

SUMMARY
Brain function is shaped by postnatal experience and vulnerable to disruption of Methyl-CpG-binding protein, Mecp2, in multiple neurodevelopmental disorders. How Mecp2 contributes to the experience-dependent refinement of specific cortical circuits and their impairment remains unknown. We analyzed vision in gene-targeted mice and observed an initial normal development in the absence of Mecp2. Visual acuity then rapidly regressed after postnatal day P35–40 and cortical circuits largely fell silent by P55-60. Enhanced inhibitory gating and an excess of parvalbumin-positive, perisomatic input preceded the loss of vision. Both cortical function and inhibitory hyperconnectivity were strikingly rescued independent of Mecp2 by early sensory deprivation or genetic deletion of the excitatory NMDA receptor subunit, NR2A. Thus, vision is a sensitive biomarker of progressive cortical dysfunction and may guide novel, circuit-based therapies for Mecp2 deficiency.
INTRODUCTION
Much of our adult behavior reflects the neural circuits actively refined by sensory experience in infancy and early childhood. Mounting evidence suggests that aberrant synaptic connections underlie many forms of neurodevelopmental disorders of human cognition (Zoghbi, 2003; Chahrour and Zoghbi, 2007). Mutations in the MECP2 gene have been reported in individuals with infantile autism, severe encephalopathy, motor abnormalities, respiratory dysfunction, mental retardation, bipolar disorders, schizophrenia, and mild learning disabilities indicating that disrupted Mecp2 expression underlies complex behavioral phenotypes of multiple human neurodevelopmental disorders (Samaco et al., 2004; Van Esch et al., 2005). While peripheral measures (e.g., respiration) are readily taken, regrettably no direct cortical biomarker is available to monitor the regression or response to treatment in Rett patients.
Rett syndrome (RTT) was first characterized in 1983 as “a progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls,” and was incorporated in the DSM-IV shortly thereafter (Amir et al., 1999; Zoghbi, 2003; Chahrour and Zoghbi, 2007). Since then, a mutation in the gene on the X chromosome encoding the transcriptional modulator protein MECP2 has been discovered to account for the vast majority of individuals diagnosed with RTT. Because of its X-linked genetics, RTT mainly affects girls, who are somatic mosaics for normal and mutant MECP2. The spatiotemporal and cellular expression of MECP2 mRNA and protein starts in basal ganglia by midgestation and extends to cortical neurons in late gestation and postnatally (Amir et al., 1999; Balmer et al., 2003; Armstrong et al., 2003). One key feature of the disorder is that the associated behavioral abnormalities are subtle at first and then progressively deviate from normal development with age. This cannot be explained simply by a pervasive defect in synapse formation (McGraw et al., 2011) but is likely to involve a disrupted process of activity-dependent neuronal circuit refinement with complex outcomes.
Mouse models of RTT, considered a gold standard of animal models due to the recapitulation of behavioral and neurobiological symptoms seen in patients, have been critical for beginning to understand the functional consequences of Mecp2 loss and gain of function. Postnatal loss of Mecp2 from neuronal and non-neuronal cells indicates that discrete features of RTT are associated with discrete circuits (Gemelli et al., 2006; Fyffe et al., 2008; Ballas et al., 2009; Samaco et al., 2009; Deng et al., 2010; Lioy et al., 2011; Derecki et al., 2012). Importantly, disruption of Mecp2 in all GABA circuits alone may manifest several aspects of Rett Syndrome, including abnormal EEG hyperexcitability, severe respiratory dysrhythmias and early lethality (Chao et al., 2010).
Mecp2 deficiency restricted to GABAergic neurons alters Gad1/2 expression and GABA neurotransmitter release, suggesting a decrease of inhibitory function while excitatory drive is grossly unaffected. Instead, global perturbation of Mecp2 expression—closer to the human condition—shifts neocortical excitatory/inhibitory (E/I) balance in favor of inhibition in vitro (Dani et al., 2005; Nelson et al., 2006; Wood et al., 2009; Wood and Shepherd, 2010), while an enhanced excitation may be found in brainstem circuits (Shepherd and Katz, 2011). Hence, synaptic dysfunctions in RTT may be diverse and highly circuit dependent. Despite these recent findings, the precise nature of cortical circuit abnormality in vivo and how sensory experience affects the maturation and maintenance of a particular brain function through Mecp2 regulation remain unclear.
To address this complex question, we have taken advantage of the well-studied visual system and specifically analyzed visual function in adult Mecp2 knockout (KO; Guy et al., 2001) and wild-type (WT) littermates, using a behavioral assay and in vivo electrophysiological recording. We discovered that vision can serve as a reliable cortical biomarker of Mecp2 disruption, which has recently been confirmed in RTT patients (G. DeGregorio, O. Khwaja, W. Kaufmann, M.F., and C.A. Nelson, unpublished data). Here, we show in KO mice that acuity initially develops normally in the absence of Mecp2 before regressing rapidly into adulthood in direct correlation with the onset of RTT phenotypes. Single-unit recordings revealed low spontaneous neuronal activity and a general silencing of cortical circuitry. Consistent with this, the propagation of neural activity in response to threshold stimulation of the white matter in visual cortical slices was strongly gated in layer 4 of the Mecp2-deficient mice.
Corresponding hyperconnectivity of perisomatic, GABAergic puncta upon excitatory neurons was already evident just after eye opening in anticipation of acuity loss. Late Mecp2 deletion only from these parvalbumin (PV) circuits instead failed to affect spontaneous activity or visual acuity, suggesting that cortical symptoms may be traced back to atypical developmental trajectories. Strikingly, early sensory deprivation or genetic deletion of NMDA receptor 2A (NR2A) subunits restored both PV connectivity and visual function. Since the brain undergoes maximal plasticity in infancy (Hensch, 2004), even minor developmental deflections could have a large impact later in life, which are vital to the design of appropriate treatments. Thus, vision may serve as a useful, nonmotor biomarker of declining cortical function that can be rescued prior to symptom onset independent of Mecp2.
RESULTS
Vision Regresses in the Absence of Mecp2
Sensory experience shapes neural circuits in the mammalian visual cortex. Optimal cortical E/I circuit balance is required for the activity-dependent development of visual properties (Hensch and Fagiolini, 2005). We first measured changes in visual acuity in response to high contrast moving gratings by an optomotor task to rapidly and reliably assess mouse vision (Prusky et al., 2004). Mecp2-deficient adults (postnatal day P55–60) exhibited lower visual acuity than littermate WT control mice (Figure 1A; p < 0.0001). This mutant line notably presents motor defects beginning around 4 weeks of age which worsen rapidly until death 4–6 weeks later (Guy et al., 2001). Yet, it is unlikely that the apparent visual defect reflected motor impairment, as Mecp2 KO mice matched their WT littermates’ abilities to track low spatial frequency stimuli (100% accuracy at 0.05 to 0.2 cycle/degrees [cpd], 30 min continuous trials).
Figure 1. Reduced Visual Acuity and Neuronal Activity of Visual Cortical Neurons in Adult Mecp2 Knockout (KO) Mice.

(A) Average acuity assessed by optomotor task (OPT; p < 0.0001, t test) or visual-evoked potential (VEP, p < 0.05, t test) in wild-type (WT; white, n = 16 and 8) or Mecp2 KO mice (black, n = 15 and 5).
(B) Visual acuity emerges normally but then regresses drastically after 40 days of age in Mecp2 KO mice. Acuity development over time for WT (○; n = 5–16) and KO (•, n = 5–15) mice.
(C) Visual acuity significantly decreases in Mecp2loxstop mutant mice after the age of P55 as the RTT phenotype emerges (6–8 mice each, left, hatched bars, p < 0.01, Mann-Whitney test). Mecp2 heterozygote females also exhibit a progressive loss of visual acuity starting around P80 and reaching a minimum around P240 (6-8 mice each, p < 0.01, Mann-Whitney test).
(D) Representative Mecp2 WT and KO spike trains and corresponding PSTH in response to two oriented gratings or a uniform gray stimulus (8 presentations each).
(E) Spontaneous and evoked neuronal activity are significantly reduced in the absence of Mecp2. Mecp2 WT: white bars, n = 47 cells; Mecp2 KO: black bars, n = 58 cells (p < 0.005, Mann-Whitney test).
(F) Signal-to-noise ratio (SNR) cumulative distribution is significantly increased in Mecp2 KO compared to WT visual cortical neurons (KS test, p < 0.01).
All data are presented as mean ± standard error. See also Figures S1–S3.
To confirm a visual rather than motor defect, we recorded visual evoked potentials (VEP) directly from the binocular region of visual cortex in anesthetized adult Mecp2 KO and WT mice. Reversing square wave gratings of low spatial frequency were presented, and visual response was acquired at several cortical depths to determine the site of maximal VEP amplitude (see Figure S1A available online). Signal strength typically decreased with increasing spatial frequency in both mutant and WT littermates. Acuity threshold was calculated as the frequency at which the cortical signal reached 0 μV (Figure S1B). Consistent with their behavioral acuity, cortical acuity in V1 was significantly reduced in the Mecp2 KO compared to WT mice (Figure 1A, p < 0.005).
To establish when the visual impairment arises, we took advantage of the optomotor task to measure acuity over the life course of the animal. Mecp2 KO mice exhibited low spatial resolution at eye opening that matured along a profile identical to that of WT animals until P30-35. While spatial acuity remained stable thereafter in adult WT mice (p > 0.1), it started to regress rapidly after P40 in Mecp2 KO animals (Figure 1B). Overall, the developmental profile of WT and KO mice was significantly different (p < 0.0001, Two-Way ANOVA). To determine whether the visual phenotype was robustly present in other Mecp2-deficient models, we measured visual acuity in the Mecp2 lox-stop line (Guy et al., 2007). These males exhibit delayed onset of RTT symptoms compared to the constitutive Mecp2 KO mice due to leakage of the lox-stop suppressor (Lioy et al., 2011).
Likewise, a decline of visual acuity began only after P60 in the Mecp2lox-stop line, reaching a minimum value around P100 (Figure 1C, left; 0.26 versus 0.4 cpd, p < 0.001, 6–8 mice each). We further found that heterozygous Mecp2 HET female mice, a closer analog of Rett patients, also exhibited significantly reduced acuity starting around P80 (0.34 versus 0.4 cpd, p < 0.05), which degraded slowly over the next months (Figure 1C, right; 0.24 cpd at P240, p < 0.001, 5–8 mice each). Mecp2 expression is therefore critical for maintaining visual function. Specifically, vision can mature normally without Mecp2 but fails to be stabilized in adulthood, reminiscent of other behavioral symptoms in RTT syndrome mouse models.
In order to evaluate neuronal activity at the level of single cells, we performed extracellular recordings in vivo across all cortical layers using multi-channel probes (Figures 1D and S2; see Experimental Procedures). The adult visual cortex was largely silent in Mecp2 KO mice compared to WT littermates, revealing a significant decrease in both spontaneous and evoked activity (Figure 1E; p < 0.005). Even among neurons with an evoked firing rate similar to that of WT cells, spontaneous activity was still affected. Consequently, the signal-to-noise ratio (SNR) was increased in neurons from all cortical layers of the mutant mice (Figures 1F and S2B; p < 0.05, KS test).
Rescuing Hyperconnected PV Circuits Lacking Mecp2
Our examination of visual physiology in vivo confirmed a shift of E/I balance in favor of inhibition as initially reported for Mecp2 KO mice in vitro (Dani et al., 2005; Wood and Shepherd, 2010). Recent studies, however, have shown that selective deletion of Mecp2 only from GABAergic cells results in a decrease of Gad1/2 and GABAergic neurotransmitter release (Chao et al., 2010).
We, therefore, examined inhibitory circuit markers in the total absence of Mecp2. Quantitative PCR of visual cortical homogenates verified a general downregulation of inhibitory markers in adult Mecp2 KO mice (Table 1), including decreased gene expression of GABA-synthetic enzyme, GAD65. GABA immunofluorescence levels were also significantly reduced, in agreement with previous reports (Chao et al., 2010). Yet not all inhibitory circuits were equally affected by total deletion of Mecp2, as the markers of three major subsets of GABAergic interneuron were regulated differentially. While mRNA of the calcium-binding proteins, calretinin and calbindin, were decreased in Mecp2 KO mice, PV levels were unexpectedly increased (Table 1).
Table 1.
Gene Expression Differences between Mecp2 WT and KO Mouse Visual Cortex
| Genes | Expression (Std. Error) | p Value |
|---|---|---|
| →GAD65 | 0.79 (0.65-0.95) | 0.023 |
| →Kv3.1 | 0.81 (0.65-1.01) | 0.028 |
| = GAD67 | 0.93 (0.69-1.21) | 0.460 |
| = VGAT | 0.92 (0.79-1.07) | 0.170 |
| = Gephyrin | 1 (0.84-1.18) | 0.983 |
| = GABAA1 | 0.98 (0.87-1.1) | 0.702 |
| →Calbindin | 0.8 (0.68-0.94) | 0.004 |
| →Calretinin | 0.6 (0.52-0.72) | < 0.0001 |
| ↑Parvalbumin | 1.38 (1.09-1.83) | 0.014 |
Relative mRNA expression levels of GABAergic inhibitory markers in homogenates of adult light-reared Mecp2 WT and KO primary visual cortex as determined by RT-qPCR (relative expression ratio, standard error, statistical significance, Mann-Whitney test). Note that despite a general decrease of GAD65, Kv3.1, calbindin, and calretinin, parvalbumin was significantly increased. See also Table S2.
An upregulation of PV immunofluorescence intensity revealed a primary effect of increased neurite complexity (Figures 2A and 2B, top) rather than a change in total PV-cell number (WT = 0.13 ± 0.06, KO = 0.11 ± 0.02 PV/DAPI-positive cells, p = 0.48, Mann-Whitney test). In particular, the number of PV-positive perisomatic boutons was increased in Mecp2 KO mice (Figure 2B, bottom). Basket type PV-cell synapses, positioned on the somata and proximal dendrites, control excitability of principal cells, adjust the gain of their integrated synaptic response (Markram et al., 2004; Atallah et al., 2012) and are particularly important for the emergence of cortical network function (Hensch, 2005; Bartos et al., 2007). Notably, sensory experience regulates the postnatal maturation of these PV circuits in visual cortex: dark-rearing from birth (DR) specifically reduces perisomatic inhibition (Katagiri et al., 2007; Sugiyama et al., 2008).
Figure 2. Reversible Parvalbumin (PV)-Circuit Hyperconnectivity and Acuity Defects in Mecp2 KO Mice.

(A) Upper panels: Mean pixel intensity of PV immunofluorescence is increased in light-reared (LR) Mecp2 KO mouse visual cortex compared to WT levels and restored to normal levels in dark-reared (DR) Mecp2 KO. Cortical layers are indicated on the left side of upper left panel. wm = white matter. Scale bar, 100 μm. Lower panels: Presynaptic PV-puncta per cell perimeter are restored to WT levels by DR. Scale bar, 10 μm.
(B) Changes in mean intensity (upper) and PV-cell innervation of pyramidal cell somata (lower) are reversibly increased in Mecp2 KO mouse compared to WT levels (WT versus Mecp2 KO, p < 0.0005; *p < 0.05, **p < 0.01, Mann-Whitney test). Error bars, mean ± SEM.
(C) Single-unit recordings from DR WT and KO adult mice revealed a significant increase in the level of spontaneous activity. Spontaneous but not evoked activity was restored to LR WT level in DR Mecp2 KO mice. Red dotted lines indicate the WT adult level of spontaneous and evoked activity.
(D) Measurements made just before and after 30 days in the dark (DR) reveal little acuity loss (n = 4, p > 0.05). Visual acuity change for LR Mecp2 KO mice or those placed in the dark from P30 (0.40 ± 0.01 and 0.39 ± 0.002 cpd) to P60 (0.22 ± 0.02 and 0.33 ± 0.10 cpd), respectively.
(E) Acuity comparison at P55-60 between Mecp2 KO (n = 15) and dark-reared (DR) KO mice from different postnatal ages, P0 (n = 6), P14–15 (n = 6), P20 (n = 4), or P24–30 (n = 8). Shaded region indicates range of normal WT acuity. Error bars, mean ± SEM.
We found that even in the absence of Mecp2, DR was sufficient to rescue PV-cell hyperconnectivity (Figures 2A and 2B), renormalizing PV levels and the number of perisomatic boutons (Figure 2 and Table S1). Firing rates of cortical pyramidal cells are homeostatically regulated (Turrigiano and Nelson, 2004) and spontaneous firing in vivo increases upon DR (Gianfranceschi et al., 2003). We confirmed an augmentation of spontaneous activity (Figure 2C; p < 0.0001) but not of evoked response (p > 0.1) in DR WT mice. Consistent with an anatomical rescue, DR restored spontaneous firing rates of Mecp2 KO mice to the same range as that of control WT cells (p > 0.1) and significantly above that of light-reared KO cells (Figure 2C; p > 0.05). No increase in evoked neural responsiveness was found, rescuing signal-to-noise ratio (SNR) across layers and cell types (Figures 2C and S2).
Concurrently, DR from birth was able to preserve visual acuity into adulthood when measured behaviorally (Figure 2E) and supported by cortical VEP recording (Figure S1C). The improvement of cortical function was specific to V1, as motor performance on rotarod and open field behavioral assay remained impaired after DR (Figure S3A). To determine when sensory experience must be removed in order to prevent progressive loss of visual function, Mecp2 KO mice were deprived of input starting just before vision regressed. Visual acuity was first measured behaviorally in a group of light-reared mutant mice at P30 (Figure 2D; p > 0.5 compared to WT littermates). A subset of these animals was then placed into total darkness, while the rest were kept in a normal light/dark cycle until adulthood (P55–60; Figure 2D). KO animals in the dark retained a significantly higher spatial acuity compared to light-reared littermates (p < 0.005). However, visual acuity was still at a lower level than that of WT light-reared mice at P60 (p < 0.005).
We, therefore, placed Mecp2 KO mice in total darkness until P55–60 from earlier stages of postnatal development (just after eye opening at P14 or at P20). All rearing paradigms preserved visual acuity into adulthood, but only those mice placed in darkness from birth or immediately after eye opening showed visual acuity in the normal range of WT animals (Figure 2E; p > 0.05). Taken together, these results surprisingly reveal that visual experience in the absence of Mecp2 has a detrimental effect on visual cortical function.
Enhanced Inhibitory Gating Precedes Vision Loss
Taken together, our results support a developmental disruption of visual cortical circuits that precedes the loss of vision. We then examined when the PV hyperconnectivity first emerges in Mecp2 KO mice. Overall PV intensity and perisomatic puncta (Figure 3A) were already significantly increased just after eye opening (P15) and well before the maturational trajectory for visual acuity deviates from normal. In contrast, decreased perisomatic GAD65 expression was not yet evident at P15 and only gradually appeared as the mice matured (>P30) (Figure 3A).
Figure 3. Enhanced Inhibitory Gating in Mecp2 KO Mouse Visual Cortex.

(A) PV-immunofluorescence is elevated at P15 in Mecp2 KO animals (3 mice each, p < 0.01, Mann-Whitney test), and this difference persists into adulthood (see also Figure 2). The density of perisomatic PV-boutons upon pyramidal cell somata is significantly increased in the absence of Mecp2 starting already at P15 and throughout life (upper right, 3–4 mice each, versus WT; *p < 0.01, **p < 0.001, Mann-Whitney test). WT mice also exhibit a significant increase in PV-puncta across development (3–4 mice each, p < 0.01, Mann-Whitney test).The level of GAD65 within PV puncta is significantly decreased starting from P30 in Mecp2 KO mice compared to WT (lower right, 3–4 mice each, versus WT; *p < 0.01, **p < 0.001, Mann-Whitney test). Scale bar, 5 μm.
(B) Propagation of neuronal activity through layer 4 at threshold stimulation is reduced in upper layers in the Mecp2 KO mouse. Schematic of recording area indicating position of the stimulating electrode (black arrowhead) in the white matter (WM) and ROIs (squares) for analysis in upper and lower layers “on beam.” Pseudocolor peak response frame from VSDI movies of WT slices 15 ms after half maximal WM stimulus, revealing strong WT response propagation to the upper layers. The upper layer response in KO slices is suppressed at threshold stimulus intensity (graph arrow). Scale bar, 250 μm. Upper/lower layer response ratio as a function of WM stimulus intensity. All results expressed as mean ± SEM (4–5 mice each); *p < 0.001, t test.
See also Figure S2.
To determine whether the early hyperconnectivity of PV puncta results in enhanced inhibitory function, we examined the spatial propagation of activity in visual cortical slices using VSDI (voltage-sensitive dye imaging; Grinvald and Hildesheim, 2004). We previously demonstrated that VSDI is sensitive to laminar changes in PV circuit reorganization (Lodato et al., 2011). Proper positioning and synaptogenesis of GABAergic cells is critical for maintaining signal propagation and E/I balance. PV circuits in particular potently gate the flow of thalamocortical activity through layer 4 (Cruikshank et al., 2007; Bagnall et al., 2011; Kirkwood and Bear, 1994; Rozas et al., 2001). We examined coronal visual cortical slices from KO and WT animals at P22–25, when PV circuits have normally reached maturity (Kuhlman et al., 2010) and prior to visual acuity loss in the Mecp2 KO mice (Figure 1B).
We monitored the spread of activity in response to a current pulse delivered to the white matter and quantified two regions (125 × 125 μm2) within supra- and infragranular layers (Lodato et al., 2011). As expected, WT mice exhibited a strong response that propagated rapidly to the upper layers “on beam” with the stimulating electrode (Figure 3B, black bars). Input-output curves of maximum fluorescence intensity revealed a gating of upper-layer response with increasing stimulus intensity (Figure 3B, right), which reflects the recruitment of inhibition in layer 4 (Lodato et al., 2011). In slices from Mecp2 KO mice, response propagation was strongly gated even at threshold stimulus intensities with layer 2/3 signals failing to reach WT levels despite normal lower layer activation (Figure 3B, red bars).
Together these results reveal an early impact of Mecp2 deficiency on inhibitory network maturation prior to cortical malfunction. To evaluate whether Mecp2 directly regulates PV expression as early as these first circuit abnormalities emerge, we performed chromatin immunoprecipitation (ChIP) experiments on homogenates of WT visual cortex at P15 followed by qPCR (Figure S4). In silico analysis of the Pvalb gene proximal promoter revealed one CpG island (Figure S4 and Table S1; see Supplemental Experimental Procedures for details). We found that one of two unbiased primers exhibited significant binding of Mecp2 upstream of the Pvalb transcription start site (TSS;1.2- to 1.5-fold enrichment over IgG, p < 0.02; Figure S4), supporting an early regulation of Pvalb transcription by Mecp2. If so, a late deletion of Mecp2 selectively from PV cells may not be sufficient to mimic deficits found in the constitutive KO mouse.
We, therefore, selectively removed Mecp2 from PV-cells after vision had fully matured. Mecp2lox/x females (Guy et al., 2001) were crossed with PV-Cre+/+ males known to express adequate amounts of Cre-recombinase in the cortex only after P30 (Hippenmeyer et al., 2005; Madisen et al., 2010; Belforte et al., 2010). Mecp2lox/y/PV-Cre−/+ (c-KO) and Mecp2+/y/PV-Cre−/+ (c-WT) littermates were generally healthy and did not exhibit any apparent behavioral phenotype as they reached adulthood. Double immunolabeling for Mecp2 and PV confirmed that Mecp2 deletion from PV cells was gradual with 90% of PV cells still expressing Mecp2 at P22 and only 8% at P90 (Figure 4A). Nevertheless, these late PV-conditional KO (c-KO) mice also exhibited an increase of PV intensity (Figure 4B, left), confirming successful Mecp2 deletion.
Figure 4. Late Mecp2 Deletion in PV Cells Induces Expression but Not Hyperconnectivity or Loss of Visual Function.

(A) Double staining for PV (green) and Mecp2 (red) shows that the majority of PV-neurons retain Mecp2 expression at P22; whereas by P90, only 8% of PV cells still express Mecp2 in Mecp2lox/y/PV-Cre−/+ (c-KO) mice compared to Mecp2+/y/PV-Cre−/+ littermates (c-WT). Scale bar, 35μm.
(B) Loss of Mecp2 expression is paralleled by an increase in PV immunoreactivity (left) but not in PV puncta density (right) in Mecp2 c-KO compared to control littermates at P90 (3 mice each, p < 0.001, Mann-Whitney test).
(C) SNR cumulative distribution is no different between late Mecp2 c-KO (solid green) and control (c-WT; dashed green) (63 and 53 cells, respectively; p = 0.88, KS test).
(D) Visual acuity is not affected in adult c-KO mice (5 mice each, p = 0.8, Mann-Whitney test).
All data are presented as mean ± standard error.
See also Figure S4.
The innervation of excitatory neurons was, however, unaffected (Figure 4B, right), as the number of perisomatic PV puncta was similar in c-KO and control mice (0.33 ± 0.01 versus 0.35 ± 0.01, p = 0.07, 3 mice each). Single-cell recordings in vivo showed a correspondingly unaltered spontaneous activity (but a decrease in evoked response) of excitatory neurons (Figure 4C, inset). SNR was not significantly different between WT and c-KO littermates. Under these conditions, visual acuity was unaffected (Figure 4D). Our results establish an early derailment of PV circuit maturation in the total absence of Mecp2, which is manifest only later as a loss of visual acuity.
NR2A Regulates Cortical Function in Mecp2KO Mice
We next searched for a molecular correlate of the developmental rescue of PV-cells. It has been reported that NMDA receptor subunit 2A (Grin2a) and NR2B (Grin2b) transcription is misregulated in the absence of Mecp2 (Asaka et al., 2006; Chahrour et al., 2008; Lee et al., 2008; McGraw et al., 2011). Indeed, we identified Mecp2 binding to the Grin2a promoter in homogenates of visual cortex at P15 by ChIP-qPCR experiments (Figure S4). Specifically, the DNA sequence 3 kb upstream and 1 kb downstream of the TSS, revealed three CpG islands (Figure S4 and Table S1; see Supplemental Experimental Procedures for details). We found that one of five unbiased primers (NR2A-1) exhibited statistically significant binding of Mecp2 downstream of the Grin2a TSS (1.3 to 3.0 fold enrichment over IgG, p = 0.027;Figure S4 and Table S1). In contrast, binding of Mecp2 to the reported enrichment site for Grin2b (Lee et al., 2008) was observed only in 3 out of 4 samples, therefore not meeting statistical significance (Figure S4).
In homogenates of Mecp2 KO mouse visual cortex, both NR2A and NR2B subunit expression were significantly decreased in adulthood (Table S2). However, NR2B disruption was more severe (Lee et al., 2008), resulting in a significant increase of NR2A/2B ratio compared to adult WT mice (Figure 5A). A greater NR2B loss (−25%) with respect to NR2A (−13%) was already evident at P30 in V1 of the Mecp2 KO mice prior to regression of visual acuity (n = 3 mice each, WT versus KO, p < 0.05 t test). Together, our results support an early regulation of NR2A expression by Mecp2.
Figure 5. Rescue of PV-Cell Hyperconnectivity by NR2A Regulation.

(A) NR2A/2B ratio in WT mice is significantly decreased by DR in visual cortex homogenates (quantified by qPCR). Mecp2 KO mice exhibit an increased ratio compared to WT which is reduced by DR to reach normal WT levels (LR WT versus LR KO: p < 0.05; LR WT versus DR WT: p < 0.05; LR KO versus DR KO: p < 0.005, one-way ANOVA).
(B) Left: Mecp2 KO/NR2A Het mice appear indistinguishable from WT or Mecp2 WT/NR2A Het mice. Double mutants exhibit regular breathing, absence of tremor and do not show hindlimb clasping phenotype. Right: Double mutant mice (blue diamond) exhibit a higher weight than Mecp2 KO mice (black circle) and in the same range as WT animals (white circle); ***p < 0.001, t test.
(C) Density of PV-positive puncta upon pyramidal cells is not significantly different between Mecp2 KO/NR2A Het and Mecp2 WT/NR2A Het across development (3–5 mice each; p = 0.9, Mann-Whitney test). Similarly, there is no difference in GAD65 intensity in PV-positive puncta between double mutant and control littermates across development (3–5 mice each; p = 0.8, Mann-Whitney test). Scale bars, 100 (upper) and 10 μm (lower).
All data are presented as mean ± standard error.
See also Figures S2–S4.
Visual experience upon eye opening directly modulates NMDA receptor subunit composition in an activity-dependent manner (Quinlan et al., 1999). DR delays the switch from NR2B to 2A-enriched receptors (Figure 5A and Table S2). We found that DR of Mecp2 KO mice was sufficient to further downregulate NR2A expression (Table S2), renormalizing NR2A/2B ratio to light-reared WT levels (Figure 5A). Moreover, selective disruption of NMDA receptors upon PV-cells in particular is known to alter cellular, network and behavioral function (Kinney et al., 2006; Belforte et al., 2010; Korotkova et al., 2010). We, therefore, examined whether direct NMDA receptor manipulation would yield a functional rescue in V1 similar to DR.
To reestablish the proper NR2A/2B ratio in Mecp2 KO mice, either NR2B should increase or NR2A decrease. We focused on NR2A-deficient mice as a potential strategy for restoring NR2A/2B ratio and, consequently, vision in Mecp2 KO mice. NR2A KO mice are viable and healthy (Fagiolini et al., 2003), as NR2A deletion is naturally restricted to a postnatal period after initial circuit formation and eye opening (Quinlan et al., 1999; Fagiolini et al., 2003). Heterozygous Mecp2 females were crossed with NR2A KO males to obtain viable first and second generation mice, respectively.
Mecp2/NR2A double-mutant offspring exhibited a strikingly healthy appearance, normal weight and absence of hindlimb clasping phenotype (Figure 5B). While only 56% of Mecp2 KO mice survived until P60 (98/174 mice), all double mutant mice reached P60 (19/19 Mecp2 KO/NR2A Het and 10/10 Mecp2 KO/NR2A KO mice). However, rotarod and open field behaviors remained partially defective in both DR and double mutant mice (Figure S3). Note that NR2A KO mice themselves exhibit some coordination defects (Kadotani et al., 1996) and Mecp2 deficiency in glial cells may largely contribute to such subcortical phenotypes (Lioy et al., 2011).
Importantly, cortical PV-cell hyperconnectivity and development were rescued (Figure 5C). Mecp2/NR2A double mutants exhibited renormalized PV intensity and density of perisomatic boutons (Figure 5C and Table S2). One consequence of reduced NR2A expression is weak orientation tuning (Fagiolini et al., 2003). NR2A deletion similarly reduced stimulus selectivity in WT and Mecp2 KO mice (Figures 6A and 6B) and was not significantly different between genotypes. Both heterozygous (Mecp2 KO/NR2A Het) and homozygous (Mecp2 KO/NR2A KO) double mutants exhibited normal spontaneous and evoked activity (Figure 6C, inset; p < 0.005) and SNR indistinguishable from WT controls (Figure 6C). Consistent with this, visual acuity was preserved (despite poor orientation tuning) in Mecp2 KO mice when combined with NR2A deletion, just as for early DR (Figure 6D; p > 0.05).
Figure 6. Rescue of Spontaneous Activity and Visual Acuity by NR2A Regulation.

(A) Examples of orientation tuning curve (insets) for high and low OSI visual cortical neurons in Mecp2 WT mice. Solid black, KO; dashed, WT; solid blue, KO/Het.
(B) Mean OSI in Mecp2 KO/NR2A Het mice is significantly decreased compared to Mecp2 WT mice (ANOVA test, Mecp2 KO/NR2A Het, n = 55 and Mecp2 WT/NR2A Het, n = 80 cells p = 0.89 and p < 0.0001 versus Mecp2 KO).
(C) Signal-to-noise ratio (SNR) cumulative distribution is not significantly different between Mecp2 KO/NR2A Het and Mecp2 WT/NR2A Het cells (KS test, p = 0.88, n = 55 and 80 cells, respectively; p > 0.05 and p < 0.0001 versus Mecp2 KO). Inset, both spontaneous and evoked activity are at WT levels following NR2A deletion (p > 0.05, Mann-Whitney test).
(D) Optomotor visual acuity comparison between Mecp2 KO/NR2A Het (blue column) and Mecp2 WT/NR2A Het (dotted column) reveals intact vision in double mutants (versus Mecp2 KO alone, p < 0.005, n = 5 mice each, Mann-Whitney test). Error bars, mean ± SEM.
DISCUSSION
We found that deletion of Mecp2 induces a direct, early upregulation of PV expression and a rapid hypermaturation of PV-cell connectivity onto cortical pyramidal neurons. This was functionally manifest in vitro as an enhanced inhibitory gate within layer 4 as early as P22. Once fully mature PV connectivity levels were finally exceeded beyond P30 in Mecp2 mutant, spontaneous activity in cortical circuits fell silent in vivo and visual acuity was lost. Far from detrimental “noise,” the spatiotemporal structure of spontaneous activity may shape neural responses during natural viewing and enable increased stimulus detection at perceptual threshold (Deco and Romo, 2008; Ringach, 2009; Schölvinck et al., 2012). In addition, the preferential reduction of spontaneous activity by GABA may normally trigger key developmental transitions in visual plasticity (T. Toyoizumi, H. Miyamoto, T.K.H., and K.D. Miller, unpublished data).
Early hyperconnectivity of perisomatic PV-circuits is well-situated to suppress spontaneous activity prematurely (Lee et al., 2012), and may underlie the cortical impact of Mecp2 dysfunction. In both developing and mature visual cortex, sensory evoked neural activity represents the modulation of ongoing circuit dynamics, rather than directly reflecting the structure of the input signal itself (Fiser et al., 2004). Moreover, the similarity between spontaneous and evoked activities develops with age (Berkes et al., 2011), suggesting that spontaneous cortical activity adapts to represent the statistics of the external world in the Bayesian view. A failure to maintain an internal model of the environment that is normally needed to interpret sensory inputs or to prepare actions is indicative of an autistic syndrome (Fox and Raichle, 2007).
Our results from the constitutive Mecp2 KO mice present the first in vivo verification of a progressive shift in cortical E/I balance favoring inhibition, which is consistent with earlier brain slice studies (Dani et al., 2005; Nelson et al., 2006; Wood et al., 2009; Wood and Shepherd, 2010). Wood and Shepherd (2010), in particular, reported a selective decrease of excitatory input onto layer 2/3 pyramidal neurons, with no change in inhibitory drive. While we cannot exclude potentially selective differences across cortical laminae, it is important to note that mIPSCs reflect all inhibitory synapses onto a given cell and fail to distinguish between input subtypes. The net result may well have appeared as no change or greater variability of mIPSCs. It would be informative to perform paired-cell recordings (from connected PV, pyramidal neurons) in these mice to ascertain the strength of individual inhibitory connections, as well as the degree of convergence from multiple PV cells onto individual layer 2–3 neurons.
The upregulation of PV circuitry in the absence of Mecp2 is unexpected, as there is a general decrease of GABA, GAD65, and calbindin/calretinin (Table 1). Neither anatomical nor functional subcircuit dissection of inhibition have previously been performed in Mecp2 KO mice. We previously showed that VSDI is sensitive to laminar changes in subtype-selective inhibition (Lodato et al., 2011). Since PV-circuit inhibition normally constitutes a “gate” in layer 4 (Cruikshank et al., 2007; Bagnall et al., 2011; Kirkwood and Bear, 1994; Rozas et al., 2001), we monitored the activity spreading upward from a white matter stimulus. This confirmed a localized strengthening of net inhibition within layer 4 when Mecp2 is lacking. Future studies should explore PV cells directly, as they control the timing of cortical critical periods (Hensch, 2005), which may be shifted in development here.
Late deletion of Mecp2 in adulthood has recently been found to impact survival and motor coordination (McGraw et al., 2011; Cheval et al., 2012; Nguyen et al., 2012). However, none of these studies have examined cortical function in detail. Moreover, humans suffering from Rett syndrome as a consequence of global Mecp2 loss of function do not have a gene deletion that is restricted to specific cell types or only at late ages. We find that developmental trajectories must be considered in detail (Figure 7). Delayed downregulation of GAD65 in the absence of Mecp2 (Table S1; Chao et al., 2010) may reflect a compensation to the early increases in perisomatic PV boutons. As GABA signaling per se promotes synapse elimination and axon pruning (Wu et al., 2012), the initial hyperconnectivity may become increasingly difficult to overcome.
Figure 7. Cortical Circuits Are Developmentally Disrupted in the Absence of Mecp2.
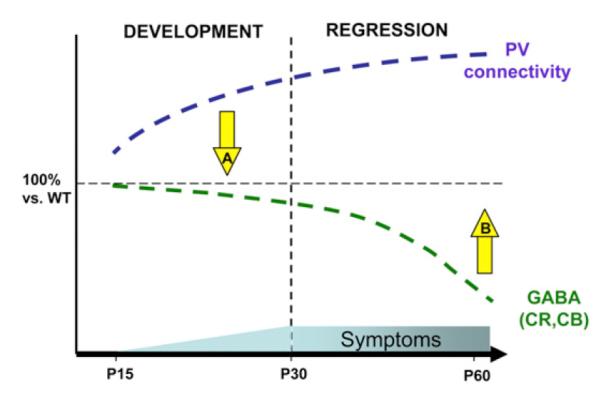
Parvalbumin (PV) intensity and puncta density onto pyramidal cells reveal a PV-circuit hyperconnectivity as early as P15 that increases throughout life, while GAD65 is not downregulated until adulthood. Circuit-based therapeutic strategies independent of Mecp2 may now be relevant. An effective strategy to prevent the delayed loss of cortical functions (such as vision or language) should focus on early interventions to dampen PV hyperconnectivity (A) rather than on acute enhancement of inhibition once regressive symptoms have emerged (B). See also Figure S4.
Implications for Therapeutic Strategies
A late reduction of GABA circuit function mirrors the delayed appearance of acute symptoms in RTT syndrome, such as seizures (Jian et al., 2006; Glaze et al., 2010; Nissenkorn et al., 2010). As a result, antiepileptic drugs have little or no beneficial effect on the cognitive aspects of RTT syndrome (Huppke et al., 2007; Nissenkorn et al., 2010). Enhancing inhibition once regressive symptoms have emerged must be tempered in view of the persistently strong PV subcircuits (Figure 7, arrow “B”). A more effective strategy to prevent the delayed loss of cortical functions reflecting a critical period (such as vision or language) may require early interventions to dampen PV hyperconnectivity (Figure 7, arrow “A”). Overlooking developmental stage or subtleties of inhibitory circuit misregulation by therapeutic approaches based on global GABAergic modulation (Chao et al., 2010) may yield counter-productive consequences for patients.
Strikingly, both an environmental (DR) and genetic (NR2A) approach could prevent the loss of cortical function in the absence of Mecp2. Imbalanced NR2A/2B subunit ratios emerged by P30 which could be rescued by DR in Mecp2 KO mice. Removing sensory experience rebalanced the ratio by retaining NR2B while preserving immature low levels of NR2A expression (Figure 3). Upon eye opening, cortical NMDA receptor subunits are typically phosphorylated in an experience-dependent manner so as to remove NR2B and insert NR2A into active synapses (Sanz-Clemente et al., 2010). Constitutive removal of NR2A, or DR started prior to P20, were most potent (Figure 2E) in preventing the early PV cell hyperconnectivity (Figure 4). Note that PV cells are exquisitely sensitive to NMDA receptor disruption (Kinney et al., 2006; Belforte et al., 2010; Korotkova et al., 2010).
Our ChIP results indicate that Mecp2 may directly regulate Pvalb and Grin2a gene expression as early as eye opening. In adult cortical tissue, MeCP2 is thought to be bound throughout the neuronal genome in a pattern similar to that of a histone protein functioning on a global scale to modulate chromatin structure (Cohen et al., 2011). Multiple CpG binding sites on the Grin2a and Grin2b promoters suggest either up- or downregulation of gene expression is possible by activity-dependent mechanisms in a cell-specific manner (Figure S4; Asaka et al., 2006; Chahrour et al., 2008; Lee et al., 2008; McGraw et al., 2011). Note that high PV expression and hyperconnectivity are present already at eye opening and that direct Mecp2 deletion only from PV cells, upregulates PV expression (Figure 4). Future work should analyze the developmental profile of activity-dependent Mecp2 binding at these discrete sites.
Identification of a specific receptor pathway within a particular cortical circuit now offers an accessible membrane target for drug intervention strategies. These would not rely upon the global re-expression of intracellular molecules, such as BDNF signaling or Mecp2 itself (Chang et al., 2006; Guy et al., 2007; Kline et al., 2010). Importantly, NR2A transcription, translation and posttranslational modifications are regulated by multiple factors, including but not limited to Mecp2 binding (Sanz-Clemente et al., 2010). For example, novel NR2A receptor antagonists (Liu et al., 2004; de Marchena et al., 2008), as well as the casein kinase pathway (Sanz-Clemente et al., 2010), can now be assayed. Ketamine—an NMDA receptor antagonist acting preferentially on PV cells (Behrens et al., 2007)—has recently been reported to reverse functional deficits in key forebrain nodes of the default mode network in Mecp2 KO mice (Kron et al., 2012). Other factors, such as the Otx2 homeoprotein, have been found to maintain PV-cells in a mature state (Beurdeley et al., 2012). Knockdown strategies regulating Otx2 content may also be fruitful in treating the Mecp2 KO mice.
Maturation of visual cortical circuits is reportedly impaired in another autism model, the Angelman syndrome mouse deficient in Ube3a (Yashiro et al., 2009), which can also be reversed by sensory deprivation. Our results indicate that ongoing endogenous neural activity may ensure the stability of cortical circuits. At a synaptic level, spontaneous transmitter release is required to maintain postsynaptic receptors (McKinney et al., 1999; Saitoe et al., 2001), while spontaneous action potentials observe spike-timing-dependent plasticity rules for synapse strengthening and maintenance of connectivity (Gilson et al., 2009; Kolodziejski et al., 2010). DR or NR2A disruption, while degrading orientation tuning even further (Figure 6), is sufficient to rescue both spontaneous neural activity and normal visual acuity in Mecp2 KO mice. Retinogeniculate circuits are instead unaffected by late DR, responding as if deprived in the Mecp2 KO mouse (Noutel et al., 2011).
Our findings ultimately reveal that vision in Rett syndrome patients may serve as a robust biomarker of both cortical status and its response to therapy. To date, visual processing and vision, in general, have never been analyzed in a systematic manner in RTT patients. Available data in the literature are limited and mixed (Saunders et al., 1995; von Tetzchner et al., 1996) and a few studies have suggested some abnormal visual processing in RTT patients (Bader et al., 1989, Stauder et al., 2006; von Tetzchner et al., 1996). This is a missed opportunity, given that eye gaze is one of the relatively well-preserved functions in non-verbal RTT girls, making vision testing feasible. Preliminary data indicate a clear correlation between visual processing and the clinical stage of RTT patients (G. DeGregorio, O. Khwaja, W. Kaufmann, M.F., and C.A. Nelson, unpublished data). Our findings thus encourage a longitudinal study of vision in Rett syndrome patients and its potential use as a biomarker of cortical function.
EXPERIMENTAL PROCEDURES
All procedures were approved by IACUC of Boston Children’s Hospital and conducted in the Mecp2 KO mouse line generated by A. Bird and colleagues (Guy et al., 2001). Double mutants for Mecp2 and NR2A were generated by crossing Mecp2 heterozygote females with NR2A KO males (originally generated by M. Mishina; Sakimura et al., 1995). fLox-STOP-Mecp2 (Guy et al., 2007) or PV-Cre × fLox-Mecp2 animals were used in some experiments (Hippenmeyer et al., 2005; Jackson Laboratories). All control animals were WT age-matched littermates of the mutant mice.
Single-Cell Electrophysiology In Vivo
Electrophysiological recordings were performed using standard techniques (Fagiolini et al., 2003). Spontaneous and evoked single-unit responses were recorded with multichannel probes (A1×16-3mm-50-177-A16, Neuronexus Technologies) in response to high contrast (100%), low spatial frequency sine wave gratings (0.025 or 0.07 cpd; 2 Hz).
Visual Evoked Potential (VEP)
Transient VEPs were recorded under nembutal/chlorprothixene anesthesia using standard techniques in mice (Fagiolini and Hensch, 2000). A tungsten electrode (1.5 MΩ, FST) was inserted into binocular V1. Receptive field was located within the visual field 20 degrees from the vertical meridian corresponding to the maximal VEP response.
Visual Behavioral Test: Optomotor Task
Behavioral threshold acuity was evaluated using the optomotor task (Prusky et al., 2004). Mice were tested every 3–5 days starting after eye opening until adulthood (P60–P240). Experimenters were blind to genotype and the animal’s previously recorded thresholds. All animals were habituated before the onset of testing by gentle handling and by placing them on the arena platform for a few minutes at a time.
In Vitro Analysis: Voltage-Sensitive Dye Imaging
Mice were decapitated under brief isoflurane anesthesia and the brain processed as detailed in Supplemental Information. A stimulating pulse (1 ms) was delivered through an ACSF-filled patch pipette to the white matter in V1. The resultant change in emitted Di-4-ANEPPS fluorescence, corresponding to a change in membrane potential, was recorded with a MiCam Ultima (Brain Vision, SciMedia) camera (at 1 frame/ms). Changes in fluorescence were averaged across ten 512 ms trials. Regions of interest (125 × 125 μm2) in the upper (150 μm below the pia) and lower layers (300 μm above the white matter) “on beam” with the stimulating electrode were analyzed for maximum change in intensity normalized to the resting intensity (DF/F).
Immunofluorescence
Primary antibodies and dilutions are detailed in the Supplemental Information. Quantitative analyses of the binocular zone of visual cortex across all layers were performed blind to genotype and treatment. Mean pixel intensity (at 1003) of the PV signal in each field (1,024 × 1,024) was measured using MacBiophotonics ImageJ software. The number of perisomatic synapses (at 100×) was determined on triple-stained images (PV, GAD65, DAPI) using the “particle analysis” function (ImageJ).
Chromatin Immunoprecipitation Analysis
ChIP experiments were performed using the MAGnify Chromatin immunoprecipitatin kit (Invitrogen) with some modifications, followed by qRT-PCR. Immunoprecipitation was performed on visual cortex homogenates using rabbit antibody against Mecp2 (Millipore) or non specific immunoglobulin IgG (Jackson Immunoresearch) as negative control. For a description of computational analysis and experimental details see Supplemental Information.
Statistical Analysis
All data are presented as mean ± standard error, with n and ages as shown in figures or stated in text. All data were first analyzed for D’Agostino & Pearson omnibus normality. The following parametric tests were used: one-way ANOVA with Tukey’s multiple comparison test for comparison of multiple groups, two-way ANOVA for comparing acuity development and unpaired t test for comparing between two groups. The following nonparametric tests were used: Mann-Whitney rank-sum test for comparing between two groups and Kruskal-Wallis analysis of variance with Dunnet’s post hoc test for comparison of multiple groups.
Supplementary Material
ACKNOWLEDGMENTS
This work is dedicated to the memory of Nonno Babbo. S.D. was supported by the International Rett Syndrome Foundation; M.F. by NIH (R21NS062277 & RO1NS070300), Harvard University Milton Fund, and Office of Faculty Development (Boston Children’s Hospital); T.K.H. by NIH (1DP1OD 003699); A.S. by JSPS (P09745). We thank S. Arber (FMI Basel) and M. Mishina (University of Tokyo) for providing original PV-Cre and NR2A KO mouse breeders, respectively; M. Greenberg (Harvard Medical School) for providing Mecp2 antibody; M. Marcotrigiano for animal care and M. Nakamura for genotyping and assistance with qPCR and ChiP-qPCR; L. Min for assistance with behavioral analysis; A.D. Hill and the Boston Children’s Hospital IDDRC (Intellectual and Developmental Disabilities Research Center) imaging core and neurodevelopmental behavioral core (supported by NIH-P30-HD-18655); and C. Chen and members of the Fagiolini and Hensch labs for helpful discussion.
Footnotes
SUPPLEMENTAL INFORMATION Supplemental Information includes four figures, two tables, and Supplemental Experimental Procedures and can be found with this article online at http://dx.doi.org/10.1016/j.neuron.2012.12.004.
REFERENCES
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Armstrong DD, Deguchi K, Antallfy B. Survey of MeCP2 in the Rett syndrome and the non-Rett syndrome brain. J. Child Neurol. 2003;18:683–687. doi: 10.1177/08830738030180100601. [DOI] [PubMed] [Google Scholar]
- Asaka Y, Jugloff DG, Zhang L, Eubanks JH, Fitzsimonds RM. Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol. Dis. 2006;21:217–227. doi: 10.1016/j.nbd.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Atallah BV, Bruns W, Carandini M, Scanziani M. Parvalbumin-expressing interneurons linearly transform cortical responses to visual stimuli. Neuron. 2012;73:159–170. doi: 10.1016/j.neuron.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader GG, Witt-Engerström I, Hagberg B. Neurophysiological findings in the Rett syndrome, II: Visual and auditory brainstem, middle and late evoked responses. Brain Dev. 1989;11:110–114. doi: 10.1016/s0387-7604(89)80078-6. [DOI] [PubMed] [Google Scholar]
- Bagnall MW, Hull C, Bushong EA, Ellisman MH, Scanziani M. Multiple clusters of release sites formed by individual thalamic afferents onto cortical interneurons ensure reliable transmission. Neuron. 2011;71:180–194. doi: 10.1016/j.neuron.2011.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballas N, Lioy DT, Grunseich C, Mandel G. Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat. Neurosci. 2009;12:311–317. doi: 10.1038/nn.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmer D, Goldstine J, Rao YM, LaSalle JM. Elevated methyl-CpG-binding protein 2 expression is acquired during postnatal human brain development and is correlated with alternative polyadenylation. J. Mol. Med. 2003;81:61–68. doi: 10.1007/s00109-002-0396-5. [DOI] [PubMed] [Google Scholar]
- Bartos M, Vida I, Jonas P. Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat. Rev. Neurosci. 2007;8:45–56. doi: 10.1038/nrn2044. [DOI] [PubMed] [Google Scholar]
- Behrens MM, Ali SS, Dao DN, Lucero J, Shekhtman G, Quick KL, Dugan LL. Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science. 2007;318:1645–1647. doi: 10.1126/science.1148045. [DOI] [PubMed] [Google Scholar]
- Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G, Li Y, Quinlan EM, Nakazawa K. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat. Neurosci. 2010;13:76–83. doi: 10.1038/nn.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkes P, Orbán G, Lengyel M, Fiser J. Spontaneous cortical activity reveals hallmarks of an optimal internal model of the environment. Science. 2011;331:83–87. doi: 10.1126/science.1195870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurdeley M, Spatazza J, Lee HH, Sugiyama S, Bernard C, Di Nardo AA, Hensch TK, Prochiantz A. Otx2 binding to perineuronal nets persistently regulates plasticity in the mature visual cortex. J. Neurosci. 2012;32:9429–9437. doi: 10.1523/JNEUROSCI.0394-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 2006;49:341–348. doi: 10.1016/j.neuron.2005.12.027. [DOI] [PubMed] [Google Scholar]
- Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, Neul JL, Gong S, Lu HC, Heintz N, et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468:263–269. doi: 10.1038/nature09582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheval H, Guy J, Merusi C, De Sousa D, Selfridge J, Bird A. Postnatal inactivation reveals enhanced requirement for MeCP2 at distinct age windows. Hum. Mol. Genet. 2012;21:3806–3814. doi: 10.1093/hmg/dds208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Gabel HW, Hemberg M, Hutchinson AN, Sadacca LA, Ebert DH, Harmin DA, Greenberg RS, Verdine VK, Zhou Z, et al. Genome-wide activity-dependent MeCP2 phosphorylation regulates nervous system development and function. Neuron. 2011;72:72–85. doi: 10.1016/j.neuron.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruikshank SJ, Lewis TJ, Connors BW. Synaptic basis for intense thalamocortical activation of feedforward inhibitory cells in neocortex. Nat. Neurosci. 2007;10:462–468. doi: 10.1038/nn1861. [DOI] [PubMed] [Google Scholar]
- Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. USA. 2005;102:12560–12565. doi: 10.1073/pnas.0506071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Marchena J, Roberts AC, Middlebrooks PG, Valakh V, Yashiro K, Wilfley LR, Philpot BD. NMDA receptor antagonists reveal age-dependent differences in the properties of visual cortical plasticity. J. Neurophysiol. 2008;100:1936–1948. doi: 10.1152/jn.90290.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deco G, Romo R. The role of fluctuations in perception. Trends Neurosci. 2008;31:591–598. doi: 10.1016/j.tins.2008.08.007. [DOI] [PubMed] [Google Scholar]
- Deng JV, Rodriguiz RM, Hutchinson AN, Kim IH, Wetsel WC, West AE. MeCP2 in the nucleus accumbens contributes to neural and behavioral responses to psychostimulants. Nat. Neurosci. 2010;13:1128–1136. doi: 10.1038/nn.2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derecki NC, Cronk JC, Lu Z, Xu E, Abbott SB, Guyenet PG, Kipnis J. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature. 2012;484:105–109. doi: 10.1038/nature10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagiolini M, Hensch TK. Inhibitory threshold for critical-period activation in primary visual cortex. Nature. 2000;404:183–186. doi: 10.1038/35004582. [DOI] [PubMed] [Google Scholar]
- Fagiolini M, Katagiri H, Miyamoto H, Mori H, Grant SG, Mishina M, Hensch TK. Separable features of visual cortical plasticity revealed by N-methyl-D-aspartate receptor 2A signaling. Proc. Natl. Acad. Sci. USA. 2003;100:2854–2859. doi: 10.1073/pnas.0536089100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiser J, Chiu C, Weliky M. Small modulation of ongoing cortical dynamics by sensory input during natural vision. Nature. 2004;431:573–578. doi: 10.1038/nature02907. [DOI] [PubMed] [Google Scholar]
- Fox MD, Raichle ME. Spontaneous fluctuations in brain activity observed with functional magnetic resonance imaging. Nat. Rev. Neurosci. 2007;8:700–711. doi: 10.1038/nrn2201. [DOI] [PubMed] [Google Scholar]
- Fyffe SL, Neul JL, Samaco RC, Chao HT, Ben-Shachar S, Moretti P, McGill BE, Goulding EH, Sullivan E, Tecott LH, Zoghbi HY. Deletion of Mecp2 in Sim1-expressing neurons reveals a critical role for MeCP2 in feeding behavior, aggression, and the response to stress. Neuron. 2008;59:947–958. doi: 10.1016/j.neuron.2008.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemelli T, Berton O, Nelson ED, Perrotti LI, Jaenisch R, Monteggia LM. Postnatal loss of methyl-CpG binding protein 2 in the forebrain is sufficient to mediate behavioral aspects of Rett syndrome in mice. Biol. Psychiatry. 2006;59:468–476. doi: 10.1016/j.biopsych.2005.07.025. [DOI] [PubMed] [Google Scholar]
- Gianfranceschi L, Siciliano R, Walls J, Morales B, Kirkwood A, Huang ZJ, Tonegawa S, Maffei L. Visual cortex is rescued from the effects of dark rearing by overexpression of BDNF. Proc. Natl. Acad. Sci. USA. 2003;100:12486–12491. doi: 10.1073/pnas.1934836100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilson M, Burkitt AN, Grayden DB, Thomas DA, van Hemmen JL. Emergence of network structure due to spike-timing-dependent plasticity in recurrent neuronal networks III: Partially connected neurons driven by spontaneous activity. Biol. Cybern. 2009;101:411–426. doi: 10.1007/s00422-009-0343-4. [DOI] [PubMed] [Google Scholar]
- Glaze DG, Percy AK, Skinner S, Motil KJ, Neul JL, Barrish JO, Lane JB, Geerts SP, Annese F, Graham J, et al. Epilepsy and the natural history of Rett syndrome. Neurology. 2010;74:909–912. doi: 10.1212/WNL.0b013e3181d6b852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinvald A, Hildesheim R. VSDI: a new era in functional imaging of cortical dynamics. Nat. Rev. Neurosci. 2004;5:874–885. doi: 10.1038/nrn1536. [DOI] [PubMed] [Google Scholar]
- Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315:1143–1147. doi: 10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensch TK. Critical period regulation. Annu. Rev. Neurosci. 2004;27:549–579. doi: 10.1146/annurev.neuro.27.070203.144327. [DOI] [PubMed] [Google Scholar]
- Hensch TK. Critical period plasticity in local cortical circuits. Nat. Rev. Neurosci. 2005;6:877–888. doi: 10.1038/nrn1787. [DOI] [PubMed] [Google Scholar]
- Hensch TK, Fagiolini M. Excitatory-inhibitory balance and critical period plasticity in developing visual cortex. Prog. Brain Res. 2005;147:115–124. doi: 10.1016/S0079-6123(04)47009-5. [DOI] [PubMed] [Google Scholar]
- Hippenmeyer S, Vrieseling E, Sigrist M, Portmann T, Laengle C, Ladle DR, Arber S. A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLoS Biol. 2005;3:e159. doi: 10.1371/journal.pbio.0030159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppke P, Köhler K, Brockmann K, Stettner GM, Gärtner J. Treatment of epilepsy in Rett syndrome. Eur. J. Paediatr. Neurol. 2007;11:10–16. doi: 10.1016/j.ejpn.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Jian L, Nagarajan L, de Klerk N, Ravine D, Bower C, Anderson A, Williamson S, Christodoulou J, Leonard H. Predictors of seizure onset in Rett syndrome. J. Pediatr. 2006;149:542–547. doi: 10.1016/j.jpeds.2006.06.015. [DOI] [PubMed] [Google Scholar]
- Kadotani H, Hirano T, Masugi M, Nakamura K, Nakao K, Katsuki M, Nakanishi S. Motor discoordination results from combined gene disruption of the NMDA receptor NR2A and NR2C subunits, but not from single disruption of the NR2A or NR2C subunit. J. Neurosci. 1996;16:7859–7867. doi: 10.1523/JNEUROSCI.16-24-07859.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katagiri H, Fagiolini M, Hensch TK. Optimization of somatic inhibition at critical period onset in mouse visual cortex. Neuron. 2007;53:805–812. doi: 10.1016/j.neuron.2007.02.026. [DOI] [PubMed] [Google Scholar]
- Kinney JW, Davis CN, Tabarean I, Conti B, Bartfai T, Behrens MM. A specific role for NR2A-containing NMDA receptors in the maintenance of parvalbumin and GAD67 immunoreactivity in cultured interneurons. J. Neurosci. 2006;26:1604–1615. doi: 10.1523/JNEUROSCI.4722-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood A, Bear MF. Hebbian synapses in visual cortex. J. Neurosci. 1994;14:1634–1645. doi: 10.1523/JNEUROSCI.14-03-01634.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline DD, Ogier M, Kunze DL, Katz DM. Exogenous brain-derived neurotrophic factor rescues synaptic dysfunction in Mecp2-null mice. J. Neurosci. 2010;30:5303–5310. doi: 10.1523/JNEUROSCI.5503-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodziejski C, Tetzlaff C, Wörgötter F. Closed-form treatment of the interactions between neuronal activity and timing-dependent plasticity in networks of linear neurons. Front. Comput. Neurosci. 2010;4:134. doi: 10.3389/fncom.2010.00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkova T, Fuchs EC, Ponomarenko A, von Engelhardt J, Monyer H. NMDA receptor ablation on parvalbumin-positive interneurons impairs hippocampal synchrony, spatial representations, and working memory. Neuron. 2010;68:557–569. doi: 10.1016/j.neuron.2010.09.017. [DOI] [PubMed] [Google Scholar]
- Kron M, Howell CJ, Adams IT, Ransbottom M, Christian D, Ogier M, Katz DM. Brain activity mapping in Mecp2 mutant mice reveals functional deficits in forebrain circuits, including key nodes in the default mode network, that are reversed with ketamine treatment. J. Neurosci. 2012;32:13860–13872. doi: 10.1523/JNEUROSCI.2159-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman SJ, Lu J, Lazarus MS, Huang ZJ. Maturation of GABAergic inhibition promotes strengthening of temporally coherent inputs among convergent pathways. PLoS Comput. Biol. 2010;6:e1000797. doi: 10.1371/journal.pcbi.1000797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Kim W, Ham BJ, Chen W, Bear MF, Yoon BJ. Activity-dependent NR2B expression is mediated by MeCP2-dependent epigenetic regulation. Biochem. Biophys. Res. Commun. 2008;377:930–934. doi: 10.1016/j.bbrc.2008.10.082. [DOI] [PubMed] [Google Scholar]
- Lee SH, Kwan AC, Zhang S, Phoumthipphavong V, Flannery JG, Masmanidis SC, Taniguchi H, Huang ZJ, Zhang F, Boyden ES, et al. Activation of specific interneurons improves V1 feature selectivity and visual perception. Nature. 2012;488:379–383. doi: 10.1038/nature11312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lioy DT, Garg SK, Monaghan CE, Raber J, Foust KD, Kaspar BK, Hirrlinger PG, Kirchhoff F, Bissonnette JM, Ballas N, Mandel G. A role for glia in the progression of Rett’s syndrome. Nature. 2011;475:497–500. doi: 10.1038/nature10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science. 2004;304:1021–1024. doi: 10.1126/science.1096615. [DOI] [PubMed] [Google Scholar]
- Lodato S, Rouaux C, Quast KB, Jantrachotechatchawan C, Studer M, Hensch TK, Arlotta P. Excitatory projection neuron subtypes control the distribution of local inhibitory interneurons in the cerebral cortex. Neuron. 2011;69:763–779. doi: 10.1016/j.neuron.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nat. Rev. Neurosci. 2004;5:793–807. doi: 10.1038/nrn1519. [DOI] [PubMed] [Google Scholar]
- McGraw CM, Samaco RC, Zoghbi HY. Adult neural function requires MeCP2. Science. 2011;333:186. doi: 10.1126/science.1206593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney RA, Capogna M, Dürr R, Gähwiler BH, Thompson SM. Miniature synaptic events maintain dendritic spines via AMPA receptor activation. Nat. Neurosci. 1999;2:44–49. doi: 10.1038/4548. [DOI] [PubMed] [Google Scholar]
- Nelson ED, Kavalali ET, Monteggia LM. MeCP2-dependent transcriptional repression regulates excitatory neurotransmission. Curr. Biol. 2006;16:710–716. doi: 10.1016/j.cub.2006.02.062. [DOI] [PubMed] [Google Scholar]
- Nguyen MV, Du F, Felice CA, Shan X, Nigam A, Mandel G, Robinson JK, Ballas N. MeCP2 is critical for maintaining mature neuronal networks and global brain anatomy during late stages of postnatal brain development and in the mature adult brain. J. Neurosci. 2012;32:10021–10034. doi: 10.1523/JNEUROSCI.1316-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissenkorn A, Gak E, Vecsler M, Reznik H, Menascu S, Ben Zeev B. Epilepsy in Rett syndrome—the experience of a National Rett Center. Epilepsia. 2010;51:1252–1258. doi: 10.1111/j.1528-1167.2010.02597.x. [DOI] [PubMed] [Google Scholar]
- Noutel J, Hong YK, Leu B, Kang E, Chen C. Experience-dependent retinogeniculate synapse remodeling is abnormal in MeCP2-deficient mice. Neuron. 2011;70:35–42. doi: 10.1016/j.neuron.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusky GT, Alam NM, Beekman S, Douglas RM. Rapid quantification of adult and developing mouse spatial vision using a virtual optomotor system. Invest. Ophthalmol. Vis. Sci. 2004;45:4611–4616. doi: 10.1167/iovs.04-0541. [DOI] [PubMed] [Google Scholar]
- Quinlan EM, Philpot BD, Huganir RL, Bear MF. Rapid, experience-dependent expression of synaptic NMDA receptors in visual cortex in vivo. Nat. Neurosci. 1999;2:352–357. doi: 10.1038/7263. [DOI] [PubMed] [Google Scholar]
- Ringach DL. Spontaneous and driven cortical activity: implications for computation. Curr. Opin. Neurobiol. 2009;19:439–444. doi: 10.1016/j.conb.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas C, Frank H, Heynen AJ, Morales B, Bear MF, Kirkwood A. Developmental inhibitory gate controls the relay of activity to the superficial layers of the visual cortex. J. Neurosci. 2001;21:6791–6801. doi: 10.1523/JNEUROSCI.21-17-06791.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoe M, Schwarz TL, Umbach JA, Gundersen CB, Kidokoro Y. Absence of junctional glutamate receptor clusters in Drosophila mutants lacking spontaneous transmitter release. Science. 2001;293:514–517. doi: 10.1126/science.1061270. [DOI] [PubMed] [Google Scholar]
- Sakimura K, Kutsuwada T, Ito I, Manabe T, Takayama C, Kushiya E, Yagi T, Aizawa S, Inoue Y, Sugiyama H, et al. Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptor epsilon 1 subunit. Nature. 1995;373:151–155. doi: 10.1038/373151a0. [DOI] [PubMed] [Google Scholar]
- Samaco RC, Nagarajan RP, Braunschweig D, LaSalle JM. Multiple pathways regulate MeCP2 expression in normal brain development and exhibit defects in autism-spectrum disorders. Hum. Mol. Genet. 2004;13:629–639. doi: 10.1093/hmg/ddh063. [DOI] [PubMed] [Google Scholar]
- Samaco RC, Mandel-Brehm C, Chao HT, Ward CS, Fyffe-Maricich SL, Ren J, Hyland K, Thaller C, Maricich SM, Humphreys P, et al. Loss of MeCP2 in aminergic neurons causes cell-autonomous defects in neurotransmitter synthesis and specific behavioral abnormalities. Proc. Natl. Acad. Sci. USA. 2009;106:21966–21971. doi: 10.1073/pnas.0912257106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz-Clemente A, Matta JA, Isaac JT, Roche KW. Casein kinase 2 regulates the NR2 subunit composition of synaptic NMDA receptors. Neuron. 2010;67:984–996. doi: 10.1016/j.neuron.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders KJ, McCulloch DL, Kerr AM. Visual function in Rett syndrome. Dev. Med. Child Neurol. 1995;37:496–504. doi: 10.1111/j.1469-8749.1995.tb12037.x. [DOI] [PubMed] [Google Scholar]
- Schölvinck ML, Friston KJ, Rees G. The influence of spontaneous activity on stimulus processing in primary visual cortex. Neuroimage. 2012;59:2700–2708. doi: 10.1016/j.neuroimage.2011.10.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd GMG, Katz DM. Synaptic microcircuit dysfunction in genetic models of neurodevelopmental disorders: focus on Mecp2 and Met. Curr. Opin. Neurobiol. 2011;21:827–833. doi: 10.1016/j.conb.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauder JE, Smeets EE, van Mil SG, Curfs LG. The development of visual- and auditory processing in Rett syndrome: an ERP study. Brain Dev. 2006;28:487–494. doi: 10.1016/j.braindev.2006.02.011. [DOI] [PubMed] [Google Scholar]
- Sugiyama S, Di Nardo AA, Aizawa S, Matsuo I, Volovitch M, Prochiantz A, Hensch TK. Experience-dependent transfer of Otx2 homeoprotein into the visual cortex activates postnatal plasticity. Cell. 2008;134:508–520. doi: 10.1016/j.cell.2008.05.054. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat. Rev. Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Van Esch H, Bauters M, Ignatius J, Jansen M, Raynaud M, Hollanders K, Lugtenberg D, Bienvenu T, Jensen LR, Gecz J, et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am. J. Hum. Genet. 2005;77:442–453. doi: 10.1086/444549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Tetzchner S, Jacobsen KH, Smith L, Skjeldal OH, Heiberg A, Fagan JF. Vision, cognition and developmental characteristics of girls and women with Rett syndrome. Dev. Med. Child Neurol. 1996;38:212–225. doi: 10.1111/j.1469-8749.1996.tb15083.x. [DOI] [PubMed] [Google Scholar]
- Wood L, Shepherd GMG. Synaptic circuit abnormalities of motor-frontal layer 2/3 pyramidal neurons in a mutant mouse model of Rett syndrome. Neurobiol. Dis. 2010;38:281–287. doi: 10.1016/j.nbd.2010.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood L, Gray NW, Zhou Z, Greenberg ME, Shepherd GMG. Synaptic circuit abnormalities of motor-frontal layer 2/3 pyramidal neurons in an RNA interference model of methyl-CpG-binding protein 2 deficiency. J. Neurosci. 2009;29:12440–12448. doi: 10.1523/JNEUROSCI.3321-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Fu Y, Knott G, Lu J, Di Cristo G, Huang ZJ. GABA signaling promotes synapse elimination and axon pruning in developing cortical inhibitory interneurons. J. Neurosci. 2012;32:331–343. doi: 10.1523/JNEUROSCI.3189-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashiro K, Riday TT, Condon KH, Roberts AC, Bernardo DR, Prakash R, Weinberg RJ, Ehlers MD, Philpot BD. Ube3a is required for experience-dependent maturation of the neocortex. Nat. Neurosci. 2009;12:777–783. doi: 10.1038/nn.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY. Postnatal neurodevelopmental disorders: meeting at the synapse? Science. 2003;302:826–830. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.