


Abstract
An important aim in the post-sequencing age of functional genomics is to translate gene sequences into protein functions. This shift of focus is particularly necessary for a very large number of human genes, referred to as novel genes, where we have no or very rudimentary information about their biochemical functions. Recently, a new method for investigating human gene functions using small interfering RNA molecules (siRNAs) has become available. siRNAs are powerful reagents for post-transcriptional silencing, where mRNA targeted by the siRNAs is degraded in vivo and the level of the encoded protein is reduced. However, the lack of antibodies against proteins encoded by novel genes restricts the general value of siRNAs as functional tools, as only the mRNA levels can be measured for these genes. We report a method that combines measurements of protein levels in cell culture for novel, exogenously expressed genes, with parallel measurements of the endogenous mRNA levels of the same genes. We find that this combinatorial approach correctly predicts siRNAs that efficiently reduce mRNA and protein levels in cultured cells. Furthermore, this method identifies proteins that have a slow turnover, which weakens the value of the RNA interference method as a tool for functional studies of such genes. The described method should prove to be valuable for large-scale functional studies of novel human genes.
INTRODUCTION
The completion of the sequencing of the human genome provides us with an opportunity to understand the molecular basis of human physiology and disease. It has been estimated, however, that at least 15 000 of the 35 000 human genes that we know exist are novel and lack functional annotations (1). To promote our basic understanding of biological processes and to provide the pharmaceutical industry with new drug targets, it is essential that we classify novel genes into functional categories and pathways. The use of RNA interference methods has dramatically affected our means to study gene functions (2,3). One version of this method involves the use of short, synthetic, small interfering RNA molecules (siRNAs) (4). With this method, the siRNAs are designed to be complementary to a 21- to 23-base region of the target mRNA. Transfection of double-stranded siRNAs into human cell culture cells leads to in vivo protein complexes processing the siRNAs into short, single-stranded siRNA molecules that bind to the target mRNA, resulting in its degradation and the subsequent loss of the encoded protein. The fact that this method relies on a mechanism endogenously active in mammalian cells strongly suggests that this approach will have significant medical use, for example in treating viral infections, cancer and other human diseases (5–7). A major technical limitation of the current approaches for employing siRNAs in cultured cells is the requirement for antibodies to monitor the endogenous levels of the protein encoded by the mRNA being targeted. In the absence of such reagents, most researchers tend to select and validate siRNAs based only on their effects on the endogenous mRNA levels (8,9). It is clear, however, that many proteins are quite stable and their concentrations in cells therefore remain relatively constant for a long period of time. We have defined and experimentally validated a strategy in cultured cells that compares the level of endogenously expressed mRNA with the protein level originating from the same gene, but derived from exogenously added plasmids and detected by an epitope tag. This combinatorial approach has been found to correctly define siRNAs that efficiently reduce both mRNA and protein levels in cultured cells. A further advantage of this method is that it also facilitates identification of those proteins that turn over slowly. This is an important experimental indication, as it is not possible to analyze the functions of such proteins in short-term RNA interference studies.
MATERIALS AND METHODS
Oligonucleotide templates for siRNA
The 21-nucleotide cDNA templates used for siRNA production were selected according to criteria described previously (10). The cDNA sequences were subjected to an siRNA Converter tool, which generates oligonucleotide templates for use with the Silencer™ siRNA Construction Kit (Ambion). Three cDNA templates for siRNA production were selected for each gene (Table 1). Sense and antisense templates were synthesized (Thermo Electron GmbH). Double-stranded, 21-nucleotide RNA (siRNA) molecules were produced by in vitro transcription with the Silencer™ siRNA Construction Kit (Ambion). A positive GAPDH siRNA was in vitro transcribed using the sense and antisense templates provided with the Silencer™ siRNA Construction Kit (Ambion).
Table 1. cDNA templates for siRNA in vitro transcription.
| NM_173475 |
| siRNA 1 template 238-AAGTCCAATGCCGAGGAGTCT-258 |
| siRNA 2 template 397-AAGTTCCAGGCTGCAACCATG-417 |
| siRNA 3 template 742-AACTTCACTCAGGTGATTGGC-762 |
| NM_020651 |
| siRNA 1 template 214-AAAGACCAGCATAGCATATCA-234 |
| siRNA 2 template 342-AACTGACACGGTTCCTGGAAG-362 |
| siRNA 3 template 995-AATGTCCTATGTGTAGGTCTG-1015 |
| NM_018109 |
| siRNA 1 template 206-AAAGACGAGAACAGGCACAGC-226 |
| siRNA 2 template 476-AACGGTCACGCGTACGGTCAA-496 |
| siRNA 3 template 1529-AACAGGAAGATACAGATCGAC-1549 |
| NM_016301 |
| siRNA 1 template 91-AACCGGTCTGTCCAAGTTGTA-111 |
| siRNA 2 template 167-AACTGATCGAGGTGGATGATG-187 |
| siRNA 3 template 716-AAAGCATGAACATTGCATTGC-736 |
| NM_006411 |
| siRNA 1 template 124-AATGGCTGGATCCTCTTCCTG-144 |
| siRNA 2 template 537-AACGAGAAACCACAATGGCTC-557 |
| siRNA 3 template 655-AAGAAGGAGCGTCGCTTCACC-675 |
| BC014077 |
| siRNA 1 template 241-AAGAAGGAAAAGACGGCCAAG-261 |
| siRNA 2 template 1237-AACAAGGCGCGGCTCCGAGTG-1257 |
| siRNA 3 template 1793-AATTCCACAGCGTGGAGGTGC-1813 |
| GFP siRNA template 672-AACAGCTGCTAGGATTACACA-692 |
Three, 21-nucleotide cDNA templates were selected according to analysis of the ORF using siRNA Converter software (Ambion). The numbers indicate the corresponding positions of the selected 21-nucleotide cDNA fragments in the ORFs.
Gene cloning
Full-length open reading frames (ORFs) representing human genes were cloned into mammalian expression vectors using the recombination-based Gateway™ system (Invitrogen). Gene-specific primers were designed to amplify coding regions for each full-length cDNA. Sense and antisense primers consisted of 18–25 nucleotides flanked by the Gateway™ recombination sites (attB sites), which comprise 31 and 30 nucleotides (forward and reverse, respectively). cDNA was transcribed from mRNA selected in the presence of oligo (dT)20 from human total RNA and reverse transcribed using the Thermoscript RT system (Invitrogen). Human total RNA isolated from human liver, fetal brain, placenta, testis, skeletal muscle and HeLa cells (Clontech) was selected for reverse transcription. Complex cDNA was pooled for use as the template for amplification by rTthDNA polymerase XL (Applied Biosystems). Polymerase chain reaction (PCR) products were obtained under the following cycle conditions: 95°C for 10 min followed by 35 cycles at 94°C for 15 s, 45–65°C for 30 s, and 60°C for 1 min, after which a final extension at 68°C for 10 min was added. Entry clones were generated by inserting attB-flanked PCR products into the pDONR201 vector in the presence of BP clonase (Invitrogen). For fusion protein expression studies, the p3xFLAG-CMV-7.1 vector (Sigma), a mammalian expression vector containing an N-terminal 3xFLAG sequence, was converted into a Gateway™-compatible vector by inserting a Gateway™ vector conversion cassette into the multiple cloning site (Invitrogen) (Fig. 1). Full-length inserts were subsequently transferred from the entry clone to the expression vector in order to generate N-terminal 3xFLAG fusions.
Figure 1.

A schematic representation of siRNA selection and validation for a set of under-characterized genes. (A) N-terminal, full-length fusion proteins are generated and plasmids are co-transfected with three unique siRNAs corresponding to the gene target and a GFP control siRNA. Protein levels are analyzed by western blot. (B) In a parallel experiment, cells are transfected with siRNAs only and the mRNA levels for the endogenous genes are analyzed by RT–PCR.
Transfection of plasmid and siRNA
Human embryonic kidney (HEK) 293 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, penicillin (100 U/ml) and streptomycin (100 µg/ml). One day before transfection, cells were trypsinized and resuspended in DMEM without antibiotics, and plated into a 24-well plate at a density of 1.5 × 105 cells per 0.6 ml per well. Co-transfection of plasmid constructs and siRNA were performed using Lipofectamine 2000 according to the manufacturer’s protocol. For a given gene, 0.4 µg of plasmid was co-transfected with each of the three gene-specific siRNAs or a control green fluorescent protein (GFP) siRNA. A 10 nM concentration of siRNA was used for all transfections. Forty-eight hours after transfection, the cells were harvested for western blot analysis of the knockdown level of the exogenous proteins by siRNA. For targeting and detection of the endogenous gene level, cells were transfected with 10 nM siRNA, with a GFP siRNA as control. After 24 h of incubation, RNA was isolated from the cells using RNAqueous™ –96 (Ambion), and the knockdown of mRNA levels was analyzed by RT–PCR.
Western blotting
Forty-eight hours after co-transfection of plasmids and siRNA, the DMEM was removed and cells were harvested in an equal volume of lysis buffer (10 mM Tris–HCl pH 7.2, 1 mM PMSF, 10 µg/ml DNase I, 5 µg/ml RNase), and incubated at room temperature for 20 min. Cell lysates were mixed with 5× loading buffer (0.3 M Tris–HCl pH 6.8, 10% SDS, 0.05% bromophenol blue, 50% glycerol) and heated for 5 min at 90°C. Gel electrophoresis was performed on a 4–20% sodium dodecyl sulfate (SDS)–polyacrylamide gradient gel (Bio-Rad). Proteins were subsequently transferred to PVDF Immobilon-P membrane (Millipore) for 1 h at 100 V. Following this, the blot membrane was incubated for 1 h in blocking buffer {5% milk powder in Tris-buffered saline (TBS)–T buffer [20 mM Tris–HCl pH 7.5, 500 mM NaCl, 0.05% (v/v) Tween 20]}. The blot membrane was then incubated with an anti-FLAG horseradish peroxidase-coupled monoclonal antibody (Sigma) in TBS–T buffer (1:5000 dilution) for 1 h at room temperature. The membrane was washed 4× 10 min in TBS–T buffer. FLAG fusion proteins were detected using an enhanced chemiluminescence assay, according to the manufacturer’s protocol (Pierce). The blot membrane was later stripped with stripping buffer (50 mM Tris–HCl pH 6.8, 100 mM β-mercaptoethanol, 1% SDS) and re-probed with anti-GAPDH (Ambion) or anti-actin (Sigma) as a loading control.
Quantitation of siRNA effect on the knockdown of proteins
The knockdown level of proteins was quantified from western blot membranes using ImageQuant software (Amersham Pharmacia Biotech). The effects of different siRNAs were calculated, with a GFP siRNA as control.
Total RNA isolation and RT–PCR analysis
Twenty-four hours after siRNA transfection, HEK 293 cells were harvested for RNA isolation. RNA was isolated using RNAqueous™ –96 (Ambion). RNA concentration was determined using a spectrophotometer, and the quality of RNA was assessed on a 1.2% denaturing agarose gel with MOPS buffer (40 mM MOPS pH 7.0, 10 mM sodium acetate, 1 mM EDTA). RT–PCR was performed with TITANIUM One-Step RT–PCR, according to the manufacturer’s instructions (BD Biosciences). Gene-specific primers were designed to flank the sequences targeted by the most effective siRNA, which was identified by western blot. Forward and reverse primers for each gene are listed in Table 2. The One-Step RT–PCR was performed under the following conditions: 50°C for 60 min, 94°C for 5 min, immediately followed by 20 cycles of 94°C for 30 s, 55°C for 30 s, and 68°C for 1 min, after which a final extension at 72°C for 2 min was added. PCR products were analyzed on a 1.5% agarose gel containing ethidium bromide.
Table 2. Forward and reverse primers designed for RT–PCR.
| NM_173475 |
| Forward primer 5′-ATGGAGCGCTTTTGCAATGACCTGTGT-3′ |
| Reverse primer 5′-TTCCCCTTCTCTTTTCCTTCGCTCCATTTC-3′ |
| NM_020651 |
| Forward primer 5′-TGGCGATAGAGGAAGGAGGAAAAGTAGGTT-3′ |
| Reverse primer 5′-GAGGGCCGGCGTCCACATAAAATCCAG-3′ |
| NM_018109 |
| Forward primer 5′-CTGAGATGCAAAATGAAAGACGAGAAC-3′ |
| Reverse primer 5′-ACTGAAATCCGGAGGCCTGGTGTGA-3′ |
| NM_016301 |
| Forward primer 5′-GCGGGCAGCGGGAAGAG-3′ |
| Reverse primer 5′-CATCAATCAGTCCACATATAGCTTTAGTCA-3′ |
| NM_006411 |
| Forward primer 5′-GATCTTGCGTCTAATGCTGCTCCACA-3′ |
| Reverse primer 5′-CCCCCAGGCTTCTTCAGATAGTCACC-3′ |
| BC014077 |
| Forward primer 5′-CCCCGAAGCAGAACGCCTCATTGTGGATTA-3′ |
| Reverse primer 5′-AGAGTTGATGAGGAGGCGGCCGGAGAGGTC-3′ |
| GAPDH |
| Forward primer 5′-ATG-GGGAAGGTGAAGGTCGGAGTC-3′ |
| Reverse primer 5′-AATGCCAGCCC-CAGCGTCAAAGGT-3′ |
RESULTS AND DISCUSSION
The goal with most RNA interference protocols is to effectively reduce protein levels in cells and to study the functional consequences of their removal. Western blot methodology using specific antibodies is the most efficient way to monitor knockdown protein levels by siRNA (11–13). Antibodies that could be used to monitor the endogenous protein levels are, however, not available for most human genes. We have developed a high-throughput method for selecting siRNAs that most effectively reduce protein levels in a cell culture assay system (Fig. 1). With this approach, cultured human cells are transfected in microtiter plates with plasmids that express different full-length proteins fused to a common N-terminal epitope tag (FLAG). These cells are also co-transfected with different siRNAs targeting mRNAs generated by the same expression plasmids. Protein levels are determined using western blots of cell lysates prepared from transfected cells. In parallel, the cultured cells are separately transfected with the same siRNAs; however, in these experiments the level of the endogenous mRNA is monitored by RT–PCR (Fig. 1). We have compared the results of the two experiments for four different genes (Fig. 2). We find that the levels of epitope-tagged proteins in most cases follow the levels of the corresponding endogenous mRNAs. This strongly suggests that the rate of degradation of endogenous mRNAs, as well as of exogenously expressed mRNA from the added plasmids, is similar in most cases. This conclusion is supported by experiments using GAPDH as a control; we found that levels of both exogenously and endogenously expressed proteins were equally affected by the transfected siRNAs (Fig. 2A). We also found that this method could be applied also to other adherent cell lines, such as HeLa cells (data not shown). This method therefore provides a route for identifying siRNAs that effectively reduce protein levels in the absence of antibodies against the endogenous protein.
Figure 2.
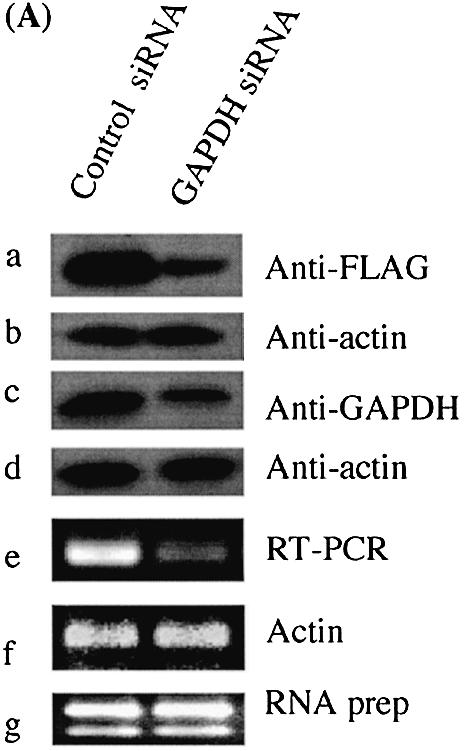


Knockdown of gene expression by siRNA. GAPDH gene was used as a control, where knockdown of GAPDH expression by siRNA was observed for both exogenously expressed FLAG-tagged GAPDH (A) and endogenously expressed GAPDH (C). Actin was used a loading control (B, D, F). (E) In co-transfection experiments monitoring knockdown of exogenous proteins by siRNA, fusion protein levels were probed by anti-FLAG. The detected bands were quantified using ImageQuant software (Amersham Pharmacia Biotech). The membrane was then re-probed with anti-GAPDH. siRNA experiments targeting the endogenous genes were analyzed by RT–PCR to monitor knockdown of mRNA levels. GAPDH was used as an internal control and total RNA was also used as a control. The data shown are from one of three independent experiments with similar results. GFP siRNA, cells transfected with a control GFP siRNA; siRNA 1, siRNA 2 and siRNA 3 represent cells transfected with three different siRNAs for each gene, respectively. The DDBJ/EMBL/GenBank accession numbers for the four genes are: (B) NM_173475; (C) NM_020651; (D) NM_018109; (E) NM_016301.
A distinctive advantage of the strategy described in Figure 1 is that it provides a method for identifying proteins that have a slow turnover and are therefore not immediately affected by the removal of the mRNAs that encode them. The genes shown in Figure 3 have endogenous mRNA levels (as determined by limiting RT–PCR) that can be seen to fluctuate dramatically depending on the efficiency of the applied siRNAs. However, the level of the exogenous protein supplied from the expression plasmid transfected into these cells does not vary according to the mRNA level (Fig. 3). No significant knockdown of these proteins was observed even after an extended incubation of cells transfected with these siRNAs for 72 h (data not shown). Similarly, increasing the siRNA concentration 2- or 4-fold did not affect the protein levels (data not shown). The best explanation for the above results is that the genes analyzed in Figure 3 encode proteins that have a long half-life.
Figure 3.

Knockdown of gene expression by siRNA is evident in RNA levels, but not in protein levels. As described in Figure 2, western blot analysis monitored knockdown of exogenous proteins by siRNA with anti-FLAG. GAPDH was probed with anti-GAPDH as a loading control. RT–PCR was performed to analyze knockdown of endogenous genes by siRNA. GAPDH was used as an internal control, and total RNA was also used as a control. The data shown represent one of three independent experiments with similar results. GFP siRNA, cells transfected with a control GFP siRNA; siRNA 1, siRNA 2 and siRNA 3 represent cells transfected with three different siRNAs for each gene, respectively. The accession numbers for the two genes are: (A) NM_006411; (B) BC014077.
So far we have applied the method described in Figure 1 to studies of 30 human genes, the majority of which are novel, and identified siRNAs that reduce RNA and protein levels. We designed three siRNAs for each gene, and found that there is considerable variation in silencing efficiency among siRNAs. This variation may be, in part, attributed to the different accessibilities of siRNAs to the target mRNA (14–17). In three cases (two shown in Fig. 3), the protein levels were not affected despite sharp reductions in the corresponding mRNA levels. One of the two cases involves a known enzyme, 1-acylglycerol-3-phosphate O-acyltransferase 1 (NM_006411), whereas the second example represents a novel gene, KIAA1533 (BC014077). In summary, we have developed a high-throughput, cell-based assay system for rapidly selecting siRNAs that effectively reduce mRNA and protein levels. The method also identifies those genes that encode proteins that due to their stability cannot be analyzed using short-term RNA interference methods.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Danielle Kemmer for providing us with the original human cDNA sequences and Claes Wahlestedt for helpful discussions. This work was supported by Pfizer, the Wenner-Gren Foundation and the Karolinska Institutet.
REFERENCES
- 1.Saha S., Sparks,A.B., Rago,C., Akmaev,V., Wang,C.J., Vogelstein,B., Kinzler,K.W. and Velculescu,V.E. (2002) Using the transcriptome to annotate the genome. Nat. Biotechnol., 20, 508–512. [DOI] [PubMed] [Google Scholar]
- 2.Paddison P.J. and Hannon,G.J. (2002) RNA interference: the new somatic cell genetics? Cancer Cell, 2, 17–23. [DOI] [PubMed] [Google Scholar]
- 3.McManus M.T. and Sharp,P.A. (2002) Gene silencing in mammals by small interfering RNAs. Nature Rev. Genet., 3, 737–747. [DOI] [PubMed] [Google Scholar]
- 4.Elbashir S.M., Harborth,J., Lendeckel,W., Yalcin,A., Weber,K. and Tuschl,T. (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature, 411, 494–498. [DOI] [PubMed] [Google Scholar]
- 5.Borkhardt A. (2002) Blocking oncogenes in malignant cells by RNA interference—new hope for a highly specific cancer treatment? Cancer Cell, 2, 167–168. [DOI] [PubMed] [Google Scholar]
- 6.Gitlin L., Karelsky,S. and Andino,R. (2002) Short interfering RNA confers intracellular antiviral immunity in human cells. Nature, 418, 430–434. [DOI] [PubMed] [Google Scholar]
- 7.Xia H., Mao,Q., Paulson,H.L. and Davidson,B.L. (2002) siRNA-mediated gene silencing in vitro and in vivo. Nat. Biotechnol., 20, 1006–1010. [DOI] [PubMed] [Google Scholar]
- 8.Sorensen D.R., Leirdal,M. and Sioud,M. (2003) Gene silencing by systemic delivery of synthetic siRNAs in adult mice. J. Mol. Biol., 327, 761–766. [DOI] [PubMed] [Google Scholar]
- 9.Hill J.A., Ichim,T.E., Kusznieruk,K.P., Li,M., Huang,X., Yan,X., Zhong,R., Cairns,E., Bell,D.A. and Min,W.P. (2003) Immune modulation by silencing IL-12 production in dendritic cells using small interfering RNA. J. Immunol., 171, 691–696. [DOI] [PubMed] [Google Scholar]
- 10.Elbashir S.M.H.J., Weber,K. and Tuschl,T. (2002) Analysis of gene function in somatic mammalian cells using small interfering RNAs. Methods, 26, 199–213. [DOI] [PubMed] [Google Scholar]
- 11.Leirdal M. and Sioud,M. (2002) Gene silencing in mammalian cells by preformed small RNA duplexes. Biochem. Biophys. Res. Commun., 295, 744–748. [DOI] [PubMed] [Google Scholar]
- 12.Burns T.F., Fei,P., Scata,K.A., Dicker,D.T. and El-Deiry,W.S. (2003) Silencing of the novel p53 target gene Snk/Plk2 leads to mitotic catastrophe in paclitaxel (taxol)-exposed cells. Mol. Cell. Biol., 23, 5556–5571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Braasch D.A., Jensen,S., Liu,Y., Kaur,K., Arar,K., White,M.A. and Corey,D.R. (2003) RNA interference in mammalian cells by chemically-modified RNA. Biochemistry, 42, 7967–7975. [DOI] [PubMed] [Google Scholar]
- 14.Holen T., Amarzguioui,M., Wiiger,M.T., Babaie,E. and Prydz,H. (2002) Positional effects of short interfering RNAs targeting the human coagulation trigger Tissue Factor. Nucleic Acids Res., 30, 1757–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kretschmer-KazemiFar R. and Sczakiel,G. (2003) The activity of siRNA in mammalian cells is related to structural target accessibility: a comparison with antisense oligonucleotides. Nucleic Acids Res., 31, 4417–4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vickers T.A., Koo,S., Bennett,C.F., Crooke,S.T., Dean,N.M. and Baker,B.F. (2003) Efficient reduction of target RNAs by small interfering RNA and RNase H-dependent antisense agents. A comparative analysis. J. Biol. Chem., 278, 7108–7118. [DOI] [PubMed] [Google Scholar]
- 17.Xu Y., Zhang,H.Y., Thormeyer,D., Larsson,O., Du,Q., Elmen,J., Wahlestedt,C. and Liang,Z. (2003) Effective small interfering RNAs and phosphorothioate antisense DNAs have different preferences for target sites in the luciferase mRNAs. Biochem. Biophys. Res. Commun., 306, 712–717. [DOI] [PubMed] [Google Scholar]