prevent graft-versus-host disease, and using alternative conditioning regimens. FA patients experience severe transplant-related toxicity and high mortality rates with standard conditioning regimens used to treat aplastic anemia in the general population.24 The use of reduced intensity conditioning regimens that remain myeloablative for FA patients has resulted in greatly improved transplant outcomes. The introduction of fludarabine, a highly immunosuppressive and myelosuppressive nucleotide analog with minimal toxicity to other organs, has facilitated the reduction or elimination of other genotoxic agents without increasing the risk of engraftment failure.
For matched sibling transplants, disease-free survival rates between 64%–89%, with improvements occurring in more recent years, have been reported (reviewed in25,26). Regimens both containing or lacking total body irradiation (TBI) have been used successfully. The risk of primary or secondary graft failure is around 5–10It is essential to test all potential sibling donors for FA regardless of clinical findings since the phenotypic variation even within a given family is broad. Careful clinical and laboratory evaluation is warranted and if any abnormalities are found suggesting underlying marrow dysfunction, FA testing should also be performed on fibroblasts from a skin biopsy to rule out the possibility of somatic mosaicism. Some transplant centers require FA skin fibroblast testing for all sibling donors.
Unrelated donor transplant outcomes were discouraging prior to the advent of fludarabine, with overall survival rates of less than 30% (reviewed in25). A review of FA patients receiving unrelated donor transplants between 1990 and 2003 found superior outcomes of fludarabine-based regimens with respect to engraftment, day100 mortality (65% without fludarabine versus 24% with fludarabine, p<0.001) and 3 year adjusted overall survival rates (13% without fludarabine versus 52% with fludarabine, P<0.001).27
Unfortunately, HSCT does not correct the non-hematological manifestations of Fanconi anemia. Solid tumor risk, particularly head and neck squamous cell carcinoma, continues to increase after transplant, particularly in FA patients experiencing severe graft versus host disease.28,29 A retrospective study comparing solid tumor risks in transplanted versus non-transplanted FA patients reported a 4.4-fold higher age-specific hazard rate of squamous cell carcinoma in patients treated with transplant. The tumors appeared at an earlier age in the transplanted cohort.8 Data on solid tumor risks in patients transplanted on the newer current regimens are as yet limited.
Patients who choose not to pursue transplant for severe cytopenias may benefit from treatment with androgens such as oxymetholone.; the usual starting dose is 2 to 5 mg/kg/day. Androgens can improve cytopenias in all three lineages, erythroid, myeloid, and platelets, but the effects are typically most pronounced for the erythoid lineage. A subset of FA patients does not respond to androgens. Some patients respond initially but later become refractory. The dose of oxymetholone should be tapered to the minimum required to sustain the blood counts. Side effects of androgens include virilization, premature epiphyseal closure, hypertension, mood swings, cholestatic jaundice, transaminitis, and peliosis hepatis. Androgens may also exacerbate the risk of liver tumors.30 Regular monitoring of hepatic function and screening liver ultrasounds are recommended. A clinical trial of danazol, which has less virilizing side effects, is currently under way.
Patients with severe or symptomatic neutropenia may benefit from granulocyte-colony stimulating factor (G-CSF), particularly if they experience recurrent or life-threatening neutropenia-related infections. Patients with severe or symptomatic anemia should be given red cell transfusions. Chronic red cell transfusions result in iron overload so timely institution of appropriate chelation therapy is essential. Bleeding secondary to thrombocytopenia is treated with platelet transfusions. The use of antifibrinolytic agents may also be helpful to control bleeding in certain situations. Consideration should be given to HSCT prior to the administration of multiple transfusions.
Patients with FA should undergo annual bone marrow aspirates, biopsies, and cytogenetic analyses, on the premise that early detection of worsening aplastic anemia, bona fide MDS or leukemia would lead to early treatment, either medical or with transplant. Annual surveillance is recommended for the major solid tumors. This includes oral examination as well as nasolaryngoscopy for HNSCC, which should begin at around age 10 (the youngest HNSCC was at age 13 in a patient who had not had a transplant), or within one year of bone marrow transplant (the youngest patient was 9, and the shortest interval following transplant was 1 year). Gynecologic surveillance should start at menarche, or age 16, whichever comes first. The vaccine for human papilloma virus (HPV) is approved in the United States for both girls and boys from ages 9 to 26, and this is appropriate for patients with FA. Although the role of HPV in tumors in FA is controversial31,32, use of the vaccine according to standard guidelines is certainly indicated.
Dyskeratosis Congenita: Clinical Features
In the evaluation of patients with aplastic anemia in which an inherited disease is suspected and FA has been ruled out, the next syndrome to consider is DC (MIM 305000, 127550, 224230). As with FA, the first descriptions involved physical findings. In fact, DC was considered a form of ectodermal dysplasia, and was called “Zinsser-Cole-Engman” syndrome after the physicians who provided the first descriptions from 1910–1930. Many case reports followed in the dermatologic literature, and only in the 1960s was an association made between DC and hematologic problems. As discussed below, the definition of DC continues to evolve.
The male:female ratio was 3.2:1 among 550 cases in the literature; it was 4.2:1 for those cases reported from 1910 through 1999, and 2.4:1 for those in the last decade (p = 0.009), reflecting the bias that DC was considered to be an X-linked recessive disorder until the recent discovery of autosomal dominant and recessive genes (see below). Seventy-five % of the cases in the literature had some physical abnormality. The diagnostic triad, defined from the beginning, includes dystrophic nails, lacy reticular pigmentation, and oral leukoplakia (Figure 8, Table 2B). These findings in literature cases comprised 70%, 67%, and 47% respectively, and 75% of reported patients had at least one of these; 46% had all 3. The next most common physical problems were constant tearing from lacrimal duct stenosis, sparse and/or early grey hair and eyebrows, poor dentition, and developmental delay. An important early sign is esophageal stenosis in 8%, requiring dilatation. Osteopenia and early hip replacement due to avascular necrosis in unusually young adults occurred in up to 10%. Important but less frequently noted problems include pulmonary fibrosis, meatal stenosis, and neurologic findings. Major ophthalmologic findings include proliferative and exudative retinopathy, both of which can lead to retinal detachment.11,21,22 Most of the physical findings in DC are age-dependent, and thus their absence in a young patient by no means eliminates DC from consideration.
Figure 8.
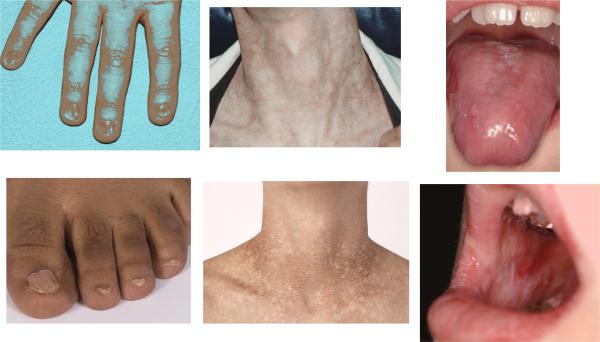
Features of the diagnostic triad in DC. Left, dystrophic nails on hands and feet. Middle, lacy reticular pigmentation on neck and upper thorax. Right, oral leukoplakia on tongue and buccal mucosa. Some of the figures are from Savage and Alter.36
There are two very severe subsets of DC. Patients with Hoyeraal-Hreidarsson (HH, MIM 300240) syndrome have cerebellar hypoplasia (Figure 9) with resultant ataxia and developmental delay, as well as microcephaly, immunodeficiency, intrauterine growth retardation, and early onset severe aplastic anemia. The diagnosis of Revesz syndrome (RS, MIM 268130) applies to young children with bilateral exudative retinopathy (similar to acquired unilateral Coats' retinopathy), intrauterine growth retardation, aplastic anemia, and central nervous system (CNS) calcifications. We have suggested that the appellation of HH requires cerebellar hypoplasia, and that of RS requires exudative (not hemorrhagic) retinopathy, in association with other features of DC. Recent discovery of mutated genes, as well as very short telomeres (see later) in these subsets of patients validates their inclusion in the DC category.23–25
Figure 9.

Cerebellar hypoplasia in the Hoyeraal-Hreidarsson variant of DC. Magnetic resonance image of brain; arrow indicates very small cerebellum.36
The median age at diagnosis of patients with DC was 14 years, range birth to 75 years, more than double the age in FA (Figure 3). The very young subset includes patients with HH or RS, while the older subset may include parents in families with autosomal dominant inheritance where the proband was a child. There was no sex difference in the age at diagnosis. Several patients were diagnosed as DC only after they had failed to respond to immunosuppressive treatment of their aplastic anemia.33 In contrast with FA, patients with DC do not have a childhood peak hazard rate for aplastic anemia, but rather a steady increase from 1% up to age 20 to almost 10% per year at age 50. Similar to FA, however, patients with DC have a cumulative incidence of severe aplastic anemia of around 50% by age 50.34
The diagnosis of DC may be suspected in the presence of features of the clinical triad, and/or other pathognomonic physical findings, with or without hematologic or neoplastic complications. It has been suggested that one of the triad, plus a hypoplastic marrow, plus any two of the other physical findings would lead to a diagnosis of DC.35 However, we have identified individuals who have none of these findings, but are family members who share the mutated DC gene of the proband, and have very short telomeres..36 These may be “silent carriers”, but warrant close observation for any of the complications that may arise in DC. There are also patients who present as adults with what appears to be acquired aplastic anemia, but who turn out to have mutations in the DC genes TERT and TERC.28,29 In addition, a subset of patients with familial pulmonary fibrosis had mutations in TERT.37
The DC equivalent of the chromosome breakage test for FA is detection of very short telomeres (less than the 1st percentile for age in a large number of normal controls) in blood leukocyte subsets (Figure 10).31 This assay has high sensitivity and specificity for identification of patients with FA, distinction of those patients from their unaffected (and mutation-negative) relatives, and from patients with any IBMFS that is not DC. In fact, very short telomeres was used as the case definition for the genetic linkage study that identified TINF2 as a new but quite common DC gene.24 At this time it appears that documentation of very short telomeres in several leukocyte subsets is the most useful “screening test” for a diagnosis of DC, although further studies may refine this suggestion. Telomere biology will be discussed below.
Figure 10.

Telomere length in blood lymphocytes according to age in patients with DC and their relatives (left), and patients with other IBMFS and their relatives (right). Vertical axis indicates telomere length in kilobases. Lines indicate the first, tenth, 50th, 90th, and 99th percentile of results from 400 normal control subjects. Left: Red circles, DC, 17 dyskeratosis congenita patients. Green triangles, HH, 4 Hoyeraal-Hreidarsson patients. Light blue diamonds, RS, 14 Revesz Syndrome patients. Dark blue square, 1 silent carrier with mutation in TERC. Open squares, 54 relatives of patients with DC. Arrows indicate 2 silent carriers initially classified as relatives, later found to have mutations in TINF2. Right: Red circles, 13 FA patients. Dark blue circle, 1 FA patient after bone marrow transplant. Light blue circles, 3 FA mosaics. Green triangle, 14 DBA patients. Black diamond, 5 SDS patients. Magenta square, 10 non-IBMFS patients. Open square, 36 relatives.39
While most patients with DC present to the hematologist with aplastic anemia, others may have MDS or AML as their first hematologic sign, and still others may have familial pulmonary fibrosis.28,30,32–36 Whatever the presentation, these individuals are at risk of any of the complications described in DC. In particular, they have a high risk for cancer, similar in order of magnitude of relative risk (11-fold compared with SEER) and in type as seen in FA.37
Bone marrow findings in patients with DC who have cytopenias are similar to those seen in FA, i.e. hypocellularity, decreased megakaryocytes, and some dyspoieses, which often may not be sufficient to make the diagnosis of MDS. Cytogenetic clones have not been a feature of the literature case reports, although we have seen them in a few patients. As in our prior experience in patients with FA38, we have observed stable or fluctuating clones over many years in patients with DC, and do not use clones alone as the determinant for SCT in DC.
The median age for survival free of cancer in cases in the literature was 68 years, much older than in FA, but the most frequent solid tumor was the same, HNSCC, and the other tumors were similar, involving the gastrointestinal and anogenital areas (Table 3, Figure 5). AML and MDS were less frequent in DC than in FA, but there is concern about whether many cases of DC are not diagnosed as such when they present as adults with no or minimal physical findings; definitive diagnosis may require analysis of telomeres, and sequencing of the known DC genes (see later).
Overall survival of cases in the literature is older than in FA, i.e. 34 years in those reported from 1910 through 1999, and 49 years for those in the past decade (Figure 6). As in FA, however, there is a recent cohort effect (p = 0.009), reflecting the combination of better medical and transplant management, as well as diagnosis of milder or even clinically healthy affected individuals. The causes of the reported deaths were similar to FA, i.e. complications of aplastic anemia, SCT, and cancer. In addition, pulmonary fibrosis was a cause of death unique to DC. Almost 90% of the patients reported recently were 18 years of age or older, i.e. adults. Unlike in FA, where there is decreased fertility in both sexes, there is no obvious problem with fertility in DC, although this has not been examined rigorously.
Dyskeratosis congenita: Molecular Features
DC is characterized by accelerated telomere shortening that results in cell loss or dysfunction.39,40 All six genes identified for DC to date function in telomere maintenance.36,41,42 (see Table 4 B). Mutations in DKC1 are associated with the X-linked form of DC. The autosomal dominant form of DC is caused by mutations in TINF2, TERC and TERT. NOP10/NOLA3 and, NHP2/NOLA2, have been identified in autosomal recessive forms of DC. Biallelic mutations in TERT have also been identified in some pedigrees, consistent with either autosomal recessive or perhaps co-dominant forms of DC.43 Recessive inheritance patterns have been described for TERT wherein the probands inherited compound heterozygous TERT mutations and exhibited early severe disease.44 Importantly, almost half of the patients with DC lack mutations in any of the known DC genes; so negative genetic testing does not rule out this diagnosis.
Five of the DC genes, DKC1, TERC, TERT, NOP10, NHP2, encode components of telomerase, an enzyme that functions in maintaining telomeres. (Figure 11). Telomeres are specialized structures at the end of chromosomes that facilitate terminal DNA replication and prevent chromosomal rearrangements resulting from free DNA ends. Telomeres are comprised of repeated tandem TTAGGG sequences that associate with a protein complex called shelterin. (Figure 11) During DNA replication, the telomerase enzyme generates telomeric repeat sequences at the 3'-hydroxyl DNA terminus using the TERC RNA template. When telomerase levels are limited, repeated cell divisions results in sequential telomere shortening. Progressive telomere shortening is a feature of aging, associated with repeated cell divisions required for tissue homeostasis and maintenance. When telomeres shorten below a critical length, replicative senescence is triggered and limits cell proliferation capacity. Tissues with a high proliferative capacity such as the hematopoietic, mucosal and epithelial systems are more often clinically affected in DC. Earlier and more severe disease manifestations, a phenomenon known as disease anticipation, were noted in successive generations with mutations in TERC45 or TERT.46 This disease anticipation resulted from the inheritance of progressively shorter telomeres in each subsequent generation.
Figure 11.
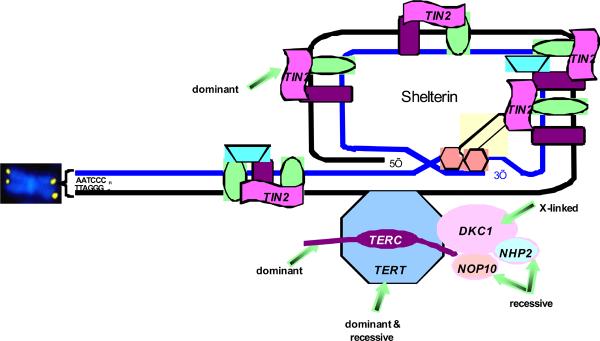
Figure 11: Telomere biology pathway with mutations in patients with DC. Telomeres are represented by the yellow dots on the ends of the chromosome (shown in blue). Telomeres are repeats of TTAGGG, added during cell replication by components of the pathway. TERT, telomerase enzyme, is a dominant. TERC, RNA template, is both dominant and recessive. DKC1, X-linked recessive gene for protein called dyskerin. NOP10/NOLA3 and NHP2/NOLA2 are autosomal recessive. TINF2 codes for the TIN2 protein, involved in maintenance of shelterin, which protects the telomere. Other proteins shown in the figure have not been found to be mutated in patients with DC. Figure courtesy of Sharon Savage.
Patients with mutations in TERC may present with aplastic anemia or MDS as the initial manifestation of their disease47 Mutations in TERT have also been identified in patients with aplastic anemia.48,49 Some patients with idiopathic pulmonary fibrosis were found to harbor mutations in TERT37 or TERC.50 Liver cirrhosis, another feature of DC, was also found in some patients with idiopathic pulmonary fibrosis and TERT mutations.51
The central role of accelerated telomere shortening in DC was further supported by the discovery of the sixth gene, TINF2, which encodes the protein TIN2.52 TIN2 associates with TRF1, TRF2, Rap1, TPP1 and POT1 to form the shelterin complex. (Figure 11). Shelterin binds and protects telomere ends to prevent telomere shortening and rearrangements. Patients with TINF2 mutations have very short telomeres.52,53TINF2 mutations were found in 6 out of 109 pediatric patients presenting with severe aplastic anemia.40TINF2 mutations and short telomeres were also found in patients with Revesz syndrome and those with Hoyeraal-Hrediarsson syndrome, confirming the clinical suspicion that these are subsets of DC.52,53
Impaired ribosome assembly and function have also been implicated in dyskeratosis congenita (reviewed in54). The dyskerin protein forms a complex with NOP10, NHP2 and GAR1. This complex associates with the H/ACA class of RNAs which includes the small nucleolar RNA (snoRNA). SnoRNA:protein complexes (snoRNPs) function in diverse cellular processes including ribosomal RNA pseudouridylation, and ribosomal RNA maturation.55 Dyskerin shares homology with pseudouridine synthases and catalyzes the isomerization of specific uridine residues within the context of the snoRNP. Pseudouridylation may affect rRNA secondary structure and binding. Pseudouridylation of rRNA is essential for ribosome function in yeast.56 Diminished rRNA pseudouridylation has been documented in patients with DC57 and in a dyskerin-deficient mouse model.58 Impaired translation from internal ribosome entry site sequences (IRES) has been observed in Dkc1m mice and in cells from X-linked DC patients wherein rRNA pseudouridylation was diminished.59
Dyskeratosis congenita: Management
Treatment for the hematologic complications of DC is very similar to the plan for FA described above. While SCT may cure the bone marrow, it does not cure other tissues in the body. In addition, the intrinsic propensity for pulmonary fibrosis in DC may be exacerbated by the preparative regimen used for the transplant. In the past, when full myeloablation was used, survival was poor.37 Unexpected problems included hepatic and pulmonary fibrosis, and late onset veno-occlusive disease, complications not seen in patients with FA.23,38–41 More than 70 patients have had transplants, and follow-up data are available for 59. The median survival interval for all DC patients who had an SCT was 7 years; it was 7 for those reported prior to 2000, and 11 years for those reported in the recent era. However, the first use of reduced intensity preparation (e.g. Fludarbine) was after the year 2000, and thus there has not been sufficient time to determine the long-term impact of this modality on survival. In our recent short-term cohort, there are 4 survivors out of 6 patients, with no hepatic or pulmonary complications to date (Dietz et al, submitted). The donor type was significant in the cases in the literature, with a 5 year survival of 31% in those with alternative donors, compared with 68% for those with matched sibling donors. By 10 years, there were no survivors in the alternative donor group, and 30% in the sibling group; however, the last patient in this group died at 20 years. Transplant-related deaths were due to graft failure, graft versus host disease, infection, pulmonary fibrosis, venoocclusive disease, and capillaritis.60
Choice of a sibling donor for a patient with DC, as in FA, requires that the donor be proven to not have DC. In the past, use of a sibling who appeared to be physically and hematologically well led to failure to engraft in rare cases; these individuals were later shown to have short telomeres and the same DC gene mutation as their sibling with aplastic anemia.61 We used short telomeres to identify a silent carrier minor child who was an HLA match to his sister with DC. At that time, the telomere assay was a research test. An ethics conference at the National Institutes of Health agreed that the child should not be a donor, and he was later found to have a mutation in TINF2 when that gene was identified as a DC gene.24,44
Medical treatment of bone marrow failure in DC is similar to the management of FA, i.e. the use of androgens, primarily oxymetholone; about half the patients respond. However, patients with DC appear to be more sensitive than those with FA, and we recommend a smaller dose of oxymetholone, such as 0.5 to 1 mg/kg/day. The side effects include virilization, behavior problems, and elevated liver enzymes. We had previously reported that the combination of erythropoietin (Epo) and G-CSF was effective in one adult with DC.62 Unfortunately, when androgens were combined with G-CSF, two patients developed splenic peliosis and rupture, and we now advise against that combination.63 Liver function and ultrasound examinations should be done at 3–4 month intervals and annually, because of the risk of liver tumors. However, so far none have been reported in DC.
Pulmonary fibrosis is a rare but very serious complication that develops in older patients who have not had an SCT. It is also a potentially fatal complication in those who have been transplanted. We recommend annual measurement of pulmonary function for all patients with DC. To our knowledge, only one patient with DC who developed pulmonary fibrosis post-SCT has received a lung transplant which has been successful in the short term; this should be considered for any patient with DC with this complication.(Alter et al, unpublished)
As shown above, patients with DC resemble those with FA in the risks for neoplasms, and thus the guidelines for FA have been applied to DC.64 Bone marrow aspirates, biopsies, and cytogenetics should be performed yearly. Oral and head and neck cancer screening was described above. Annual screening should also be offered for gynecologic and male anal cancers. These recommendations are for patients with clinical DC. However, we also consider asymptomatic individuals with mutations in any of the genes in the telomere biology pathway to be at risk for complications associated with DC, and recommend counseling and surveillance.
Diamond-Blackfan Anemia: Clinical Features
While patients with FA or DC were initially recognized because of a combination of physical findings and aplastic anemia (FA), or physical findings alone (DC), DBA (MIM 105650) was first identified only because of pure anemia, which was present at birth or soon thereafter, and required early treatment. Close to 1000 cases have now been reported in the literature, as well as several large case series including a total of another 1000 or so.48–50 Approximately 25% of the patients in those series and in the literature reports had at least one birth defect, but these were much milder than in FA or DC (Table 2). The male:female ratio was 1.1, as expected for a disorder with autosomal dominant inheritance. The most frequent observation was short stature, but it is not clear whether that was genetic, or iatrogenic in patients whose anemia was treated with corticosteroids (see below). The next most frequently reported finding was an abnormality of the thumb, range from triphalangeal, to bifid, to subluxed, to only subtle flattening of the thenar muscles. Unlike FA, the radius is normal in DBA. Most of the other systems described as abnormal represent were reported in less than 5% of the patients. Distinct from FA and DC, 4% of patients with DBA were reported to have cleft palate and/or cleft lip. Characteristic facial features and short neck were described, albeit infrequently. There is some overlap with features more characteristic of FA, such as the thumb anomalies, abnormal eyes, and structural renal anomalies.
The median age for diagnosis in DBA was 3 months, with the range from birth to 64 years of age; there was no difference between males and females (Figure 3). More than 98% were identified within the first year. Diagnoses in the older patients were because of late recognition of chronic anemia with DBA-characteristic physical findings such as abnormal thumbs65, anemia only during pregnancy, or silent carriers of mutations found in symptomatic family members. Unlike FA and DC, with chromosome breakage and telomere length serving as highly sensitive screening tests, the diagnosis of DBA is not so clear-cut; it is reviewed elsewhere.50 Blood counts usually demonstrate macrocytic anemia with reticulocytopenia, often with and Hb F, as well as elevated red cell adenosine deaminase in more than 85% of patients. Bone marrow has normal or reduced cellularity, with erythroblastopenia and usually normal myeloid and megakaryocytic lineages.To our knowledge, clonal cytogenetics have not been a feature of the bone marrow of patients with DBA. The major differential diagnosis is between DBA and transient erythroblastopenia of childhood (TEC). However, the age for TEC is usually above 2 years, and the majority of patients recover spontaneously after at most 1 or 2 transfusions.
The prognosis for DBA is brighter than for FA or DC. Although many DBA patients respond to corticosteroids, or require transfusions, or occasionally SCT (see below), the risk for malignancy is low, and for aplastic anemia is very low. While the crude rate for cancer was cited as 4%50, and was 3% in the literature cases, the 30 reported cases of leukemia or a tumor (Table 3) warranted a time-dependent analysis (Figure 5), which demonstrated that more than half the patients survived and were free of cancer beyond age 40. The neoplastic cases included 10 with AML, 1 with ALL, 6 patients with osteosarcomas, 3 with Hodgkin disease, 2 with breast cancer, 2 with hepatomas, and 1 each with other cancers. It should be pointed out that 4 of the patients with osteosarcoma had received prior treatment for short stature with human growth hormone. There were also 3 cases of with MDS alone, 1 with MDS followed by AML, and 1 with MDs after chemotherapy for Hodgkin disease. In our prospective cohort, we have found no cases of leukemia among more than 60 patients with DBA (Alter et al, submitted).
The median overall survival in DBA was around 40 years, and did not differ in the pre- or post-2000 reporting era. This may be due to less change in treatment over time (when compared with FA and DC), as well as an apparent “spontaneous” remission rate of around 25%. Similar to FA and DC, more than 85% of DBA patients survived past age 18 years, i.e. into adulthood. The major causes of death were anemia, sepsis, and iron overload, as well as leukemia and other cancers. Pregnancy in women with DBA may lead to a transient worsening of their anemia, due to the expanded blood volume required for the fetus and placenta; there may be fetal loss, preeclampsia, preterm deliveries, and intrauterine deaths.52,53
Diamond-Blackfan Anemia: Molecular Features
DBA is caused by heterozygous mutations in genes encoding the protein components of either the small 40S (RPS19, RPS17, RPS24) or large 60S (RPL35A, RPL5, RPL11) ribosomal subunits.66 (see Table 4C, Figure 12) Mutations in many of these genes have been shown to affect ribosomal RNA processing. Around 50% of DBA patients however lack identifiable genetic mutations so additional genes likely remain to be identified.
Figure 12.

Pathways involved in ribosomal synthesis, linking DC, DBA, and SDS.
The genes encoding the ribosomal protein components RPS19, RPS17, RPS24, RPL5, RPL11, and RPL35A are mutated in DBA. Mutations in these genes affect 40S and 60S ribosome biogenesis. The DKC1 gene encodes the dyskerin protein which has been implicated in ribosomal RNA pseudouridylation (ψ). The SBDS protein appears to be involved in the joining of the 40S and 60S ribosomal subunits to form the mature 80S ribosome.
The original Rps19−/− mouse model had an embryonic lethal phenotype and heterozygous mice lacked a hematological phenotype.67 A recent mouse model with mutations in Rps19 was reported with a hypoproliferative anemia and small size.68 Zebrafish models using antisense morpholinos targeting rps19 and other ribosomal genes also manifested impaired erythropoiesis and developmental malformations.69,70 These phenotypes in both mice and zebrafish could be at least partially rescued by knocking out p53. A model has been proposed whereby disruption of ribosomal biogenesis activates stress signaling pathways such as p53 to result in apoptosis or cell cycle arrest of erythroid progenitors or in the developing embryo (reviewed in71,72).
Ribosome assembly is normally a highly regulated and stoichiometrically balanced process. Disruption of the balance of ribosomal proteins as a result of ribosomal gene mutations may lead to the accumulation of free unassembled ribosomal proteins. One potential pathway connecting ribosomal stress to p53 activation involves the interaction of specific ribosomal proteins (RPL5, RPL11, RPL23) with MDM2. MDM2 binds p53 and targets p53 for proteosomal degradation. The binding of these ribosomal proteins to MDM2 results in the release and stabilization of p53.73,74–77 Another potential mechanism was raised by studies from the Thomas laboratory which demonstrated that haploinsufficiency of RPS6 leads to increased translation of 5'TOP mRNAs such as RPL11,78 Elevated levels of RPL11 resulted in p53 activation. These models do not yet fully explain how mutations in RPL5 and RPL11 result in DBA. Investigations of additional nucleolar stress signaling pathways are warranted.
Further support for the role of ribosomal proteins in hematopoiesis and malignant transformation came from the identification of the critical role of the RPS14 gene in 5q- MDS.79 Knockdown of RPS14 in human CD34+ cells resulted in impaired erythroid differentiation and restoration of RPS14 expression rescued this defect in patient-derived marrow cells. As in DBA, haploinsufficiency of RPS14 resulted in a block in pre-ribosomal RNA processing. How mutations affecting ribosomes result in red cell aplasia and cancer predisposition remain unclear.
Diamond-Blackfan Anemia: Management
As for the disorders discussed above, SCT is the only current modality for cure of the hematopoietic defect. The choice for SCT for a syndrome that affects only red cell production and not other lineages is difficult, and depends on the trade-offs of steroids, transfusions, and possible treatment-free remissions (see below). The post-SCT survival probability for the approximately 100 patients reported leveled off at 70% by 5 years. While era of publication did not matter, there was a significant difference according to the type of donor. Matched sibling donor transplants had an 81% survival plateau by 2 years, while procedures using alternative donors reached a plateau at 4 years of only 48% survival (p=0.007). The age of the recipient was not significantly different; the survival plateau was 71% for those patients less than 10 years of age, compared with 66% for older patients (p=0.5). The impact of more recent nonmyelablative preparative regimens is unknown, since the number of transplants reported with that modality is very small. The causes of death following transplant were not specific to DBA, and included graft rejection, graft versus host disease, infection, and bleeding. No cases were reported to develop leukemia or solid tumors following SCT.
The traditional treatment for DBA is corticosteroids, which have been used successfully for more than 50 years. The majority of the patients respond to an initial prednisone dose of 2 mg/kg/day, although some patients then may lose their response, or may require a high dose which leads to unacceptable side effects, including hypertension, weight gain, diabetes mellitus, etc. Details with regard to management and tapering of steroids are provided in the consensus guidelines.50 Some hematologists prefer to avoid steroids during the first year of life because of growth and neuromotor side effects, while others use steroids if an effective but tolerable dose can be identified; there was no consensus on this. If the dose of steroid is sufficiently high to lead to adrenal or immunosuppression, prophylaxis may be provided against Pneumocystis carinii. The aim with steroid treatment is to move at a conservative pace to tapering of the dose, and then administration on alternate days, to reduce side effects.
Transfusions are the mainstay of management until the diagnosis is clearly defined as DBA. For those who continue transfusions throughout infancy, or those who decline to take or fail to respond to adequate doses of steroids, the main considerations are inconvenience, venous access, transfusions risks (alloimmunization, or viral transmissions), and eventually iron overload, which becomes clinically significant after 1–2 years of transfusions. The major sites of iron toxicity are the liver, heart, and endocrine organs, and iron overload may be fatal. The traditional method for iron chelation has been subcutaneous desferrioxamine through an infusion pump, for 8–12 hours 4–6 nights per week, at 40 mg/kg/day, as is traditional in patients with beta thalassemia.80 One study found that half of 31 patients with DBA receiving transfusions had elevated liver iron, despite chelation (which was often inadequate due to late onset or poor compliance). The correlation between ferritin levels and liver iron was poor, indicating a need for noninvasive evaluation by SQUID (superconductive quantum interference device) or MRI (magnetic resonance imaging) R2 and T2* of liver and heart.81 More recently, deferasirox, an oral chelator, was shown to be safe and effective at 20–30 mg/kg/day in reducing liver iron in a group of 30 patients with DBA.56
About 20–25% of patients with DBA may eventually attain a remission, in which they may have an adequate hemoglobin level without continuing to take steroids or require transfusions, at no specific age. At this time there is no way to predict who these will be; perhaps future genotype data will help with this prognosis. In addition, since some of these remitters may relapse and require subsequent treatment, they need their blood counts monitored at reasonable intervals (at least annually). DBA patients in remission from anemia may still remain at risk for malignancies.
Several alternative therapies have been used in the past, or proposed for the future. The ones used previously have had initial high expectations, but ultimately in controlled trials only limited or no efficacy. These include androgens, high dose corticosteroids, Epo, interleukin-3, cyclosporine with or without prednisone, metoclopromide, valproic acid, and leucine, as well as others50.
Screening for silent carriers should be performed in DBA families, including hemoglobin, MCV, Hb F, and red cell ADA, as well as genetic testing in those families in which a mutated ribosomal gene has been identified. All individuals who are then labeled “DBA”, whether symptomatic or not, should be offered regular hematologic follow-up with blood counts at least annually. Screening annual bone marrow examinations should be considered. The only solid tumor which was reported with any frequency was osteosarcoma, and since 4 of those 6 patients had received growth hormone, we do not recommend the use of that hormone in patients with DBA in the absence of growth hormone deficiency. There are no specific guidelines for cancer surveillance.
Shwachman-Diamond Syndrome: Clinical Features
The first description of Shwachman-Diamond Syndrome (SDS, MIM 260400) was motivated by the observation that several children with malabsorption due to pancreatic insufficiency also had neutropenia.82 As in FA and DC, the initial clue to a syndrome was non-hematologic. Although the inheritance is autosomal recessive, there is a statistically significant excess of male case reports (male:female 1:48:1, p<0.001). Characteristic physical abnormalities have also been noted in more than half of the 560 cases described (Table 2D). The most frequent is short stature in 50%, followed by radiographic evidence of metaphyseal dysostosis in 25%. Thoracic abnormalities and delayed development are the next most common, and other findings in the table were reported less often.
The median age at which malabsorption was reported was 2 weeks, ranging from birth to 11 years (Figure 3), although the reported age may have been retrospective and thus inaccurate. Neutropenia was often noted somewhat later, and thus the actual age at “diagnosis” may have been delayed. The current diagnosis of SDS relies on the combination of documented exocrine pancreatic dysfunction plus signs of bone marrow involvement.83 Pancreatic malfunction is suspected because of frequent fatty, greasy stools, and can be demonstrated by ultrasound or CT imaging of a fatty pancreas, or measurement of duodenal enzymes, or stool content; the current recommendation is documentation of low serum levels of trypsinogen and pancreatic isoamylase.59 The hematopoietic component requires demonstration of neutropenia on more than one occasion, sometimes associated with macrocytosis, elevated Hb F, and sometimes anemia and/or thrombocytopenia. Bone marrow examination usually shows hypocellularity, often due to a decrease in myeloid precursors. Confirmation of the diagnosis rests on demonstration of biallelic mutations in the SBDS gene (see later).
Patients with SDS may evolve beyond neutropenia to anemia and/or thrombocytopenia, with bone marrow evidence of aplastic anemia; this was reported in 20% of patients at a median of 3 years of age (range birth to 35). Rare patients with SDS may have aplastic anemia as their initial presentation, with biallelic mutations in the SBDS gene proving the diagnosis. Less clear is the pathogenic role of heterozygosity in SBDS in patients who appear to have acquired aplastic anemia.60
The most serious hematologic complication in SDS is evolution of the marrow to MDS and AML. The diagnosis of MDS was suggested in more than 70 patients, but 25 of those had a marrow cytogenetic clone but without morphologic dyspoieses characteristic of MDS (Table 3E). The median age was 8 years (range 6 mo – 42 years), and there was an excess of males in this group. Close to 40 patients had leukemia, primarily AML, at a median age of 19 years (range 2 – 43). The male:female ratio in all the cases of SDS in the literature was 1.5:1, and appeared to rise to 2.0:1 in MDS and 2.9:1 in those with leukemia, but this trend was not significant. The median age for remaining free from leukemia was 37 years (Figure 5). MDS was not included with leukemia, because of the unclear definition of “MDS” in the context of SDS (see next).
More than 15% of the case reports described marrow clonal cytogenetics. The most frequent was i(7q), found in 35 patients with either refractory anemia (RA) or a clone without morphologic MDS (Table 3F), and, of special note, in none of the cases with leukemia. On the other hand, monosomy 7 or der(7), the second clone category in frequency, was found in 7 cases of AML, 12 RA, and 10 with a clone without clinical MDS. The third clone, del(20), was seen in 14 patients with a clone without MDS, 3 with RA, and also in 2 with AML. It is interesting to note that the chromosome locus for SBDS is at the centromere of 7, the region that is duplicated in i(7q). There is no clear explanation for the striking absence i(7q) among the SDS patients with leukemia, or those who had MDS prior to leukemia. Nevertheless, the data suggest that this specific clonal cytogenetic finding may not have a bad prognosis.
The most frequent causes of death were infection (sepsis or pneumonia), AML, and myocardial necrosis. The latter were in 8 of 16 Finnish patients in a single report, and this complication has not been important in other reports.84 There is a trend toward improved survival in cases in the last decade (Figure 6), mostly due to better survival in younger patients. The recent median is 36 years of age. In this decade, 87% of the cases survived to adulthood (age 18 and older). There is essentially no information regarding fertility or pregnancies in SDS; our own case had complications requiring a C-section due to thrombocytopenia.85
Shwachman-Diamond Syndrome: Molecular Features
The majority of SDS patients (>90%) harbor biallelic mutations in the SBDS gene (Table 4D).86SBDS is highly conserved across eukaryotes and archaea and widely expressed across different tissues.86 Abrogation of SBDS gene expression in mouse models results in early embryonic lethality indicating that it is an essential gene.87 Consistent with these findings, no patients have been identified with homozygous null mutations in SBDS. SBDS encodes a protein whose crystal structure lacks any apparent homology to known protein functional domains.88,89 SBDS is emerging as a multifunctional protein. Current data support a direct role for SBDS in ribosomal maturation and in mitotic spindle stabilization.
SBDS co-sediments with the 60S large ribosomal subunit in sucrose gradients.90 The SBDS protein associates with proteins from the large ribosomal subunit,91 rRNA processing factors92 and with 28S ribosomal RNA,90 which is a component of the large ribosomal subunit. (Figure 12). Altered expression profiles of genes involved in ribosome biogenesis and RNA processing have been noted in bone marrow cells from SDS patients.93 Yeast deficient in the SBDS orthologue, Sdo1, grow poorly. Mutations in Tif6 were found to suppress the slow growth phenotype of Sdo−/− yeast. Tif6 is the yeast orthologue of EIF6, which functions in 60S ribosomal subunit maturation94 and prevents the joining of the 60S to the 40S subunit to form the mature 80S ribosome.95 A model has been proposed whereby Sdo1 functions to promote the release of Tif6 from the nascent 60S subunit. Thus, in contrast to the situation in DBA where assembly of the ribosomal subunits is impaired, SBDS affects ribosome biogenesis at a later stage with different cellular consequences.96
Cells from SDS patients are sensitive to nocodazole, a microtubule-destabilizing agent, and resistant to taxol, which stabilizes microtubules. Purified recombinant SBDS protein binds isolated microtubules in vitro and promotes microtubule polymerization, supporting a direct role of SBDS in this process.97 SBDS co-localizes with the spindle in mitotic cells.97,98 SBDS has also been observed at centrosomes.98 These data suggest that the genomic instability characteristic of SDS99 might arise, at least in part, from aberrant mitotic spindle function.
Hematopoietic cells from SDS patients exhibit cell intrinsic defects in proliferation and differentiation.100 This was confirmed in a mouse model where Sbds knockdown in hematopoietic stem cell populations resulted in reduced ability to repopulate an irradiated wildtype recipient mouse.101 SBDS loss is associated with increased apoptosis and Fas hypersensitivity.102 SBDS-depletion results in hypersensitivity to multiple types of DNA damage as well as endoplasmic reticulum stress.103 SDS marrow stromal cells exhibit an impaired ability to support hematopoiesis of hematopoietic progenitor cells from healthy controls.100 Abnormal B and T cell numbers and function have also been observed in some SDS patients,104 and lymphocyte abnormalities have been reported in Sbds-deficient mouse models.101 F-actin polymerization/depolymerization kinetics are altered during chemotaxis of SDS patient-derived neutrophils.105 Since SBDS colocalizes with F-actin, a role for SBDS in actin polymerization has been suggested.
Shwachman-Diamond Syndrome: Management
The leading causes of mortality in SDS are the hematological complications of marrow failure and malignancy. Patients with severe neutropenia are at increased risk for infections. Neutrophil functional abnormalities and immunologic abnormalities may compound the risk of infection in some patents. In contrast to disorders of neutrophil chemotaxis, SDS patients maintain the ability to localize neutrophils to sites of infection and form abscesses.106 Neutropenic patients with recurrent or severe infections may benefit from treatment with G-CSF. Data on the prophylactic use of G-CSF based on neutrophil counts alone are scarce in SDS patients. Limited data suggest that the use of G-CSF per se does not increase the risk for leukemia in patients with SDS.107,108,107 Supportive care for severe or symptomatic anemia or thrombocytopenia consists of red cell or platelet transfusions.
A hematopoietic SCT is indicated for marrow failure with severe or symptomatic cytopenias, MDS, or AML. For all cases reported in the literature, the survival was 58% at 2 years, and did not differ according to the donor type. The reports prior to 2000 had a plateau of 45% survival at 1 year, while the more recently reported cases leveled off at 64% at 2 years. As with most of the other marrow failure syndromes, most of the published literature consists of small or individual case reports of diverse transplant regimens (reviewed in 109). Lower survival rates were noted in patients who had developed overt leukemia.110,111 A high incidence of transplant-related organ toxicity, particularly cardiac, hepatic, and pulmonary, was noted in SDS patients. Two recent reports of reduced-intensity conditioning regimens sparing the use of cyclophosphamide and total body irradiation showed promising results.110,112 100% donor chimerism could be achieved with low regimen-related toxicities.
Severe Congenital Neutropenia: Clinical Features
Patients with severe congenital neutropenia (SCN, MIM 202700) do not have any significant birth defects that would provide a clue to their diagnosis. They present early in infancy with severe infections, such as abscesses or pneumonia. The neutrophil count is well below the normal value of 1.5×109/L, often less than 0.5×109/L, on multiple occasions, while the hemoglobin and platelet count are usually normal. Bone marrow examination reveals an arrest at the promyelocyte/myelocyte stage, with cellularity that is normal or slightly reduced, and other cell lineages have normal maturation. 113 The major differential diagnosis is with cyclic neutropenia CN), and the usual method is to obtain white blood cell counts and differentials 2 to 3 times a week for 6 weeks, in order to encompass the equivalent of two 21 day cycles. Mutations in ELA2/ELANE do not clearly distinguish SCN from CN, since the same gene is involved in both (see below).
Severe Congenital Neutropenia: Molecular Features
Congenital neutropenia is emerging as a heterogenous disorder arising from a variety of genetic mutations affecting multiple diverse molecular pathophysiologic pathways (Table 4E) (Reviewed in 114). All of the molecular pathways share in common an increased propensity to activate apoptosis. In some cases, characteristic phenotypic findings have been associated with specific genes or mutations.
Heterozygous mutations in the ELA2/ELANE gene are found in approximately 50% of patients with severe congenital neutropenia arising in an autosomal dominant or sporadic fashion.115 Cyclic neutropenia is also associated with mutations in ELA2, and there is some overlap in the mutations causing these two syndromes. ELA2 encodes the enzyme neutrophil elastase, a serine protease component of the primary azurophilic granules of neutrophils and, to a lesser extent, monocytes. ELA2 mutations exhibit variable effects on elastase enzymatic activity and intracellular localization. Recent data support a model whereby mutations in ELA2 cause protein misfolding within the endoplasmic reticulum to trigger the unfolded protein response (UPR) resulting in apoptosis.116–118
The original autosomal recessive form of SCN first described by Kostmann is caused by biallelic mutations in HAX1.119HAX1 is a mitochondrial protein with homology to other proteins in the BCL2 family. HAX1 deficiency results in a reduction of the inner mitochondrial membrane potential. This leads to a pro-apoptotic state with engagement of BAX, release of cytochrome c and activation of the apoptotic caspase cascade. There are two HAX1 splice variants. One isoform resulting in partial excision of exon 2 is expressed in neuronal cells. Mutations affecting both splice isoforms are associated with neurological impairment or seizures in addition to neutropenia.
An X-linked form of congenital neutropenia is caused by mutations affecting the autoinhibitory domain of the WAS protein120,121 resulting in increased actin polymerization.122 Variable effects on lymphocyte number and function may also be seen. In contrast, WASP mutations causing loss of function result in Wiskott-Aldrich syndrome.123
Heterozygous dominant-negative mutations in the transcriptional repressor GFI1 have been reported in two patients with neutropenia and monocytosis.124 ELA2 is a transcriptional target of GFI1. Neutropenia is also noted in Gfi1-deficient mouse models.125,126 Lymphopenia was also a clinical feature associated with GFI1 mutations. GFI1 controls the expression of a number of hematopoietic genes including HOXA9, PBX1, MEIS1, BAX, C/EBPalpha and C/EBP epsilon (reviewed in 127,128). GFI1 also affects expression of the micoRNAs miR-21 and miR-196b which regulate myeloid maturation.129
Mutations in G6PC3, a gene involved in the glucose-6-phosphate pathway, were recently associated with congenital neutropenia.130 These patients also manifested developmental anomalies of the cardiac and genitourinary systems. G6PC3 localizes to the endoplasmic reticulum. G6PC3 deficiency results in a lowered threshold of activation of the unfolded protein response and decreased GSK3beta activity resulting in increased apoptosis.
Acquired mutations in the gene encoding the G-CSF receptor, CSF3R, were noted in a subset of patients with SCN. These truncating mutations resulted in the deletion of the terminal negative autoregulatory region of the G-CSF receptor. Cells harboring this mutation manifested increased proliferation and resistance to apoptosis. In some patients, the CSF3R mutations were found to precede the initiation of G-CSF treatment, indicating that G-CSF administration was not necessary for the development of these mutations.131 Acquisition of the CSF3R mutation may be associated with an increased risk of progression to AML in some patients, though the molecular mechanisms contributing to leukemia progression remain unclear. Of note, some patients with CSF3R mutations have not progressed to leukemia over many years, suggesting that CSF3R mutations alone are not sufficient for leukemia progression. Interestingly, AML arising in SCN patients manifests a different spectrum of mutations from those arising in the general population. MDS and AML arising in SCN patients frequently harbored CSF3R mutations, whereas mutations in genes common to de novo AML, such as NPM1, FLT3, c-KIT, and CEBPα, were not found.132 Mutated ELA2 was not predictive of AML, since the risk was similar in those with mutated and those with wild type ELA2.107,108
Other genes that are mutated in patients who present with neutropenia are summarized in Table 4E, and other syndromes in which neutrpenia is prominent are listed in Table 4F and reviewed by Boztug and Klein.133
Severe Congenital Neutropenia: Management
Prior to the availability of G-CSF, prognosis was poor with early mortality from bacterial infections. A randomized phase III trial of G-CSF therapy for SCN patients with neutrophil counts <0.5 × 109/L (500/microliter) demonstrated that 90% of patients receiving G-CSF increased their neutrophil counts to >1.5 × 109/L.134 The incidence and duration of infections were significantly reduced. Cases of MDS and AML were reported in SCN patients prior to the advent of G-CSF treatment, suggesting an inherent malignant predisposition. A recent study of 374 patients with SCN on long-term G-CSF reported the cumulative incidence of mortality of 8% from sepsis and 21% for MDS/AML.107 A high-risk subgroup was identified among patients requiring >8mcg/kg/day of G-CSF without achieving the median absolute neutrophil count of 2.188 × 109/L (2188/microliter). In this high-risk group, the 10-year cumulative incidence of MDS/AML was 40% while 14% of patients died of sepsis. This was 3 to 4-fold higher than the risks of MDS/AML or sepsis in patients with a good ANC response to lower doses of G-CSF.
Hematopoietic stem cell transplant is reserved for those patients with a poor response to G-CSF or who develop MDS/AML. Transplant outcomes are largely surmised from reports of individual patients or small case reports with diverse transplant regimens. SCT can cure the marrow abnormalities in these patients. Outcomes are best when the transplant donor is an HLA-matched sibling and the transplant is initiated prior to leukemic transformation.135 A retrospective study of 6 patients with SCN transplanted after the development of MDS (2 cases) or AML (4 cases) reported poor transplant outcomes for AML. One donor was an HLA matched sibling; the other 5 donors were unrelated. Only one unrelated donor was matched at 10/10 loci by high-resolution typing. The remaining patients were matched at 8/8 loci for class I by intermediate DNA typing and for class II by high-resolution typing. The conditioning regimen was busulfan, cytarabine and cyclophosphamide for 5 patients while one patient received cyclophosphamide and total body irradiation. The two patients transplanted for MDS without prior chemotherapy were alive at 2.7 and 4 years from transplant. All four patients transplanted for AML received induction chemotherapy and were in remission at the time of transplant. None of the patients with AML survived. Induction chemotherapy was associated with prolonged pancytopenia and severe mucositis in all patients and was complicated by life-threatening infections in two patients. Cause of death was chronic graft-versus-host disease for two patients and primary graft failure for two patients receiving single-antigen-mismatched unrelated donor stem cells.
Other less Common Inherited Bone Marrow Failure Syndromes Amegakaryocytic Thrombocytopenia
Patients with amegakaryocytic thrombocytopenia (Amega or CAMT, MIM 604498) do not have characteristic birth defects. They usually present in infancy with petechiae or more serious hemorrhages, although they may sometimes evolve to aplastic anemia or even MDS or AML without the preceding thrombocytopenia being recognized. Although more than 100 cases have been reported, this is an underestimate of the frequency of this condition. It may be under-recognized, and is certainly under-reported. Since many such patients have mutations in the MPL gene (see below), it is worth considering this diagnosis during the evaluation of a child with apparently acquired aplastic anemia.136 Bone marrow cellularity early in the course of Amega may be normal, with decreased or absent megakaryocytes, although cellularity may decrease as the disease progresses.
The majority of patients with Amega harbor biallelic mutations in the MPL gene which encodes the receptor for thrombopoietin (TPO, Table 4G).137 TPO is an essential regulator of megakaryocytopoiesis and platelet production.138,139 TPO also plays an important role in early hematopoietic progenitor or stem cells. Mpl−/− mice have a severe reduction of multipotent hematopoietic progenitor cells and low platelet counts.140,141 The importance of TPO in hematopoiesis is further supported by the development of aplastic anemia affecting all hematopoietic lineages in patients with MPL mutations.
MPL mutations that completely disrupt TPO receptor signaling, such as nonsense or frameshift mutations Type I), are generally associated with a more severe disease phenotype and early development of pancytopenia.142 Missense mutations in MPL that result in diminished but residual receptor function are generally associated with a milder disease phenotype with rising platelet counts in the first year of life and milder marrow failure. (Type II).136,143 This generalization does not hold true for all patients and the causes contributing to clinical phenotype variability are likely complex.144 There has been insufficient long term follow-up of genotype patients to determine whether the eventual risk for AA, MDs, or AML depends on the specific genetic mutation.
Hematopoietic stem cell transplant remains the only curative therapy for this disorder (reviewed in145,146). Unlike many of the disorders discussed above, it appears that the standard protocols used for acquired aplastic anemia are successful in patients with Amega. No increased rate of transplant-related toxicity has been reported in these patients. The optimal timing of HSCT in these patients remains unclear. The major supportive care treatment for bleeding is platelet transfusion. Anti-fibrinolytic therapies such as aminocaproic acid or tranexamic acid are also useful, particularly for mucous membrane bleeding. A clinical trial of recombinant IL-3 and GM-CSF in 5 patients resulted in only transient improvements in platelet counts in 2 patients.147
Thrombocytopenia Absent Radii
Patients with thrombocytopenia absent radii (TAR, MIM 274000) present as newborns, with thrombocytopenia, and characteristic physical findings of bilateral absent radii (unilateral in about 2% of cases), with thumbs present albeit abnormal in appearance. They are distinguished from those with FA by the presence of thumbs in TAR, which are absent in patients with FA if radii are absent (Figure 13). FA should be ruled out by testing chromosome breakage (see above). More than 100 cases have been reported. TAR patients may have other birth defects as well, including hypoplastic ulnae, hypoplastic humeri, phocomelia, abnormal shoulders, bowed legs, hip dysplasias, abnormal knees, abnormal facies, renal malformations, and other findings.146 Gastroenteritis and cow's milk intolerance are frequent but not well understood. The thrombocytopenia may be severe, and marrow examination reveals absent or small and abnormal megakaryocytes; the other lineages are normal.
Figure 13.
Comparison of radial ray anomalies in TAR and FA. Left, TAR. Right, FA. TAR patient has absent radii, but thumbs are present, albeit not normal in shape or position. FA patient has an absent radius, but the thumb is also absent, and the fingers are abnormal.64,153
The specific gene for TAR has not been identified, and the inheritance is thought (although not proven) to be autosomal recessive or complex (Table 4G). A 200 kb microdeletion at 1q21.1 was described in one allele of patients, but this was insufficient to explain the condition, since one parent had a microdeletion without the syndrome, and the other parent was normal in that region.65 The deleted region is large, and does not contain any true candidate genes in which mutations have been found on the other allele. Thus the deletion is necessary but not sufficient for the diagnosis of TAR.
Early management of TAR is aimed at addressing the major symptom, bleeding. Platelet transfusions are given as needed, and usually are no longer required beyond the first year of life, when the platelet count improves, albeit not necessarily to normal levels. Early deaths are from bleeding, with a plateau of survival in the literature of 80% by age 1–2 years. SCT is rarely required, but may be curative.66,67 Four cases were reported who developed AML or ALL, which suggests (but without a denominator does not prove) that TAR patients have an increased risk for leukemia. Similarly, rare reports of patients with TAR and solid tumors may be coincidences.
Radioulnar Synostosis
Radioulnar synostosis (RUS, MIM 605432) is a very rare disorder, in which patients may come to medical attention from infancy to adulthood, with thrombocytopenia or aplastic anemia, and be noted to have limited pronation/supination of the arms due to proximal radioulnar synostosis.68,69 Other physical findings included clinodactyly, syndactyly, hip dysplasia, and sensorineural hearing loss. The disorders to be considered in the differential diagnosis include FA, Amega, and TAR (see above). Bone marrow examination when the presentation was thrombocytopenia revealed absent megakaryoctyes with normal cellularity and normal maturation of the other lineages, consistent with a form of amegakaryocytic thrombocytopenia. After evolution to aplastic anemia, the marrow was consistent with that diagnosis.
The inheritance is autosomal dominant, with incomplete penetrance, and the mutated gene was found to be HOXA11 (Table 4G). Two families had the same single bp deletion in exon 2, leading to a truncated protein with a dominant negative effect. This defect was then shown in vitro to inhibit megakaryocyte differentiation.70,71
Transfusion support was used as needed, and bone marrow transplantation was reported to cure the hematopoietic defect in 2 children.148
Pearson Syndrome
Patients with Pearson Syndrome (PS, MIM 557000), also called “Pearson marrow-pancreas syndrome”, were first described with malabsorption and aregenerative anemia in 1979. They were distinguished from SDS because of normal marrow cellularity, vacuoles in marrow precursors, ringed sideroblasts, and anemia, as well as fibrotic rather than fatty pancreas.72 Subsequently, the phenotypic description focused on the hematologic and metabolic components, namely refractory sideroblastic anemia and metabolic and lactic acidosis, and PS was added to the differential diagnosis of childhood MDS.73 There are less than 100 cases reported, with a male:female ratio of 0.7. Representative bone marrow fields are shown in Figure 14, with cytoplasmic vacuoles in myeloid and erythroid precursors, and ringed sideroblasts.
Figure 14.
Bone marrow morphology in Pearson Syndrome. Left, vacuoles in myeloid precursor. Middle, vacuoles in erythroid precursor. Right, ringed sideroblast.
The clinical spectrum was found to be due to abnormalities in mitochondrial DNA, leading to heteroplasmic deletions of various sizes, which included the respiratory enzymes.73 Mitochondrial DNA is circular, 16.5 kb, containing 13 genes for respiratory enzymes, 22 transfer RNAs, and genes for 12s and 16s ribosomal RNAs. The deletions have ranged in size from around 3 kb to 8 kb, with the most frequent deletion 4977 bp in almost half the patients. The proportion of abnormal mitochondria varies within tissues, organs, and patients. All cases appear to be sporadic, although mitochondrial inheritance is maternal, since mitochondria are present in the cytoplasm of ova, while sperm lack cytoplasm.
Treatment includes supportive transfusions with blood and platelets as needed. Epo has been used in rare cases with renal insufficiency, as well as G-CSF for severe neutropenia.149,150 In several cases, the bone marrow function appeared to improve, perhaps due to a selective growth advantage of stem cells with a lower load of mutant mitochondria. Two patients were reported to have unrelated donor SCTs. The first rejected the first graft, received a second SCT, had acute and chronic GVHD, then AML of recipient origin with del(7q) and trisomy 8, and died of progressive disease.76 The second was transplanted because of MDS with del(7q) as well, and was hematologically normal at 3 years after cord blood SCT.151
However, the function of non-hematologic organs usually declines, and the outcome is poor from a hepatic, renal, and neurologic perspective. Those who survive childhood may develop symptoms consistent with Kearns-Sayre Syndrome. Treatment of the metabolic disorder is often attempted with combinations of vitamins and cofactors, including riboflavin, thiamine, folic acid, CoQ10, l-carnitine, and creatine, but the disorder usually progresses despite this.152 Treatment has limited utility; the oldest reported patient was 20 years of age, and most deaths were prior to age 5.
Conclusions
In summary, the classical phenotypes originally described for the inherited marrow failure syndromes are now recognized as the more severe end of the highly variable clinical spectrum of these disorders. Early recognition of these disorders allows appropriate medical management and surveillance. An understanding of the indications and limitations of the growing number of available laboratory tests for these disorders is essential. These syndromes offer unique insights into global molecular pathways regulating hematopoiesis and malignant transformation.
Table 2.
Physical Findings Reported in the Literature
D) Shwachman-Diamond Syndrome*
| Any Physical Abnormality 55%. Male:female 1.5:1. |
|---|
| Short (50%) |
| Metaphyseal Dysostosis (25%) |
| Low Birth Weight (10%) |
| Abnormal thorax (8%): asphyxiating thoracic dystrophy (Jeune syndrome), small thorax, narrow chest, pectus carinatum |
| Delayed Development (7%): mental retardation, developmental delay |
| Eyes (2%): hypertelorism, retinitis pigmentosum, esotropia |
| Abnormal Facies (2%): dysmorphic |
| Cardiac (1%): cardiomegaly, patent ductus arteriosus, right aortic arch, transposition of the great vessels, myocardial fibrosis |
| Legs (1%): dysplastic hips, bow legs, short limbs, Legg Calve Perthes |
| Head (1%): microcephaly, macrocephaly, hydrocephalus |
| Neck (1%): short neck |
| Cleft lip/palate (<1%): cleft lip, cleft palate |
Listed in approximate order of frequency. Percent is from 560 cases reported in the literature from 1949 to 2009. Frequencies are very approximate, since many reports did not mention physical descriptions.
Table 3.
Cancer in the Major IBMFS
A) Fanconi Anemia, not Transplanted
| Type | Subtype | Number* | Male | Female | Median Age, Yrs (Range) |
|---|---|---|---|---|---|
| Leukemia | Total Leukemias | 175 | 86 | 71 | 13 (0.1–49) |
| AML + AL | 165 | 83 | 65 | 13 (0.1–49) | |
| ALL | 6 | 3 | 3 | 5 (1–10) | |
| CMML | 4 | 0 | 3 | 11,20 | |
| MDS | MDS*** | 110 | 56 | 51 | 14 (2–49) |
| Tumors | Total Tumors | 124 | 42 | 76 | 23 (0.2–56) |
| HNSCC | 43 | 17 | 26 | 29 (13–56) | |
| Esophagus | 14 | 3 | 11 | 29 (20–50) | |
| Vulva, anus | 21 | 0 | 21 | 27 (14–38) | |
| Cervix | 6 | 0 | 6 | 22 (3.7–25) | |
| Brain | 24 | 9 | 11 | 3 (0.5–11) | |
| Breast | 7** | 0 | 7 | 37 (26–45) | |
| Lung | 4 | 4 | 0 | 30 (23–34) | |
| Stomach | 3 | 3 | 0 | 21, 22, 35 | |
| Renal, including Wilms | 17 | 9 | 6 | 1 (0.5–36) | |
| Lymphoma | 2 | 1 | 1 | 0.3, 2.5 | |
| Retinoblastoma | 1 | 0 | 1 | 0.3 | |
| Osteosarcoma | 1 | 0 | 1 | 7 | |
| Bladder | 1 | 2 | 0 | 38 | |
| Neuroblastoma | 6 | 4 | 1 | 0.8 (0.2–1.4) | |
| Dermatofibroma | 1 | 0 | 1 | 20 | |
| Liver | Total Liver Tumors | 46 | 27 | 19 | 13 (5–50) |
| Hepatoma | 30 | 20 | 10 | 14 (5–50) | |
| Adenoma | 16 | 7 | 9 | 11 (8–48) |
Number of cancers. Total number of patients without transplant with any cancer = 320. 25 patients had multiple solid tumors (22 had 2, 3 had 3). 14 with solid tumors also had leukemia and 6 had liver tumors; 6 with leukemia also had liver tumors.
One patient had 2 breast cancers. Ages were not reported for some cancers. AML, acute myeloid leukemia. AL, acute leukemia. ALL, acute lymphocytic leukemia. CMML, chronic myelomonocytic leukemia. MDS, myelodysplastic syndrome. HNSCC, head and neck squamous cell carcinoma.
MDS not otherwise specified in most reports.
Table 3.
Cancer in the Major IBMFS
B) Fanconi Anemia, after Stem Cell Transplantion
| Type | Subtype | Number* | Male | Female | Median Age, Yrs (Range) |
|---|---|---|---|---|---|
| Solid Tumors | HNSCC* | 41 | 22 | 19 | 22 (9–34) |
| Leukemia | ALL | 1 | 1 | 0 | 18 |
| AML | 7 | 4 | 5, 7, 10 |
Abbreviations as in (A).
Most were tongue or other oral cancers.
Table 3.
Cancer in the Major IBMFS
C) Dyskeratosis Congenita, not Transplanted
| Type | Subtype | Number* | Male | Female | Median Age, Yrs (Range) |
|---|---|---|---|---|---|
| Leukemia | AML | 1 | 1 | 0 | 29 |
| MDS | Total MDS | 4 | 4 | 0 | 12 (8–20) |
| Monosomy 7 | 2 | 2 | 0 | 8,20 | |
| MDS | 2 | 2 | 0 | 9,15 | |
| Tumors | Total Tumors | 50 | 30 | 8 | 28 (1.5–68) |
| HNSCC | 19 | 13 | 6 | 32 (17–49) | |
| Esophagus | 2 | 2 | 0 | 25, 41 | |
| Nasopharnyx | 2 | 1 | 1 | 42, 49 | |
| Stomach | 4 | 4 | 0 | 23 (16–28) | |
| Anorectal | 6 | 6 | 0 | 28 (17–49) | |
| Pancreas | 1 | 1 | 0 | 29 | |
| Lung | 3 | 3 | 0 | 56 (49–68) | |
| Liver adenoma | 1 | 1 | 0 | 32 | |
| Hodgkin | 2 | 2 | 0 | 23, 28 | |
| Cervix | 1 | 0 | 1 | 31 | |
| Colon | 1 | 1 | 0 | 25 | |
| Retinoblastoma | 1 | 1 | 0 | 1.5 | |
| Skin SCC | 6 | 5 | 1 | 18 (4–34) | |
| Non Hodgkin Lympoma | 1 | 1 | 0 | 43 |
Abbreviations as in (A). Total number of patients with any neoplasm = 42. 6 patients had multiple tumors (4 had 2, 1 had 3, 1 had 4).1 rectal cancer occurred 14 months after bone marrow transplantation. Ages were not reported for some neoplasms.
Table 3.
Cancer in the Major IBMFS
D) Diamond-Blackfan Anemia, not Transplanted
| Type | Subtype | Number | Male | Female | Median Age, Yrs (Range) |
|---|---|---|---|---|---|
| MDS/Leukemia | AML | 10 | 6 | 2 | 22 (1.2–44) |
| ALL | 1 | 0 | 1 | 13 | |
| MDS* | 5 | 5 | 0 | 21 (15–45) | |
| Tumors | Total Tumors | 19 | 10 | 9 | 23 (4–36) |
| Osteosarcoma | 6 | 2 | 4 | 12 (4–23) | |
| Soft tissue sarcoma | 1 | 1 | 0 | 30 | |
| Breast | 2 | 0 | 2 | 26, 27 | |
| Hodgkin | 3 | 3 | 0 | 7,15,23 | |
| Hepatoma | 2 | 2 | 0 | 24,26 | |
| Stomach | 1 | 1 | 0 | 26 | |
| Melanoma | 1 | 0 | 1 | 36 | |
| Colon | 1 | 0 | 1 | 34 | |
| Fibrohistiocytoma | 1 | 1 | 0 | 23 | |
| Non Hodgkin Lymphoma | 1 | 0 | 1 | 15 |
Abbreviations as in (A). Thirty patients had cancer.
1 MDS evolved to AML. I MDs followed chemotherapy for Hodgkin lymphoma.
Table 3.
Cancer in the Major IBMFS
E) Shwachman-Diamond Syndrome
| Type | Subtype | Number* | Male | Female | M:F | Median Age, Yrs (Range) |
|---|---|---|---|---|---|---|
| Leukemia | Total leukemias | 38 | 26 | 9 | 2.89 | 19 (2–43) |
| AML | 31 | 19 | 9 | 21 (3–43) | ||
| ALL | 5 | 5 | 0 | 14 (2–14) | ||
| JCML | 1 | 1 | 0 | 8 | ||
| AML after BMT | 1 | 1 | 0 | 21 | ||
| MDS | Total MDS | 72 | 47 | 23 | 2.04 | 8 (0.5–42) |
| RA | 36 | 23 | 13 | 8 (0.8–35) | ||
| RARS | 1 | 1 | 0 | 23 | ||
| RAEB | 5 | 5 | 0 | 19 (4–30) | ||
| RAEBT | 3 | 3 | 0 | 6, 8, 42 | ||
| Clone alone | 25 | 15 | 10 | 10 (0.5–25) |
11 patients had MDS prior to leukemia. One of these had RA pre-BMT, followed by AML post-BMT. 25 patients are included as MDS if they had only a clone without MDS morphology. Gender was missing in 3 cases. JCML, juvenile chronic myelogenous leukemia. BMT, bone marrow transplant. RA, refractory anemia. RARS, refractory anemia with ringed sideroblasts. RAEB, refractory anemia with excess blasts. RAEBT, refractory anemia with excess blasts in transformation.
Table 3.
Cancer in the Major IBMFS
F) Clones in Shwachman-Diamond Syndrome
| Type | Subtype | Mono7 or der (7) | i(7q) | del(20) | Not 7 or 20 | At least one clone |
|---|---|---|---|---|---|---|
| All patients | 28 | 35 | 19 | 26 | 87 | |
| Leukemia | Total Leukemia | 7 | 0 | 2 | 16 | 17 |
| AML | 6 | 0 | 2 | 14 | 15 | |
| ALL | 0 | 0 | 0 | 1 | 1 | |
| JCML | 0 | 0 | 0 | 0 | 0 | |
| AML after BMT | 1 | 0 | 0 | 1 | 1 | |
| MDS | Total MDS | 24 | 35 | 17 | 15 | 76 |
| RA | 12 | 16 | 3 | 8 | 31 | |
| RARS | 0 | 0 | 0 | 0 | 0 | |
| RAEB | 1 | 0 | 0 | 2 | 2 | |
| RAEBT | 1 | 0 | 0 | 2 | 2 | |
| Clone alone | 10 | 19 | 14 | 3 | 41 |
Abbreviations as in (E). 4 patients had leukemia following the diagnosis of MDS (1 was post-transplant), and thus the total number of patients exceeds the sum of those with leukemia plus those with MDS. 21 patients had multiple clones.
Table 4.
Mutated Genes in the Major IBMFS
A) Fanconi Anemia*
| Gene | Locus | Mutations | % of Patients | Genetics |
|---|---|---|---|---|
| FANCA | 16q24.3 | 355 | 60 | AR |
| FANCB | Xp22.31 | 14 | 2 | XLR |
| FANCC | 9q22.3 | 54 | 14 | AR |
| FANCD1/BRCA2 | 13q12.3 | 35 | 3 | AR |
| FANCD2 | 3p25.3 | 33 | 3 | AR |
| FANCE | 6p21.3 | 24 | 3 | AR |
| FANCF | 11p15 | 14 | 2 | AR |
| FANCG/XRCC9 | 9p13 | 49 | 10 | AR |
| FANCI | 15q25-26 | 20 | 1 | AR |
| FANCJ/BACH1BRIP1 | 17q22.3 | 12 | 2 | AR |
| FANCL | 2p16.1 | 3 | 0.2 | AR |
| FANCM | 14q21.3 | 3 | 0.2 | AR |
| FANCN/PALB2 | 16p12.1 | 15 | 0.7 | AR |
Leiden Open Variation Database154
Table 4.
Mutated Genes in the Major IBMFS
B) Dyskeratosis Congenita*
| Gene | Locus | Mutations | ~% of Patients | Genetics |
|---|---|---|---|---|
| DKC1 | Xq28 | ~40 | 35% | XLR |
| TINF2 | 14q11.2 | ~15 | 10–20% | AD |
| TERC | 3q26.3 | ~5 | 10% | AD, AR |
| TERT | 5p15.53 | ~10 | 5% | AD, AR |
| NOP10/NOLA3 | 15q14-q15 | 1 | <1% | AR |
| NHP2/NOLA2 | 5q35.5 | 1 | <1% | AR |
Savage et al36
Table 4.
Mutated Genes in the Major IBMFS
C) Diamond-Blackfan Anemia
| Gene | Locus | Mutations | ~% of Patients | Genetics |
|---|---|---|---|---|
| RPS19 | 19q13.3 | 50 | 25 | AD |
| RPS24 | 10q22-23 | 3 | 2 | AD |
| RPS17 | 15q25 | 1 | Rare | AD |
| RPL5 | 1p22.1 | 18 | 9 | AD |
| RPL11 | 1p36.1-35 | 13 | 6 | AD |
| RPL35A | 3q29-qter | 2 | 2 | AD |
Table 4.
Mutated Genes in the Major IBMFS
D) Shwachman-Diamond Syndrome
| Gene | Locus | Mutations | ~% of Patients | Genetics |
|---|---|---|---|---|
| SBDS | 7 centromere | 258+2T>C; 183−184TA>CT; 183−184TA>CT, 258+2T>C on one allele | >90 | AR |
Table 4.
Mutated Genes in the Major IBMFS
E) Severe Congenital Neutropenia
| Gene | MIM #* | Locus | Mutations | ~% of Patients | Genetics |
|---|---|---|---|---|---|
| ELA-2/ELANE | 202700 | 19p13.3 | 75 | 60 | AD |
| GFI1 | 613107 | 1p22 | 2 | <1 | AD |
| HAX1 | 610738 | 1q21.3 | ~10 | 1 | AR |
| WAS | 300299 | Xp11.33–11.22 | 3 | <1 | XLR |
| G6PC3 | 612541 | 17q21.31 | 4 | <1 | AR |
OMIM155
Table 4.
Mutated Genes in the Major IBMFS
F) Other Rare Disorders associated with Neutropenia*
| Syndrome | MIM# | Gene | Genetics, locus | Features |
|---|---|---|---|---|
| Reticular dysgenesis | 267500 | AK2 | AR, 1p34 | Severe combined immunodeficiency; hearing loss; mitochondrial functon |
| WHIM Syndrome | 193670 | CXCR4 | AD, 2q21 | Warts, hypogammaglobulinemia, immunodeficiency, myelokathexis |
| Glycogen Storage Disease 1b | 232220 | SLC37A4 | AR, 11q23 | Glycogen metabolism |
| Barth Syndrome | 302060 | TAZ | XLR, Xq28 | Dilated cardiac and skeletal myopathy, increased 3-methylglutaconic aciduria |
| Cartilage Hair Hypoplasia | 250250 | RMRP | AR, 9p21-9p12 | Immunodeficiency, abnormal hair, skeletal hypoplasia |
| Chediak-Higashi Syndrome | 214500 | LYST | AR, 1q42.1-42.2 | Partial albinism, neurologic, lymphoma |
| Griscelli Syndrome, type 1 | 214450 | MY05A | AR, 15q21 | Partial albinism, neurologic |
| Griscelli Syndrome, type 2 | 603868 | RAB27A | AR, 15q21 | Partial albinism, neurologic, hemophagocytic syndrome |
| Hermansky-Pudlak Syndrome | 608233 | AP3B1 | AR, 5q14.1 | Oculocutaneous albinism, immunodeficiency, abnormal platelets |
| P14-deficiency | 610389 | MAPBPIP | AR, 1q22 | Growth failure, hypopigmentation, immunodeficiency |
| Cohen Syndrome | 216550 | VPS13B/COH1 | AR, 8q22-q23 | Retardation, skeletal anomalies, pigmented retinopathy |
| Charcot-Marie-Tooth Disease, type 2 | 606482 | DNM2 | AD, 19p13.2 | Axonal demyelinating neuropathy |
Table 4.
Mutated Genes in the Major IBMFS
G) Other Rare Syndromes with Marrow Failure
| Syndrome | Gene | Locus | Mutations | ~% of Patients | Genetics |
|---|---|---|---|---|---|
| Amegakaryocytic Thrombocytopenia | MPL | 1p34 | 15 | 100 | AR |
| Thrombocytopenia Absent Radii | del 1q21 | 1q21 | - | 100 | AR |
| Radioulnar synostosis | HOXA11 | 7p15-14.2 | - | - | AD |
| Pearson Syndrome | Mitochondrial deletion | Mitochondrial DNA, respiratory enzymes | ~5 | 100 | Maternal |
Acknowledgements
This work was supported in part by the Intramural Program of the National Institutes of Health and the National Cancer Institute (B.P.A.) and a grant from the National Heart Lung and Blood Institute of the National Institutes of Health (A.S.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement None to declare.
REFERENCES
- 1.Bagby GC, Meyers G. Bone marrow failure syndromes. Preface. Hematol Oncol Clin North Am. 2009;23:xiii–xiv. doi: 10.1016/j.hoc.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 2.Tsilou ET, Giri N, S W, et al. Ocular and orbital manifestations of the inherited bone marrow failure syndromes: Fanconi anemia and dyskeratosis congenita. Ophthalmology. 2009 doi: 10.1016/j.ophtha.2009.08.023. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson MA, Olson S, Alter BP, Giri N, Hogan WJ, Richards CS. An unusual case of Fanconi Anemia with adult onset, mosaicism in an asymptomatic sibling, and a possible molecular explanation. 2009 [Google Scholar]
- 4.Auerbach AD, Adler B, Chaganti RS. Prenatal and postnatal diagnosis and carrier detection of Fanconi anemia by a cytogenetic method. Pediatrics. 1981;67:128–135. [PubMed] [Google Scholar]
- 5.Ameziane N, Errami A, Leveille F, Fontaine C, de Vries Y, van Spaendonk RM, de Winter JP, Pals G, Joenje H. Genetic subtyping of Fanconi anemia by comprehensive mutation screening. Hum Mutat. 2008;29:159–166. doi: 10.1002/humu.20625. [DOI] [PubMed] [Google Scholar]
- 6.Alter BP, Joenje H, Oostra AB, Pals G. Fanconi anemia: adult head and neck cancer and hematopoietic mosaicism. Arch Otolaryngol Head Neck Surg. 2005;131:635–639. doi: 10.1001/archotol.131.7.635. [DOI] [PubMed] [Google Scholar]
- 7.Rosenberg PS, Huang Y, Alter BP. Individualized risks of first adverse events in patients with Fanconi anemia. Blood. 2004;104:350–355. doi: 10.1182/blood-2004-01-0083. [DOI] [PubMed] [Google Scholar]
- 8.Rosenberg PS, Socie G, Alter BP, Gluckman E. Risk of head and neck squamous cell cancer and death in patients with Fanconi anemia who did and did not receive transplants. Blood. 2005;105:67–73. doi: 10.1182/blood-2004-04-1652. [DOI] [PubMed] [Google Scholar]
- 9.Alter BP, Rosenberg PS, Brody LC. Clinical and molecular features associated with biallelic mutations in FANCD1/BRCA2. J Med Genet. 2007;44:1–9. doi: 10.1136/jmg.2006.043257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alter B, Frissora C, et al. Fanconi's anemia and pregnancy. Br J Haematol. 1991;77:410–418. doi: 10.1111/j.1365-2141.1991.tb08593.x. [DOI] [PubMed] [Google Scholar]
- 11.Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet. 2007;8:735–748. doi: 10.1038/nrg2159. [DOI] [PubMed] [Google Scholar]
- 12.de Winter JP, Joenje H. The genetic and molecular basis of Fanconi anemia. Mutat Res. 2009;668:11–19. doi: 10.1016/j.mrfmmm.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 13.Green AM, Kupfer GM. Fanconi anemia. Hematol Oncol Clin North Am. 2009;23:193–214. doi: 10.1016/j.hoc.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moldovan GL, D'Andrea AD. How the Fanconi Anemia Pathway Guards the Genome. Annu Rev Genet. 2009 doi: 10.1146/annurev-genet-102108-134222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andreassen PR, D'Andrea AD, Taniguchi T. ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev. 2004;18:1958–1963. doi: 10.1101/gad.1196104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taniguchi T, Garcia-Higuera I, Xu B, Andreassen PR, Gregory RC, Kim ST, Lane WS, Kastan MB, D'Andrea AD. Convergence of the Fanconi anemia and ataxia telangiectasia signaling pathways. Cell. 2002;109:459–472. doi: 10.1016/s0092-8674(02)00747-x. [DOI] [PubMed] [Google Scholar]
- 17.Nakanishi K, Taniguchi T, Ranganathan V, New HV, Moreau LA, Stotsky M, Mathew CG, Kastan MB, Weaver DT, D'Andrea AD. Interaction of FANCD2 and NBS1 in the DNA damage response. Nat Cell Biol. 2002;4:913–920. doi: 10.1038/ncb879. [DOI] [PubMed] [Google Scholar]
- 18.Meetei AR, Sechi S, Wallisch M, Yang D, Young MK, Joenje H, Hoatlin ME, Wang W. A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Mol Cell Biol. 2003;23:3417–3426. doi: 10.1128/MCB.23.10.3417-3426.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pichierri P, Franchitto A, Rosselli F. BLM and the FANC proteins collaborate in a common pathway in response to stalled replication forks. Embo J. 2004;23:3154–3163. doi: 10.1038/sj.emboj.7600277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fagerlie S, Lensch MW, Pang Q, Bagby GC., Jr. The Fanconi anemia group C gene product: signaling functions in hematopoietic cells. Exp Hematol. 2001;29:1371–1381. doi: 10.1016/s0301-472x(01)00755-x. [DOI] [PubMed] [Google Scholar]
- 21.Bagby GC, Alter BP. Fanconi anemia. Semin Hematol. 2006;43:147–156. doi: 10.1053/j.seminhematol.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 22.Tonnies H, Huber S, Kuhl JS, Gerlach A, Ebell W, Neitzel H. Clonal chromosomal aberrations in bone marrow cells of Fanconi anemia patients: gains of the chromosomal segment 3q26q29 as an adverse risk factor. Blood. 2003;101:3872–3874. doi: 10.1182/blood-2002-10-3243. [DOI] [PubMed] [Google Scholar]
- 23.Shimamura A. Treatment of Hematologic Abnormalities in Fanconi Anemia. In: Eiler ME, Frohnmayer D, Frohnmayer L, Larsen K, Owen J, editors. Fanconi Anemia Guidelines for Diagnosis and Management. 3rd Edition Fanconi Anemia Research Fund; Eugene: 2008. pp. 49–75. [Google Scholar]
- 24.Gluckman E, Devergie A, Schaison G, Bussel A, Berger R, Sohier J, Bernard J. Bone marrow transplantation in Fanconi anaemia. Br J Haematol. 1980;45:557–564. doi: 10.1111/j.1365-2141.1980.tb07178.x. [DOI] [PubMed] [Google Scholar]
- 25.Gluckman E, Wagner JE. Hematopoietic stem cell transplantation in childhood inherited bone marrow failure syndrome. Bone Marrow Transplant. 2008;41:127–132. doi: 10.1038/sj.bmt.1705960. [DOI] [PubMed] [Google Scholar]
- 26.Myers KC, Davies SM. Hematopoietic stem cell transplantation for bone marrow failure syndromes in children. Biol Blood Marrow Transplant. 2009;15:279–292. doi: 10.1016/j.bbmt.2008.11.037. [DOI] [PubMed] [Google Scholar]
- 27.Wagner JE, Eapen M, MacMillan ML, Harris RE, Pasquini R, Boulad F, Zhang MJ, Auerbach AD. Unrelated donor bone marrow transplantation for the treatment of Fanconi anemia. Blood. 2007;109:2256–2262. doi: 10.1182/blood-2006-07-036657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deeg HJ, Socie G, Schoch G, Henry-Amar M, Witherspoon RP, Devergie A, Sullivan KM, Gluckman E, Storb R. Malignancies after marrow transplantation for aplastic anemia and fanconi anemia: a joint Seattle and Paris analysis of results in 700 patients. Blood. 1996;87:386–392. [PubMed] [Google Scholar]
- 29.Guardiola P, Socie G, Li X, Ribaud P, Devergie A, Esperou H, Richard P, Traineau R, Janin A, Gluckman E. Acute graft-versus-host disease in patients with Fanconi anemia or acquired aplastic anemia undergoing bone marrow transplantation from HLA-identical sibling donors: risk factors and influence on outcome. Blood. 2004;103:73–77. doi: 10.1182/blood-2003-06-2146. [DOI] [PubMed] [Google Scholar]
- 30.Velazquez I, Alter BP. Androgens and liver tumors: Fanconi's anemia and non-Fanconi's conditions. Am J Hematol. 2004;77:257–267. doi: 10.1002/ajh.20183. [DOI] [PubMed] [Google Scholar]
- 31.Kutler DI, Wreesmann VB, Goberdhan A, Ben-Porat L, Satagopan J, Ngai I, Huvos AG, Giampietro P, Levran O, Pujara K, Diotti R, Carlson D, Huryn LA, Auerbach AD, Singh B. Human papillomavirus DNA and p53 polymorphisms in squamous cell carcinomas from Fanconi anemia patients. J Natl Cancer Inst. 2003;95:1718–1721. doi: 10.1093/jnci/djg091. [DOI] [PubMed] [Google Scholar]
- 32.van Zeeburg HJ, Snijders PJ, Wu T, Gluckman E, Soulier J, Surralles J, Castella M, van der Wal JE, Wennerberg J, Califano J, Velleuer E, Dietrich R, Ebell W, Bloemena E, Joenje H, Leemans CR, Brakenhoff RH. Clinical and molecular characteristics of squamous cell carcinomas from Fanconi anemia patients. J Natl Cancer Inst. 2008;100:1649–1653. doi: 10.1093/jnci/djn366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Al-Rahawan MM, Giri N, Alter BP. Intensive immunosuppression therapy for aplastic anemia associated with dyskeratosis congenita. Int J Hematol. 2006;83:275–276. doi: 10.1532/IJH97.06030. [DOI] [PubMed] [Google Scholar]
- 34.Rosenberg PS, Giri N, Savage SA, BP A. Cancer Epidemiology in the National Cancer Institute Inherited Bone Marrow Failure Syndromes Cohort: First Report. American Society of Hematology: American Society of Hematology. 2008 [Google Scholar]
- 35.Vulliamy TJ, Marrone A, Knight SW, Walne A, Mason PJ, Dokal I. Mutations in dyskeratosis congenita: their impact on telomere length and the diversity of clinical presentation. Blood. 2006;107:2680–2685. doi: 10.1182/blood-2005-07-2622. [DOI] [PubMed] [Google Scholar]
- 36.Savage SA, Alter BP. Dyskeratosis congenita. Hematol Oncol Clin North Am. 2009;23:215–231. doi: 10.1016/j.hoc.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Armanios MY, Chen JJ, Cogan JD, Alder JK, Ingersoll RG, Markin C, Lawson WE, Xie M, Vulto I, Phillips JA, 3rd, Lansdorp PM, Greider CW, Loyd JE. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356:1317–1326. doi: 10.1056/NEJMoa066157. [DOI] [PubMed] [Google Scholar]
- 38.Alter BP, Caruso JP, Drachtman RA, Uchida R, Velagaleti GVN, Elghetany MT. Fanconi Anemia: Myelodysplasia as a predictor of outcome. Cancer Genet Cytogeneti. 2000;117:125–131. doi: 10.1016/s0165-4608(99)00159-4. [DOI] [PubMed] [Google Scholar]
- 39.Alter BP, Baerlocher GM, Savage SA, Chanock SJ, Weksler BB, Willner JP, Peters JA, Giri N, Lansdorp PM. Very short telomere length by flow fluorescence in situ hybridization identifies patients with dyskeratosis congenita. Blood. 2007;110:1439–1447. doi: 10.1182/blood-2007-02-075598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Du HY, Pumbo E, Ivanovich J, An P, Maziarz RT, Reiss UM, Chirnomas D, Shimamura A, Vlachos A, Lipton JM, Goyal RK, Goldman F, Wilson DB, Mason PJ, Bessler M. TERC and TERT gene mutations in patients with bone marrow failure and the significance of telomere length measurements. Blood. 2009;113:309–316. doi: 10.1182/blood-2008-07-166421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walne AJ, Dokal I. Advances in the understanding of dyskeratosis congenita. Br J Haematol. 2009;145:164–172. doi: 10.1111/j.1365-2141.2009.07598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kirwan M, Dokal I. Dyskeratosis congenita, stem cells and telomeres. Biochim Biophys Acta. 2009;1792:371–379. doi: 10.1016/j.bbadis.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marrone A, Walne A, Tamary H, Masunari Y, Kirwan M, Beswick R, Vulliamy T, Dokal I. Telomerase reverse-transcriptase homozygous mutations in autosomal recessive dyskeratosis congenita and Hoyeraal-Hreidarsson syndrome. Blood. 2007;110:4198–4205. doi: 10.1182/blood-2006-12-062851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Du HY, Pumbo E, Manley P, Field JJ, Bayliss SJ, Wilson DB, Mason PJ, Bessler M. Complex inheritance pattern of dyskeratosis congenita in two families with 2 different mutations in the telomerase reverse transcriptase gene. Blood. 2008;111:1128–1130. doi: 10.1182/blood-2007-10-120907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vulliamy T, Marrone A, Szydlo R, Walne A, Mason PJ, Dokal I. Disease anticipation is associated with progressive telomere shortening in families with dyskeratosis congenita due to mutations in TERC. Nat Genet. 2004;36:447–449. doi: 10.1038/ng1346. [DOI] [PubMed] [Google Scholar]
- 46.Armanios M, Chen JL, Chang YP, Brodsky RA, Hawkins A, Griffin CA, Eshleman JR, Cohen AR, Chakravarti A, Hamosh A, Greider CW. Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita. Proc Natl Acad Sci U S A. 2005;102:15960–15964. doi: 10.1073/pnas.0508124102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamaguchi H, Baerlocher GM, Lansdorp PM, Chanock SJ, Nunez O, Sloand E, Young NS. Mutations of the human telomerase RNA gene (TERC) in aplastic anemia and myelodysplastic syndrome. Blood. 2003;102:916–918. doi: 10.1182/blood-2003-01-0335. [DOI] [PubMed] [Google Scholar]
- 48.Vulliamy TJ, Walne A, Baskaradas A, Mason PJ, Marrone A, Dokal I. Mutations in the reverse transcriptase component of telomerase (TERT) in patients with bone marrow failure. Blood Cells Mol Dis. 2005;34:257–263. doi: 10.1016/j.bcmd.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 49.Yamaguchi H, Calado RT, Ly H, Kajigaya S, Baerlocher GM, Chanock SJ, Lansdorp PM, Young NS. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N Engl J Med. 2005;352:1413–1424. doi: 10.1056/NEJMoa042980. [DOI] [PubMed] [Google Scholar]
- 50.Tsakiri KD, Cronkhite JT, Kuan PJ, Xing C, Raghu G, Weissler JC, Rosenblatt RL, Shay JW, Garcia CK. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci U S A. 2007;104:7552–7557. doi: 10.1073/pnas.0701009104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alder JK, Chen JJ, Lancaster L, Danoff S, Su SC, Cogan JD, Vulto I, Xie M, Qi X, Tuder RM, Phillips JA, 3rd, Lansdorp PM, Loyd JE, Armanios MY. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A. 2008;105:13051–13056. doi: 10.1073/pnas.0804280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Savage SA, Giri N, Baerlocher GM, Orr N, Lansdorp PM, Alter BP. TINF2, a component of the shelterin telomere protection complex, is mutated in dyskeratosis congenita. Am J Hum Genet. 2008;82:501–509. doi: 10.1016/j.ajhg.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Walne AJ, Vulliamy T, Beswick R, Kirwan M, Dokal I. TINF2 mutations result in very short telomeres: analysis of a large cohort of patients with dyskeratosis congenita and related bone marrow failure syndromes. Blood. 2008;112:3594–3600. doi: 10.1182/blood-2008-05-153445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shimamura A. Inherited bone marrow failure syndromes: molecular features. Hematology Am Soc Hematol Educ Program. 2006:63–71. doi: 10.1182/asheducation-2006.1.63. [DOI] [PubMed] [Google Scholar]
- 55.Meier UT. The many facets of H/ACA ribonucleoproteins. Chromosoma. 2005;114:1–14. doi: 10.1007/s00412-005-0333-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.King TH, Liu B, McCully RR, Fournier MJ. Ribosome structure and activity are altered in cells lacking snoRNPs that form pseudouridines in the peptidyl transferase center. Mol Cell. 2003;11:425–435. doi: 10.1016/s1097-2765(03)00040-6. [DOI] [PubMed] [Google Scholar]
- 57.Mochizuki Y, He J, Kulkarni S, Bessler M, Mason PJ. Mouse dyskerin mutations affect accumulation of telomerase RNA and small nucleolar RNA, telomerase activity, and ribosomal RNA processing. Proc Natl Acad Sci U S A. 2004;101:10756–10761. doi: 10.1073/pnas.0402560101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ruggero D, Grisendi S, Piazza F, Rego E, Mari F, Rao PH, Cordon-Cardo C, Pandolfi PP. Dyskeratosis congenita and cancer in mice deficient in ribosomal RNA modification. Science. 2003;299:259–262. doi: 10.1126/science.1079447. [DOI] [PubMed] [Google Scholar]
- 59.Yoon A, Peng G, Brandenburg Y, Zollo O, Xu W, Rego E, Ruggero D. Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science. 2006;312:902–906. doi: 10.1126/science.1123835. [DOI] [PubMed] [Google Scholar]
- 60.Berthou C, Devergi A, D'Agay MF, Sonsino E, Scrobohaci ML, Loirat C, E. G. Late vascular complications after bone marrow transplantation for dyskeratosis congenita. British J of Haematology. 1991;79:335–336. doi: 10.1111/j.1365-2141.1991.tb04543.x. [DOI] [PubMed] [Google Scholar]
- 61.Fogarty PF, Yamaguchi H, Wiestner A, Baerlocher GM, Sloand E, Zeng WS, Read EJ, Lansdorp PM, Young NS. Late presentation of dyskeratosis congenita as apparently acquired aplastic anaemia due to mutations in telomerase RNA. Lancet. 2003;362:1628–1630. doi: 10.1016/S0140-6736(03)14797-6. [DOI] [PubMed] [Google Scholar]
- 62.Alter BP, Gardner EH, Hall RE. Treatment of dyskeratosis congenita with granulocyte macrophage colony-stimulating factor and erythropoietin. British J Haematol. 1997;97:309–311. doi: 10.1046/j.1365-2141.1997.622717.x. [DOI] [PubMed] [Google Scholar]
- 63.Giri N, Pitel PA, Green D, Alter BP. Splenic peliosis and rupture in patients with dyskeratosis congenita on androgens and granulocyte colony-stimulating factor. Br J Haematol. 2007;138:815–817. doi: 10.1111/j.1365-2141.2007.06718.x. [DOI] [PubMed] [Google Scholar]
- 64.Fanconi Anemia Research Fund I . Guidelines for Diagnosis and Management. Fanconi Anemia Research Fund, Inc.; Eugene, OR: 2008. [Google Scholar]
- 65.Balaban EP, Buchanan GR, Graham M, Frenkel EP. Diamond-Blackfan syndrome in adult patients. Am J Med. 1985;78:533–538. doi: 10.1016/0002-9343(85)90352-3. [DOI] [PubMed] [Google Scholar]
- 66.Lipton JM, Ellis SR. Diamond-Blackfan anemia: diagnosis, treatment, and molecular pathogenesis. Hematol Oncol Clin North Am. 2009;23:261–282. doi: 10.1016/j.hoc.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Matsson H, Davey EJ, Draptchinskaia N, Hamaguchi I, Ooka A, Leveen P, Forsberg E, Karlsson S, Dahl N. Targeted disruption of the ribosomal protein S19 gene is lethal prior to implantation. Mol Cell Biol. 2004;24:4032–4037. doi: 10.1128/MCB.24.9.4032-4037.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McGowan KA, Li JZ, Park CY, Beaudry V, Tabor HK, Sabnis AJ, Zhang W, Fuchs H, de Angelis MH, Myers RM, Attardi LD, Barsh GS. Ribosomal mutations cause p53-mediated dark skin and pleiotropic effects. Nat Genet. 2008;40:963–970. doi: 10.1038/ng.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Danilova N, Sakamoto KM, Lin S. Ribosomal protein S19 deficiency in zebrafish leads to developmental abnormalities and defective erythropoiesis through activation of p53 protein family. Blood. 2008;112:5228–5237. doi: 10.1182/blood-2008-01-132290. [DOI] [PubMed] [Google Scholar]
- 70.Uechi T, Nakajima Y, Chakraborty A, Torihara H, Higa S, Kenmochi N. Deficiency of ribosomal protein S19 during early embryogenesis leads to reduction of erythrocytes in a zebrafish model of Diamond-Blackfan anemia. Hum Mol Genet. 2008;17:3204–3211. doi: 10.1093/hmg/ddn216. [DOI] [PubMed] [Google Scholar]
- 71.Dianzani I, Loreni F. Diamond-Blackfan anemia: a ribosomal puzzle. Haematologica. 2008;93:1601–1604. doi: 10.3324/haematol.2008.000513. [DOI] [PubMed] [Google Scholar]
- 72.Ellis SR, Lipton JM. Diamond Blackfan anemia: a disorder of red blood cell development. Curr Top Dev Biol. 2008;82:217–241. doi: 10.1016/S0070-2153(07)00008-7. [DOI] [PubMed] [Google Scholar]
- 73.Jin A, Itahana K, O'Keefe K, Zhang Y. Inhibition of HDM2 and activation of p53 by ribosomal protein L23. Mol Cell Biol. 2004;24:7669–7680. doi: 10.1128/MCB.24.17.7669-7680.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dai MS, Zeng SX, Jin Y, Sun XX, David L, Lu H. Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Mol Cell Biol. 2004;24:7654–7668. doi: 10.1128/MCB.24.17.7654-7668.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Marechal V, Elenbaas B, Piette J, Nicolas JC, Levine AJ. The ribosomal L5 protein is associated with mdm-2 and mdm-2-p53 complexes. Mol Cell Biol. 1994;14:7414–7420. doi: 10.1128/mcb.14.11.7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lohrum MA, Ludwig RL, Kubbutat MH, Hanlon M, Vousden KH. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell. 2003;3:577–587. doi: 10.1016/s1535-6108(03)00134-x. [DOI] [PubMed] [Google Scholar]
- 77.Zhang Y, Wolf GW, Bhat K, Jin A, Allio T, Burkhart WA, Xiong Y. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol Cell Biol. 2003;23:8902–8912. doi: 10.1128/MCB.23.23.8902-8912.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fumagalli S, Di Cara A, Neb-Gulati A, Natt F, Schwemberger S, Hall J, Babcock GF, Bernardi R, Pandolfi PP, Thomas G. Absence of nucleolar disruption after impairment of 40S ribosome biogenesis reveals an rpL11-translation-dependent mechanism of p53 induction. Nat Cell Biol. 2009;11:501–508. doi: 10.1038/ncb1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ebert BL, Pretz J, Bosco J, Chang CY, Tamayo P, Galili N, Raza A, Root DE, Attar E, Ellis SR, Golub TR. Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature. 2008;451:335–339. doi: 10.1038/nature06494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Olivieri NF, Brittenham GM. Iron-chelating therapy and the treatment of thalassemia. Blood. 1997;89:739–761. [PubMed] [Google Scholar]
- 81.Roggero S, Quarello P, Vinciguerra T, Longo F, Piga A, Ramenghi U. Severe iron overload in Blackfan-Diamond anemia: a case-control study. Am J Hematol. 2009;84:729–732. doi: 10.1002/ajh.21541. [DOI] [PubMed] [Google Scholar]
- 82.Shwachman H, Diamond LK, Oski FA, Khaw KT. The Syndrome of Pancreatic Insufficiency and Bone Marrow Dysfunction. J Pediatr. 1964;65:645–663. doi: 10.1016/s0022-3476(64)80150-5. [DOI] [PubMed] [Google Scholar]
- 83.Rothbaum R, Perrault J, Vlachos A, Cipolli M, Alter BP, Burroughs S, Durie P, Elghetany MT, Grand R, Hubbard V, Rommens J, Rossi T. Shwachman-Diamond syndrome: report from an international conference. J Pediatr. 2002;141:266–270. doi: 10.1067/mpd.2002.125850. [DOI] [PubMed] [Google Scholar]
- 84.Savilahti E, Rapola J. Frequent myocardial lesions in Shwachman's syndrome. Eight fatal cases among 16 Finnish patients. Acta Paediatr Scand. 1984;73:642–651. doi: 10.1111/j.1651-2227.1984.tb09989.x. [DOI] [PubMed] [Google Scholar]
- 85.Alter BP, Kumar M, Lockhart LL, Sprinz PG, Rowe TF. Pregnancy in bone marrow failure syndromes: Diamond-Blackfan anaemia and Shwachman-Diamond syndrome. Br J Haematol. 1999;107:49–54. doi: 10.1046/j.1365-2141.1999.01668.x. [DOI] [PubMed] [Google Scholar]
- 86.Boocock GR, Morrison JA, Popovic M, Richards N, Ellis L, Durie PR, Rommens JM. Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat Genet. 2003;33:97–101. doi: 10.1038/ng1062. [DOI] [PubMed] [Google Scholar]
- 87.Zhang S, Shi M, Hui CC, Rommens JM. Loss of the mouse ortholog of the shwachman-diamond syndrome gene (sbds) results in early embryonic lethality. Mol Cell Biol. 2006;26:6656–6663. doi: 10.1128/MCB.00091-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Savchenko A, Krogan N, Cort JR, Evdokimova E, Lew JM, Yee AA, Sanchez-Pulido L, Andrade MA, Bochkarev A, Watson JD, Kennedy MA, Greenblatt J, Hughes T, Arrowsmith CH, Rommens JM, Edwards AM. The Shwachman-Bodian-Diamond syndromeprotein family is involved in RNA metabolism. J Biol Chem. 2005;280:19213–19220. doi: 10.1074/jbc.M414421200. [DOI] [PubMed] [Google Scholar]
- 89.Shammas C, Menne TF, Hilcenko C, Michell SR, Goyenechea B, Boocock GR, Durie PR, Rommens JM, Warren AJ. Structural and mutational analysis of the SBDS protein family: insight into the leukemia-associated shwachman-diamond syndrome. J Biol Chem. 2005;280:19221–19229. doi: 10.1074/jbc.M414656200. [DOI] [PubMed] [Google Scholar]
- 90.Ganapathi KA, Austin KM, Lee CS, Dias A, Malsch MM, Reed R, Shimamura A. The human Shwachman-Diamond syndrome protein, SBDS, associates with ribosomal RNA. Blood. 2007;110:1458–1465. doi: 10.1182/blood-2007-02-075184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ng CL, Waterman DG, Koonin EV, Walters AD, Chong JP, Isupov MN, Lebedev AA, Bunka DH, Stockley PG, Ortiz-Lombardia M, Antson AA. Conformational flexibility and molecular interactions of an archaeal homologue of the Shwachman-Bodian-Diamond syndrome protein. BMC Struct Biol. 2009;9:32. doi: 10.1186/1472-6807-9-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Luz JS, Georg RC, Gomes CH, Machado-Santelli GM, Oliveira CC. Sdo1p, the yeast orthologue of Shwachman-Bodian-Diamond syndrome protein, binds RNA and interacts with nuclear rRNA-processing factors. Yeast. 2009;26:287–298. doi: 10.1002/yea.1668. [DOI] [PubMed] [Google Scholar]
- 93.Rujkijyanont P, Adams SL, Beyene J, Dror Y. Bone marrow cells from patients with Shwachman-Diamond syndrome abnormally express genes involved in ribosome biogenesis and RNA processing. Br J Haematol. 2009;145:806–815. doi: 10.1111/j.1365-2141.2009.07692.x. [DOI] [PubMed] [Google Scholar]
- 94.Basu U, Si K, Warner JR, Maitra U. The Saccharomyces cerevisiae TIF6 gene encoding translation initiation factor 6 is required for 60S ribosomal subunit biogenesis. Mol Cell Biol. 2001;21:1453–1462. doi: 10.1128/MCB.21.5.1453-1462.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ceci M, Gaviraghi C, Gorrini C, Sala LA, Offenhauser N, Marchisio PC, Biffo S. Release of eIF6 (p27BBP) from the 60S subunit allows 80S ribosome assembly. Nature. 2003;426:579–584. doi: 10.1038/nature02160. [DOI] [PubMed] [Google Scholar]
- 96.Moore JBt, Farrar JE, Arceci RJ, Liu JM, Ellis SR. Distinct ribosome maturation defects in yeast models of Diamond Blackfan anemia and Shwachman Diamond syndrome. Haematologica. 2009 doi: 10.3324/haematol.2009.012450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Austin KM, Gupta ML, Coats SA, Tulpule A, Mostoslavsky G, Balazs AB, Mulligan RC, Daley G, Pellman D, Shimamura A. Mitotic spindle destabilization and genomic instability in Shwachman-Diamond syndrome. J Clin Invest. 2008;118:1511–1518. doi: 10.1172/JCI33764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Orelio C, Verkuijlen P, Geissler J, van den Berg TK, Kuijpers TW. SBDS expression and localization at the mitotic spindle in human myeloid progenitors. PLoS One. 2009;4:e7084. doi: 10.1371/journal.pone.0007084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Maserati E, Pressato B, Valli R, Minelli A, Sainati L, Patitucci F, Marletta C, Mastronuzzi A, Poli F, Lo Curto F, Locatelli F, Danesino C, Pasquali F. The route to development of myelodysplastic syndrome/acute myeloid leukaemia in Shwachman-Diamond syndrome: the role of ageing, karyotype instability, and acquired chromosome anomalies. Br J Haematol. 2009;145:190–197. doi: 10.1111/j.1365-2141.2009.07611.x. [DOI] [PubMed] [Google Scholar]
- 100.Dror Y, Freedman MH. Shwachman-Diamond syndrome: An inherited preleukemic bone marrow failure disorder with aberrant hematopoietic progenitors and faulty marrow microenvironment. Blood. 1999;94:3048–3054. [PubMed] [Google Scholar]
- 101.Rawls A, Gregory A, Wolosznek J, Liu F, Link D. Lentiviral-mediated RNAi inhibition of Sbds in murine hematopoietic progenitors impairs their hematopoietic potential. Blood. 2007;110:2414–2422. doi: 10.1182/blood-2006-03-007112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Watanabe K, Ambekar C, Wang H, Ciccolini A, Schimmer AD, Dror Y. SBDS-deficiency results in specific hypersensitivity to Fas stimulation and accumulation of Fas at the plasma membrane. Apoptosis. 2009;14:77–89. doi: 10.1007/s10495-008-0275-9. [DOI] [PubMed] [Google Scholar]
- 103.Ball HL, Zhang B, Riches JJ, Gandhi R, Li J, Rommens JM, Myers JS. Shwachman-Bodian Diamond syndrome is a multi-functional protein implicated in cellular stress responses. Hum Mol Genet. 2009;18:3684–3695. doi: 10.1093/hmg/ddp316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dror Y, Ginzberg H, Dalal I, Cherepanov V, Downey G, Durie P, Roifman CM, Freedman MH. Immune function in patients with Shwachman-Diamond syndrome. Br J Haematol. 2001;114:712–717. doi: 10.1046/j.1365-2141.2001.02996.x. [DOI] [PubMed] [Google Scholar]
- 105.Orelio C, Kuijpers TW. Shwachman-Diamond syndrome neutrophils have altered chemoattractant-induced F-actin polymerization and polarization characteristics. Haematologica. 2009;94:409–413. doi: 10.3324/haematol.13733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Grinspan ZM, Pikora CA. Infections in Patients with Shwachman-Diamond Syndrome. Pediatr Infect Dis J. 2005;24:179–181. doi: 10.1097/01.inf.0000151042.90125.f6. [DOI] [PubMed] [Google Scholar]
- 107.Rosenberg PS, Alter BP, Bolyard AA, Bonilla MA, Boxer LA, Cham B, Fier C, Freedman M, Kannourakis G, Kinsey S, Schwinzer B, Zeidler C, Welte K, Dale DC. The incidence of leukemia and mortality from sepsis in patients with severe congenital neutropenia receiving long-term G-CSF therapy. Blood. 2006;107:4628–4635. doi: 10.1182/blood-2005-11-4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rosenberg PS, Alter BP, Link DC, Stein S, Rodger E, Bolyard AA, Aprikyan AA, Bonilla MA, Dror Y, Kannourakis G, Newburger PE, Boxer LA, Dale DC. Neutrophil elastase mutations and risk of leukaemia in severe congenital neutropenia. Br J Haematol. 2008;140:210–213. doi: 10.1111/j.1365-2141.2007.06897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Burroughs L, Woolfrey A, Shimamura A. Shwachman-Diamond syndrome: a review of the clinical presentation, molecular pathogenesis, diagnosis, and treatment. Hematol Oncol Clin North Am. 2009;23:233–248. doi: 10.1016/j.hoc.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cesaro S, Oneto R, Messina C, Gibson BE, Buzyn A, Steward C, Gluckman E, Bredius R, Boogaerts M, Vermylen C, Veys P, Marsh J, Badell I, Michel G, Gungor T, Niethammer D, Bordigoni P, Oswald C, Favre C, Passweg J, Dini G. Haematopoietic stem cell transplantation for Shwachman-Diamond disease: a study from the European Group for blood and marrow transplantation. Br J Haematol. 2005;131:231–236. doi: 10.1111/j.1365-2141.2005.05758.x. [DOI] [PubMed] [Google Scholar]
- 111.Donadieu J, Michel G, Merlin E, Bordigoni P, Monteux B, Beaupain B, Leverger G, Laporte JP, Hermine O, Buzyn A, Bertrand Y, Casanova JL, Leblanc T, Gluckman E, Fischer A, Stephan JL. Hematopoietic stem cell transplantation for Shwachman-Diamond syndrome: experience of the French neutropenia registry. Bone Marrow Transplant. 2005;36:787–792. doi: 10.1038/sj.bmt.1705141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bhatla D, Davies SM, Shenoy S, Harris RE, Crockett M, Shoultz L, Smolarek T, Bleesing J, Hansen M, Jodele S, Jordan M, Filipovich AH, Mehta PA. Reduced-intensity conditioning is effective and safe for transplantation of patients with Shwachman-Diamond syndrome. Bone Marrow Transplant. 2008 doi: 10.1038/bmt.2008.151. [DOI] [PubMed] [Google Scholar]
- 113.Dale DC, Bolyard AA, Schwinzer BG, Pracht G, Bonilla MA, Boxer L, Freedman MH, Donadieu J, Kannourakis G, Alter BP, Cham BP, Winkelstein J, Kinsey SE, Zeidler C, Welte K. The Severe Chronic Neutropenia International Registry: 10-Year Follow-up Report. Support Cancer Ther. 2006;3:220–231. doi: 10.3816/SCT.2006.n.020. [DOI] [PubMed] [Google Scholar]
- 114.Welte K, Zeidler C. Severe congenital neutropenia. Hematol Oncol Clin North Am. 2009;23:307–320. doi: 10.1016/j.hoc.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 115.Dale DC, Person RE, Bolyard AA, Aprikyan AG, Bos C, Bonilla MA, Boxer LA, Kannourakis G, Zeidler C, Welte K, Benson KF, Horwitz M. Mutations in the gene encoding neutrophil elastase in congenital and cyclic neutropenia. Blood. 2000;96:2317–2322. [PubMed] [Google Scholar]
- 116.Kollner I, Sodeik B, Schreek S, Heyn H, von Neuhoff N, Germeshausen M, Zeidler C, Kruger M, Schlegelberger B, Welte K, Beger C. Mutations in neutrophil elastase causing congenital neutropenia lead to cytoplasmic protein accumulation and induction of the unfolded protein response. Blood. 2006;108:493–500. doi: 10.1182/blood-2005-11-4689. [DOI] [PubMed] [Google Scholar]
- 117.Grenda DS, Murakami M, Ghatak J, Xia J, Boxer LA, Dale D, Dinauer MC, Link DC. Mutations of the ELA2 gene found in patients with severe congenital neutropenia induce the unfolded protein response and cellular apoptosis. Blood. 2007;110:4179–4187. doi: 10.1182/blood-2006-11-057299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Xia J, Link DC. Severe congenital neutropenia and the unfolded protein response. Curr Opin Hematol. 2008;15:1–7. doi: 10.1097/MOH.0b013e3282f13cd2. [DOI] [PubMed] [Google Scholar]
- 119.Klein C, Grudzien M, Appaswamy G, Germeshausen M, Sandrock I, Schaffer AA, Rathinam C, Boztug K, Schwinzer B, Rezaei N, Bohn G, Melin M, Carlsson G, Fadeel B, Dahl N, Palmblad J, Henter JI, Zeidler C, Grimbacher B, Welte K. HAX1 deficiency causes autosomal recessive severe congenital neutropenia (Kostmann disease) Nat Genet. 2007;39:86–92. doi: 10.1038/ng1940. [DOI] [PubMed] [Google Scholar]
- 120.Devriendt K, Kim AS, Mathijs G, Frints SG, Schwartz M, Van Den Oord JJ, Verhoef GE, Boogaerts MA, Fryns JP, You D, Rosen MK, Vandenberghe P. Constitutively activating mutation in WASP causes X-linked severe congenital neutropenia. Nat Genet. 2001;27:313–317. doi: 10.1038/85886. [DOI] [PubMed] [Google Scholar]
- 121.Ancliff PJ, Blundell MP, Cory GO, Calle Y, Worth A, Kempski H, Burns S, Jones GE, Sinclair J, Kinnon C, Hann IM, Gale RE, Linch DC, Thrasher AJ. Two novel activating mutations in the Wiskott-Aldrich syndrome protein result in congenital neutropenia. Blood. 2006;108:2182–2189. doi: 10.1182/blood-2006-01-010249. [DOI] [PubMed] [Google Scholar]
- 122.Moulding DA, Blundell MP, Spiller DG, White MR, Cory GO, Calle Y, Kempski H, Sinclair J, Ancliff PJ, Kinnon C, Jones GE, Thrasher AJ. Unregulated actin polymerization by WASp causes defects of mitosis and cytokinesis in X-linked neutropenia. J Exp Med. 2007;204:2213–2224. doi: 10.1084/jem.20062324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ochs HD, Thrasher AJ. The Wiskott-Aldrich syndrome. J Allergy Clin Immunol. 2006;117:725–738. doi: 10.1016/j.jaci.2006.02.005. quiz 739. [DOI] [PubMed] [Google Scholar]
- 124.Person RE, Li FQ, Duan Z, Benson KF, Wechsler J, Papadaki HA, Eliopoulos G, Kaufman C, Bertolone SJ, Nakamoto B, Papayannopoulou T, Grimes HL, Horwitz M. Mutations in proto-oncogene GFI1 cause human neutropenia and target ELA2. Nat Genet. 2003;34:308–312. doi: 10.1038/ng1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Karsunky H, Zeng H, Schmidt T, Zevnik B, Kluge R, Schmid KW, Duhrsen U, Moroy T. Inflammatory reactions and severe neutropenia in mice lacking the transcriptional repressor Gfi1. Nat Genet. 2002;30:295–300. doi: 10.1038/ng831. [DOI] [PubMed] [Google Scholar]
- 126.Hock H, Hamblen MJ, Rooke HM, Traver D, Bronson RT, Cameron S, Orkin SH. Intrinsic requirement for zinc finger transcription factor Gfi-1 in neutrophil differentiation. Immunity. 2003;18:109–120. doi: 10.1016/s1074-7613(02)00501-0. [DOI] [PubMed] [Google Scholar]
- 127.Hock H, Orkin SH. Zinc-finger transcription factor Gfi-1: versatile regulator of lymphocytes, neutrophils and hematopoietic stem cells. Curr Opin Hematol. 2006;13:1–6. doi: 10.1097/01.moh.0000190111.85284.8f. [DOI] [PubMed] [Google Scholar]
- 128.Kazanjian A, Gross EA, Grimes HL. The growth factor independence-1 transcription factor: new functions and new insights. Crit Rev Oncol Hematol. 2006;59:85–97. doi: 10.1016/j.critrevonc.2006.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Velu CS, Baktula AM, Grimes HL. Gfi1 regulates miR-21 and miR-196b to control myelopoiesis. Blood. 2009;113:4720–4728. doi: 10.1182/blood-2008-11-190215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Boztug K, Appaswamy G, Ashikov A, Schaffer AA, Salzer U, Diestelhorst J, Germeshausen M, Brandes G, Lee-Gossler J, Noyan F, Gatzke AK, Minkov M, Greil J, Kratz C, Petropoulou T, Pellier I, Bellanne-Chantelot C, Rezaei N, Monkemoller K, Irani-Hakimeh N, Bakker H, Gerardy-Schahn R, Zeidler C, Grimbacher B, Welte K, Klein C. A syndrome with congenital neutropenia and mutations in G6PC3. N Engl J Med. 2009;360:32–43. doi: 10.1056/NEJMoa0805051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Germeshausen M, Ballmaier M, Welte K. Incidence of CSF3R mutations in severe congenital neutropenia and relevance for leukemogenesis: Results of a long-term survey. Blood. 2007;109:93–99. doi: 10.1182/blood-2006-02-004275. [DOI] [PubMed] [Google Scholar]
- 132.Link DC, Kunter G, Kasai Y, Zhao Y, Miner T, McLellan MD, Ries RE, Kapur D, Nagarajan R, Dale DC, Bolyard AA, Boxer LA, Welte K, Zeidler C, Donadieu J, Bellanne-Chantelot C, Vardiman JW, Caligiuri MA, Bloomfield CD, DiPersio JF, Tomasson MH, Graubert TA, Westervelt P, Watson M, Shannon W, Baty J, Mardis ER, Wilson RK, Ley TJ. Distinct patterns of mutations occurring in de novo AML versus AML arising in the setting of severe congenital neutropenia. Blood. 2007;110:1648–1655. doi: 10.1182/blood-2007-03-081216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Boztug K, Klein C. Novel genetic etiologies of severe congenital neutropenia. Curr Opin Immunol. 2009;21:472–480. doi: 10.1016/j.coi.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 134.Dale DC, Bonilla MA, Davis MW, Nakanishi AM, Hammond WP, Kurtzberg J, Wang W, Jakubowski A, Winton E, Lalezari P, et al. A randomized controlled phase III trial of recombinant human granulocyte colony-stimulating factor (filgrastim) for treatment of severe chronic neutropenia. Blood. 1993;81:2496–2502. [PMC free article] [PubMed] [Google Scholar]
- 135.Zeidler C, Welte K, Barak Y, Barriga F, Bolyard AA, Boxer L, Cornu G, Cowan MJ, Dale DC, Flood T, Freedman M, Gadner H, Mandel H, O'Reilly RJ, Ramenghi U, Reiter A, Skinner R, Vermylen C, Levine JE. Stem cell transplantation in patients with severe congenital neutropenia without evidence of leukemic transformation. Blood. 2000;95:1195–1198. [PubMed] [Google Scholar]
- 136.Germeshausen M, Ballmaier M, Welte K. MPL mutations in 23 patients suffering from congenital amegakaryocytic thrombocytopenia: the type of mutation predicts the course of the disease. Hum Mutat. 2006;27:296–301. doi: 10.1002/humu.9415. [DOI] [PubMed] [Google Scholar]
- 137.Ihara K, Ishii E, Eguchi M, Takada H, Suminoe A, Good RA, Hara T. Identification of mutations in the c-mpl gene in congenital amegakaryocytic thrombocytopenia. Proc Natl Acad Sci U S A. 1999;96:3132–3136. doi: 10.1073/pnas.96.6.3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kaushansky K, Lok S, Holly RD, Broudy VC, Lin N, Bailey MC, Forstrom JW, Buddle MM, Oort PJ, Hagen FS, et al. Promotion of megakaryocyte progenitor expansion and differentiation by the c-Mpl ligand thrombopoietin. Nature. 1994;369:568–571. doi: 10.1038/369568a0. [DOI] [PubMed] [Google Scholar]
- 139.Bartley TD, Bogenberger J, Hunt P, Li YS, Lu HS, Martin F, Chang MS, Samal B, Nichol JL, Swift S, et al. Identification and cloning of a megakaryocyte growth and development factor that is a ligand for the cytokine receptor Mpl. Cell. 1994;77:1117–1124. doi: 10.1016/0092-8674(94)90450-2. [DOI] [PubMed] [Google Scholar]
- 140.Gurney AL, Carver-Moore K, de Sauvage FJ, Moore MW. Thrombocytopenia in cmpl-deficient mice. Science. 1994;265:1445–1447. doi: 10.1126/science.8073287. [DOI] [PubMed] [Google Scholar]
- 141.Alexander WS, Roberts AW, Nicola NA, Li R, Metcalf D. Deficiencies in progenitor cells of multiple hematopoietic lineages and defective megakaryocytopoiesis in mice lacking the thrombopoietic receptor c-Mpl. Blood. 1996;87:2162–2170. [PubMed] [Google Scholar]
- 142.King S, Germeshausen M, Strauss G, Welte K, Ballmaier M. Congenital amegakaryocytic thrombocytopenia: a retrospective clinical analysis of 20 patients. Br J Haematol. 2005;131:636–644. doi: 10.1111/j.1365-2141.2005.05819.x. [DOI] [PubMed] [Google Scholar]
- 143.Ballmaier M, Germeshausen M, Schulze H, Cherkaoui K, Lang S, Gaudig A, Krukemeier S, Eilers M, Strauss G, Welte K. c-mpl mutations are the cause of congenital amegakaryocytic thrombocytopenia. Blood. 2001;97:139–146. doi: 10.1182/blood.v97.1.139. [DOI] [PubMed] [Google Scholar]
- 144.Savoia A, Dufour C, Locatelli F, Noris P, Ambaglio C, Rosti V, Zecca M, Ferrari S, di Bari F, Corcione A, Di Stazio M, Seri M, Balduini CL. Congenital amegakaryocytic thrombocytopenia: clinical and biological consequences of five novel mutations. Haematologica. 2007;92:1186–1193. doi: 10.3324/haematol.11425. [DOI] [PubMed] [Google Scholar]
- 145.Ballmaier M, Germeshausen M. Advances in the understanding of congenital amegakaryocytic thrombocytopenia. Br J Haematol. 2009;146:3–16. doi: 10.1111/j.1365-2141.2009.07706.x. [DOI] [PubMed] [Google Scholar]
- 146.Geddis AE. Congenital amegakaryocytic thrombocytopenia and thrombocytopenia with absent radii. Hematol Oncol Clin North Am. 2009;23:321–331. doi: 10.1016/j.hoc.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Guinan EC, Lee YS, Lopez KD, Kohler S, Oette DH, Bruno E, Kozakewich H, Nathan DG, Hoffman R. Effects of interleukin-3 and granulocyte-macrophage colony- stimulating factor on thrombopoiesis in congenital amegakaryocytic thrombocytopenia. Blood. 1993;81:1691–1698. [PubMed] [Google Scholar]
- 148.Thompson AA, Woodruff K, Feig SA, Nguyen LT, Schanen NC. Congenital thrombocytopenia and radio-ulnar synostosis: a new familial syndrome. Br J Haematol. 2001;113:866–870. doi: 10.1046/j.1365-2141.2001.02834.x. [DOI] [PubMed] [Google Scholar]
- 149.Oblender MG, Richardson CJ, BP A. Pearson syndrome (PS) presenting as nonimmune hydrops fetalis. Clin Res. 1993;41 [Google Scholar]
- 150.Fleming WH, Trounce I, N K, et al. Cytokine treatment improves the hematologic manifestations of Pearson's syndrome. Blood Cells Mol Dis. 1994;84 [Google Scholar]
- 151.Hoyoux C, Dresse MF, Robinet S, Forget P, Pieltain C, Ketelslegers O, Beguin Y. Cord blood transplantation in a child with Pearson's disease. Pediatr Blood Cancer. 2008;51:566. doi: 10.1002/pbc.21615. [DOI] [PubMed] [Google Scholar]
- 152.DiMauro S, Mancuso M. Mitochondrial diseases: therapeutic approaches. Biosci Rep. 2007;27:125–137. doi: 10.1007/s10540-007-9041-4. [DOI] [PubMed] [Google Scholar]
- 153.Alter BP. Inherited Bone Marrow Failure Syndromes. In: Nathan DG, Orkin SH, Ginsburg D, Look AT, editors. Nathan and Oski's Hematology of Infancy and Childhood. Vol. 1. W.B. Saunders; Philadelphia: 2003. pp. 280–365. [Google Scholar]
- 154.Leiden Open Variation Database: Fanconi anemia database: Leiden University Medical Center. 2009. [Google Scholar]
- 155.Online Mendelian Inheritance in Man, OMIM (TM) ®: McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University, National Center for Biotechnology Information, National Library of Medicine. 2009. [Google Scholar]