

SUMMARY
Mammalian telomeres repress DNA damage activation at natural chromosome ends by recruiting specific inhibitors of the DNA damage machinery that form a protective complex termed shelterin. Within this complex, TRF2 plays a crucial role in end-protection as it is required to suppress ATM activation and the formation of end-to-end chromosome fusions1, 2. Here, we address the molecular properties of TRF2 that are both necessary and sufficient to protect chromosome ends. Our data support a two-step mechanism for TRF2-mediated end protection. First, the dimerization domain of TRF2 is required to inhibit ATM activation, the key initial step involved in activation of a DNA damage response. Next, TRF2 independently suppresses the propagation of DNA damage signaling downstream of ATM activation. This novel modulation of the DNA damage response at telomeres occurs at the level of the E3 ubiquitin ligase RNF168 3. Inhibition of RNF168 at telomeres involves the de-ubiquitinating enzyme BRCC3 and the ubiquitin ligase UBR5 and is sufficient to suppress chromosome end-to-end fusions. This two-step mechanism for TRF2-mediated end protection helps to explain the apparent paradox of frequent localization of DNA damage response proteins at functional telomeres without concurrent induction of detrimental DNA repair activities.
Keywords: telomere, genomic stability, ATM, DNA damage, NHEJ, TRF2, RNF168, Brca1
In mammalian cells, protection of chromosome ends requires TRF2 4. When telomeres become critically short, insufficient recruitment of TRF2 leads to telomere deprotection and initiation of a DNA damage response at chromosome termini. Indeed, TRF2-depleted telomeres elicit the same response as critically short telomeres, such as recruitment of DNA damage response factors (e.g. MDC1, RNF8, and 53BP1) 2,5,6, activation of a cell cycle checkpoint, and subsequent repair activities resulting in end-to-end chromosome fusions 1,7. Amongst the telomere binding proteins, TRF2 is unique in its ability to suppress the ATM-dependent DNA damage response (DDR) pathway and the non-homologous end joining (NHEJ) pathway 2,8.
To define the unique molecular properties of TRF2 that are involved in end protection, we performed a domain-swapping approach between TRF2 and the structurally similar but functionally divergent telomere binding protein, TRF1 9. TRF2 and TRF1 are both composed of 4 domains: a C-terminal Myb Domain required for binding to double stranded telomeric DNA (TTAGGG), a flexible Hinge domain involved in protein-protein interaction, a TRFH domain required for homodimerization 10, and a divergent N-terminal domain (Fig. 1a). The N-terminal domain of TRF2 is rich in basic residues (basic domain), while the TRF1 N-terminal domain is composed of acidic residues (acidic domain) (Fig.1a). We generated a set of chimeric Telomere Repeat Factors (TRFc) in which the corresponding domains in TRF1 replace TRF2 domains. The resulting alleles were tested for their ability to complement for the loss of endogenous TRF2 using TRF2 conditional MEFs (mouse embryo fibroblasts) 1. To ensure synchronous and complete TRF2 depletion we used TRF2F/F cells carrying an inducible CRE recombinase (R26-CreERT2) that can be activated by 4-hydroxytamoxifen (OHT) (Supplemental Fig. 1). All the TRFc alleles analyzed showed the expected telomere localization in the presence or absence of endogenous TRF2 (Supplemental Fig. 2a-d). Importantly, ectopic TRF1 expression cannot complement for the loss of TRF2 as frequent DNA damage foci at telomeres, termed Telomere Induced Foci (TIFs), and chromosome end-to-end fusions still appear (Fig. 1b-d). On the contrary, a TRF2 allele lacking the N-terminal basic domain (TRF2ΔB) is able to suppress TIF formation and end-to-end chromosome fusions (Fig. 1b-d). Similarly, an allele of TRF2 in which the DNA binding domain was replaced with TRF1 Myb domain (TRFcM) can complement for loss of TRF2 (Fig. 1b-d). These data show that, in vivo, the specific ability of TRF2 to protect chromosome ends cannot be explained by a specificity of its DNA binding domain, or by its unique N-terminal basic domain. In contrast, both the TRFH domain and the Hinge domain are required to prevent the initial steps in the DNA damage response pathway as assessed by γH2AX localization at TRFcT and TRFcH -bound telomeres (Fig. 1b-c). Interestingly, we found that localization of the TRFcT allele at telomeres is sufficient to inhibit recruitment of key mediators of the DNA damage response pathway downstream of γH2AX such as 53BP1 (Fig. 1c, Supplemental Fig. 3a-c and Supplemental Fig. 8). In agreement with this observation, the TRFcT allele can also prevent chromosome fusions (Fig. 1d, Supplemental Fig. 7, Supplemental Table 1), a process that requires 53BP1 recruitment 11,12. We exclude that the TRFcT has a dominant negative effect since its expression in TRF2 proficient cells does not result in DNA damage induction (Supplemental Fig. 5). In contrast, in cells expressing the TRFcH both γH2AX and 53BP1 localize to telomeres (Fig. 1b-c, Supplemental Fig. 3a-c). Interestingly, despite levels of TIFs that are comparable to those observed in TRF2-/- cells, telomeres are partially protected from chromosome fusions (Fig. 1d, Supplemental Fig. 7, Supplemental Table 1). In agreement with what observed in TRF2 -/- cells2 we found that the DNA damage response initiated at TRFcT or TRFcH -bound telomeres is ATM-dependent (Supplemental Fig. 6). Our finding suggests that the TRFH domain is required to prevent the initial step of the DDR response, but other portions of TRF2 can independently suppress propagation of this signal to downstream effectors.
Figure 1. Critical role for the TRFH domain and the Hinge domain of TRF2 in end-protection.
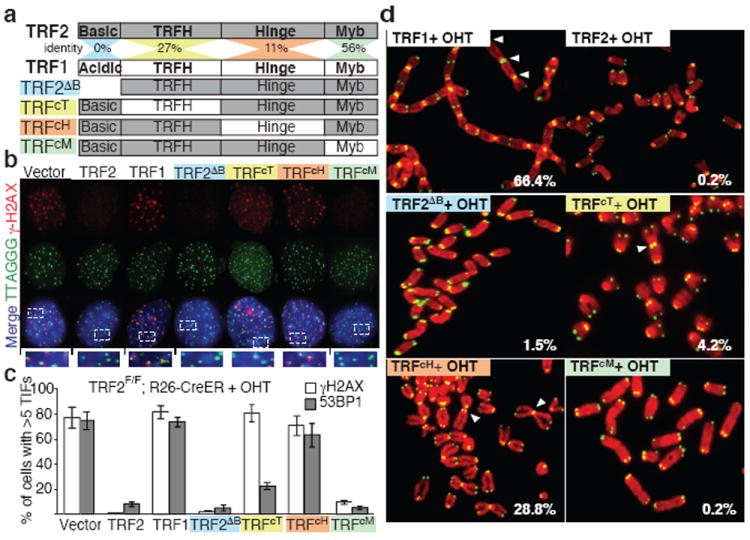
a Schematic representation of the TRF alleles. b The indicated MEFs were treated with 4-hydroxytamoxifen (+OHT) and stained for γH2AX, telomere DNA, and DAPI (blue). c Quantification of cells with 5 or more 53BP1 (or γH2AX) foci at telomeres, s.d. is derived from three experiments. d Metaphase spreads of MEFs treated as in c were stained for telomere DNA (green) and DAPI (red). Percentages of chromosomes fusions are indicated.
Next, we addressed what step in the DDR response is inhibited by TRF2 by testing the localization of MDC1, RNF8, and RNF168 DNA damage factors that are downstream of γH2AX and upstream of 53BP113. Upon TRF2 depletion, γH2AX, MDC1 and, RNF8 localize to telomeres in cells expressing TRFcT, confirming that the TRF2 TRFH domain is required to prevent the initial steps in the DNA damage response pathway (Fig. 2a,b, and d). Similarly, we did not detect defects in SUMO1 accumulation at telomeres (data not shown). In contrast, the ubiquitin ligase RNF168 does not localize to TRFcT-bound telomeres (Fig. 2c and d). We exclude that this is due to a general inhibition of RNF168 activity in these cells as they readily form RNF168 irradiation induced foci (IRIFs) (Supplemental Fig. 9). Recruitment of RNF168 at sites of damage is required for efficient 53BP1 recruitment 3,14, which in turn promotes chromosome fusions12.
Figure 2. Inhibition of RNF168 recruitment at chromosome ends.
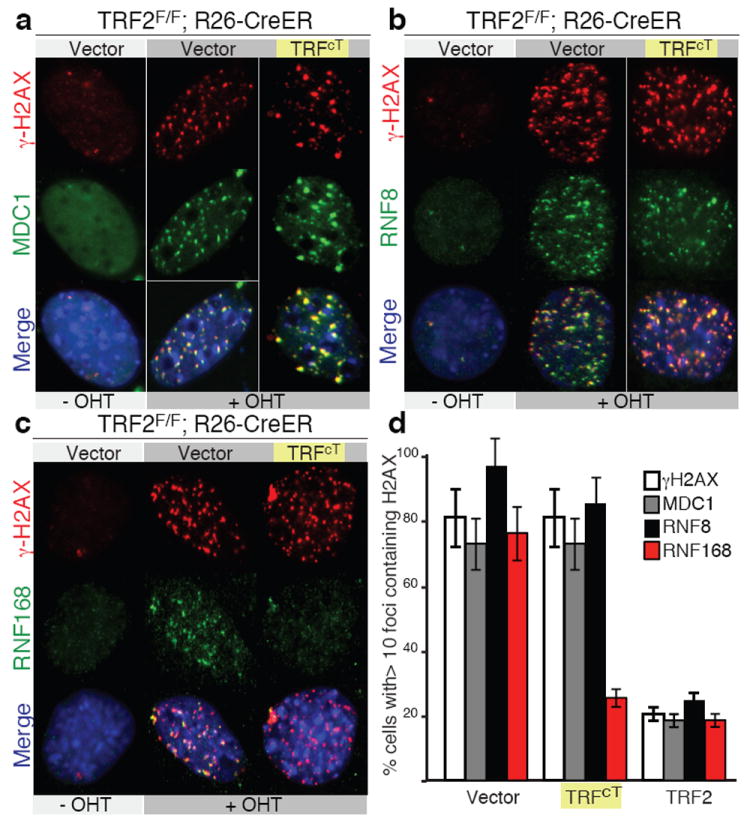
a and b MEFs infected with the indicated constructs and expressing MDC1-GFP (panel a) or RNF8 GFP (panel b) were treated with OHT and stained for γH2AX, GFP and, DAPI (blue). c MEFs infected with the indicated constructs were stained for γH2AX, RNF168 and, DAPI (blue). d Quantification of data presented in panels a to c, median values and s.d. are derived from 3 independent experiments.
To identify the critical region of TRF2 involved in the suppression of RNF168 recruitment, we focused on the Hinge domain given the high frequency of 53BP1 TIFs observed in cells expressing the TRFcH (Fig. 1d). The main function attributed to this domain to date is the interaction with RAP1 and TIN2, two members of the shelterin complex that have been implicated in end protection 15,16,17. We excluded a role for RAP1 in inhibiting RNF168 recruitment since deletion of the RAP1 interaction motif (aa 286-299) 18 in the context of the TRFcT allele did not result in 53BP1 localization to telomeres (Fig. 3a-b and, Supplemental Fig. 11b). Similarly, deletion of the TIN2 interaction motif (aa 352-367) 19 resulted only in a minor induction of 53BP1 accumulation, thus excluding a critical role for this interaction in the suppression of RNF168 at telomeres (Fig. 3 a-b, Supplemental Fig. 11b). This implicated the C- terminal portion of the Hinge domain (aa 407-431), a region that by sequence alignment shows a high degree of conservation between species (Supplemental Fig. 10). Deletion of this region, that we have termed the Inhibitor of DNA Damage Response (iDDR) region, in the context of the TRFcT allele, resulted in levels of 53BP1 that are comparable to those observed in TRF2-/- cells (Fig. 3a-b, Supplmental Fig. 11). To further validate this finding and to test whether this region is also sufficient to prevent DDR activation, we expressed the iDDR region in the context of TRF1 (Fig. 3a). Strikingly, the resulting TRF1iDDR allele can complement the phenotypes associated with TRF2 loss with significant inhibition of 53BP1 and RNF168 localization and chromosome fusions (Fig. 3b-c and Supplemental Fig. 11b, f and Supplemental Fig. 12). In contrast, expression of the TRF1iDDR allele could not fully complement loss of TRF2 in regard to γH2AX localization at telomeres (Fig. 3b and Supplemental Fig. 11c) a result that is in agreement with our data indicating a critical role for the TRFH domain of TRF2 in suppressing the initial activation of the DNA damage response.
Figure 3. The C-terminal portion of TRF2’s Hinge domain is necessary and sufficient to prevent 53BP1 localization at telomeres.
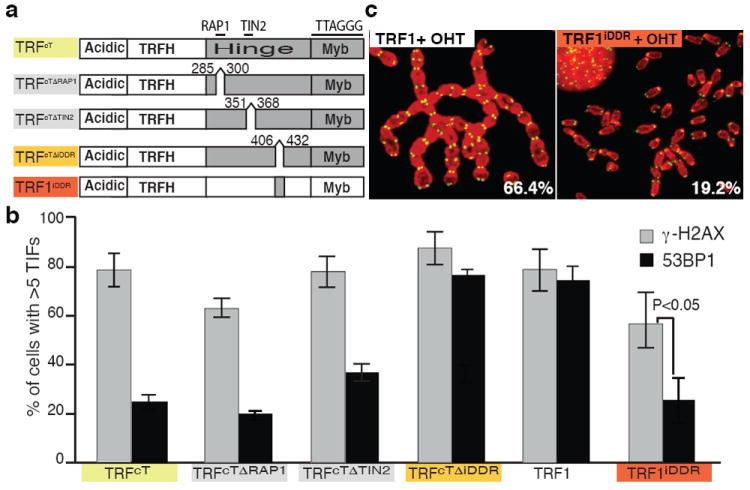
a Schematic representation of the alleles used to define the role of the Hinge domain in end protection. b Quantification of cells that shows 5 or more 53BP1 (or γH2AX) foci colocalizing at telomeres, s.d. is derived from three experiments. Cells expressing the indicated constructs were treated with OHT to deplete endogenous TRF2. c Metaphase spreads of MEFs infected with either TRF1 or TRF1iDDR and treated with OHT were stained for telomere DNA (green) and DAPI (red). Percentages of chromosomes with fusions are indicated.
We then addressed the mechanism for the iDDR-dependent inhibition of RNF168 recruitment to dysfunctional telomeres. RNF168 is a ubiquitin ligase that is recruited to damaged chromatin by ubiquitin chains generated by the RNF8-UBC13 complex 3,14. Ubiquitinated proteins can be detected at dysfunctional telomeres with an antibody raised against conjugated ubiquitin (FK2, Supplemental Fig. 13e). In contrast, we did not detect conjugated ubiquitin at TRFcT-bound telomeres (Supplemental Fig. 13e) suggesting a defective ubiquitin-mediated signaling in this context. Two mechanisms can explain this phenotype; inhibition of RNF8 activity, or recruitment of deubiquitinating (DUB) enzymes. Since RNF8 is localized at telomeres in cells expressing the TRFcT allele (Fig. 2b), we focused on the two DUB enzymes that have been shown to counteract the action of RNF8-UBC13 at sites of DNA damage: OTUB1 and BRCC3 20,21. We reasoned that if either of these DUBs plays a role in TRF2-mediated end protection, then reducing their levels should result in 53BP1 recruitment to telomeres in cells expressing the TRFcT allele. Efficient (>90%) shRNA-mediated depletion of OTUB1 did not result in 53BP1 foci formation (Supplemental Fig. 13a-b). In contrast, two independent shRNA constructs directed against BRCC3 (knockdown efficiency 95% and 98% respectively, Supplemental Fig. 13c-d) resulted in the accumulation of 53BP1 and ubiquitin chains at telomeres in cells expressing the TRFcT allele (Fig. 4a and Supplemental Fig. 13e). Moreover, BRCC3 depletion in these cells resulted in levels of end-to-end chromosome fusions that are comparable to what observed in TRF2-/- cells (Supplemental Figure 14). This latter result further corroborates the previous observation suggesting a critical role for 53BP1 localization for efficient DNA repair events.
Figure 4. Mechanism of TRF2-mediated inhibition of RNF168.
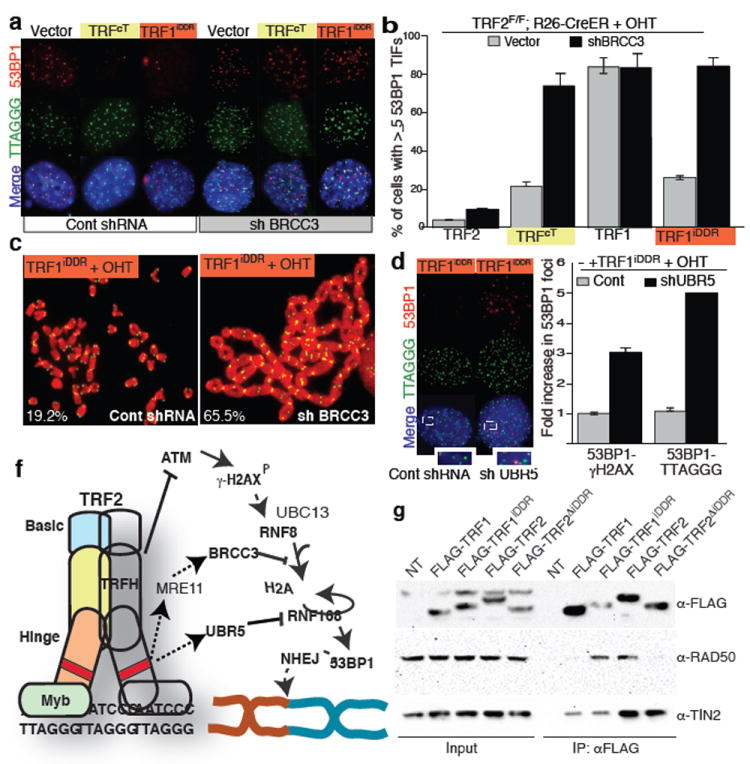
a Cells were infected with a BRCC3 shRNA (or a control), treated with OHT and stained for 53BP1 and telomere DNA. b Quantification of panel a. c Metaphases of TRF1iDDR expressing cells treated as described in a were stained for telomere DNA (green) and DAPI (red). Percentages of chromosome fusions are indicated. d TRF1iDDR expressing MEFs infected with UBR5 shRNA or a control shRNA, were treated as described in panel a. e Quantification of cells with 53BP1 co-localizing with γH2AX (or telomere DNA (TTAGGG)). Data are normalized to the control samples. f Schematic of the proposed model for TRF2 mediated chromosome-end protection, dashed lines indicate that additional factors may be involved. g HEK293 cells were transfected with the indicated constructs. Cell extracts were immunoprecipitated (IP) with anti-FLAG resin and immunoblotted as indicated.
To verify whether BRCC3-mediated suppression of the DNA damage response is associated with the iDDR region of TRF2 we tested whether TRF2-/- cells expressing the TRF1iDDR allele require BRCC3 expression to ensure end protection. Indeed, shRNA mediated downregulation of BRCC3 in these cells abolishes the protective role of the TRF1iDDR allele resulting in levels of 53BP1 localization and chromosome fusions that are comparable to the ones observed in TRF2-/- cells (Fig. 4a-c).
To identify proteins that can be recruited at telomeres by the iDDR region of TRF2, FLAG-tagged TRF1iDDR was immunopurified and analyzed by mass spectrometry (Supplemental Fig.17 and Supplemental Table 3). As a negative control, we used FLAG-TRF1. The members of the MRE11 complex (MRE11, NBS1, and RAD50) and the ubiquitin ligase UBR5 were identified in TRF1iDDR immunoprecipitates but not in TRF1 immunoprecipitates in 3 independent experiments (Supplemental Fig. 17). We confirmed that the iDDR region of TRF2 is required and sufficient to interact with RAD50 (Fig. 4g). Interestingly, the MRE11 complex has been shown to interact directly with BRCA1 22,23. We therefore hypothesized that members of the MRE11 complex could recruit BRCC3. Indeed, we found that NBS1 interacts with BRCC3 in co-IP experiments and thus can provide a physical link between TRF2 and BRCC3 (Supplemental Fig. 17f). Based on these data we propose a model in which TRF2, by its established interaction with the MRE11 complex 24, is able to recruit the BRCC3 enzyme at telomeres, which in turn can suppress RNF168 recruitment. However, we cannot exclude that additional unknown mechanisms are involved in BRCC3 recruitment to telomeres. In addition, we found that the ubiquitin ligase UBR5 is required to mediate the iDDR-mediated end protection observed in TRF2-/- cells expressing the TRF1iDDR allele (Fig. 4 d,e and Supplemental Fig. 18). Recent reports have shown that UBR5 targets RNF168 for degradation 25. We therefore propose a model in which RNF168 recruitment at telomeres is opposed by the action of BRCC3 and UBR5 (Fig 4f).
In summary, we found that two distinct regions within TRF2 are required to prevent activation of the DNA damage response pathway at chromosome ends (Fig. 4f). Our data indicate that the TRFH domain of TRF2 can prevent activation of the ATM pathway independently from other regions of TRF2. Together with previous observations, this suggests that this region might be involved in t-loop formation, a structural conformation of chromosome ends that has been proposed to hide the ends of chromosomes from the DNA damage machinery 26. In addition, we have identified a novel function for TRF2 downstream of ATM activation and dependent on a portion of the Hinge domain that we named inhibition of the DNA damage response (iDDR). TRF2 can sever the DNA damage signaling cascade at the level of RNF168, preventing 53BP1 localization and, consequently chromosome fusions. This finding provides a model to explain the apparent paradox of frequent localization of DNA damage proteins at functional telomeres 24,27. Interestingly, in S. pombe inhibition of 53BP1 recruitment at telomeres involves modulation of the methylation status of Histone H4 28. Our data suggest that in mammalian cells a similar effect is achieved by inhibiting ubiquitin-dependent signaling at chromosome ends. This novel end protection role for TRF2 is mediated by the BRCA1 complex through its associated DUB enzyme BRCC3 and by the ubiquitin ligase UBR5. In support of a critical role for the BRCA1 complex in chromosome end protection and in agreement with previous reports 29,30 we show that inhibition of BRCA1, RAP80, or BRCC3 results in partial loss of end protection (Supplemental Fig 16). The identification of BRCC3 as a critical factor involved in TRF2-dependent telomere protection suggests that an important physiological function of the BRCA1 complex is to maintain genomic stability aiding telomere associated proteins in maintaining telomere integrity.
Methods
Mice and MEFs
Rosa26 CRE-ER Mice (Jackson) and mice carrying a conditional TRF2 allele 1 were crossed to generate mouse embryo fibroblasts (MEFs). MEFs were immortalized with pBabeSV40LT, and treated 4-hydroxytamoxifen (0.6 μM) to induce CRE-mediated recombination.
Constructs, Plasmids and viral infections
TRF chimera constructs were generated by PCR amplification using as templates pBabe-myc-TRF1 or pBabe-myc-TRF2 constructs (primers used are listed in Supplemental Table 2). pLDT-GFP-RNF8, pLDT-GFP-RNF168 and, GFP-MDC1, were a gift from Matthew D. Weitzman (U. Penn). pOZ-FH-BRCC3 was obtained from Addgene (#27496). pcDNA-Myc-Mre11 and pcDNA-Myc-NBS1 were a gift from Dr. Xiaohua Wu.
IF, FISH and ChIP
Immunoflourescence, FISH and ChIP experiments were performed as described previously2. The following antibodies were used: Myc (9B11, Cell signaling), CHK2 (BD biosciences), hRad50 (Novus, NB100-154SS), hUBR5 (EDD) (Santa Cruz, sc-9562), FLAG (sigma, F7425) or HA (Covance, 16B12), γH2AX (Millipore, JBW301), 53BP1 (Novus, NB 100-304), GFP (Invitrogen, A6455), BRCA1 (a gift from Dr. Xiaochun Yu), RNF168 (a gift from Dr. Daniel Durocher), FK2 (Millipore, 04-263). For IP the following antibodies were used: Myc (9B11, Cell signaling), FLAG M2 affinity gel (Sigma, A2220). Quantification of IF experiments was performed counting at least 200 cells/ condition. Data from three independent experiments were used to calculate median value and standard deviation.
shRNA
pLKO lentiviral vectors were used to express shRNAs directed against the following targeting sequences: GCTCAGTATTTACCAAGAATT (BRCC3), GTCCATCCAAGTGGAGTACAT (OTUB1), CCCATTCAGTATCCTGGCTT (RAP80), AACCAGATGTCTGTACTAAGG (BRCA1).
Purification of protein interacting with the iDDR region
HEK293 cells were transfected with FLAG–tagged TRF1 or FLAG–tagged TRF1iDDR. Cells were lysed (50 mM Tris–HCl at pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.5% TritonX-100) and immunopurified with anti-FLAG agarose resin (Sigma). After washing, proteins were eluted by competition with FLAG peptide (Sigma). For mass spectrometry analysis samples were denatured, reduced and alkylated prior to an overnight digestion with trypsin. Peptide mixtures were analyzed by nanoflow liquid chromatography mass spectrometry using an Eksigent nanopump (Dublin, CA) and LTQ-Orbitrap mass spectrometer (Thermo Scientific, Bremen, Germany) using a 7 step MudPIT separation. MS/MS spectra were collected in a data dependent fashion and resulting spectra were extracted using RawXtract. Protein identification was done with Integrated Proteomics Pipeline (IP2) by searching against UniProt Human database and filtering to 1% false positive at the spectrum level using DTASelect. IPs between FLAG-tagged TRFs and RAD50 were performed on nuclear extracts.
Supplementary Material
Acknowledgments
We thank Titia de Lange, Matthew D. Weitzman, Dan Durucher, Xiaochun Yu and, Xiaohua Wu for providing critical reagents. We are grateful to Agnel Sfeir, Travis Stracker, Kyle Miller, and Claire Attwooll for critical reading of the manuscript. This work was supported by a Pew Scholars Award (E.L.D.), the Novartis Advanced Discovery Institute (E.L.D.), NIH AG038677 (E.L.D.), National Center for Research Resources (5P41RR011823-17) (J.R.Y.) and National Institute of General Medical Sciences (8 P41 GM103533-17) (J.R.Y.).
Footnotes
Contribution
E.L.D. and K.O. conceived the experimental design. K.O., C.B., I.O. and E.L.D. performed the experiments and analyzed the data. J.K.D. and J.R.Y. performed the mass spectrometry analysis. E.L.D. wrote the manuscript.
References
- 1.Celli GB, de Lange T. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat Cell Biol. 2005;7:712–718. doi: 10.1038/ncb1275. [DOI] [PubMed] [Google Scholar]
- 2.Denchi EL, de Lange T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature. 2007;448:1068–1071. doi: 10.1038/nature06065. [DOI] [PubMed] [Google Scholar]
- 3.Stewart GS, et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell. 2009;136:420–434. doi: 10.1016/j.cell.2008.12.042. [DOI] [PubMed] [Google Scholar]
- 4.de Lange T. Protection of mammalian telomeres. Oncogene. 2002;21:532–540. doi: 10.1038/sj.onc.1205080. [DOI] [PubMed] [Google Scholar]
- 5.di Fagagna FD, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 6.Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol. 2003;13:1549–1556. doi: 10.1016/s0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]
- 7.van Steensel B, Smogorzewska A, de Lange T. TRF2 protects human telomeres from end-to-end fusions. Cell. 1998;92:401–413. doi: 10.1016/s0092-8674(00)80932-0. [DOI] [PubMed] [Google Scholar]
- 8.Smogorzewska A, Karlseder J, Holtgreve-Grez H, Jauch A, de Lange T. DNA ligase IV-dependent NHEJ of deprotected mammalian telomeres in G1 and G2. Curr Biol. 2002;12:1635–1644. doi: 10.1016/s0960-9822(02)01179-x. [DOI] [PubMed] [Google Scholar]
- 9.Broccoli D, Smogorzewska A, Chong L, de Lange T. Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2. Nat Genet. 1997;17:231–235. doi: 10.1038/ng1097-231. [DOI] [PubMed] [Google Scholar]
- 10.Broccoli D, Smogorzewska A, Chong L, deLange T. Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2. Nature Genetics. 1997;17:231–235. doi: 10.1038/ng1097-231. [DOI] [PubMed] [Google Scholar]
- 11.Difilippantonio S, et al. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature. 2008;456 doi: 10.1038/nature07476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dimitrova N, Chen YC, Spector DL, de Lange T. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature. 2008;456:524–528. doi: 10.1038/nature07433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lukas J, Lukas C, Bartek J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat Cell Biol. 13:1161–1169. doi: 10.1038/ncb2344. [DOI] [PubMed] [Google Scholar]
- 14.Doil C, et al. RNF168 Binds and Amplifies Ubiquitin Conjugates on Damaged Chromosomes to Allow Accumulation of Repair Proteins. Cell. 2009;136:435–446. doi: 10.1016/j.cell.2008.12.041. [DOI] [PubMed] [Google Scholar]
- 15.Sfeir A, et al. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. 2009;138:90–103. doi: 10.1016/j.cell.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sarthy J, Bae NS, Scrafford J, Baumann P. Human RAP1 inhibits non-homologous end joining at telomeres. Embo J. 2009;28:3390–3399. doi: 10.1038/emboj.2009.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takai KK, Kibe T, Donigian JR, Frescas D, de Lange T. Telomere Protection by TPP1/POT1 Requires Tethering to TIN2. Molecular Cell. 44:647–659. doi: 10.1016/j.molcel.2011.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sfeir A, Kabir S, van Overbeek M, Celli GB, de Lange T. Loss of Rap1 induces telomere recombination in the absence of NHEJ or a DNA damage signal. Science. 327:1657–1661. doi: 10.1126/science.1185100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Y, et al. A shared docking motif in TRF1 and TRF2 used for differential recruitment of telomeric proteins. Science. 2008;319:1092–1096. doi: 10.1126/science.1151804. [DOI] [PubMed] [Google Scholar]
- 20.Nakada S, et al. Non-canonical inhibition of DNA damage-dependent ubiquitination by OTUB1. Nature. 466:941–946. doi: 10.1038/nature09297. [DOI] [PubMed] [Google Scholar]
- 21.Shao G, et al. The Rap80-BRCC36 de-ubiquitinating enzyme complex antagonizes RNF8-Ubc13-dependent ubiquitination events at DNA double strand breaks. Proc Natl Acad Sci U S A. 2009;106:3166–3171. doi: 10.1073/pnas.0807485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y, et al. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 2000;14:927–939. [PMC free article] [PubMed] [Google Scholar]
- 23.Chen L, Nievera CJ, Lee AY, Wu X. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J Biol Chem. 2008;283:7713–7720. doi: 10.1074/jbc.M710245200. [DOI] [PubMed] [Google Scholar]
- 24.Zhu XD, Kuster B, Mann M, Petrini JH, de Lange T. Cell-cycle-regulated association of RAD50/MRE11/NBS1 with TRF2 and human telomeres. Nat Genet. 2000;25:347–352. doi: 10.1038/77139. [DOI] [PubMed] [Google Scholar]
- 25.Gudjonsson T, et al. TRIP12 and UBR5 Suppress Spreading of Chromatin Ubiquitylation at Damaged Chromosomes. Cell. 150:697–709. doi: 10.1016/j.cell.2012.06.039. [DOI] [PubMed] [Google Scholar]
- 26.Griffith JD, et al. Mammalian telomeres end in a large duplex loop. Cell. 1999;97:503–514. doi: 10.1016/s0092-8674(00)80760-6. [DOI] [PubMed] [Google Scholar]
- 27.Verdun RE, Crabbe L, Haggblom C, Karlseder J. Functional human telomeres are recognized as DNA damage in G2 of the cell cycle. Mol Cell. 2005;20:551–561. doi: 10.1016/j.molcel.2005.09.024. [DOI] [PubMed] [Google Scholar]
- 28.Carneiro T, et al. Telomeres avoid end detection by severing the checkpoint signal transduction pathway. Nature. 467:228–U124. doi: 10.1038/nature09353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Al-Wahiby S, Slijepcevic P. Chromosomal aberrations involving telomeres in BRCA1 deficient human and mouse cell lines. Cytogenetic and Genome Research. 2005;109:491–496. doi: 10.1159/000084208. [DOI] [PubMed] [Google Scholar]
- 30.McPherson JP, et al. A role for Brca1 in chromosome end maintenance. Hum Mol Genet. 2006;15:831–838. doi: 10.1093/hmg/ddl002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.