

Summary
Neurotransmitter spillover represents a form of neural transmission not restricted to morphologically defined synaptic connections. Communication between climbing fibers (CFs) and molecular layer interneurons (MLIs) in the cerebellum is mediated exclusively by glutamate spillover. Here, we show how CF stimulation functionally segregates MLIs based on their location relative to glutamate release. Excitation of MLIs that reside within the domain of spillover diffusion coordinates inhibition of MLIs outside the diffusion limit. CF excitation of MLIs is dependent on extrasynaptic NMDA receptors that enhance the spatial and temporal spread of CF signaling. Activity mediated by functionally segregated MLIs converges onto neighboring Purkinje cells (PCs) to generate a long-lasting biphasic change in inhibition. These data demonstrate how glutamate release from single CFs modulates excitability of neighboring PCs, thus expanding the influence of CFs on cerebellar cortical activity in a manner not predicted by anatomical connectivity.
Introduction
Mapping neural circuits to establish the pathways of information transfer not only requires a physical representation of connectivity but also an understanding of communication not easily inferred from structure, such as neuro-glial interactions and volume or extrasynaptic transmission (Lichtman et al., 2008; DeFelipe, 2010; Sporns, 2011). While monoamine and peptide signaling are accepted to occur through volume transmission (Fuxe and Agnati, 1991; but see Beckstead et al., 2004), rapid glutamatergic transmission to postsynaptic receptors is largely restricted to morphologically defined synapses. Nevertheless, glutamate can escape from the synaptic cleft (Asztely et al., 1997; for review see Kullmann, 2000) in concentrations sufficient to activate extrasynaptic receptors (Carter and Regehr, 2000; Mitchell and Silver, 2000; Brasnjo and Otis, 2001; Diamond, 2001; Arnth-Jensen et al., 2002; Chen and Diamond, 2002; Wadiche and Jahr, 2005). In theory, extrasynaptic neurotransmitter spillover degrades the capacity for computation due to a loss of ‘synapse specificity’ (Kullmann, 2000; Barbour, 2001), but transmitter spillover has also been shown to synchronize neuronal output (Isaacson, 1999) and improve transmission efficacy (DiGregorio et al., 2002; Sargent et al., 2005).
In the cerebellum, a single climbing fiber (CF) makes hundreds of individual contacts with one Purkinje cell (PC; Palay and Chan-Palay, 1974). CF activation evokes large excitatory postsynaptic currents (EPSCs) due to the numerous synaptic sites and the release of multiple vesicles from each site, a process termed multivesicular release (MVR; Wadiche and Jahr, 2001; Rudolph et al., 2011). Although MVR generates high synaptic glutamate concentration transients, the likelihood of glutamate spillover between sites is minimized because each CF release site is encased by glial membranes that express high densities of glutamate transporters (Lehre and Danbolt, 1998; Xu-Friedman et al., 2001; Tzingounis and Wadiche, 2007). Although there is no evidence for spillover to neighboring CF-PC synaptic receptors (Wadiche and Jahr, 2001), CF stimulation results in spillover-mediated activation of glutamate receptors located on presynaptic terminals (Satake et al., 2000), perisynaptic membranes (Brasnjo and Otis, 2001; Wadiche and Jahr, 2005), and glia (Bergles et al., 1997). CF-dependent glutamate spillover has also been reported to reach molecular layer interneurons (MLIs) in vivo (Jörntell and Ekerot, 2003) and in vitro (Szapiro and Barbour, 2007) despite the absence of presynaptic or postsynaptic specializations at these junctions (Kollo et al., 2006; Brown et al., 2012).
In principle, glutamate spillover could engage local microcircuits not predicted by conventional anatomical mapping as occurs with multiple CF stimulation (Mathews et al., 2012). Here we show that MLIs excited by spillover from a single CF inhibits MLIs outside the spillover limit resulting in a functional dissociation of MLI activity based on proximity to the active CF. Consistent with the role of glutamate transporters limiting spillover (Bergles et al., 1997; Brasnjo and Otis, 2001; Dzubay and Otis, 2002; Wadiche and Jahr, 2005; Szapiro and Barbour, 2007; Tsai et al., 2012), MLI excitation and subsequent inhibition are robustly enhanced by blocking glutamate transporters. Yet even with uptake intact, glutamate spillover activates AMPA and NMDA receptors (NMDARs) on MLIs to promote spiking. The slow time course of spillover transmission enhances the temporal spread of CF-mediated feed-forward inhibition to PCs and other MLIs. The functional segregation of MLIs excited and inhibited by CF spillover enables single CFs to both decrease and increase simple spiking of neighboring PCs, similar to a phenomenon previously demonstrated in vivo (Bloedel et al., 1983).
Results
Climbing fiber glutamate spillover to molecular layer interneurons
We recorded from MLIs (basket and stellate cells) located in the inner two thirds of the molecular layer of acute cerebellar slices maintained near physiological temperature (~ 32°C). Following the work by Szapiro and Barbour (2007), we first isolated putative CF inputs in the presence of the GABAA receptor antagonist SR95531 (5 μM). We used the following criteria to distinguish CF inputs from conventional parallel fiber (PF) inputs onto MLIs: 1) The stimulating electrode position and intensity was adjusted to evoke an all-or-none response with little fluctuation in peak amplitude (Figure 1A and 1B), unlike responses following PF stimulation that were variable and graded (Konnerth et al., 1990); 2) Using an inter-stimulus interval of 50 ms, the paired-pulse ratio (PPR) of CF-MLI responses showed marked depression (EPSC2/EPSC1 = 0.14 ± 0.01, n = 67; Figure 1A) in contrast to PF responses that facilitated (1.37 ± 0.08, n = 22; Figure 1C); 3) The EPSC kinetics were slower (rise: 0.7 ± 0.02 ms, decay: 4.2 ± 0.2 ms, n = 67) than those from PF synaptic connections (rise: 0.3 ± 0.02 ms, decay time constant: 1.4 ± 0.09 ms, n = 22, P < 0.0001 for both measures); 4) The glutamate transporter antagonist, TBOA (50 μM) potentiated the peak amplitude of CF EPSCs by 322 ± 44% (n = 21; Figure 1D top), but did not affect EPSCs following PF stimulation (103 ± 6.0%, n = 8, P = 0.45; Figure 1D bottom). Together, these data recapitulate previous results (Szapiro and Barbour, 2007) and establish the criteria we used to unambiguously distinguish CF stimulation from PF stimulation in subsequent experiments.
Figure 1. Glutamate spillover results in all-or-none feed-forward (FF) IPSCs.
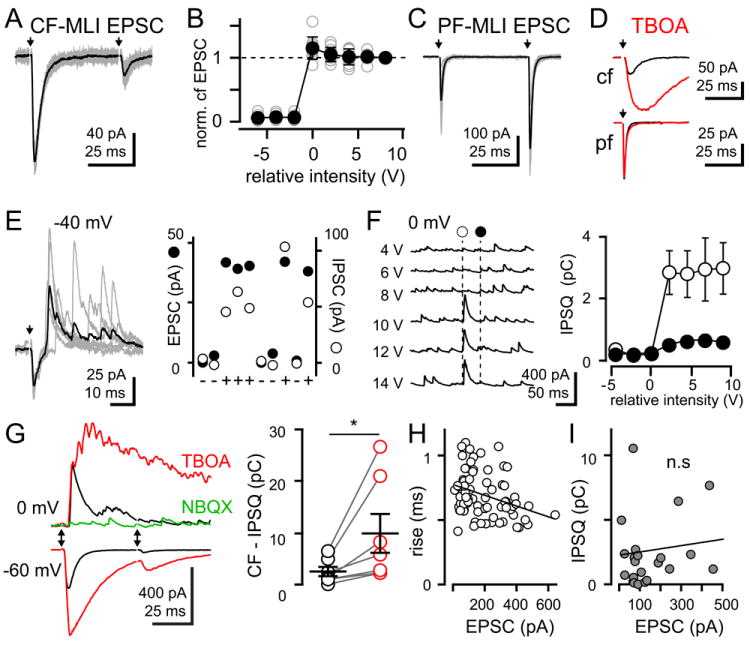
(A) Ten superimposed (grey) and averaged (black) CF-MLI EPSCs (-65 mV) in response to paired-pulse stimulation (50 ms). Arrows denote stimulation.
(B) Plot of CF-MLIs EPSCs normalized to peak amplitude with increasing stimulus intensity. Individual experiments are displayed by grey open symbols and solid circles represent the averaged response ± SEM (n = 9). Recordings in panels A-D are in the presence of SR95531 (5 μM).
(C) PF-MLI EPSCs (-65 mV) in response to paired-pulse stimulation (50 ms).
(D) Blockade of glutamate uptake (50 μM TBOA; red) increases the amplitude and time course of CF- (top), but not PF- (bottom) MLI EPSCs.
(E) Left: CF-MLI biphasic responses (-40 mV with inhibition intact) display outward currents with variable onset latencies. Outward currents were blocked by SR95531 (not shown). Right: Successes (+) and failures (-) of IPSCs and EPSCs are correlated.
(F) Left: Increasing intensity paired stimuli recruits all-or-none CF-MLI IPSCs (0 mV) with marked depression. Dashed lines denote synaptic stimulation with intensity indicated on left. Right: Plot of CF-MLI IPSQs for the first (open circles) and second (filled circles) IPSCs ± SEM.
(G) Representative averaged EPSCs (-60 mV) and IPSCs (0 mV) following CF-stimulation show paired-pulse depression (black) and sensitivity to TBOA (red). Both EPSCs (not shown) and IPSCs are blocked by NBQX (0 mV, green). Right: CF-mediated IPSCs were quantified by measuring the outward charge (IPSQ for 50 ms) before (black circles) and in TBOA (red symbols). Horizontal lines are mean values ± SEM for six cells (each shown with open symbols).
(H) Correlation of EPSC rise times (20 - 80%) versus peak amplitude suggests that larger EPSCs are closer to CF release sites. Solid line is the linear regression fit. Pearson correlation coefficient r2 = 0.12 (P = 0.003).
(I) No correlation between EPSC amplitude and feed-forward inhibitory charge (IPSQ) suggests that interneuron connectivity is uniformly organized. Pearson correlation coefficient r2 = 0.004 (P = 0.8).
To assess spillover at near-physiological [Ca2+], CF-MLI EPSCs were measured in a 1 mM extracellular [Ca2+] solution. On average, responses in 1 mM [Ca2+] were 55.0 ± 3.0 % smaller than those in 2.5 mM [Ca2+] (n = 6, P = 0.01) and showed less pair-pulse depression (0.28 ± 0.03, n = 6, P = 0.03), suggesting that spillover transmission to MLIs occurs at near-physiological release probability.
Spillover currents trigger feed-forward inhibition between molecular layer interneurons
We next asked if CF-mediated glutamate spillover was sufficient to trigger feed-forward inhibition (FFI) from MLIs. Since multiple CF inputs can be detected in a single MLI (Szapiro and Barbour, 2007), we reasoned that spillover from a single CF may also reach several MLIs to trigger APs. The high input resistance and membrane time constants of MLIs assure that even small synaptic inputs will produce large changes in the membrane potential sufficient to elicit firing (Carter and Regehr, 2002).
To identify FFI, we evoked CF-mediated responses in MLIs held at -40 mV, a membrane potential between the EPSC and IPSC reversal potentials. Indeed, FFI was present in our recordings as evidenced by the timing of evoked inward and outward currents following CF-stimulation (Figure 1E, left). While the onset of EPSCs was relatively invariant, outward currents sensitive to inhibition by SR95531 (5 μM, data not shown; n = 16) were measured at varying latencies suggestive of FFI. Accordingly, IPSC failures correlated with EPSC failures suggesting that both required activation of the same CF (Figure 1E, right).
We next recorded at the EPSC reversal potential (~ 0 mV) to verify that the IPSCs originated from CFs rather than from PFs. Since CF stimulation often evoked multiple IPSCs, we quantified the current-time integral of IPSCs (IPSQ) rather than their peak amplitude (50 ms bins). First, IPSCs responded in an all-or-none fashion (Figure 1F). Consistent with a CF-evoked response, the IPSQ depressed with paired-pulse stimulation (IPSQ2/IPSQ1 = 0.14 ± 0.03, n = 8). Furthermore, the average onset latency of the first IPSC was 5.0 ± 0.4 ms (n = 15; Figure 1G black), significantly slower than the EPSC latency recorded at the GABAA receptor reversal potential (~ -60 mV; 2.3 ± 0.2 ms; n = 15, P < 0.0001). CF-MLI signaling was not regulated by GABABRs or cannabinoid receptors as neither EPSCs nor IPSQs were affected by a cocktail of 2 μM CGP55845 and 5 μM AM251 (not shown, n = 4, P = 0.56 for EPSCs and IPSQs). Subsequent inhibition of glutamate transporters with TBOA potentiated both the EPSC peak amplitude and the IPSQ (Figure 1G red) from 2.8 ± 0.9 to 10.1 ± 3.7 pC (n = 7, P = 0.04; Figure 1G right). Finally, NBQX application (Figure 1G green) blocked the CF-IPSC (by 91.7 ± 2.1 %, n = 12) confirming that IPSCs were due to FFI.
For comparison, we recorded conventional feed-forward IPSCs evoked following PF stimulation that were also inhibited by either SR95531 (n = 6) or NBQX (n = 6; Figure S1; Mittmann et al., 2005). Feed-forward PF-IPSCs were readily distinguishable from CF-IPSCs because PF-IPSCs facilitated with paired-pulse stimulation (IPSQ2/IPSQ1 = 1.39 ± 0.25, n = 6) and the PF-IPSC charge (IPSQ) was not significantly altered by TBOA (1.4 ± 0.6 to 1.3 ± 0.5 pC, n = 7, P = 0.87; Figure S1). Together these data show that CF-dependent glutamate spillover recruits FFI between neighboring MLIs to engage unconventional microcircuits.
The glutamate concentration that results from spillover is lower than from conventional synapses (Szapiro and Barbour, 2007) and is expected to be proportional to the distance from CF release sites. The number of glutamate receptors activated and their glutamate binding rate are also proportional to concentration (Patneau and Mayer, 1990; Jonas and Sakmann, 1992). Therefore, if the concentration generated by spillover is in the linear range, EPSC rise times will be inversely proportional to peak amplitude since concentration will determine both the number and rate of receptor activation. Indeed, larger amplitude EPSCs had faster rise times than smaller EPSCs (n = 78; Figure 1H). Variability in CF-MLI EPSC amplitude is less likely to indicate clustering of extrasynaptic receptors, since the same glutamate concentration acting at large or small receptor clusters will affect the amplitude but not the rise time of responses. We also found that the distance between MLIs and the active CF (assayed by the postsynaptic PC) was inversely correlated with the CF-MLI EPSC amplitude (n = 8 pairs; Figure S2). Together, these results indicate that the CF EPSC amplitude in MLIs primarily reflects the extracellular glutamate concentration and, due to dilution of glutamate with increasing distance, the proximity from CF release sites.
In contrast, the amplitude of CF EPSCs, and thus proximity to CF release sites, did not correlate to the quantity of FFI (n = 22; Figure 1I) suggesting that interneuron connectivity is uniformly organized throughout the molecular layer. Together these results suggest that CF release generates spillover EPSCs in MLIs that depend on their proximity to the active CF, with feedforward IPSCs distributed across MLIs independent of their proximity to the active CF.
Spillover-mediated feed-forward inhibition is long-lasting
The CF-EPSC was sensitive to NBQX (10 μM), indicating that AMPA/kainate receptors mediate the majority of the excitatory spillover response. However in 21 out of 26 MLIs, an NBQX-insensitive current remained that was blocked by AP5 (100 μM, 95.5 ± 1.6 % inhibition, n = 4), indicating that NMDA receptors (NMDARs) also contribute to the spillover EPSC. The NMDAR EPSC had characteristic voltage dependence and slow decay time (Τdecay 98.5 ± 10.3 ms, n = 9; Figure 2A). Interestingly, a significant NMDAR response was measured at -50 mV, near the MLI resting potential (EPSC-50mV/EPSC+40mV = 24.1 ± 3.0 %, n = 11; see Chavas and Marty, 2003), suggesting that glutamate released from a single CF is sufficient to evoke NMDAR-responses at physiologically relevant membrane potentials. Thus, we wondered if MLI NMDARs participate in the recruitment of FFI. To test this idea we first isolated CF responses near -60 mV and then stepped the voltage to ~ 0 mV (as shown in Figures 1F - 1G) to measure spillover-mediated IPSCs. CF-stimulation (dotted line) increased the frequency of IPSCs for a prolonged duration (~100 ms) above the background spontaneous activity (black traces; Figure 2B). We quantified IPSQs by generating a latency histogram (in 10 ms bins) that is a measure of the inhibitory conductance (black histogram; Figure 2C). Using this measure, inhibition increased by 839.0 ± 129.4 % (n = 24) following CF stimulation (dotted line) and decayed back to baseline levels with a time course described by the sum of two exponentials: 8.0 ± 0.3 ms (82 ± 2%) and 117 ± 8 ms (n = 24). Blocking NMDARs abolished the slow component of the IPSQs without altering the fast component (821.1 ± 200.4%, n = 12, P = 0.8; Figures 2B and 2C). The time course of the latency histogram followed a single exponential decay of 8.9 ± 0.6 ms (orange histogram, n = 12; Figure 2C) in the presence of AP5, similar to the time course of inhibition recruited by PF stimulation (7.3 ± 0.3 ms, n = 7, P = 0.3, Figure S1C). Thus, CF-mediated FFI has a fast component mediated by AMPAR activation and a slow component mediated by NMDARs. Using the relative weights of the fast and slow time constants, we estimate that approximately 76 ± 5% (n = 23) of the total FFI following CF stimulation in MLIs is due to NMDAR activation.
Figure 2. Long-lasting FF IPSCs are driven by NMDARs.
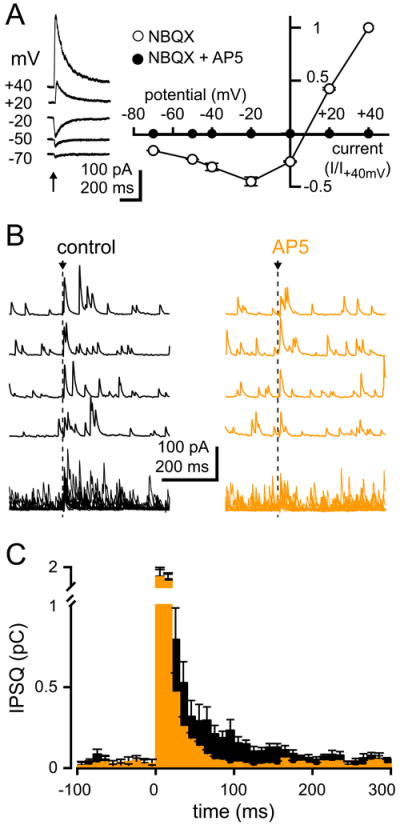
(A) CF-MLI EPSCs recorded in 10 μM NBQX. The current-voltage relationship in NBQX (open circles, n = 9) and in NBQX+AP5 (closed circles, n = 4). Arrow indicates synaptic stimulation.
(B) Individual (top) and superimposed (bottom) MLI-IPSCs following CF-stimulation at 0 mV before (black) and in the presence of AP5 (orange). Arrow and dotted line indicates synaptic stimulation.
(C) Average peri-stimulus histogram of the inhibitory charge (IPSQ, 10 ms bins) with CF stimulation (at 0 ms) in the absence (black, n = 24) or presence of AP5 (orange, n = 12).
Climbing fiber mediated excitation of molecular layer interneurons
The robust and long-lasting increase in IPSCs suggests that MLIs experience a prolonged period of NMDAR-dependent excitability. We tested this directly by measuring the effect of CF-stimulation on spontaneous action potentials (APs) that occurred with a baseline probability of 0.08 ± 0.01 (n = 14; 10 ms bins). CF connectivity was first verified in voltage clamp before switching to current clamp configuration. As shown in Figures 3 and 4, CF stimulation led to a transient and robust increase in the AP frequency evident in raw traces, the raster plots, and peri-stimulus probability histograms (PSH; Figure 3Ai and 3Aii). On average, CF stimulation increased the peak AP probability to 1.24 ± 0.12 (n = 14; Figure 3Aii). Probabilities > 1 reflect multiple APs in each time bin. To measure the net spike output in response to CF stimulation we integrated the PSH to yield the cumulative spike probability, that was then corrected for the spontaneous spike rate (see Experimental Procedures and Mittmann et al., 2005; Figure 3Aii inset). Inhibition of NMDARs by AP5 strongly decreased the number of additional spikes evoked with CF stimulation (from 2.6 ± 0.3 to 1.2 ± 0.2, n = 14, P < 0.001), to a quantity similar to PF stimulation (0.95 ± 0.2 additional APs, n = 3, P = 0.45; Figure S3). Consistent with the idea that CF-mediated increased excitability is due to glutamate spillover, TBOA dramatically increased the peak probability of APs to 2.1 ± 0.14, significantly greater than CF-stimulation alone (0.98 ± 0.02, n = 7 each, P < 0.001; Figure 3Bii). TBOA also enhanced the excitability that is reflected in the PSH and in the cumulative spike probability plot (8.6 ± 1.4 additional APs in TBOA, n = 7, P < 0.01; Figure 3B).
Figure 3. Increased MLI excitability follows CF-stimulation.

(Ai) Examples of spontaneous APs (top) and raster plots (bottom) in the absence (black) or presence of AP5 (orange). Arrows indicate CF stimulation.
(Aii) Peri-stimulus probability histogram (10 ms bins) of APs and cumulative spike probability plot (inset) in the absence (black) or presence of AP5 (orange). Dotted line is the average baseline firing probability. AP5 did not affect the spontaneous AP probability (n = 14, P > 0.05).
(Bi) Examples of spontaneous APs (top) and raster plots (bottom) in the absence (black) or presence of TBOA (red).
(Bii) Peri-stimulus probability histogram (10 ms bins) of APs and cumulative spike probability plot (inset) in the absence (black) or presence of TBOA (red). TBOA did not affect the spontaneous AP probability (n = 7, P > 0.05).
Figure 4. CF-mediated excitation of MLIs.
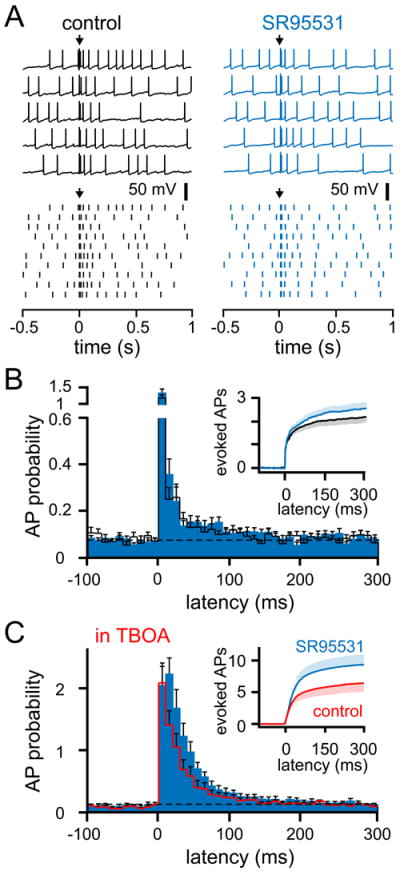
(A) Examples of spontaneous APs (top) and raster plots (bottom) in the absence (black) or presence of SR95531 (blue). Arrows indicate CF stimulation.
(B) Peri-stimulus probability histogram (10 ms bins) of APs and cumulative spike probability plot (inset) in the absence (black) or presence of SR95531 (blue). Dotted line is the average baseline firing probability. In SR95531, the baseline firing frequency was adjusted (see Experimental Procedures).
(C) Peri-stimulus probability histogram of APs and cumulative spike probability plot (inset) in the presence of TBOA (50 μM red) or with SR95531 (blue). Dotted line is the average baseline firing probability.
Because it was necessary to confirm CF- and lack of PF-mediated transmission, the experiments described above were performed in the whole-cell configuration. However, we also verified the influence of CF spillover on spike activity with noninvasive cell-attached recordings. Similar to the results in whole cell configuration, stimulation of CFs transiently increased the peak probability and number of evoked APs to 1.0 ± 0.06 (from 0.12 ± 0.01) and 1.5 ± 0.2, respectively (n = 3). TBOA application further increased the peak AP probability (1.8 ± 0.1) and the number of additional APs (5.7 ± 0.6, n = 3; Figure S4). At the conclusion of each experiment, the membrane patch was ruptured to verify the presence of CF-mediated spillover currents and a lack of PF-mediated transmission. Thus, the cell-attached experiments replicated the whole-cell results, ruling out the possibility that the intracellular ionic composition affected our whole-cell results.
Climbing fiber mediated reciprocal inhibition of excited molecular layer interneurons
Since the time course of IPSQs and AP probability following CF stimulation are similar (Figures 2 and 3), we reasoned that inhibition between MLIs limits excitation. To test this idea directly, we measured the effect of blocking inhibition on the probability, duration and quantity of APs. In addition to increasing the spontaneous AP frequency (see Experimental Procedures; Häusser and Clark, 1997), SR95531 increased the CF-evoked peak AP probability (from 1.2 ± 0.09 to 1.35 ± 0.1, n = 17, P < 0.05; Figure 4A and 4B), and decreased the latency of the first evoked AP from 2.9 ± 0.1 ms to 2.7 ± 0.06 ms, n = 17, P < 0.05). Blocking inhibition also slightly increased the number of added APs (from 2.15 ± 0.15 to 2.53 ± 0.26, n = 17, P < 0.05; Figure 4B inset).
The surprisingly small effect of blocking inhibition may result from several non-mutually exclusive mechanisms. First, we considered whether the quantity of inhibition was too small to robustly affect CF-mediated excitation since CF-mediated FFI is highly variable with some MLIs receiving essentially no FFI (note several cells with ~100 pA EPSCs but almost no inhibition; see Figure 1I). Because different intracellular solutions are required to measure AP probability and IPSC amplitude, we were unable to directly correlate these two measures in the same cells. We, therefore, tested the effects of inhibition in the presence of TBOA that generates a large increase in IPSQ (see Figure 1G). In TBOA, blocking inhibition broadened the AP probability distribution, slightly increased the peak AP probability (from 2.1 ± 0.2 to 2.5 ± 0.2, n = 9, P < 0.05; Figure 4C), and robustly added ~3 APs with each CF stimulation (from 6.3 ± 1.4 to 9.2 ± 1.6, n = 9, P < 0.01; Figure 4C inset). These data support the idea that the size of the inhibition contributes to variability in responsiveness of CF-induced spiking in SR95531.
We also considered whether the effect of SR95531 depends on the location of the EPSP compared to the IPSP. Typically, somatic shunting inhibition strongly inhibits AP firing triggered by dendritic excitation. However, recent work shows that CFs preferentially localize to MLI cell bodies (Brown et al., 2012) suggesting that CF-excitation is generated at the soma and thus may be relatively resistant to shunting inhibition distributed across MLI dendrites (Palay and Chan-Palay, 1974; p. 191). We used dynamic-clamp recordings (Robinson and Kawai, 1993; Sharp et al., 1993) to assess the potential influence of somatic inhibition on CF-evoked APs by imposing an inhibitory conductance while pharmacologically blocking synaptic inhibition. Somatic conductance injections limited CF-MLI spiking demonstrating that somatic inhibition can robustly block somatic excitation (Figure S5; also see Carter and Regehr, 2002). By inference, somatic CF-mediated excitation in combination with dendritic shunting inhibition may contribute to the small effect of CF-inhibition on spiking (Figure 4A and B) although other mechanisms may be at work.
Lastly, although blocking inhibition increased CF-evoked spiking in most cells (12/17), SR95531 application decreased or did not affect spiking in a fraction of cells (5/17), suggesting that [Cl-] concentration at inhibitory synaptic sites may vary. Since dendritic [Cl-] is not clamped by the [Cl-] of the pipette during whole cell recordings (Pearce, 1993; Jarolimek et al., 1999; Khirug et al., 2005), variations in [Cl-] concentration at inhibitory synaptic sites can contribute to the direction and magnitude of the SR95531 block. This variability is consistent with a previous report showing that GABAergic synaptic input triggers spiking in 30% of MLIs (Chavas and Marty, 2003; but see Carter and Regher, 2002). Together our results suggest that reciprocal inhibition between MLIs has only a minor role in limiting spillover-mediated spiking, perhaps due to variability in the magnitude, sub-cellular location and reversal potential of inhibitory conductances.
Spillover-evoked feed-forward inhibition pauses molecular interneuron spiking
That we failed to detect robust regulation of MLI spiking by reciprocal inhibition led us to consider the influence of inhibition outside the limits of glutamate spillover. We explored this idea by identifying MLIs that receive inhibition without excitation following CF-stimulation. First, a MLI that received a typical excitatory CF input was voltage clamped at - 40 mV to directly monitor CF activation (green traces; Figure 5A or see Figure 1E). We then searched for a second MLI (up to 150 μm away) without a spillover-mediated EPSC but with time-locked IPSCs (5.0 ± 0.3 ms, latency range: 3.4 - 7.5 ms, n = 15 out of 25 cells; Figure 5A black traces). These ‘pause-MLIs’ with exclusively CF-mediated FFI had IPSCs with paired-pulse depression that succeeded or failed coincidentally with CF EPSCs in the first MLI (not shown). Our selection criteria was not restricted to synaptically connected MLI pairs because we simply used the first MLI as a readout for CF input. We then switched to current clamp to test the influence of CF stimulation on spontaneous APs. The first MLI responded with increased spiking (as in Figures 3 and 4). The second MLI, however, responded with a delay in spontaneous spiking. Delayed spiking was quantified by aligning the last AP preceding CF stimulation and measuring the first inter-spike interval (ISI). We validated this methodology by comparing the average ISI during a one-second long period (baseline: 99.4 ± 9.5 ms, n = 15) to the ISI of the AP preceding the aligned spike (no stim: 99.9 ± 11.0 ms, n = 15, P = 0.9; Figure 5Bi and 5Bii). CF stimulation increased the ISI to 166.8 ± 23.5 ms (or 204.4 ± 23.7% of control, n = 8, P < 0.001, ANOVA), and this delay was partially blocked by AP5 application (AP5: 126.2 ± 23.7 ms or to 146.6 ± 11.3% of control; n = 8, P < 0.05, ANOVA; Figure 5Bi and 5Bii). In a separate group of cells we tested whether the ISI increase was sensitive to inhibition of glutamate uptake. In voltage clamp, we confirmed that TBOA application did not uncover a CF-mediated EPSC suggesting that the cells tested were located well beyond the spillover limit. In current clamp, TBOA increased the ISI to 232.8 ± 13.3 ms (or to 243.4 ± 17.9 % of control, TBOA, n = 8, P < 0.01, ANOVA; Figure 5Ci and 5Cii) presumably by prolonging spike activity in MLIs receiving spillover excitation (see Figure 3) or by recruiting additional MLIs to spike in response to CF-excitation. Finally, we blocked inhibition with SR95531 to confirm that the CF-dependent delay in spiking results from feed-forward GABAergic circuitry (ISI in SR95531: 114.6 ± 14.3 or 96.3 ± 2.5 % of control, n = 3, P > 0.05, ANOVA; Figure 5Cii). Together, these results indicate that CF-stimulation functionally segregates MLIs depending on their proximity to the climbing fiber; MLIs within the limit of glutamate spillover are excited despite reciprocal inhibition whereas MLIs outside of limit of glutamate spillover are strongly inhibited. It is important to note that these results do not exclude the possibility that some MLIs are excited by GABA because ‘pause-MLIs’ were selected by their outward IPSCs.
Figure 5. Spillover FFI without excitation pauses MLI firing.
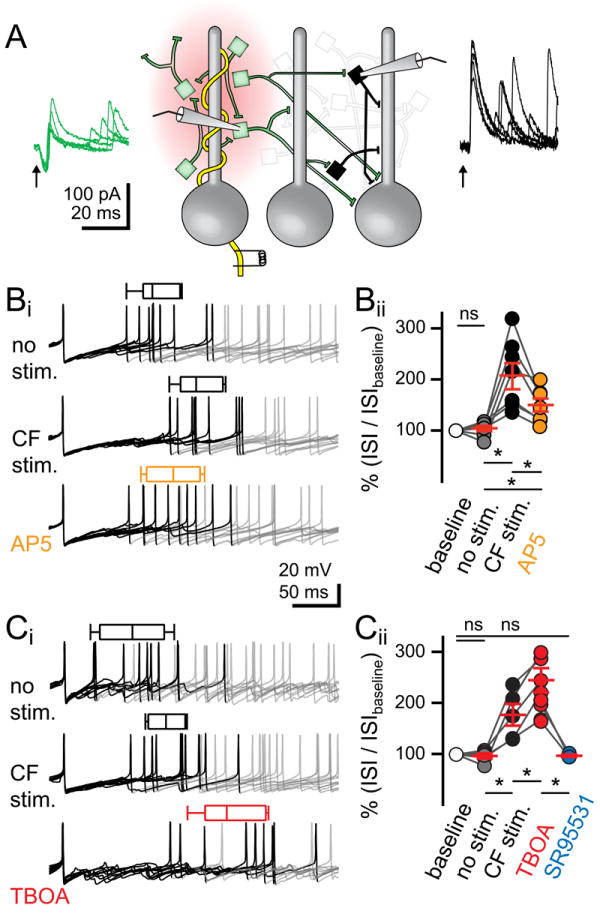
(A) Schematic of paired recording configuration. The first MLI was voltage clamped at -40 mV to monitor CF input (superimposed green traces and cells) within the glutamate spillover limit (red shaded area). A second MLI (black cell, -40 mV) with CF-mediated feed-forward IPSCs without EPSCs (black traces) is outside the glutamate spillover limit. Arrows indicates CF stimulation.
(Bi) Superimposed voltage recordings aligned to the AP preceding CF stimulation. The inter-spike interval (ISI) is the time between the aligned spike and either the preceding spike (no stim.), the subsequent spike following CF stimulation (CF stim.), or the subsequent spike following CF stimulation in AP5 (orange). Box-and-whiskers represent mean, 95% confidence interval of the mean, and SEM for traces in the representative cell. APs following the AP used for the ISI measure have been greyed for clarity.
(Bii) Summary of % change in ISI in each condition normalized to the baseline ISI. Red horizontal bars are the mean ± SEM.
(Ci) Superimposed voltage recordings aligned to the AP preceding CF stimulation for the preceding spike (no stim.), subsequent spike following CF stimulation (CF stim.), subsequent spike following CF stimulation in TBOA (red).
(Cii) Summary of % change in the ISI in each condition.
We also tested whether CF-FFI regulates PF-evoked spiking in MLIs. PF stimulation intensity was set to trigger APs in ~ 50% of trials from MLIs that were hyperpolarized to prevent spontaneous or CF-evoked spiking (0.48 ± 0.05, n = 5; Figure S6 filled circles). CF-triggered FFI that preceded PF stimulation by 5 - 20 ms robustly decreased spike probability (0.17 ± 0.07, n = 5, ANOVA, Figure S6 open circles). The probability of firing a second spike (stimulated at a 5 ms interval) was not altered by CF stimulation (0.56 ± 0.05 compared to 0.64 ± 0.08, n = 5, P > 0.05, ANOVA; Figure S6 filled and open squares, respectively) presumably due to several factors including PF-mediated FFI, PF-mediated paired-pulse facilitation, and a refractory period. These results show that CF stimulation generates robust time-dependent inhibition of PF-mediated spiking and reveals a potential physiological function of CF-FFI in the control of PF-excitation of MLIs.
Spillover feed-forward inhibition pauses Purkinje cell simple spikes
The results presented above establish that CF stimulation can either increase or decrease MLI spike probability, but it is unclear how the aggregate MLI activity will affect downstream Purkinje cells (PCs). We approached this question by using simultaneous dual recordings to test how synaptic CF input to a PC affects excitability of a neighboring PC. We stimulated CF input to the first PC, resulting in a large all-or-none EPSC while simultaneously recording simple spikes from a second, nearby PC (Figure 6A). Peri-stimulus spike probability histograms revealed that CF stimulation (supra-threshold) decreased simple spike probability from 0.08 ± 0.02 to 0.03 ± 0.01, an effect that recovered in ~ 30 ms. In the presence of TBOA, CF stimulation reduced the simple spike probability to 0.02 ± 0.01 for ~ 70 ms (n = 9, Figure 6B). As in Figure 5, we used the first cell as a readout for CF input and analyzed the data from the second PC by aligning the first AP preceding CF stimulation and measuring the first ISI. The ISI of the AP preceding the aligned spike was not significantly different from the average ISI during a one-second long baseline period, thus validating this methodology for PC recordings (baseline: 66.6 ± 7.2 ms and no stimulus: 67.1 ± 7.5 ms, n = 27 each, P >0.05, ANOVA; Figure 6C and 6D). Supra-threshold CF stimulation (measured in first PC) increased the ISI of the subsequent spike to 127.1 ± 6.7 % of control (supra-threshold: 80.7 ± 17.0 ms), significantly more than when the stimulus failed to evoke CF-EPSCs (sub-threshold: 102.7 ± 1.5 % or 71.0 ± 13.2 ms, n = 9, P < 0.01, ANOVA). Consistent with glutamate spillover-activation of MLIs, the ISI increase was sensitive to glutamate uptake inhibition (supra-threshold + TBOA: 164.3 ± 7.6 % or 116.1 ± 25.8 ms, n = 9, P < 0.001, ANOVA) and blocked by GABAAR antagonists (supra-threshold + SR955331: 99.4 ± 4.3 % or 67.0 ± 33 ms, n = 9, P > 0.05, ANOVA). These results indicate that CF-dependent stimulation of MLIs is sufficient to delay the timing of simple spike activity in PCs that are not the postsynaptic target of the CF.
Figure 6. Spillover FFI delays PC spiking.
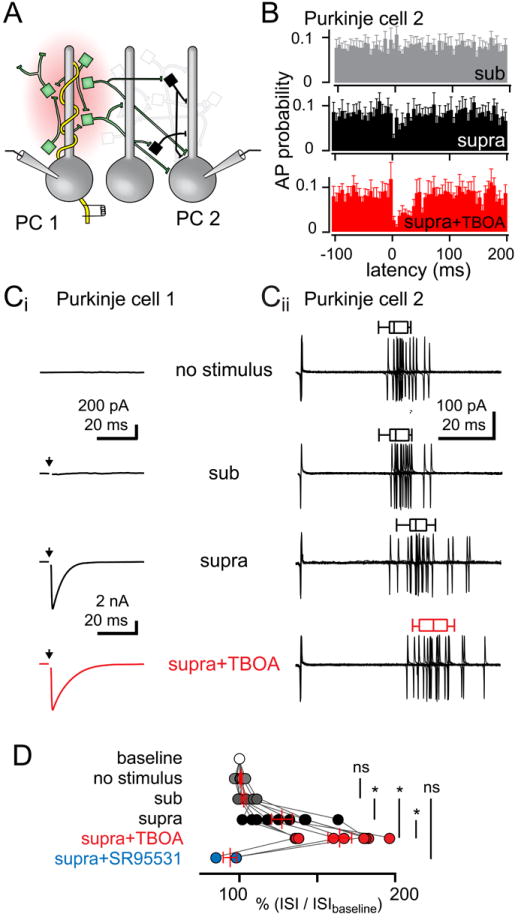
(A) Schematic of paired PC recording configuration. The first PC was voltage clamped to monitor direct CF synaptic input (PC1 with yellow CF) while a second PC was monitored in the cell-attached configuration.
(B) Peri-stimulus probability histogram (5 ms bins) of APs with sub-threshold stimulation (at 0 ms), supra-threshold stimulation or supra-threshold CF stimulation in TBOA.
(Ci) Whole cell currents for PC1 (-10 mV) with no stimulus, sub-threshold stimulation, supra-threshold stimulation, or supra-threshold stimulation in TBOA. Arrows indicate CF stimulation.
(Cii) Cell-attached recordings for PC2 corresponding to conditions in (Ci) aligned to the AP preceding CF stimulation. The inter-spike interval (ISI) is the time between the aligned spike and the preceding spike (no stim.) or the subsequent spike following CF stimulation. Box-and-whiskers represent mean, 95% confidence interval of the mean, and SEM for the representative cell. Current recordings following the AP used for ISI measure were blanked for clarity.
(D) Summary of % change in ISI in each condition normalized to the baseline ISI. Vertical bars are the mean ± SEM.
Climbing fiber stimulation generates biphasic change in inhibition of neighboring Purkinje cells
The pause in PC simple spikes is consistent with excitation of MLIs following CF stimulation (Figure 6), but our data also shows that MLIs located outside the limits of spillover delay their firing in response to CF stimulation (as in Figure 5). We thus predicted that CF stimulation would produce both an increase and decrease in inhibition to neighboring PCs. To test the potential influence of ‘pause-MLIs’ on PCs, we again turned to paired PC recordings and used the large all-or-none CF-PC EPSC as a readout of single CF activation. In a neighboring PC, we first confirmed the lack of CF or PF EPSC and then monitored spillover-mediated feed-forward inhibition with IPSC recordings (Figure 7A and 7B). PCs receive a high frequency of spontaneous IPSCs that contribute to the signal-averaged inhibition (Konnerth et al., 1990; Figure 7B middle and bottom) that were unaffected by sub-threshold CF stimulation (sub-threshold; 110.8 ± 6.4%, n = 24, P > 0.05; Figure 7B). Supra-threshold CF-stimulation evoked phasic all-or-none IPSCs in 22 of 46 paired recordings (supra-threshold; Figure 7B) with an onset latency similar to that measured in MLIs (3.9 ± 0.2 ms, n = 22, P > 0.05). Interestingly, supra-threshold CF stimulation also led to the reduction of spontaneous IPSCs, evident in both the individual traces (middle) and the signal-averaged responses (bottom traces). Time-locked and spontaneous IPSCs were quantified by plotting the inhibitory charge (in 5 ms bins) and generating a latency histogram (Figure 7C). CF-evoked all-or-none phasic inhibition was brief (7.2 ± 0.6 ms half-width, n = 22) and resulted in an increase of charge above spontaneous inhibition (583.6 ± 93.3%, n = 22, P < 0.05). Following phasic inhibition, CF stimulation reduced the charge of spontaneous IPSCs by 91.5 ± 2.8 % (n = 24, P < 0.01), for a duration of 79.9 ± 10.0 ms (half-width, n = 22; Figures 7B and 7Ci). The biphasic change in inhibition persisted in conditions more similar to those occurring in vivo (1.5 mM extracellular Ca2+ and 37°C, Figure S7; Borst, 2010). TBOA application subsequently increased the evoked inhibition in all 9 cell pairs tested, as well as unmasked a CF-evoked IPSC in two additional cell pairs (by 1115.1 ± 422.9 %, n = 11, P < 0.05; and for 14.3 ± 1.8 ms half-width, n = 11). TBOA also prolonged the disinhibition period (115.6 ± 10.8 ms, n = 11, P < 0.05), suggesting that inhibition and disinhibition are generated by CF-spillover to MLIs located near and far away from the stimulated CF, respectively (Figure 7B and 7Cii). Supporting this idea, NBQX application blocked both CF-mediated inhibition and disinhibition, demonstrating that feed-forward circuits are necessary to engage surrounding PCs (109.9 ± 8.4 %, n = 24, P > 0.05; Figure 7B and 7Ciii). Furthermore, AP5 reduced the increase of charge (by 40.6 ± 7.3 %, n = 13, P < 0.05) and the quantity and duration of disinhibition (63.5 ± 11.6 % and 44.7 ± 14.0 ms, n = 13 for each, P < 0.001 and P < 0.005, respectively; Figure 7Civ), illustrating the prominent role of NMDAR activation following CF-evoked activation of MLIs.
Figure 7. Spillover FFI to neighboring PCs generates biphasic change in IPSQ.
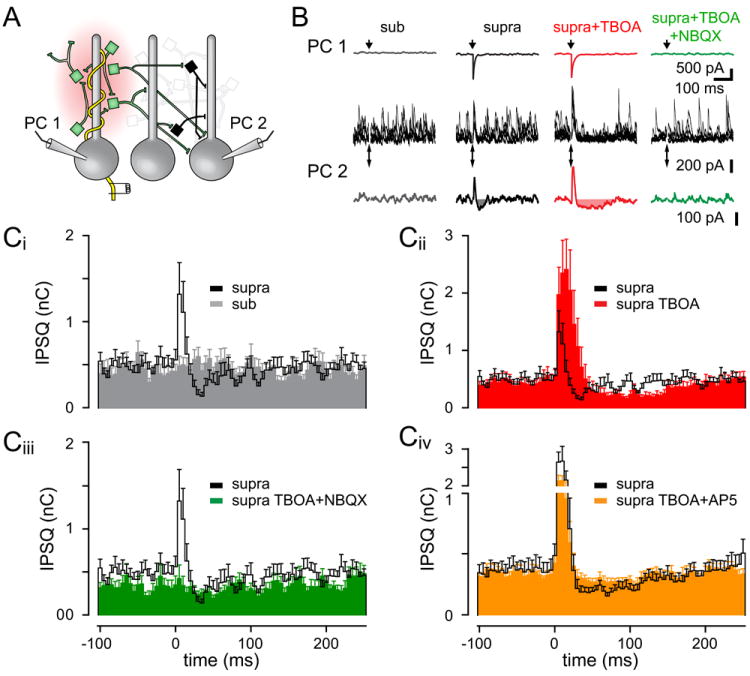
(A) Schematic of paired PC recording configuration. PC1 was voltage clamped to monitor direct CF synaptic input (with yellow CF) while PC2 was voltage clamped at the EPSC reversal potential to monitor IPSCs. Green MLIs are poised to inhibit and the black MLIs will disinhibit PC2.
(B) Top: whole cell currents for PC1 (-20 mV) with either sub-threshold stimulation, supra-threshold stimulation, supra-threshold stimulation in TBOA (red), or supra-threshold stimulation in TBOA+NBQX (green). Middle and bottom: simultaneous individual and averaged IPSCs recorded in PC2. Shaded region denotes disinhibition.
(Ci) Peri-stimulus histogram of the inhibitory charge (IPSQ, 5 ms bins) with sub- (gray) or supra-threshold (black) CF stimulation at 0 ms (n = 9).
(Cii) Peri-stimulus histogram of the inhibitory charge with supra-threshold stimulation in the absence (black) or presence of TBOA (red; n = 9).
(Ciii) Peri-stimulus histogram of the inhibitory charge with supra-threshold stimulation in the absence (black) or presence of NBQX (green; n = 9).
(Civ) Peri-stimulus histogram of the inhibitory charge with supra-threshold stimulation in the absence (black) or presence of AP5 (orange: in the presence of TBOA; n = 13).
Climbing fiber-spillover inhibition regulates parallel fiber-evoked firing probability
Our results show that CF stimulation causes an increase and subsequent decrease in inhibitory charge in PCs not receiving direct CF input. We next performed two sets of complementary experiments designed to study how CF feed-forward activity regulates PC evoked spiking. We used dynamic clamp to test how simulated CF-mediated inhibition controls PF-evoked excitation, and to test how CF-mediated inhibition controls simulated PF excitation. Using dynamic clamp to simulate inhibition or excitation allowed those components to be isolated from other potential stimulus-evoked circuit effects.
First, we simulated a steady state inhibitory conductance that approximates the spontaneous afferent inhibition onto PCs. The probability of PF-mediated spiking during steady state inhibition (PFtest) was compared to spiking during simulated increases and decreases in inhibition (PFinhibition and PFdisinhibition, respectively) modeled after CF-evoked biphasic activity (Figure 8A and red traces in 8Bi). Current was injected to prevent spontaneous spiking and PF stimulation intensity was set to trigger PC spiking in ~50% of trials during steady-state inhibition (PFtest). PF-evoked spiking at the peak of the simulated inhibition was dramatically decreased (from 0.52 ± 0.06 to 0.04 ± 0.02) whereas PF-evoked spiking at the trough of the disinhibition was dramatically increased (from 0.57 ± 0.04 to 0.92 ± 0.04, n = 5 each, P < 0.05 for both measures, paired t-tests; Figure 8Bi and 8Bii). We repeated these experiments in the same neurons with no holding current, allowing PCs to fire spontaneously. Under these conditions, the probability of PF-evoked spiking also decreased with phasic inhibition and trended to increase with disinhibition to 0.11 ± 0.04 and 0.67 ± 0.06; respectively from a control evoked-spiking probability of 0.51 ± 0.06 (n = 5; P < 0.05 and P > 0.05, ANOVA; data not shown). Thus, a simulated CF-mediated biphasic change in inhibition regulates PF evoked PC excitability.
Figure 8. Regulation of PC evoked excitability probability.
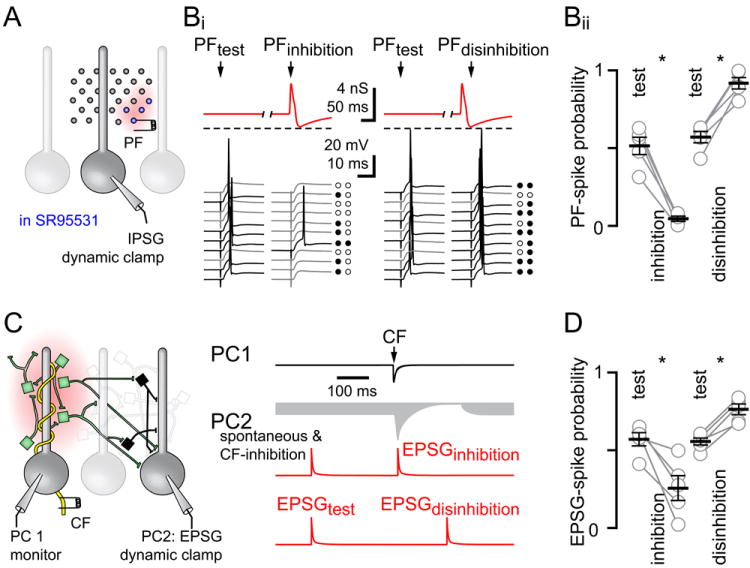
(A and Bi top) Schematic of dynamic clamp recordings. PF induced-spiking was tested during a steady-state inhibitory conductance injection (steady state: 2 nS red traces; PFtest) and during phasic inhibition (PFinhibition: 4 nS; 1 ms rise and 7 ms half-width) or subsequent disinhibition (PFdisinhibition: 0.2 nS; 10 ms rise and 90 ms half-width) that mimicked experimental measures (spontaneous: 2.3 ± 0.3 nS, n = 11; inhibition: 4.5 ± 0.8 nS, n = 33; disinhibition: 0.2 nS = spontaneous * 8.5 ± 2.8 %, n = 24). Dotted lines indicate 0 inhibitory conductance.
(Bi bottom) Representative PC spiking in response to PF stimulation during steady-state IPSG (PFtest), and during simulated CF-mediated inhibition (PFinhibition) or disinhibition (PFdisinhibition). Black and grey traces indicate spike success and failures, also shown as filled and empty circles.
(Bii) Summary of PF-spike probability during simulated inhibition and disinhibition. PF stimulation intensity was set to evoke spiking in ~50% of trials for PFtest. Each point represents individual experiments and black horizontal traces are the mean values ± SEM.
(C) Schematic of paired PC recording configuration. PC1 was voltage clamped to monitor direct CF synaptic input (black trace) with simultaneous dynamic clamp simulation of PF excitation of PC2 (red trace). Green MLIs will inhibit while black MLIs will disinhibit PC2. An EPSG was injected either before (-700 ms) or after (+10 ms) CF stimulation to determine spike probability during inhibition (EPSGinhibition) or before (-700 ms) or after (+90 ms) CF stimulation to determine spike probability during disinhibition (EPSGdisinhibition). The gray shaded region represents CF-mediated inhibition and disinhibition superimposed on spontaneous inhibition. The magnitude of the EPSGtest was set to evoke spiking in ~50% of trials.
(D) Summary of EPSGtest-spike probability. Each data point represents individual experiments and black horizontal traces are the mean values ± SEM.
Injection of somatic conductances, however, could overestimate the consequences of CF-mediated inhibition (as suggested from our MLI experiments, Figures 4 and S5). Thus, in the second set of experiments, PF input was mimicked with conductance injection (EPSG red traces; Figure 8C) into one PC (PC2) while CF stimulation on a nearby PC cell (PC1) triggered spillover inhibition and disinhibition (Figure 8C grey area). We adjusted the simulated EPSG amplitude so that PC2 spiked in ~ 50% of trials with spontaneous inhibition (Figure 8D, EPSGtest). The probability of EPSG-spiking was significantly reduced when the excitatory conductance was injected 10 ms after CF stimulation, a time that coincided with the peak of spillover inhibition (from 0.57 ± 0.04 to 0.26 ± 0.08, n = 5, P < 0.05, paired t-test; EPSGinhibition, Figure 8C and 8D). Conversely, PC2 spiking probability increased when the EPSG was injected during CF-spillover disinhibition (CF + 90 ms; from 0.55 ± 0.02 to 0.76 ± 0.03; n = 5, P <0.01, paired t-test; EPSGdisinhibition, Figure 8C and 8D). Together, these data show that glutamate spillover following single CF activation regulates MLI inhibition to alter PC excitability in a biphasic manner.
Discussion
Here, we detail how synaptic signaling exclusively from glutamate spillover engages neural circuits not previously predicted by anatomical mapping. First, we show that CF-mediated glutamate spillover affects the excitability of close and distantly located MLIs. MLIs excited by spillover inhibit MLIs outside the spillover limit resulting in segregated activity based on proximity to the active CF. Single CF stimulation recruits AMPARs and NMDARs on MLIs within the spillover limit to trigger spiking, and thus mediate a long-lasting component of spillover-mediated FFI to MLIs outside the spillover limit. Concerted activity of MLIs within and outside the spillover limit converges on neighboring PCs to generate a biphasic change in inhibitory synaptic tone that initially decreases and subsequently increases evoked spike probability. These results demonstrate a novel pathway for information transfer in the cerebellar cortex that extends the influence of CFs beyond the conventional one-to-one relationship with postsynaptic PCs.
Spillover transmission generates feed-forward inhibition
Synaptic transmission can be divided into fast and slow forms based on the kinetics of the postsynaptic response (Isaacson et al., 1993). Slow synaptic transmission is mediated by transmitters that act at diffusely distributed receptors located outside synapses (Fuxe and Agnati, 1991; but see Beckstead et al., 2004), whereas fast transmission is typically confined to synapses. In this view, spillover can be considered as an intermediate form of transmission, where traditional fast neurotransmitters act at receptors distant from release sites. Spillover is not only associated with indirect modulation of fast transmission through G-protein coupled receptors (i.e. Isaacson et al., 1993; Scanziani et al., 1997; Mitchell and Silver, 2000), but also with direct signaling through activation of ionotropic receptors (i.e. Isaacson, 1999; DiGregorio et al., 2002; Rancz et al., 2007; Scimemi et al., 2009). Direct spillover-mediated transmission improves efficacy and reliability of point-to-point transmission at some specialized synapses (DiGregorio et al., 2002; Sargent et al., 2005; Rancz et al., 2007) and has recently been implicated in the ability of synaptic inputs to generate nonlinear responses mediated by NMDARs (NMDA spikes; Chalifoux and Carter, 2011). In a few cases, synaptic signaling between neurons occurs solely via spillover in the absence of morphologically identified synaptic contacts (Isaacson, 1999; Szapiro and Barbour, 2007; Szmajda and DeVries, 2011). Although there is debate about the prevalence of spillover at typical small glutamatergic synapses, our demonstration that spillover-mediated signaling recruits local microcircuits supports its functional significance.
Spillover of the fast neurotransmitter glutamate was first postulated as an explanation for the different quantal content sensed by high affinity, putative extrasynaptic NMDARs compared to lower affinity synaptic AMPARs (Kullmann, 1994; Kullmann and Asztely, 1998). Subsequently it has been established that high affinity NMDARs are a common target for spillover-mediated signaling (i.e. Asztely et al., 1997; Isaacson, 1999; Overstreet et al., 1999; Carter and Regehr, 2000; Scimemi et al., 2004). At PF-MLI synapses, NMDARs activation is only detected during high frequency or high intensity molecular layer stimulation, indicating that NMDARs are located outside the postsynaptic density (Carter and Regehr, 2000; Clark and Cull-Candy, 2002). Such stimulation protocols produce synchronous activation of a high density of local fibers, generating extrasynaptic signaling that may be rare in vivo during physiological stimuli (Arnth-Jensen et al., 2002; Marcaggi and Attwell, 2005). We found that spillover from a single CF generates both AMPAR and NMDAR-mediated depolarization of MLIs, suggesting that CF and PF stimulation activates different sets of receptors. In contrast to FFI mediated by PFs (Figure S3 and Mittmann et al., 2005), CF stimulation generates in a long-lasting (~ 100 ms) component of inhibition to MLIs that contributes to the long-lasting component of disinhibition to PCs (Figure 7). The persistent NMDAR-mediated component thus expands both inhibition and disinhibition to PCs, potentially enhancing the contrast between areas of active and inactive PCs.
Feed-forward inhibition regulates of CF-mediated excitation in MLIs
Typical FFI narrows the window for synaptic integration by providing a rapid increase in principal cell inhibition that provides balanced regulation of excitation (Pouille and Scanziani, 2001; Wehr and Zador, 2003; Mittmann et al., 2005; House et al., 2011). Thus we were surprised that blocking GABAARs had only small, variable effects on the number of CF-evoked APs in individual cells (Figure 4). We considered three potential factors that could produce variability in the effectiveness of CF-FFI, including the magnitude of FFI, the location of FFI relative to CF-mediated excitation, and the potential for a fraction of MLI inputs to promote MLI excitability (Chavas and Marty, 2003). Since CF-mediated inhibition of PF-evoked spiking was robust (Figure S6) and somatic inhibitory conductance injection effectively decreased CF excitation of MLIs, we predict that the locations of excitatory and inhibitory conductances could promote the transmission of somatic CF-mediated excitation (Brown et al., 2012) despite reciprocal inhibition. Although MLIs are generally thought to be electronically compact because of their high input resistance and short dendrites, their thin dendrites behave as passive cables that filter synaptic responses resulting in sub-linear integration (Abrahamsson et al., 2012). This suggests that shunting that depends on location (i.e.: Gulledge and Stuart, 2003) may be important for MLI inhibition. Although further studies will be required to understand the relative insensitivity of CF-mediated inhibition between MLIs, we speculate that this insensitivity allows CF excitation to alter the activity of neighboring PCs via FFI inhibition and disinhibition (Figures 7 and 8).
Implications for cerebellar circuit processing
Jorntell and Ekerot (2002) used in vivo recordings of sensory-evoked activity to show that dual CF/PF activation enlarges MLI receptive fields whereas PF stimulation alone reduces MLI receptive fields. Subsequent work (Jörntell and Ekerot, 2003) showed that the sensory-evoked CF response in MLIs that triggers the robust plasticity in MLI receptive fields is a slow and long-lasting depolarization. Together, these two studies show that CF-mediated excitation of MLIs profoundly alters PF receptive fields. Our results show how CF activity is transmitted into long-lasting NMDAR-receptor mediated depolarization of MLIs that may be a signal driving CF-mediated plasticity of receptive fields described in vivo.
Our results also provide a potential circuit-level mechanism for in vivo observations that CF activation can alter spiking in PCs not directly targeted by the active CF. CF regulation of target and neighboring PC simple spike firing has been documented in vivo in rats (Schwarz and Welsh, 2001), rabbits (Barmack and Yakhnitsa, 2003), and mice (Bosman et al., 2010; Barmack and Yakhnitsa, 2011). CF activation in vivo is associated with increased responsiveness of PF-induced spiking in PCs not targeted by the CF, with stimulus-induced simple spiking either increased or decreased by CF activation (Bloedel et al., 1983; Ebner et al., 1983; Ebner and Bloedel, 1984). Our experiments in acute slices show that single CF activation can increase or decrease neighboring PC spiking (Figure 8). We thus propose that the functional segregation of excited and inhibited MLIs following glutamate spillover from CFs could contribute to the in vivo observation of CF-dependent gain control of simple spiking in neighboring PCs (Bloedel et al., 1983). A recent study illustrated that optogenetic activation of multiple CFs produces robust inhibition of neighboring PCs (Mathews et al., 2012). Our results using single CF stimulation reveal that CF spillover also engages MLI circuits to generate disinhibition of neighboring PCs. We speculate that there is a temporal and spatial organization of PC inhibition and disinhibition since MLIs nearest the active CF are likely to generate initial inhibition to nearby PCs whereas the persistent disinhibition may extend to more distant PCs. However, defining the significance of CF-mediated spillover in the intact brain will require additional studies given potential differences in tortuosity as well as the complex spatial and temporal organization of CF activity (Ozden et al., 2009; Schultz et al., 2009; De Zeeuw et al., 2011).
Together our results show a significant role for glutamate spillover in fast signal transmission, and further establish a pathway by which single CFs can alter the dynamics of local inhibition in the cerebellar network.
Experimental Procedures
All experiments were conducted with protocols approved by the Institutional Animal Care and Use Committee of UAB.
Slice Preparation
Parasagittal cerebellar slices were prepared from C57BL/6 mice aged 16–21 days. Animals were anesthetized by isoflurane inhalation and decapitated. The cerebellar vermis was dissected and glued to the stage of a slicer (Leica VT1200, Leica Instruments) in a solution containing (in mM) 110 CholineCl, 7 MgCl2, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 25 Glucose, 11.5 Na-Ascorbate, 3 Na-pyruvate, 25 NaHCO3, bubbled with 95% O2–5% CO2. Slices of 270 μm thickness were cut and incubated in (mM): 125 NaCl, 2.5 KCl, 1 NaH2PO4, 26.2 NaHCO3, 11 Glucose, 2.5 CaCl2, 1.3 MgCl2 at 35°C for 30 min before use.
Electrophysiology
Recordings were made at ~32°C or ~37°C maintained with an inline heating device (Warner Instruments). Cells were visualized using infrared contrast optics on an Olympus BX51WI upright microscope (Olympus). Recordings were made from identified PCs and MLIs with high input resistances located in the inner and middle thirds of the molecular layer. Recorded cells were located well below the slice surface so that diffusion and connectivity more closely resembled that of intact tissue. Responses were measured by a Multiclamp 700B amplifier (pClamp software, Molecular Devices), filtered at 2 - 5 kHz and digitized at 15 - 50 kHz (Digidata 1440). Patch pipettes (BF150-110 or BF150-086, Sutter Inst.) were pulled with a P-97 horizontal puller (Sutter Inst.) to resistances between 2.5–4 MΩ for MLIs and between 1–2 MΩ for PCs. The series resistance (Rs), as measured by an instantaneous current response to a 1–5 mV step with the pipette capacitance canceled, was always less than 10 MΩ for PC recordings and compensated ~80%, and less than 20 MΩ for MLI recordings and uncompensated. Data was discarded if Rs changed significantly (>20%). The intracellular pipette solution for voltage clamp recordings contained (mM): 125 CsMeSO3, 15 CsCl, 10 HEPES, 10 EGTA, 4 MgATP, 0.4 NaGTP, 5 QX314 (omitted for cell-attached experiments). The intracellular pipette solution for current clamp or dynamic clamp experiments contained (mM): 130 KGluconate, 15 KCl, 10 HEPES, 0.5 EGTA, 4 MgATP, 0.4 NaGTP. The intracellular [Cl-] was based on Chavas and Marty (2003; but see Carter and Regehr, 2002). In paired PC experiments, the “monitor” PC with direct CF input was voltage clamped at -10 mV and filled with (mM): 35 CsF, 100 CsCl, 10 EGTA, 10 HEPES, 5 QX314. Dynamic clamp recordings were made at 40 kHz using a digital signal processing board (P25M, Innovative Integration) run with SM-2 digital conductance software (Cambridge Conductance). For these recordings, ECl- was set at -60 mV for MLIs and -80 mV for PCs.
Single climbing fibers were stimulated (1-20 V, 100 μs) with a theta glass pipette filled with bath solution placed near the PC layer. The pipette was repositioned, and the stimulus intensity was adjusted until the voltage required to elicit an all-or-none response was minimized to eliminate PF activation or direct depolarization of neighboring MLIs. In a subset of cells, we measured the SR95531-dependent increase of spontaneous APs (from 7.4 ± 0.6 to 12.66 ± 1.2 Hz, n = 7, see Häusser and Clark, 1997) that we adjusted with DC current (7.4 ± 0.5 pA) to match the observed rate in control conditions.
Anatomical reconstructions
Experiments were performed using internal solutions with AlexaFluor 488 or 568 hydrazide (100 μM; Life Technologies) or 0.2 % biocytin. Slices were fixed in 4% paraformaldehyde for 1 h and mounted with anti-fade reagent (ProLong Gold, Life Technologies), or incubated with streptavidin-conjugated AlexaFluor 647 prior to mounting. Digital images were acquired using a 20X (NA 0.85) oil immersion objective on an Olympus FluoView 300 confocal microscope. Images were reconstructed in Neurolucida (MicroBrightField, Inc).
Data Analysis
Data was analyzed using AxoGraphX software. Changes to basal spontaneous action potential rate were quantified as in Mittmann et al. (2005). Briefly, peri-stimulus histograms (PSH) were computed and integrated. A linear fit to the baseline of the integral was extrapolated over the entire sweep and subtracted from the integral to yield the cumulative spike probability plot. We averaged between 300–400 ms period after stimulation to measure the number of spikes evoked by the input.
Statistical Analysis
Data are displayed as means ± SEM, and significance was analyzed with two-tailed Student’s t-tests (Microsoft Excel and GraphPad Prism). N values indicate number of cells. Spearman or Pearson correlations were used depending on the normality of the data. ANOVAs were followed by Bonferroni’s multiple comparison test unless noted.
Drugs
SR95531 (GABAAR antagonist, 5 μM), NBQX (AMPAR antagonist, 10 μM), AP5 (NMDAR antagonist, 100 μM), and QX314 (Na+-channel blocker, 5 mM) were obtained from Abcam Inc. DL-TBOA (50 μM) was purchased from Tocris Bioscience. All other chemicals and compounds were obtained from Sigma or Fisher Scientific.
Supplementary Material
Highlights.
glutamate spillover to interneurons (MLIs) triggers feed-forward inhibition
CF stimulation activates extrasynaptic NMDARs to extend the duration of inhibition
closely located MLIs coordinate inhibition to MLIs outside the diffusion limit
Purkinje cells experience a biphasic change in excitability due to MLI activity
Acknowledgments
This work was supported by NIH NS064025 (LO-W) and NS065920 (JIW). We thank Kamran Khodakhah, Ming-Chi Tsai, Anastassios Tzingounis, and members of the Wadiche labs for discussions and reading the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrahamsson T, Cathala L, Matsui K, Shigemoto R, Digregorio DA. Thin dendrites of cerebellar interneurons confer sublinear synaptic integration and a gradient of short-term plasticity. Neuron. 2012;73:1159–1172. doi: 10.1016/j.neuron.2012.01.027. [DOI] [PubMed] [Google Scholar]
- Arnth-Jensen N, Jabaudon D, Scanziani M. Cooperation between independent hippocampal synapses is controlled by glutamate uptake. Nat Neurosci. 2002;5:325–331. doi: 10.1038/nn825. [DOI] [PubMed] [Google Scholar]
- Asztely F, Erdemli G, Kullmann D. Extrasynaptic glutamate spillover in the hippocampus: Dependence on temperature and the role of active glutamate uptake. Neuron. 1997;18:281–293. doi: 10.1016/s0896-6273(00)80268-8. [DOI] [PubMed] [Google Scholar]
- Barbour B. An evaluation of synapse independence. J Neurosci. 2001;21:7969–7984. doi: 10.1523/JNEUROSCI.21-20-07969.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barmack NH, Yakhnitsa V. Cerebellar climbing fibers modulate simple spikes in purkinje cells. J Neurosci. 2003;23:7904–7916. doi: 10.1523/JNEUROSCI.23-21-07904.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barmack NH, Yakhnitsa V. Microlesions of the inferior olive reduce vestibular modulation of purkinje cell complex and simple spikes in mouse cerebellum. J Neurosci. 2011;31:9824–9835. doi: 10.1523/JNEUROSCI.1738-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckstead MJ, Grandy DK, Wickman K, Williams JT. Vesicular dopamine release elicits an inhibitory postsynaptic current in midbrain dopamine neurons. Neuron. 2004;42:939–946. doi: 10.1016/j.neuron.2004.05.019. [DOI] [PubMed] [Google Scholar]
- Bergles D, Dzubay J, Jahr C. Glutamate transporter currents in bergmann glial cells follow the time course of extrasynaptic glutamate. Proc Natl Acad Sci USA. 1997;94:14821–14825. doi: 10.1073/pnas.94.26.14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloedel JR, Ebner TJ, Yu QX. Increased responsiveness of purkinje cells associated with climbing fiber inputs to neighboring neurons. J Neurophysiol. 1983;50:220–239. doi: 10.1152/jn.1983.50.1.220. [DOI] [PubMed] [Google Scholar]
- Borst JGG. The low synaptic release probability in vivo. Trends Neurosci. 2010;33:259–266. doi: 10.1016/j.tins.2010.03.003. [DOI] [PubMed] [Google Scholar]
- Bosman LW, Koekkoek SK, Shapiro J, Rijken BF, Zandstra F, van der Ende B, Owens CB, Potters JW, de Gruijl JR, Ruigrok TJ, De Zeeuw CI. Encoding of whisker input by cerebellar purkinje cells. J Physiol. 2010;588:3757–3783. doi: 10.1113/jphysiol.2010.195180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasnjo G, Otis T. Neuronal glutamate transporters control activation of postsynaptic metabotropic glutamate receptors and influence cerebellar long-term depression. Neuron. 2001;31:607–616. doi: 10.1016/s0896-6273(01)00377-4. [DOI] [PubMed] [Google Scholar]
- Brown KM, Sugihara I, Shinoda Y, Ascoli GA. Digital morphometry of rat cerebellar climbing fibers reveals distinct branch and bouton types. J Neurosci. 2012;32:14670–14684. doi: 10.1523/JNEUROSCI.2018-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter A, Regehr W. Prolonged synaptic currents and glutamate spillover at the parallel fiber to stellate cell synapse. J Neurosci. 2000;20:4423–4434. doi: 10.1523/JNEUROSCI.20-12-04423.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter A, Regehr W. Quantal events shape cerebellar interneuron firing. Nat Neurosci. 2002;5:1309–1318. doi: 10.1038/nn970. [DOI] [PubMed] [Google Scholar]
- Chalifoux JR, Carter AG. Glutamate spillover promotes the generation of nmda spikes. J Neurosci. 2011;31:16435–16446. doi: 10.1523/JNEUROSCI.2777-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavas J, Marty A. Coexistence of excitatory and inhibitory gaba synapses in the cerebellar interneuron network. J Neurosci. 2003;23:2019–2031. doi: 10.1523/JNEUROSCI.23-06-02019.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Diamond JS. Synaptically released glutamate activates extrasynaptic nmda receptors on cells in the ganglion cell layer of rat retina. J Neurosci. 2002;22:2165–2173. doi: 10.1523/JNEUROSCI.22-06-02165.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark B, Cull-Candy S. Activity-dependent recruitment of extrasynaptic nmda receptor activation at an ampa receptor-only synapse. J Neurosci. 2002;22:4428–4436. doi: 10.1523/JNEUROSCI.22-11-04428.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Zeeuw CI, Hoebeek FE, Bosman LWJ, Schonewille M, Witter L, Koekkoek SK. Spatiotemporal firing patterns in the cerebellum. Nat Rev Neurosci. 2011;12:327–344. doi: 10.1038/nrn3011. [DOI] [PubMed] [Google Scholar]
- DeFelipe J. From the connectome to the synaptome: An epic love story. Science. 2010;330:1198–1201. doi: 10.1126/science.1193378. [DOI] [PubMed] [Google Scholar]
- Diamond J. Neuronal glutamate transporters limit activation of nmda receptors by neurotransmitter spillover on ca1 pyramidal cells. J Neurosci. 2001;21:8328–8338. doi: 10.1523/JNEUROSCI.21-21-08328.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGregorio D, Nusser Z, Silver R. Spillover of glutamate onto synaptic ampa receptors enhances fast transmission at a cerebellar synapse. Neuron. 2002;35:521–533. doi: 10.1016/s0896-6273(02)00787-0. [DOI] [PubMed] [Google Scholar]
- Dzubay J, Otis T. Climbing fiber activation of metabotropic glutamate receptors on cerebellar purkinje neurons. Neuron. 2002;36:1159–1167. doi: 10.1016/s0896-6273(02)01052-8. [DOI] [PubMed] [Google Scholar]
- Ebner TJ, Bloedel JR. Climbing fiber action on the responsiveness of purkinje cells to parallel fiber inputs. Brain Res. 1984;309:182–186. doi: 10.1016/0006-8993(84)91027-8. [DOI] [PubMed] [Google Scholar]
- Ebner TJ, Yu QX, Bloedel JR. Increase in purkinje cell gain associated with naturally activated climbing fiber input. J Neurophysiol. 1983;50:205–219. doi: 10.1152/jn.1983.50.1.205. [DOI] [PubMed] [Google Scholar]
- Fuxe K, Agnati LF. Volume transmission in the brain (Raven Pr) 1991 [Google Scholar]
- Gulledge A, Stuart G. Excitatory actions of gaba in the cortex. Neuron. 2003;37:299–309. doi: 10.1016/s0896-6273(02)01146-7. [DOI] [PubMed] [Google Scholar]
- Häusser M, Clark B. Tonic synaptic inhibition modulates neuronal output pattern and spatiotemporal synaptic integration. Neuron. 1997;19:665–678. doi: 10.1016/s0896-6273(00)80379-7. [DOI] [PubMed] [Google Scholar]
- House DR, Elstrott J, Koh E, Chung J, Feldman DE. Parallel regulation of feedforward inhibition and excitation during whisker map plasticity. Neuron. 2011;72:819–831. doi: 10.1016/j.neuron.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson J, Solís J, Nicoll R. Local and diffuse synaptic actions of gaba in the hippocampus. Neuron. 1993;10:165–175. doi: 10.1016/0896-6273(93)90308-e. [DOI] [PubMed] [Google Scholar]
- Isaacson JS. Glutamate spillover mediates excitatory transmission in the rat olfactory bulb. Neuron. 1999;23:377–384. doi: 10.1016/s0896-6273(00)80787-4. [DOI] [PubMed] [Google Scholar]
- Jarolimek W, Lewen A, Misgeld U. A furosemide-sensitive k+-cl-cotransporter counteracts intracellular cl- accumulation and depletion in cultured rat midbrain neurons. J Neurosci. 1999;19:4695–4704. doi: 10.1523/JNEUROSCI.19-12-04695.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas P, Sakmann B. Glutamate receptor channels in isolated patches from ca1 and ca3 pyramidal cells of rat hippocampal slices. J Physiol (Lond) 1992;455:143–171. doi: 10.1113/jphysiol.1992.sp019294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jörntell H, Ekerot C-F. Reciprocal bidirectional plasticity of parallel fiber receptive fields in cerebellar purkinje cells and their afferent interneurons. Neuron. 2002;34:797–806. doi: 10.1016/s0896-6273(02)00713-4. [DOI] [PubMed] [Google Scholar]
- Jörntell H, Ekerot C-F. Receptive field plasticity profoundly alters the cutaneous parallel fiber synaptic input to cerebellar interneurons in vivo. J Neurosci. 2003;23:9620–9631. doi: 10.1523/JNEUROSCI.23-29-09620.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khirug S, Huttu K, Ludwig A, Smirnov S, Voipio J, Rivera C, Kaila K, Khiroug L. Distinct properties of functional kcc2 expression in immature mouse hippocampal neurons in culture and in acute slices. Eur J Neurosci. 2005;21:899–904. doi: 10.1111/j.1460-9568.2005.03886.x. [DOI] [PubMed] [Google Scholar]
- Kollo M, Holderith NB, Nusser Z. Novel subcellular distribution pattern of a-type k+ channels on neuronal surface. J Neurosci. 2006;26:2684–2691. doi: 10.1523/JNEUROSCI.5257-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konnerth A, Llano I, Armstrong C. Synaptic currents in cerebellar purkinje cells. Proc Natl Acad Sci USA. 1990;87:2662–2665. doi: 10.1073/pnas.87.7.2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann DM. Amplitude fluctuations of dual-component epscs in hippocampal pyramidal cells: Implications for long-term potentiation. Neuron. 1994;12:1111–1120. doi: 10.1016/0896-6273(94)90318-2. [DOI] [PubMed] [Google Scholar]
- Kullmann DM. Spillover and synaptic cross talk mediated by glutamate and gaba in the mammalian brain. Prog Brain Res. 2000;125:339–351. doi: 10.1016/S0079-6123(00)25023-1. [DOI] [PubMed] [Google Scholar]
- Kullmann DM, Asztely F. Extrasynaptic glutamate spillover in the hippocampus: Evidence and implications. Trends Neurosci. 1998;21:8–14. doi: 10.1016/s0166-2236(97)01150-8. [DOI] [PubMed] [Google Scholar]
- Lehre K, Danbolt N. The number of glutamate transporter subtype molecules at glutamatergic synapses: Chemical and stereological quantification in young adult rat brain. J Neurosci. 1998;18:8751–8757. doi: 10.1523/JNEUROSCI.18-21-08751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman JW, Livet J, Sanes JR. A technicolour approach to the connectome. Nat Rev Neurosci. 2008;9:417–422. doi: 10.1038/nrn2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcaggi P, Attwell D. Endocannabinoid signaling depends on the spatial pattern of synapse activation. Nat Neurosci. 2005;8:776–781. doi: 10.1038/nn1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews PJ, Lee KH, Peng Z, Houser CR, Otis TS. Effects of climbing fiber driven inhibition on purkinje neuron spiking. J Neurosci. 2012;32:17988–17997. doi: 10.1523/JNEUROSCI.3916-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell S, Silver R. Glutamate spillover suppresses inhibition by activating presynaptic mglurs. Nature. 2000;404:498–502. doi: 10.1038/35006649. [DOI] [PubMed] [Google Scholar]
- Mittmann W, Koch U, Häusser M. Feed-forward inhibition shapes the spike output of cerebellar purkinje cells. J Physiol (Lond) 2005;563:369–378. doi: 10.1113/jphysiol.2004.075028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet L, Kinney G, Liu Y, Billups D, Slater N. Glutamate transporters contribute to the time course of synaptic transmission in cerebellar granule cells. J Neurosci. 1999;19:9663–9673. doi: 10.1523/JNEUROSCI.19-21-09663.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozden I, Sullivan MR, Lee HM, Wang SS-H. Reliable coding emerges from coactivation of climbing fibers in microbands of cerebellar purkinje neurons. J Neurosci. 2009;29:10463–10473. doi: 10.1523/JNEUROSCI.0967-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palay SL, Chan-Palay V. Cerebellar cortex: Cytology and organization. 1974:348. [Google Scholar]
- Patneau DK, Mayer ML. Structure-activity relationships for amino acid transmitter candidates acting at n-methyl-d-aspartate and quisqualate receptors. J Neurosci. 1990;10:2385–2399. doi: 10.1523/JNEUROSCI.10-07-02385.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce RA. Physiological evidence for two distinct gabaa responses in rat hippocampus. Neuron. 1993;10:189–200. doi: 10.1016/0896-6273(93)90310-n. [DOI] [PubMed] [Google Scholar]
- Pouille F, Scanziani M. Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science. 2001;293:1159–1163. doi: 10.1126/science.1060342. [DOI] [PubMed] [Google Scholar]
- Rancz E, Ishikawa T, Duguid I, Chadderton P, Mahon S, Häusser M. High-fidelity transmission of sensory information by single cerebellar mossy fibre boutons. Nature. 2007;450:1245–1248. doi: 10.1038/nature05995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson HP, Kawai N. Injection of digitally synthesized synaptic conductance transients to measure the integrative properties of neurons. J Neurosci Methods. 1993;49:157–165. doi: 10.1016/0165-0270(93)90119-c. [DOI] [PubMed] [Google Scholar]
- Rudolph S, Overstreet-Wadiche L, Wadiche JI. Desynchronization of multivesicular release enhances purkinje cell output. Neuron. 2011;70:991–1004. doi: 10.1016/j.neuron.2011.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargent P, Saviane C, Nielsen T, DiGregorio D, Silver R. Rapid vesicular release, quantal variability, and spillover contribute to the precision and reliability of transmission at a glomerular synapse. J Neurosci. 2005;25:8173–8187. doi: 10.1523/JNEUROSCI.2051-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satake S, Saitow F, Yamada J, Konishi S. Synaptic activation of ampa receptors inhibits gaba release from cerebellar interneurons. Nat Neurosci. 2000;3:551–558. doi: 10.1038/75718. [DOI] [PubMed] [Google Scholar]
- Scanziani M, Salin P, Vogt K, Malenka R, Nicoll R. Use-dependent increases in glutamate concentration activate presynaptic metabotropic glutamate receptors. Nature. 1997;385:630–634. doi: 10.1038/385630a0. [DOI] [PubMed] [Google Scholar]
- Schultz SR, Kitamura K, Post-Uiterweer A, Krupic J, Häusser M. Spatial pattern coding of sensory information by climbing fiber-evoked calcium signals in networks of neighboring cerebellar purkinje cells. J Neurosci. 2009;29:8005–8015. doi: 10.1523/JNEUROSCI.4919-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz C, Welsh J. Dynamic modulation of mossy fiber system throughput by inferior olive synchrony: A multielectrode study of cerebellar cortex activated by motor cortex. J Neurophysiol. 2001;86:2489–2504. doi: 10.1152/jn.2001.86.5.2489. [DOI] [PubMed] [Google Scholar]
- Scimemi A, Fine A, Kullmann DM, Rusakov DA. Nr2b-containing receptors mediate cross talk among hippocampal synapses. J Neurosci. 2004;24:4767–4777. doi: 10.1523/JNEUROSCI.0364-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scimemi A, Tian H, Diamond JS. Neuronal transporters regulate glutamate clearance, nmda receptor activation, and synaptic plasticity in the hippocampus. J Neurosci. 2009;29:14581–14595. doi: 10.1523/JNEUROSCI.4845-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AA, O’Neil MB, Abbott LF, Marder E. Dynamic clamp: Computer-generated conductances in real neurons. J Neurophysiol. 1993;69:992–995. doi: 10.1152/jn.1993.69.3.992. [DOI] [PubMed] [Google Scholar]
- Sporns O. The human connectome: A complex network. Ann N Y Acad Sci. 2011;1224:109–125. doi: 10.1111/j.1749-6632.2010.05888.x. [DOI] [PubMed] [Google Scholar]
- Szapiro G, Barbour B. Multiple climbing fibers signal to molecular layer interneurons exclusively via glutamate spillover. Nat Neurosci. 2007;10:735–742. doi: 10.1038/nn1907. [DOI] [PubMed] [Google Scholar]
- Szmajda BA, DeVries SH. Glutamate spillover between mammalian cone photoreceptors. J Neurosci. 2011;31:13431–13441. doi: 10.1523/JNEUROSCI.2105-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai M-C, Tanaka K, Overstreet-Wadiche L, Wadiche JI. Neuronal glutamate transporters regulate glial excitatory transmission. J Neurosci. 2012;32:1528–1535. doi: 10.1523/JNEUROSCI.5232-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzingounis AV, Wadiche JI. Glutamate transporters: Confining runaway excitation by shaping synaptic transmission. Nat Rev Neurosci. 2007;8:935–947. doi: 10.1038/nrn2274. [DOI] [PubMed] [Google Scholar]
- Wadiche JI, Jahr CE. Multivesicular release at climbing fiber-purkinje cell synapses. Neuron. 2001;32:301–313. doi: 10.1016/s0896-6273(01)00488-3. [DOI] [PubMed] [Google Scholar]
- Wadiche JI, Jahr CE. Patterned expression of purkinje cell glutamate transporters controls synaptic plasticity. Nat Neurosci. 2005;8:1329–1334. doi: 10.1038/nn1539. [DOI] [PubMed] [Google Scholar]
- Wehr M, Zador AM. Balanced inhibition underlies tuning and sharpens spike timing in auditory cortex. Nature. 2003;426:442–446. doi: 10.1038/nature02116. [DOI] [PubMed] [Google Scholar]
- Xu-Friedman M, Harris K, Regehr W. Three-dimensional comparison of ultrastructural characteristics at depressing and facilitating synapses onto cerebellar purkinje cells. J Neurosci. 2001;21:6666–6672. doi: 10.1523/JNEUROSCI.21-17-06666.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.