

Abstract
Background
S. erythraea is a Gram-positive filamentous bacterium used for the industrial-scale production of erythromycin A which is of high clinical importance. In this work, we sequenced the whole genome of a high-producing strain (E3) obtained by random mutagenesis and screening from the wild-type strain NRRL23338, and examined time-series expression profiles of both E3 and NRRL23338. Based on the genomic data and transcriptpmic data of these two strains, we carried out comparative analysis of high-producing strain and wild-type strain at both the genomic level and the transcriptomic level.
Results
We observed a large number of genetic variants including 60 insertions, 46 deletions and 584 single nucleotide variations (SNV) in E3 in comparison with NRRL23338, and the analysis of time series transcriptomic data indicated that the genes involved in erythromycin biosynthesis and feeder pathways were significantly up-regulated during the 60 hours time-course. According to our data, BldD, a previously identified ery cluster regulator, did not show any positive correlations with the expression of ery cluster, suggesting the existence of alternative regulation mechanisms of erythromycin synthesis in S. erythraea. Several potential regulators were then proposed by integration analysis of genomic and transcriptomic data.
Conclusion
This is a demonstration of the functional comparative genomics between an industrial S. erythraea strain and the wild-type strain. These findings help to understand the global regulation mechanisms of erythromycin biosynthesis in S. erythraea, providing useful clues for genetic and metabolic engineering in the future.
Keywords: S. erythraea, Erythromycin biosynthesis, Functional comparative genetics, Regulation mechanism
Background
Saccharopolyspora erythraea, formerly identified as Streptomyces erythraeus, is a Gram-positive filamentous bacterium. It has been used for the industrial-scale production of erythromycin A, a broad-spectrum macrolide antibiotic against pathogenic Gram-positive bacteria [1]. The antimicrobial spectrum of erythromycin A is similar to that of penicillin, and Erythromycin A is often prescribed as an alternative for patients with an allergy to penicillin. Moreover, a series of derivatives of erythromycin, derived by chemical and biotechnological transformation, have been shown to have antiparasitic, antineoplastic, immunosuppressant, neurotrophic, anti-inflammatory, and gastroenteric therapeutic activities [2]. In view of the high clinical importance of erythromycin and its derivatives, extensive efforts have been devoted to increase the erythromycin production in S. erythraea which has even been studied as a model system for antibiotic production [3,4].
Over the past 50 years, the S. erythraea strain improvement has been carried out mainly by multiple rounds of random mutagenesis and selection [2,5,6]. Meanwhile, erythromycin productivity has also been enhanced by the optimization of fermentation process [7,8]. Since 1990s, genetic studies have provided insights into the genes involved in erythromycin biosynthesis, and the erythromycin gene cluster was found to contain 20 genes arranged in four major polycistronic units [9-11], which facilitated strain improvements by genetic and metabolic engineering [12-15]. Recently, BldD, a key developmental regulator in actinomycetes [16,17], was identified to positively regulate the synthesis of erythromycin at the transcriptional level [18], which opened the possibility of enhancing the erythromycin production by modifying the regulation mechanism. However, bldD was later reported to exhibit an opposite gene expression pattern with respect to most of ery genes in a higher-producing strain [6], which made the regulation mechanism underlying erythromycin production complicated and confusing. As a result, the engineering works have still been focusing on erythromycin biosynthesis pathway [19] and feeder pathway [15,20].
Noticing that genetic modifications targeting to crucial genes are sometimes lethal, people realized that omics data may pave the way for the productivity optimization at the whole-genome scale from the perspective of systems biology [21]. In recent years, the development of high-throughput technologies has revived efforts to increase strain productivity [22-26]. For example, a new regulator of avermectin biosynthesis in Streptomyces avermitilis, HrdB, was identified through transcriptome profiling in our previous study, and increased productivity was achieved by modifying the hrdB gene [27].
The complete genome of S. erythraea strain NRRL23338 was sequenced in 2007 [1], which allows more rational improvement of strains to achieve high-titer erythromycin production. Several expression microarray experiments based on the wild-type strain and mutant strains including both overproducing and defected strains were later on released [6,28-30]. It was found that genes involved in erythromycin biosynthesis cluster and feeder pathway were up-regulated at the transcriptional level in overproducing strains. Even though, the global genetic basis for erythromycin biosynthesisis far from full elucidation.
In this work, we sequenced the whole genome of a high-producing strain (E3) obtained from random mutagenesis and screening, and examined time-series expression profiles of both E3 and the wild-type strain NRRL23338. A large number of genomic variations, including 60 insertions, 46 deletions and 584 single nucleotide variations (SNV) were identified in E3 in comparison with NRRL23338; the genes involved in erythromycin biosynthesis and feeder pathways were significantly up-regulated during the 60 hours time-course. According to our data, bldD did not show any positive correlations with the expression of ery cluster, implying alternative regulation mechanisms of erythromycin synthesis in S. erythraea, and/or the combinatorial effects of various regulation mechanisms. By integrating comparative genomic data to comparative transcriptomic data, we proposed several regulators which are potentially associated with erythromycin production. This functional comparative genomics work represents an important step towards understanding the over-producing mechanism of S. erythraea strain E3 on a genomic scale, and provided useful clues to strain engineering for improved production of erythromycin.
Results
S. erythraea E3 strain genome features and comparisons with NRRL23338 strain
The main features of the genome sequence of high-producing strain E3 are schematically represented in Figure 1, and summarized in comparison with wild-type strain NRRL23338 in Table 1.
Figure 1.
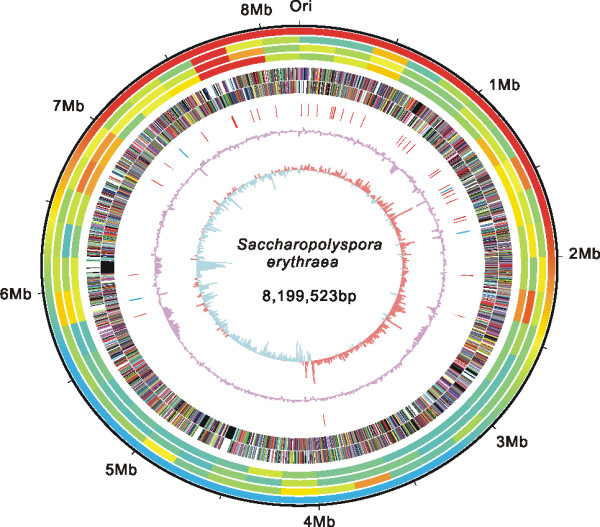
Schematic representation of the S. erythraea E3 genome. The outer scale is numbered in megabases from the origin of replication (ori) and the first circle indicates the core (red) and noncore (blue) regions, and the boundaries between them are mark by gradient colors. Circles 2, 3 and 4 (from the outside in), whole genome alignment (by Mummer) between S. erythraea and M. tuberculosis, N. farcinica, S. coelicolor, respectively (conservation of sequence regions are marked by gradient colors: blue, yellow, red. Most conserved regions are in red.). Circles 5 and 6, all genes (forward and reverse strand, respectively) colored corresponding to the COG functional assignment. Circle 7, tRNAs (red) and rRNAs (skyblue). Circle 8, GC content; circle 9, GC skew ((G – C/G + C), plum indicates values >1, lightblue values <1).
Table 1.
General features of the S. erythraea E3 and NRRL23338 genome
| Component of the genome | NRRL23338 | E3 |
|---|---|---|
|
Length |
8,212,805 bp |
8,199,523 bp |
|
G + C content |
71.10% |
71.00% |
|
Coding sequences |
7,264 |
7,257 |
|
rRNA |
6* |
12 |
|
tRNA |
50 |
50 |
| CDSs | 7,198 | 7,195 |
CDS, protein-coding sequence. *attributed to a statistical error of the previous literature.
The genome of strain E3 is comprised of a single circular chromosome of 8,199,523 bp, which is 0.16% smaller than that of NRRL23338 (8,212,805 bp) [1]. The average GC content of the E3 genome is 71.0%, almost the same as NRRL23338 (71.1%) [1]. The replication origin of E3 genome was determined by homology mapping to NRRL23338, which was identified based on GC skew and the position of the replication origin oriC. In strain E3, there is a definite GC skew inversion at oriC and also on the opposite side of the chromosome to oriC. The initiation codon of the dnaA gene, adjacent to oriC, was set as the starting point for numbering the coding sequences.
The overall features of E3 genome are highly similar to NRRL23338 (Table 1). In summary, E3 contains a total of 7,257 predicted coding sequences, including 7195 protein-coding sequences (CDSs), 50 tRNA genes, and 12 rRNA genes in four copies of 16S-23S-5S rRNA operons. The coding density is 84.9%, identical to strain NRRL 23338. The 50 tRNA genes encode all tRNAs required for protein biosynthesis. The rRNA gene number of E3 strain, 12, is different from that of NRRL23338 reported earlier, 16 [1]. To address this doubt, we carefully compared the rRNA coding regions of two strains, and found that their sequences are completely identical. We then noticed that there are actually only 12 rRNA coding gene records of NRRL23338, but not the claimed 16, deposited in NCBI. All these observations suggest that there should be a total of 12 rRNA genes in both E3 and NRRL23338. Among the 7195 CDSs, a putative function could be ascribed to 4783 (66.5%) of these. Of the rest, 829 (11.5%) showed similarity to hypothetical proteins in other genomes, and 1,583 (22.0%) had no substantial similarity to predicted proteins in public databases.
Similar to the NRRL23338 strain, the core region of the genome, extending either side of the oriC and ending in the regions of markedly lower GC content (Figure 1), accounts for a total of 4.3 Mbp. In comparison with the regions outside the core, the core region shows substantial conservation when comparing with other actinobacteria (e.g. M. tuberculosis, N. farcinica, S. coelicolor, Figure 1) [1].
The genomic changes in E3 relative to NRRL23338 involve 60 insertions, 46 deletions and 584 single nucleotide variations (SNVs) (see Additional file 1: Table S1). Most of the variations occur in intragenic regions: 40 out of 60 insertions, 28 out of 46 deletions and 511 out of the 584 SNVs are located in intragenic regions, and the other 20 insertions, 18 deletions and 73 SNVs are located in intergenic regions (Additional file 1: Table S1). Based on the intragenic variations, which are expected to contribute to the altered phenotype, we identified 139 proteins whose amino acid sequence is changed by SNVs, 28 proteins and 32 proteins which are affected by deletions and insertions respectively (Additional file 2:Table S2, Additional file 3: Table S3, Additional file 4: Table S4). These altered proteins are overrepresented in the COG categories of “replication, recombination and repair” with p-value of 4.20E-08, and “signal transduction mechanisms” with p-value of 0.005.
The largest variation between E3 and NRRL23338 genomes is an 11 Kb deletion which spans 11 genes including a predicted integrase and a predicted aminoglycoside phosphotranserase as well as 9 hypothetical proteins (Additional file 3: Table S3). It is likely to be a prophage (integrated plasmid) because its gene content and organization are very similar to the isolated plasmids pSE101 and pSE211 previously identified in strain NRRL23338 [1]. Moreover, E3 genome are found to contain integrated pSE101 and pSE211, one copy for each plasmid, which include14 and 27 candidate protein-encoding genes respectively, spanning 10.9 kb and 17.3 kb [1]. It is noticeable that the integrated plasmid regions in strain E3 and the isolated pSE101 and pSE211 in strain NRRL23338 share an integrase (recombinase) gene.
Comparative transcriptome analysis between E3 and NRRL23338 strains
Beyond the whole genome sequencing of two S. erythraea strains, high-producing E3 and the wild-type NRRL23338, we designed a time-course microarray experiment to investigate the gene expression profiles of E3 and NRRL23338. Six time points 10 h, 16 h, 24 h, 36 h, 48 h, 60 h were chosen according to the growth curve [28] and erythromycin production curve (Additional file 5: Figure S1) of S. erythraea.
A total of 1500 genes (approximately 20% of all S. erythraea genes) were identified to be differentially expressed (DE) in terms of between-time series, which emphasizes on the expression variation at the same time point between two strains. The differentially expressed genes (DEGs) were then clustered into 3 groups according to the pattern of expression change: up-regulated in strain E3, down-regulated in strain E3, different expression pattern across time between two strains (Figure 2 and Additional file 6: Table S5).
Figure 2.
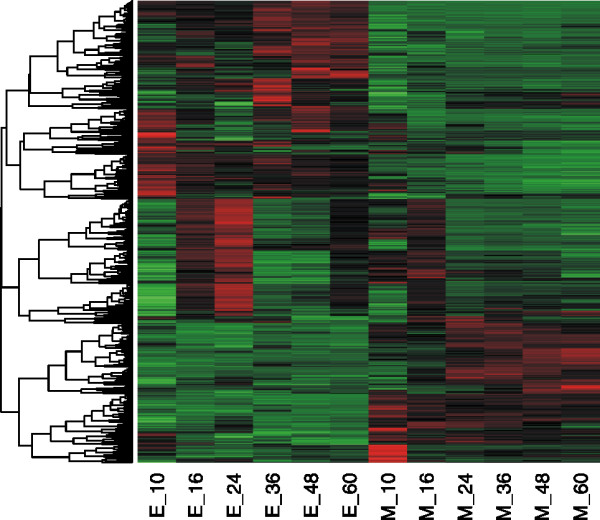
Heatmap of the 1500 differentially expressed genes between NRRL23338 and E3. Red = up-regulated, Green = down-regulated, the dendrogram on the left is the clustering result, the sample names (M: model strain NRRL23338, E: high-producing strain E3, the number represents the time point) are list on the bottom.
According to the functional enrichment analysis results based on COG category annotation [31] (Additional file 7: Table S6), [C] energy production and conversion and [Q] Secondary metabolites biosynthesis, transport and catabolism are up-regulated in strain E3; [O] posttranslational modification, protein turnover, chaperones and [E] amino acid transport and metabolism are over-represented both in up-regulated genes and down-regulated genes in strain E3, indicating that these functions present different activities in two strains, and some of the related genes are up-regulated in E3 while some are down-regulated (Additional file 7: Table S6). Additionally, [L] replication, recombination and repair are enriched in the genes with different expression profile across time between two strains. Among them, we carefully scrutinized the specific pathways related to fermentation physiology, including erythromycin biosynthesis, feeder pathway, transporters, energy production and secondary metabolite synthesis.
Erythromycin biosynthesis and feeder pathways
Noticeably, all genes (SACE_0712-0721, SACE_0723-0734) in erythromycin biosynthesis cluster (ery cluster) were dramatically up-regulated (Mean: 5.8-fold, Max: 30.8-fold) for the duration of 60 hours time-course in the overproducing strain E3 in comparison with the wild-type NRRL23338 (Additional file 8: Table S7), consistent with previous observations that the expression of ery cluster was correlated to erythromycin production [11,32]. Moreover, the feeder pathway was also activated in E3 (Additional file 6: Table S5, Additional file 8: Table S7). In many streptomycetes, at least four pathways have been characterized to be connected to the methylmalonyl-CoA pool (precursor of erythromycin biosynthesis): MCM pathway which catalyzes the reversible isomerization of succinyl-CoA and methymalonyl-CoA, CCR pathway utilizing crotonyl-CoA reductase or adenosylcobalamin-dependent isobutyryl-CoA mutase, MeaA pathway from acetoacetyl-CoA, and PCC pathway through carboxylation of propionyl-CoA by proprionyl-CoA carboxylase. It was found that S. erythraea has no CCR pathway and MeaA pathway for the precursor supply [1]. Genome sequence analysis suggests that PCC pathway may play a role in precursor flow in S. erythraea. In S. erythraea at least five genetic loci (SACE_0026-0028; SACE_3241-3242; SACE_3398-3400; SACE_3856/6509; SACE_4237) might encode biotin-dependent carboxylases catalyzing carboxylation of propionyl-CoA to methylmalonyl-CoA. So far, it is still unclear which gene set makes a contribution to erythromycin biosynthesis. Comparative transcriptome analysis found that only SACE_4237 gene displayed obvious up-regulation in high-producing strain E3, indicating that over-expression of SACE_4237 may enhance supply of methylmalonyl-CoA through carboxylation of propionyl-CoA. Further experiments are required to validate this assumption.
The expression of genes encoding key enzymes of carbon and fatty acid metabolisms was also significantly changed in turn affecting the flux of metabolites through erythromycin feeder pathways. The industrial strain E3 exhibited an impressive activation of fatty acid catabolic pathway, glycolysis/citrate cycle pathways, valine, leucine and isoleucine degradation pathway that supplies propionyl-CoA and methymalonyl-CoA for biosynthesis of 6-deoxyerythronolide B. The genes coding for key enzymes of valine, leucine and isoleucine catabolic pathway (fadE1, SACE_4125 coding for acyl-CoA dehydrogenase; echA7, SACE_4571 coding for enoyl-CoA hydratase/isomerase-like activity; pksG, SACE_4570 encoding hydroxymethylglutaryl-CoA synthase; mmsA1, SACE_4672 encoding methylmalonate-semialdehyde dehydrogenase) were strongly over-expressed in industrial strain E3. Up-regulation of genes involved in fatty acid catabolic pathway, including SACE_4038 (fadD) coding for long-chain acyl-CoA synthetase, SACE_4125, SACE_4571, SACE_6363 (fadA) encoding acetyl-CoA acetyltransferase, maybe also lead to the increased propionyl-CoA in strain E3. The glycolysis/citrate cycle pathways are connected to the feeder pathway of erythromycin biosynthesis via succinyl-CoA, an important metabolite of the Krebs cycle. Transcription of gap, pgk, tpiA and eno in glycolysis was moderately elevated in strain E3. Most genes of TCA cycle, including SACE_1638 (sucB), SACE_3674 (mdh), SACE_3811 (acn), SACE_3926/SACE_3927 (korA), SACE_3952 (pdhA2), SACE_3953 (pdhB1), SACE_3954 (bkdC2), SACE_4581 (citE3), SACE_6118 (pyc), SACE_6636 (isocitrate dehydrogenase) and SACE_6668/6669 (sucC), showed higher expression level, as compared to wild-type strain. It was found that the variations affected three genes of the Krebs cycle, including SACE_0633 (citrate synthase), SACE_6636 and SACE_6668 (Additional file 2: Table S2, Additional file 3: Table S3, and Additional file 4: Table S4).
These results demonstrate that enhanced level of erythromycin biosynthesis in strain E3 is likely to be attributed to the alterations in many pathways, and the up-regulation of erythromycin biosynthesis and feeder pathway seemed to be crucial events.
Transporters
Differential expression analysis showed that genes encoding phosphate transport system (SACE_7097-7099, SACE_6643-6645), Iron ion uptake system (SACE_4076-4077), D-methionine transport system (SACE_0804-0806), and sulfonate/nitrate/taurine transport system (SACE_0556, SACE_1672-1674) were significantly activated, among which the transport of D-methionine (SACE_0806) was most activated (5.9 folds). PhoP-regulated phn operon (SACE_6643-6645) was induced by 5.5 folds. Moreover, several genes involved in nitrogen sources intake and assimilation were inhibited in E3 strain, including the oligopeptide transporter operons (SACE_0257-0260, SACE_0844-0848), amino acid transporter operon (SACE_6266-6269), glutamate transporter operon (SACE_1743-1746), glutamine transporter operon (SACE_7284-7286), and ammonium transporter gene (amt, SACE_6062). The most repressed gene was amt (12.5 folds). It was observed that the genes related to phosphate and nitrogen transport were differentially expressed between two strains. Compared with NRRL23338, E3 showed phosphate starvation response and repressive nitrogen-metabolism through the entire time-course. These observations indicated that P-N metabolism balance may play an important role in the overproducing strain E3.
Energy production
Generally speaking, the expression of genes involved in bioenergetics and oxidative phosphorylation was moderately enhanced in industrial E3 strain, which may contribute to energy supply for erythromycin production. The most important members of this group are nuo operon encoding NADH dehydrogenase I (nuoN-A, SACE_6889-6902) and atp operon coding for ATP synthases (atpDGAHFEB, SACE_6280-6286). This is consistent with the observation that the nuo genes exhibited higher expression level in mutant rif1 (higher producer) than wild type and mutant rif6 strains (lower producer) [6].
Secondary metabolite synthesis
There are 25 clusters (involving a total of 202 genes) for the biosynthesis of polyketides, terpenes and nonribosomally synthesized peptides in the S. erythraea genome. Three clusters (tpc2, tpc3, and tpc4) of the six terpene synthase gene clusters, three PKS gene clusters (pks3, pke, and pks6), and one gene cluster encoding nonribosomal peptide synthetase (nrps1) were activated in strain E3 in comparison with NRRL23338 (Figure 3). Among these up-regulated gene clusters, pks6 (SACE_4567-4577) showed the most significantly induced expression (Mean: about 5.7 folds; Max: about 21 folds). In contrast, only two gene clusters for production of secondary metabolites (nrps6 and rpp) were inhibited in E3. This indicated the general enhancement of secondary metabolite synthesis in high producing strain.
Figure 3.
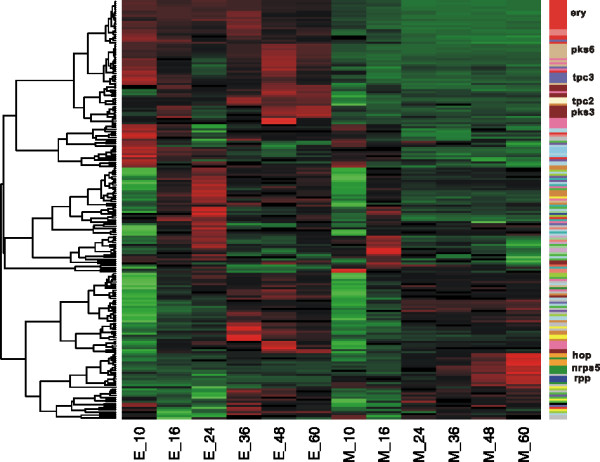
Heatmap of secondary metabolite synthesis gene clusters between NRRL23338 and E3. Red = up-regulated, Green = down-regulated, the dendrogram on the left is the clustering result, the sample names (M: model strain NRRL23338, E: high-producing strain E3, the number represents the time point) are list on the bottom. Genes in the same gene cluster are marked in the same color, the well clustered gene cluster ery, tpc2, tpc3, pks3, pks6, nrps5 and hopp and rpp clusters are marked on the left.
Since differential coexpression analysis (DCEA) is much more useful than traditional differential expression analysis (DEA) for investigating the global transcriptional mechanisms underlying the associated phenotypic changes [33,34], we then ranked genes according to its differential coexpression with the others in the coexpression network by using DCp method [35] (Additional file 8: Table S7). Noticeably, Ure operon, nar operon, nir operon, glnA2, glnB), involved in nitrogen metabolism, were differentially coexpressed, implying an altered coordination of nitrogen metabolism with the other functions in strain E3 compared with NRRL23338.
Functional genomics analysis
After separately analyzing the genomic data and transcriptomic data, we set out to explore the links between genomic variations and transcriptomic variations, which provide an opportunity to identify causative mutations accounting for the high productivity, and eventually elucidate the regulation mechanisms underlying the erythromycin synthesis.
We first focused on the ery cluster genes, including the promoter regions, and found that they were identical at the genomic level in both strains, but significantly up-regulated at the transcriptional level in high-producing strain E3. We then resorted to the upstream regulatory factors of ery cluster. The coordinate alteration of the ery cluster gene expression suggested that the ery cluster may be co-regulated by the regulator(s), whose differences at the sequence level in two strains may lead to the expression difference of ery cluster. The transcriptional regulator, bldD (SACE_2077), which was reported to positively regulate the ery cluster [18], however, showed lower expression level in overproducing strain E3, opposite to the ery genes. This was consistent with the data reported more recently [6]. It seems that the regulatory effect of of bldD on ery cluster needs further investigation. At least, there must be other regulatory factors involved in the transcriptional regulation of ery genes.
We manually checked the annotation information of these genomic variant genes, and found a total of 18 potential regulators (Additional file 9: Table S8 and Additional file 10: Dataset S1). According to the comparison to the sequence of orthologous genes in closely related species, 7 genes were supposed to be mutated in E3 and 2 genes were supposed to be mutated in NRRL23338; 11 genes were uncertain as no orthologues of them were found. We focused on the two regulators, MtrA and MoxR, which were mutated in the functional domain regions in strain E3.
In S. erythraea, SACE_6447-SACE_6445 operon encodes proteins that are homologous to the products of the mtrAB-lpqB genes of Mycobacterium tuberculosis. MtrA is a member of the OmpR/PhoB family of response regulators, forming a two-component system (TCS) with its cognate sensor kinase (MtrB). According to NCBI conserved domain database (CDD, http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml), Pfam (http://pfam.janelia.org), and the three-dimensional structure drawn from Mycobacterium tuberculosis[36], MtrA possesses two domains, an N-terminal regulatory domain displaying the classic α/β fold and a C-terminal DNA-binding domain with a winged helix-turn-helix. MtrA has been related to the regulation of diverse cellular functions [37-40]. In S. coelicolor, SCO3011-SCO3013 operon appears to be an orthologue of the mtrAB-lpqB operon. The function of this operon is unclear but null mutations in mtrB and lpqB affect normal septation and sporulation. Attempts to disrupt mtrA have so far been unsuccessful, suggesting this may also be an essential gene in Streptomyces[41]. Analysis of the limited experimental research available on MtrAB suggests that the primary role of MtrAB is in regulating cell cycle progression, the composition and function of the cell envelope or, perhaps more likely, in responding to general stresses that could ultimately inhibit cell division. However, many questions remain about the MtrAB two-component system in Streptomyces, such as: how does MtrAB govern growth and MtrAB govern growth and development? How does MtrAB play a role in antibiotic biosyntheses? To date, no experimental work has been performed on this operon, so information about its function in the cell is not available. Recently the direct repeats of GTCACAgcg-like consensus sequences (MtrA-Box) recognized by MtrA were identified in M. tuberculosis[42], but we failed to find it in S. erythraea genome, indicating that S. erythraea has distinct MtrA binding site upstream of the target genes [43]. Although both mtrA and mtrB werenot differentially expressed between two S. erythraea strains, MtrAofE3 strain had two amino acids deleted (H196 and V197), compared to M. tuberculosis and S. erythraea NRRL23338. The deletion was located in the C-terminal domain, and specifically, next to the DNA-binding site, V195 (Additional file 9: Table S8 and Additional file 10: Dataset S1).
We therefore applied homology modeling to study the variation sites of E3 MtrA in the structural context. It was found that the deleted sites in the M. tuberculosis model was in an α-helix (Additional file 11: Figure S2), which may lead to the distortion of the related helix or even more. This probably interfere its DNA binding activity, and thus the regulatory activity of MtrA. Follow-up functional experiments of MtrA in S. erythraea will help to identify its regulatory role in antibiotic synthesis.
We also observed another interesting regulator, MoxR (SACE_3795). The MoxR family is a subset of AAA + ATPases, a large, diverse group of ATPases associated with various cellular activities including cell-cycle regulation, DNA repair and replication, protein proteolysis and disaggregation, and so on. MoxR is often observed in close proximity to Von Willebrand Factor Type A (VWA) proteins and are likely to function with them to form a chaperone system. More importantly, MoxR also acts as DNA helicases and transcription factors [44]. In S. erythraea, the moxR gene is immediately adjacent to two genes (SACE_3794 and SACE_3793) encoding VWA proteins. A single-nucleotide variation (N133D) was identified in E3 moxR gene (Additional file 11: Table S2 and Additional file 12: Figure S3), compared with NRRL23338 and other related species, such as Actinosynnema mirum DSM 43827. When searching NCBI CDD, we found this protein was a putative member of the AAA + (ATPases Associated with a wide variety of cellular Activities) superfamily. The sequence alignment showed that the variation N133D was located in the very vicinity of ATP-binding site (two residues away) and Walker B motif (related to Mg++ binding, one residue away), suggesting that the substitution may perturb its activity of ATP and Mg++ binding, although the homology modeling result did not provide further evidence. Additionally, of the 18 regulator genes, moxR gene was mostly strongly over-expressed in industrial strain E3 compared with NRRL23338 (about 3-fold).
Discussion
Erythromycin and its semi-synthetic derivatives are widely used in the clinic, and thus improved producers are highly sought after. In recent years, the availability of the entire genome sequence of S. erythraea has opened the possibility of defining the erythromycin biosynthesis mechanisms by using global approaches [1,28]. These high-throughput approaches have led to the discovery that BldD, a key developmental regulator in actinomycetes [16,17], which activates the synthesis of erythromycin at the transcriptional level [18]. Meanwhile, there is evidence that increasing the flux through feeder metabolic pathways strongly influences the erythromycin yields, which has been recently obtained by engineering the methylmalonyl-CoA metabolite node in S. erythraea and in A. erythreum, a non-filamentous erythromycin A producer [15,45,46].
In our present work, the comparative analysis of S. erythraea overproducing E3 and wild-type NRRL23338, at both genomic and transcriptomic level, indicate that ery clustering for erythromycin synthesis is up-regulated in E3 as expected, whereas E3 and NRRL23338 have identical ery genomic sequences (Figure 3). Furthermore, the feeder pathways were also activated in E3 strain. This could elevate the concentrations of propionyl-CoA and methymalonyl-CoA, the precursor metabolites of erythromycin production (starter unit and extender unit respectively), and thus improve the erythromycin yield. Indeed, increased supply of methylmalonyl-CoA has been demonstrated to increase erythromycin production, which was achieved by altering the metabolic flux distribution of its different precursors through genetic manipulation [15]. The extender unit, methymalonyl-CoA, can be derived from different pathways such as carboxylation of propionyl-CoA and rearrangement of succinyl-CoA. The mutA and mutB genes (SACE_5638, 5639) encode methylmalonyl-CoA mutase (MCM), which catalyzes the reversible isomerization of succinyl-CoA and methymalonyl-CoA. Reeves et al. have proposed a metabolic model where in carbohydrate-based fermentations MCM acts as a drain on the methylmalonyl-CoA metabolite pool, and in oil-based fermentations, MCM acts in the reverse direction to fill the methylmalonyl-CoA pool [45]. The induction or repression of mutAB may elevate or reduce concentrations of methymalonyl-CoA to affect the erythromycin synthesis rate depending on the medium. Therefore, overproduction of erythromycin was achieved by inactivating the mutB in a carbohydrate-based medium [15], or by duplication of the MCM operon (mutA, mutB, meaB and mutR) in an oil-based fermentation medium [20]. In this study, the carbon flow under industrial medium condition may be from succinyl-CoA to methylmalonyl-CoA. Improvement of erythromycin production in E3 strain could be at least partly attributed to the increase of methylmalonyl-CoA pool as a result of the overexpression of mutAB genes.
More interestingly, bldD has the same sequences in two strains, and is down-regulated in the overproducing E3, which is contrary to the observation reported by the BldD finder [18], while consistent with a more recent literature [6]. Other possible regulation mechanisms of erythromycin synthesis in S. erythraea are therefore expected.
According to our data, the differential expression between overproducer E3 and wild-type NRRL23338 significantly involved transport of phosphate and nitrogen, and phosphate starvation response and repressive nitrogen-metabolism was observed in E3 through entire time-course compared with NRRL2338 strain. This suggests that the high-level expression of erythromycin biosynthesis pathway probably result from the dysfunction of global regulators sensing stress or nutritional starvation signal, as is known in the signaling system of PhoP-AfsR-AfsS in S. coelicolor A3(2), controlling expression of genes involved in P-N nutrition stress response and secondary metabolism [47]. AfsS, a 63-amino-acid sigma-like regulatory protein, was found in streptomycetes including S. coelicolor, S. dividans, S. griseus, and S. noursei. In S. coelicolor, the activation of AfsS by AfsR can further activate the transcription of genes coding for pathway-specific transcription factors, e.g. actII-ORF4 and Moreover, AfsS, as a crucial master regulator of both antibiotic synthesis and nutritional stress response, also regulates the expression of genes involved in phosphate transport and nitrogen metabolism. The global network of signal transduction cascades and cross-talk of PhoP and AfsR-AfsS was modeled recently, which controls gene expression involved in P-N balance and secondary metabolism in S. coelicolor[48-50]. PhoP repressed the transcription of nitrogen genes by binding to the promoter of glnR, the major nitrogen regulator, to the promoters of glnA and glnII, two glutamine synthetases, and to the promoter of the amtB-glnK-glnD operon, encoding an ammonium transporter [51]. It is interesting that the S. erythraea orthologs of these genes and other GlnR-regulon genes were significantly down-regulated in strain E3, such as glnR (SACE_7101), glnB (SACE_6061) encoding the nitrogen regulatory protein PII, amt (SACE_6062), glnA-1 (SACE 1623) coding for the glutamine synthetase, ure operons (SACE_0634-0636, SACE_2526-2527) coding for the urease, narK-nir operon (SACE_3799-3803) encoding the assimilatory nitrite reductase and nitrite extrusion protein. The previous works also demonstrated that many genes related to nitrogen metabolism showed lower expression in S. erythraea overproducing strain rif1 with S444F mutation in rpoB than in impaired strain rif6 with Q426R mutation [6]. In order to investigate if these nitrogen metabolism genes are under the control of PhoP in S. erythraea, we searched these genes for PHO boxes using the model based on the alignment of 25 PHO DRu compiled by Blanco et al. [52], and detected one candidate PHO box (two DRus: GTTCGCCTTCTGTTCACAATTG) in the glnR promoter region, and one (two DRus: CTTCCCGTGCCGTTCAGCAACG) in the afsR promoter. However, we failed to find sequences coding AfsS-like protein in S. erythraea genome. So far the signaling system of PhoP-AfsR-AfsS, controlling expression of genes involved in P-N nutrition stress response and secondary metabolism in S. erythraea has not been built. Our findings suggest the existence of a similar regulation mechanism underlying the erythromycin biosynthesis in S. erythraea (Figure 4).
Figure 4.
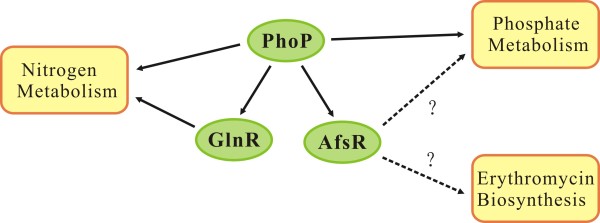
Putative regulatory roles of PhoP, GlnR, and AfsR in phosphate, nitrogen metabolism and erythromycin biosynthesis in S. erythraea.
Taken together, it seems that the regulatory mechanism of erythromycin biosynthesis is much more complicated than expected, probably involving more than one global regulator. Further function experiments, such as the identification of the regulators and their target genes, the identification of PhoP-AfsR-AfsS signaling system, will help to elucidate the precise mechanisms. Furthermore, the accumulation of genomic and transcriptomic data for more over-producing strains will also help to find out the causal variations which directly contribute to antibiotic production. Actually, a comparative genomics and transciptomic profiles of another erythromycin over-producing strain Px and NRRL2338 was released very recently [53]. It was found that the genomic variations affect a total of 227 proteins of Px strain and a quite number of mutations alter key enzymes in the central carbon and nitrogen metabolism and in the biosynthesis of secondary metabolites, which probably results in the redirection of common precursors for erythromycin biosynthesis [53]. Similar to the current report, it is difficult to sort out the mutations which are directly related to the high production of erythromycin. Besides experimental verification, next efforts on the comparison of different overproducers by using advanced data integration techniques will help to generate more insights on the regulation mechanism of erythromycin biosynthesis, and eventually provide valuable information on new strategies for strain improvement.
Conclusions
In this work, we sequenced the whole genome of a high-producing S. erythraea strain E3 obtained by random mutagenesis and screening from the wild-type strain NRRL23338, and examined time-series expression profiles of both E3 and NRRL23338. Based on the genomic data and transcriptpmic data of these two strains, we carried out comparative analysis of high-producing strain and wild-type strain at both the genomic level and the transcriptomic level. A large number of genomic variations, including 60 insertions, 46 deletions and 584 single nucleotide variations (SNV) were identified in E3 in comparison with NRRL23338, and the analysis of time series transcriptomic data indicated that the genes involved in erythromycin biosynthesis and feeder pathways were significantly up-regulated during the 60 hours time-course. According to our data, BldD, a previously identified ery cluster regulator, did not show any positive correlations with the expression of ery cluster, suggesting the existence of alternative regulation mechanisms of erythromycin synthesis in S. erythraea. Several potential regulators were then proposed by integration analysis of genomic and transcriptomic data. This functional comparative genomics work represents an important step towards understanding the over-producing mechanism of S. erythraea strain E3 on a genomic scale, and provided useful clues to strain engineering for improved production of erythromycin.
Methods
Bacterial strains and culture conditions
Two S. erythraea strains, the wild-type NRRL23338 and a high-producing strain E3, were used in this study. S. erythraea strains were grown on agar plates of the medium [10 g cornstarch, 10 g corn steep liquor, 3 g NaCl, 3 g (NH4)2SO4, 2 g CaCO3, and 2 g agar per liter of distilled H2O, pH 7.2] at 34°C for sporulation. An agar piece about 1 cm2 was inoculated into a 500 ml flask containing 50 ml of the seed medium [50 g/L cornstarch, 18 g/L soybean flour, 13 g/L corn steep liquor, 3 g/L NaCl, 1 g/L (NH4)2SO4, 1 g/L NH4NO3, 5 g/L soybean oil, and 6 g/L CaCO3, pH 6.8 to 7.0] and grown for 48 h at 34°C and 200 rpm, then added 5 ml of the seed culture to a 500 ml flask containing 60 ml of the fresh industrail fermentation medium, incubation was continued at 34°C and 200 rpm for 4 days. The industrail medium consists of 40 g/L cornstarch, 30 g/L soybean flour, 30 g/L dextrin, 2 g/L (NH4)2SO4, 10 g/L soybean oil, and 6 g/L CaCO3 (pH 7.0 to 7.2). All media types were sterilized by autoclaving at 121°C for 30 min. Both supernatant and cell pellet samples (1 ml) were harvested at six different time points (10 h, 16 h, 24 h, 36 h, 48 h, and 60 h) during 4 days culture period, and erythromycin titers and biomass were determined. Mycelium for RNA isolation were immediately quenched in liquid nitrogen, and stored at −80°C until further use. Two independently cultured replicates were performed for the two strains fermentation experiment.
Erythromycin titer measurements
The amount of the erythromycin produced in the fermentation broth was determined by means of biological method. Bioassays for erythromycin were performed using a large plate double-agar layer system. The bottom agar layer consisted of the test medium (5 g peptone, 3 g beef extract, 3 g K2HPO4, and 15 g agar per liter of distilled H2O) without Bacillus pumilus culture. Once the bottom layer was solidified, a top agar layer (contains B. pumilus culture) was poured. After the upper layer was solidified at room temperature, fermentation supernatant (260 μl) was added to stainless steel cylinders on agar plates. The bioassay plates were incubated overnight at 37°C. After incubation, the erythromycin production was estimated by measuring the diameters of the inhibition zones and calculated according to the calibration curve made by using the commercially available erythromycin as a control. Whereas two strains grew at similar rates, the E3 strain produced about 30-fold more erythromycin (600 mg/L) than the NRRL23338 strain (20 mg/L) (Additional file 5: Figure S1).
Genome sequencing and assembly
The nucleotide sequence of the S. erytheraea strain E3 genome was determined by using a massively parallel pyrosequencing technology (Roche 454 GS FLX). A total of 273 contigs (>500 bp) with a total size of 8.2 Mb were assembled from 876,307 reads (average length of 238 bp) using Newbler software of the 454 suite package, providing a 25-fold coverage. Relationship among contigs was determined using the genome of S. erytheraea NRRL 23338 as reference, and validated by PCR. Gaps between contigs were filled by sequencing PCR products. The final sequence assembly was carried out using phred/phrap/consed package (http://www.phrap.org/phredphrapconsed.html), and all the low sequence quality regions, including homopolymeric sites, were resequenced using PCR Sanger sequencing. The final sequencing accuracy of genome was 99.9965%.
Genome annotation and analysis
CDSs were predicted by using Genemark (http://exon.biology.gatech.edu/) [54] and Glimmer (http://ccb.jhu.edu/software/glimmer/index.shtml) [55], and manually curated. Functional annotation of CDSs was performed through comparisons to NCBI nr database using BLASTP (2.2.14) [56] and classified according to COGs (http://www.ncbi.nlm.nih.gov/COG/) [31] and Gene Ontology (http://www.geneontology.org/) [57]. Transfer RNA genes were predicted with tRNAscan-SE [58]. Whole genome alignment between strain E3 and NRRL23338 were performed by using BLASTN (2.2.14) [56].
Microarray construction and transcript profiling
The S. erythraea DNA microarrays (SER v1.0) were customized using Agilent eArray 6.0 according to the manufacturer’s recommendations (https://earray.chem.agilent.com/earray/).
Each customized microarray (8x15K) contained spots in duplicate with 7,198 gene-specific 60-mer oligonucleotide probes interrogating the 7,198 predicted ORFs in S. erythraea (as reported for the S. erythraea genome at http://131.111.43.95/gnmweb/index.html). RNA was extracted from mycelium pellets deriving from two independent culture samples using the Column Plant RNAout (TIANDZ) according to the standard protocols. The RNAs were subsequently purified by QIAGEN RNeasy Mini Kit. The quality and quantity were determined by nanodrop UV spectroscopy (Ocean Optics) and analysis on a RNA 6000 Nano LabChip (Agilent Technologies) using a 2100 bioanalyzer (Agilent Technologies). RNA samples were processed and hybridized to the customized chips SER v1.0.
Data processing and analysis
Data quality assessment and normalization were performed by using limma package in Bioconductor [59]. The differentially expressed genes were identified with timecourse package [60] in terms of between- time series by comparing time-course mean profiles with multivariate empirical Bayes model [61].
A recently published differential coexpression analysis method (‘DCp’) was applied to explore differentially coexpressed genes (DCGs) [35]. When constructing the coexpression networks for two contrastive strains, the correlation cutoff was set as 0.8 to filter coexpression links. The differentially coexpressed genes (DCGs) were ranked according to their p values.
Protein structure modeling
Homology modeling was applied to predict three-dimensional structures of the interested proteins. We first searched PDB (http://www.rcsb.org) for putative structural templates using NCBI BLAST [62] with sequence identity greater than 30%. After that, only the best template was kept according to the alignment coverage, sequence identity and structural resolution in combination. MODELLER was then adopted to concisely align the interested protein to its template, and build the three-dimensional homology model automatically [63]. DOPE score, as suggested by the manual of MODELLER, was used to sort out the best structure model from all those generated. The selected models were analyzed in PyMOL (http://www.pymol.org/).
Motif search method
The positional weight matrix (PWM) was generated according to the literature [64]. Upstream 300 bp and downstream 50 bp from the gene start positions were scanned by Patser to find potential target regulated genes and TFBSs [65].
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YWB, LH and ZY carried out the molecular genetics experiments. CX and LYY performed the data analysis. YZQ, YH and LBH also contribute to the data analysis. LYX, YBC and LYY conceived of the study, and participated in its design and coordination. LYY, YBC and CX draft the manuscript. ZSL contributed to the coordination. All authors read and approved the final manuscript.
Supplementary Material
All nucleotide variations detected in E3 compared with NRRL23338.
Single amino acid substitution detected in E3 compared with NRL23338.
Amino acid deletion detected in E3 compared with NRRL23338.
Amino acid insertion detected in E3 compared with NRRL23338.
Erythromycin production curve during the time-course for the industrial S. erythraea strain and the wild-type NRRL23338 strain. E1 and E2 are two replicates for the industrial strain; M1 and M2 are two replicates for the wild-type strain.
{kind=link}
Genome annotation of S. erythraea E3 strain.
COG classification of differentially expressed genes.
Gene expression data.
Regulators involving amino acid variation.
Sequence alignment results of 18 regulators.
Structural Models of MtrA (V197 H198 deletion): SACE_6447 and ETHR_6255. His197 and Val198 of SACE_6447 (A), deleted in ETHR_6255 (B), are located in the center of an α-helix. The deletions are supposed to break the helix and to perturb the DNA-binding function of Val196 in the active state. In addition, His197 contributes to the inter-domain interactions in the inactive state, and its deletion would thus affect the stability of ETHR_6255.
Structural Models of MoxR (N133D): SACE_3795 and ETHR_3703. The Asn133 in SACE_3795 (A), substituted by Asp133 in ETHR_6255 (B), is located in the vicinity of Asp130 and Asn172, both of which were annotated as ATP-binding sites according to NCBI CDD. The Asn133 may thereby be implicated in the ATP-binding through the proxy of Asp130 and Asn172, or even through direct interaction with ATP. In addition, the nearby residues 128–131 are also putative ‘Walker B motif’, which is important for interacting with Mg2+ cation. So the substitution of Asn133 to Asp133, resulting in negative charge on the side chain, may affect the ATP-binding or Mg2+ cation-binding of the protein and thus its catalytic capability.
Contributor Information
Yuan-Yuan Li, Email: yyli@scbit.org.
Xiao Chang, Email: roarchang@gmail.com.
Wen-Bang Yu, Email: xiaolidu@mail.ecust.edu.cn.
Hao Li, Email: lihaoexe@yahoo.com.cn.
Zhi-Qiang Ye, Email: yezq@pkusz.edu.cn.
Hui Yu, Email: yuhui@scbit.org.
Bao-Hong Liu, Email: bhliu@scbit.org.
Yan Zhang, Email: saenda@126.com.
Si-Liang Zhang, Email: siliangz@ecust.edu.cn.
Bang-Ce Ye, Email: bcye@ecust.edu.cn.
Yi-Xue Li, Email: yxli@sibs.ac.cn.
Acknowledgements
This work was financially supported by grants from National key basic research program (2012CB316501), National Key Technologies R&D Program (2012AA022101), National Natural Science Foundation of China (31171268, 31000380, and 21276079), and SRFDP (No. 20120074110009).
References
- Oliynyk M, Samborskyy M, Lester JB, Mironenko T, Scott N. et al. Complete genome sequence of the erythromycin-producing bacterium Saccharopolyspora erythraea NRRL23338. Nat Biotechnol. 2007;25:447–453. doi: 10.1038/nbt1297. [DOI] [PubMed] [Google Scholar]
- Mironov VA, Sergienko OV, Nastasiak IN, Danilenko VN. Biogenesis and regulation of biosynthesis of erythromycins in Saccharopolyspora erythraea: a review. Prikl Biokhim Mikrobiol. 2004;40:613–624. [PubMed] [Google Scholar]
- McDaniel R, Licari P, Khosla C. Process development and metabolic engineering for the overproduction of natural and unnatural polyketides. Adv Biochem Eng Biotechnol. 2001;73:31–52. doi: 10.1007/3-540-45300-8_3. [DOI] [PubMed] [Google Scholar]
- Leadlay PF. Combinatorial approaches to polyketide biosynthesis. Curr Opin Chem Biol. 1997;1:162–168. doi: 10.1016/S1367-5931(97)80005-1. [DOI] [PubMed] [Google Scholar]
- Adrio JL, Demain AL. Genetic improvement of processes yielding microbial products. FEMS Microbiol Rev. 2006;30:187–214. doi: 10.1111/j.1574-6976.2005.00009.x. [DOI] [PubMed] [Google Scholar]
- Carata E, Peano C, Tredici SM, Ferrari F, Tala A. et al. Phenotypes and gene expression profiles of Saccharopolyspora erythraea rifampicin-resistant (rif) mutants affected in erythromycin production. Microb Cell Fact. 2009;8:18. doi: 10.1186/1475-2859-8-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Enshasy HA, Mohamed NA, Farid MA, El-Diwany AI. Improvement of erythromycin production by Saccharopolyspora erythraea in molasses based medium through cultivation medium optimization. Bioresour Technol. 2008;99:4263–4268. doi: 10.1016/j.biortech.2007.08.050. [DOI] [PubMed] [Google Scholar]
- Zou X, Hang HF, Chu J, Zhuang YP, Zhang SL. Enhancement of erythromycin A production with feeding available nitrogen sources in erythromycin biosynthesis phase. Bioresour Technol. 2009;100:3358–3365. doi: 10.1016/j.biortech.2009.01.064. [DOI] [PubMed] [Google Scholar]
- Donadio S, Staver MJ, McAlpine JB, Swanson SJ, Katz L. Modular organization of genes required for complex polyketide biosynthesis. Science. 1991;252:675–679. doi: 10.1126/science.2024119. [DOI] [PubMed] [Google Scholar]
- Weber JM, Leung JO, Maine GT, Potenz RH, Paulus TJ. et al. Organization of a cluster of erythromycin genes in Saccharopolyspora erythraea. J Bacteriol. 1990;172:2372–2383. doi: 10.1128/jb.172.5.2372-2383.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves AR, English RS, Lampel JS, Post DA, Vanden Boom TJ. Transcriptional organization of the erythromycin biosynthetic gene cluster of Saccharopolyspora erythraea. J Bacteriol. 1999;181:7098–7106. doi: 10.1128/jb.181.22.7098-7106.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel R, Thamchaipenet A, Gustafsson C, Fu H, Betlach M. et al. Multiple genetic modifications of the erythromycin polyketide synthase to produce a library of novel “unnatural” natural products. Proc Natl Acad Sci USA. 1999;96:1846–1851. doi: 10.1073/pnas.96.5.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer BA, Admiraal SJ, Gramajo H, Cane DE, Khosla C. Biosynthesis of complex polyketides in a metabolically engineered strain of E. coli. Science. 2001;291:1790–1792. doi: 10.1126/science.1058092. [DOI] [PubMed] [Google Scholar]
- Kealey JT, Liu L, Santi DV, Betlach MC, Barr PJ. Production of a polyketide natural product in nonpolyketide-producing prokaryotic and eukaryotic hosts. Proc Natl Acad Sci USA. 1998;95:505–509. doi: 10.1073/pnas.95.2.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves AR, Cernota WH, Brikun IA, Wesley RK, Weber JM. Engineering precursor flow for increased erythromycin production in Aeromicrobium erythreum. Metab Eng. 2004;6:300–312. doi: 10.1016/j.ymben.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Elliot M, Damji F, Passantino R, Chater K, Leskiw B. The bldD gene of Streptomyces coelicolor A3(2): a regulatory gene involved in morphogenesis and antibiotic production. J Bacteriol. 1998;180:1549–1555. doi: 10.1128/jb.180.6.1549-1555.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliot MA, Bibb MJ, Buttner MJ, Leskiw BK. BldD is a direct regulator of key developmental genes in Streptomyces coelicolor A3(2) Mol Microbiol. 2001;40:257–269. doi: 10.1046/j.1365-2958.2001.02387.x. [DOI] [PubMed] [Google Scholar]
- Chng C, Lum AM, Vroom JA, Kao CM. A key developmental regulator controls the synthesis of the antibiotic erythromycin in Saccharopolyspora erythraea. Proc Natl Acad Sci USA. 2008;105:11346–11351. doi: 10.1073/pnas.0803622105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Deng W, Wu J, Qian J, Chu J. et al. Genetic modulation of the overexpression of tailoring genes eryK and eryG leading to the improvement of erythromycin A purity and production in Saccharopolyspora erythraea fermentation. Appl Environ Microbiol. 2008;74:1820–1828. doi: 10.1128/AEM.02770-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves AR, Brikun IA, Cernota WH, Leach BI, Gonzalez MC. et al. Engineering of the methylmalonyl-CoA metabolite node of Saccharopolyspora erythraea for increased erythromycin production. Metab Eng. 2007;9:293–303. doi: 10.1016/j.ymben.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephanopoulos G, Kelleher J. Biochemistry. How to make a superior cell. Science. 2001;292:2024–2025. doi: 10.1126/science.1062556. [DOI] [PubMed] [Google Scholar]
- Bro C, Nielsen J. Impact of ‘ome’ analyses on inverse metabolic engineering. Metab Eng. 2004;6:204–211. doi: 10.1016/j.ymben.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Lee SY, Lee DY, Kim TY. Systems biotechnology for strain improvement. Trends Biotechnol. 2005;23:349–358. doi: 10.1016/j.tibtech.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Stafford DE, Stephanopoulos G. Metabolic engineering as an integrating platform for strain development. Curr Opin Microbiol. 2001;4:336–340. doi: 10.1016/S1369-5274(00)00214-9. [DOI] [PubMed] [Google Scholar]
- van der Werf MJ, Jellema RH, Hankemeier T. Microbial metabolomics: replacing trial-and-error by the unbiased selection and ranking of targets. J Ind Microbiol Biotechnol. 2005;32:234–252. doi: 10.1007/s10295-005-0231-4. [DOI] [PubMed] [Google Scholar]
- Askenazi M, Driggers EM, Holtzman DA, Norman TC, Iverson S. et al. Integrating transcriptional and metabolite profiles to direct the engineering of lovastatin-producing fungal strains. Nat Biotechnol. 2003;21:150–156. doi: 10.1038/nbt781. [DOI] [PubMed] [Google Scholar]
- Zhuo Y, Zhang W, Chen D, Gao H, Tao J. et al. Reverse biological engineering of hrdB to enhance the production of avermectins in an industrial strain of Streptomyces avermitilis. Proc Natl Acad Sci U S A. 2010;107:11250–11254. doi: 10.1073/pnas.1006085107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peano C, Bicciato S, Corti G, Ferrari F, Rizzi E. et al. Complete gene expression profiling of Saccharopolyspora erythraea using GeneChip DNA microarrays. Microb Cell Fact. 2007;6:37. doi: 10.1186/1475-2859-6-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum AM, Huang J, Hutchinson CR, Kao CM. Reverse engineering of industrial pharmaceutical-producing actinomycete strains using DNA microarrays. Metab Eng. 2004;6:186–196. doi: 10.1016/j.ymben.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Chang X, Liu S, Yu YT, Li YX, Li YY. Identifying modules of coexpressed transcript units and their organization of Saccharopolyspora erythraea from time series gene expression profiles. PLoS One. 2010;5:e12126. doi: 10.1371/journal.pone.0012126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B. et al. The COG database: an updated version includes eukaryotes. BMC Bioinforma. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve L, Baumberg S. Physiological controls of erythromycin production by Saccharopolyspora erythraea are exerted at least in part at the level of transcription. Biotechnol Lett. 1998;20:585–589. doi: 10.1023/A:1005357930000. [DOI] [Google Scholar]
- de la Fuente A. From ‘differential expression’ to ‘differential networking’ - identification of dysfunctional regulatory networks in diseases. Trends Genet. 2010;26:326–333. doi: 10.1016/j.tig.2010.05.001. [DOI] [PubMed] [Google Scholar]
- Yu H, Liu BH, Ye ZQ, Li C, Li YX. et al. Link-based quantitative methods to identify differentially coexpressed genes and gene pairs. BMC Bioinforma. 2011;12:315. doi: 10.1186/1471-2105-12-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu BH, Yu H, Tu K, Li C, Li YX. et al. DCGL: an R package for identifying differentially coexpressed genes and links from gene expression microarray data. Bioinformatics. 2010;26:2637–2638. doi: 10.1093/bioinformatics/btq471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedland N, Mack TR, Yu M, Hung LW, Terwilliger TC. et al. Domain orientation in the inactive response regulator Mycobacterium tuberculosis MtrA provides a barrier to activation. Biochemistry. 2007;46:6733–6743. doi: 10.1021/bi602546q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocker M, Bott M. Evidence for activator and repressor functions of the response regulator MtrA from Corynebacterium glutamicum. FEMS Microbiol Lett. 2006;264:205–212. doi: 10.1111/j.1574-6968.2006.00456.x. [DOI] [PubMed] [Google Scholar]
- Zahrt TC, Deretic V. An essential two-component signal transduction system in Mycobacterium tuberculosis. J Bacteriol. 2000;182:3832–3838. doi: 10.1128/JB.182.13.3832-3838.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cangelosi GA, Do JS, Freeman R, Bennett JG, Semret M. et al. The two-component regulatory system mtrAB is required for morphotypic multidrug resistance in Mycobacterium avium. Antimicrob Agents Chemother. 2006;50:461–468. doi: 10.1128/AAC.50.2.461-468.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fol M, Chauhan A, Nair NK, Maloney E, Moomey M. et al. Modulation of Mycobacterium tuberculosis proliferation by MtrA, an essential two-component response regulator. Mol Microbiol. 2006;60:643–657. doi: 10.1111/j.1365-2958.2006.05137.x. [DOI] [PubMed] [Google Scholar]
- Hoskisson PA, Hutchings MI. MtrAB-LpqB: a conserved three-component system in actinobacteria? Trends Microbiol. 2006;14:444–449. doi: 10.1016/j.tim.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Rajagopalan M, Dziedzic R, Al Zayer M, Stankowska D, Ouimet MC. et al. Mycobacterium tuberculosis origin of replication and the promoter for immunodominant secreted antigen 85B are the targets of MtrA, the essential response regulator. J Biol Chem. 2010;285:15816–15827. doi: 10.1074/jbc.M109.040097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch MT, Hughes TR. Dramatic changes in transcription factor binding over evolutionary time. Genome Biol. 2010;11:122. doi: 10.1186/gb-2010-11-6-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider J, Houry WA. MoxR AAA + ATPases: a novel family of molecular chaperones? J Struct Biol. 2006;156:200–209. doi: 10.1016/j.jsb.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Reeves AR, Brikun IA, Cernota WH, Leach BI, Gonzalez MC. et al. Effects of methylmalonyl-CoA mutase gene knockouts on erythromycin production in carbohydrate-based and oil-based fermentations of Saccharopolyspora erythraea. J Ind Microbiol Biotechnol. 2006;33:600–609. doi: 10.1007/s10295-006-0094-3. [DOI] [PubMed] [Google Scholar]
- Reeves A, Haldenwang WG. Isolation and characterization of dominant mutations in the Bacillus subtilis stressosome components RsbR and RsbS. J Bacteriol. 2007;189:1531–1541. doi: 10.1128/JB.01649-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian W, Jayapal KP, Charaniya S, Mehra S, Glod F. et al. Genome-wide transcriptome analysis reveals that a pleiotropic antibiotic regulator, AfsS, modulates nutritional stress response in Streptomyces coelicolor A3(2) BMC Genomics. 2008;9:56. doi: 10.1186/1471-2164-9-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JF, Liras P. Engineering of regulatory cascades and networks controlling antibiotic biosynthesis in Streptomyces. Curr Opin Microbiol. 2010;13:263–273. doi: 10.1016/j.mib.2010.02.008. [DOI] [PubMed] [Google Scholar]
- Martin JF. Phosphate control of the biosynthesis of antibiotics and other secondary metabolites is mediated by the PhoR-PhoP system: an unfinished story. J Bacteriol. 2004;186:5197–5201. doi: 10.1128/JB.186.16.5197-5201.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Beneit F, Rodriguez-Garcia A, Sola-Landa A, Martin JF. Cross-talk between two global regulators in Streptomyces: PhoP and AfsR interact in the control of afsS, pstS and phoRP transcription. Mol Microbiol. 2009;72:53–68. doi: 10.1111/j.1365-2958.2009.06624.x. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Garcia A, Sola-Landa A, Apel K, Santos-Beneit F, Martin JF. Phosphate control over nitrogen metabolism in Streptomyces coelicolor: direct and indirect negative control of glnR, glnA, glnII and amtB expression by the response regulator PhoP. Nucleic Acids Res. 2009;37:3230–3242. doi: 10.1093/nar/gkp162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco AG, Sola M, Gomis-Ruth FX, Coll M. Tandem DNA recognition by PhoB, a two-component signal transduction transcriptional activator. Structure. 2002;10:701–713. doi: 10.1016/S0969-2126(02)00761-X. [DOI] [PubMed] [Google Scholar]
- Peano C, Tala A, Corti G, Pasanisi D, Durante M. et al. Comparative genomics and transcriptional profiles of Saccharopolyspora erythraea NRRL 2338 and a classically improved erythromycin over-producing strain. Microb Cell Fact. 2012;11:32. doi: 10.1186/1475-2859-11-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukashin AV, Borodovsky M. GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res. 1998;26:1107–1115. doi: 10.1093/nar/26.4.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999;27:4636–4641. doi: 10.1093/nar/27.23.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK. In: Bioinformatics and Computational Biology Solutions using R and Bioconductor. Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W, editor. New York: Springer; 2005. Limma: linear models for microarray data; pp. 397–420. [Google Scholar]
- Tai YC. timecourse: Statistical Analysis for Develepmental Microarray Time Course Data. 2007. http://wwwbioconductororg.
- Tai YC, Speed TP. A multivariate empirical Bayes statistic for replicated microarray time course data. Ann Statist. 2006;34:2387–2412. doi: 10.1214/009053606000000759. [DOI] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Hartmann H, Guthöhrlein EW, Siebert M, Luehr S, Söding J. P-value-based regulatory motif discovery using positional weight matrices. Genome Res. 2013;23:181–194. doi: 10.1101/gr.139881.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz GZ, Stormo GD. Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics. 1999;15:563–577. doi: 10.1093/bioinformatics/15.7.563. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
All nucleotide variations detected in E3 compared with NRRL23338.
Single amino acid substitution detected in E3 compared with NRL23338.
Amino acid deletion detected in E3 compared with NRRL23338.
Amino acid insertion detected in E3 compared with NRRL23338.
Erythromycin production curve during the time-course for the industrial S. erythraea strain and the wild-type NRRL23338 strain. E1 and E2 are two replicates for the industrial strain; M1 and M2 are two replicates for the wild-type strain.
Genome annotation of S. erythraea E3 strain.
COG classification of differentially expressed genes.
Gene expression data.
Regulators involving amino acid variation.
Sequence alignment results of 18 regulators.
Structural Models of MtrA (V197 H198 deletion): SACE_6447 and ETHR_6255. His197 and Val198 of SACE_6447 (A), deleted in ETHR_6255 (B), are located in the center of an α-helix. The deletions are supposed to break the helix and to perturb the DNA-binding function of Val196 in the active state. In addition, His197 contributes to the inter-domain interactions in the inactive state, and its deletion would thus affect the stability of ETHR_6255.
Structural Models of MoxR (N133D): SACE_3795 and ETHR_3703. The Asn133 in SACE_3795 (A), substituted by Asp133 in ETHR_6255 (B), is located in the vicinity of Asp130 and Asn172, both of which were annotated as ATP-binding sites according to NCBI CDD. The Asn133 may thereby be implicated in the ATP-binding through the proxy of Asp130 and Asn172, or even through direct interaction with ATP. In addition, the nearby residues 128–131 are also putative ‘Walker B motif’, which is important for interacting with Mg2+ cation. So the substitution of Asn133 to Asp133, resulting in negative charge on the side chain, may affect the ATP-binding or Mg2+ cation-binding of the protein and thus its catalytic capability.