

Abstract
We report here the biochemical characterization of the deafness-associated mitochondrial tRNASer(UCN) T7511C mutation, in conjunction with homoplasmic ND1 T3308C and tRNAAla T5655C mutations using cybrids constructed by transferring mitochondria from lymphoblastoid cell lines derived from an African family into human mtDNA-less (ρ°) cells. Three cybrids derived from an affected matrilineal relative carrying the homoplasmic T7511C mutation, exhibited ∼75% decrease in the tRNASer(UCN) level, compared with three control cybrids. This amount of reduction in the tRNASer(UCN) level is below a proposed threshold to support a normal rate of mitochondrial protein synthesis in lymphoblastoid cell lines. This defect is likely a primary contributor to ∼52% reduction in the rate of mitochondrial protein synthesis and marked defects in respiration and growth properties in galactose-containing medium. Interestingly, the T5655C mutation produces ∼50% reduction in the tRNAAla level in mutant cells. Strikingly, the T3308C mutation causes a significant decrease both in the amount of ND1 mRNA and co-transcribed tRNALeu(UUR) in mutant cells. Thus, mitochondrial dysfunctions caused by the T5655C and T3308C mutations may modulate the phenotypic manifestation of the T7511C mutation. These observations imply that a combination of the T7511C mutation with two mtDNA mutations accounts for the high penetrance of deafness in this family.
INTRODUCTION
Mutations in mitochondrial DNA (mtDNA) have been found to be associated with both syndromic and nonsyndromic forms of sensorineural hearing impairment (1,2). Of these, the homoplasmic A1555G mutation in the 12S rRNA gene has been shown to be one of the most prominent causes of aminoglycoside-induced and nonsyndromic deafness in families of different ethnic backgrounds (3–6). Recently, the homoplasmic C1494T mutation in the same gene has also been found to be associated with aminoglycoside induced and nonsyndromic deafness in a large Chinese family (7). The mitochondrial tRNASer(UCN) gene appears to be another hot spot for mutations associated with nonsyndromic deafness, as four mutations: A7445G (8–11), 7472insC (12,13), T7510C (14,15) and T7511C (16–18), have been identified in this gene. Unlike other pathogenic mtDNA mutations such as the MELAS A3243G mutation in the tRNALeu(UUR) gene (19), the deafness-associated mutations in the tRNASer(UCN) gene often occur in homoplasmy or in high levels of heteroplasmy, indicating a high threshold for pathogenicity. The primary defect in tRNASer(UCN) mutations appears to be a failure in tRNA metabolism, thereby leading to a decrease in the steady-state levels of affected tRNAs. Subsequently, a shortage of the tRNASer(UCN) is responsible for the reduced rate of mitochondrial protein synthesis and the respiration defects, as in the cases of cell lines carrying the A7445G or 7472insC mutation (20–22).
The T7511C mutation has been identified to be associated with nonsyndromic deafness in several families from different ethnic groups, including African (16), French (17) and Japanese (18). The T7511C mutation often exists in homoplasmy in most matrilineal relatives of these pedigrees and in a high level of heteroplasmy in some matrilineal relatives of those families, but not in the controls (16–18). However, the levels of homoplasmy and heteroplasmy did not correlate with the severity and age-of-onset of deafness (16–18). Despite sharing some common features, including bilateral, symmetric and sensorineural hearing impairment, matrilineal relatives of intra-families or inter-families carrying the T7511C mutation exhibited the variable severity, age-of-onset and progression in hearing impairment (16–18,23). Strikingly, these pedigrees differ considerably in the penetrance of the T7511C mutation (17,18,23). In particular, only a small portion of matrilineal relatives in two French pedigrees developed hearing impairment (43 and 30% penetrance, respectively) (17), whereas 14 of 21 matrilineal relatives in the Japanese family suffered from hearing impairment (18). In contrast, 36 of 43 matrilineal relatives in the large African pedigree exhibited hearing impairment (16,23). The occurrence of the T7511C mutation in these genetically unrelated pedigrees affected by sensorineural hearing impairment is clear evidence that this mutation is responsible for the pathogenesis. However, this mutation is apparently not sufficient to produce the clinical phenotype since some individuals carrying the mutation have normal hearing. Thus, other factors including other mtDNA mutations/polymorphisms and/or nuclear backgrounds modulate the phenotypic variability and penetrance of deafness associated with the T7511C mutation.
To investigate the pathogenic mechanism of the T7511C mutation in the large African family, cybrid cell lines were constructed by transferring mitochondria from lymphoblastoid cell lines derived from an affected matrilineal relative with mtDNA mutations and from a control individual lacking mtDNA mutations, into human mtDNA-less (ρ°) cells (24,25). These cybrid cell lines were first examined for the presence and degree of mtDNA mutations. These cell lines were then assessed for the effects of the mtDNA mutations on the expression of mitochondrial tRNAs, including tRNASer(UCN), tRNAAla and tRNALeu(UUR), mitochondrial mRNAs, the rate of mitochondrial protein synthesis, the endogenous respiration and substrate-dependent respiration, and the growth properties in glucose and galactose medium.
MATERIALS AND METHODS
Cell lines and culture conditions
Immortalized lymphoblastoid cell lines derived from one affected matrilineal relative (III-27) of the African family and one genetically unrelated African control individual with comparable age (16) were grown in RPMI 1640 medium with 10% fetal bovine serum (FBS). The bromodeoxyuridine (BrdU)-resistant 143B.TK– cell line was grown in DMEM (containing 4.5 mg of glucose and 0.11 mg pyruvate per ml), supplemented with 100 µg of BrdU per ml and 5% FBS. The mtDNA-less ρ°206 cell line, derived from 143B.TK– (24) was grown under the same conditions as the parental line, except for the addition of 50 µg of uridine/ml. All cybrid cell lines constructed with enucleated lymphoblastoid cell lines were maintained in the same medium as the 143B.TK– cell line or the DMEM medium containing 0.9 mg galactose per ml and 0.5 mg pyruvate per ml (20,26,27) (hereafter referred to as special DMEM-galactose), supplemented with 5% dialyzed FBS.
Mitochondria-mediated ρ°206 cell transformation
Immortalized lymphoblastoid cell lines derived from one affected member of the African family (III-27) and one African control individual were used for the generation of cybrid cell lines. Transformation by cytoplasts of mtDNA-less ρ°206 cells was performed as described elsewhere (24–26).
Mitochondrial DNA analysis
An analysis for the presence and level of the T7511C mutation in the tRNASer(UCN) gene, the T3308C mutation in the ND1 gene and the T5655C mutation in the tRNAAla gene was carried out as previously described (16). The quantification of mtDNA copy numbers from different cybrids was performed by slot blot hybridization as detailed elsewhere (27).
Mitochondrial tRNA analysis
Total mitochondrial RNA were obtained using TOTALLY RNA™ kit (Ambion) from mitochondria isolated from cybrid cell lines (∼4.0 × 107 cells), as described previously (28); 5 µg of total mitochondrial RNA were electrophoresed through a 10% polyacrylamide/7 M urea gel in Tris–borate–EDTA buffer (TBE) (after heating the sample at 65°C for 10 min), and then electroblotted onto a positively charged nylon membrane (Roche) for the hybridization analysis with oligodeoxynucleotide probes. For the detection of tRNASer(UCN), tRNAAla, tRNALeu(UUR), tRNALys and tRNAGlu, the following non-radioactive DIG-labeled oligodeoxynucleotides specific for each RNA were used: 5′-CAAG CCAACCCCATGGCCTC-3′ (tRNASer(UCN)); 5′-GCATCAA CTGAACGCAATTC-3′ (tRNAAla); 5′-TCACTGTAAAGA GGTGTTGG-3′ (tRNALys); 5′-TGTTAAGAAGAGGAAT TGAA-3′ (tRNALeu(UUR)); 5′-TATTCTCGCACGGACTA CAA-3′ (tRNAGlu). DIG-labeled oligodeoxynucleotides were generated by using the DIG oligonucleotide Tailing kit (Roche).
The hybridization was performed in DIG Easy Hyb (Roche) hybridization solution for 16 h at 37°C. After hybridization, the blots were washed twice for 5 min in 2× SSC, 0.1% SDS at room temperature and then in 0.5× SSC, 0.1% SDS twice for 15 min at 37°C. The DIG-labeled probes were detected immunologically with DIG Wash and Block buffer, Anti-digoxigenin-AP, Fab fragments and CDP-Star (Roche). Quantification of density in each band was made as detailed elsewhere (20,29).
Mitochondrial mRNA analysis
Equal amounts (2 µg) of total mitochondrial RNA were fractionated by electrophoresis through a 1.8% agarose-formaldehyde gel, transferred onto a positively charged membrane (Roche), and initially hybridized with the ND6 (20) or ND1 specific RNA probe. Probes were synthesized on the corresponding restriction enzyme-linearized plasmid using a DIG RNA Labeling kit (Roche). RNA blots were then stripped and re-hybridized with the DIG-labeled ND4, COX1 and 12S rRNA probes, respectively. The plasmids used for RNA probes were constructed by PCR-amplifying fragments of the ND1 (positions 3506–3839), ND4 (positions 9966–10859), COX1 (positions 7346–7625) and 12S rRNA (positions 616–1606) (30) and cloning the fragments into the pCRII-TOPO vector carrying SP6 and T7 promoters (Clontech).
Analysis of mitochondrial protein synthesis
Pulse-labeling of the cell lines for 30 min with [35S]methionine–[35S]cysteine in methionine-free DMEM in the presence of emetine, electrophoretic analysis of the translation products, and quantification of radioactivity in the whole electrophoretic patterns or in individual well-resolved bands were carried out as detailed previously (20,27,31).
O2 consumption measurements
Rates of O2 consumption in intact cells were determined with a YSI 5300 oxygraph (Yellow Springs Instruments) on samples of 5 × 106 cells in 1.5 ml of special DMEM-glucose lacking glucose, and supplemented with 10% dialyzed FBS (24). Polarographic analysis of digitonin-permeabilized cells, using different respiratory substrates and inhibitors, to test the activity of the various respiratory complexes, was carried out as detailed elsewhere (32).
Growth measurements
These were made by plating samples of 105 cells on 10-cm Petri dishes in 10 ml of DMEM or special DMEM-galactose, supplemented with 5% dialyzed FBS, incubating them at 37°C for 3 days, and performing cell counts at daily intervals. The population doubling times (DTs) of the cell lines were determined from the growth curves or by using the following formula: DT = (t – t0) log2/(log N – log N0), where DT is the doubling time, t and t0 are the times at which the cells were counted, and N and N0 are the cell numbers at times t and t0, respectively (27).
Computer analysis
Statistical analysis was performed by the unpaired, two-tailed Student’s t-test contained in Microsoft Excel for Macintosh (version 5). Correlation analysis was performed using the curve fitting routine in CA-Cricket Graph III™ for Macintosh (version 1.5.2).
RESULTS
The African pedigree and mtDNA mutation sites
A large African pedigree with maternally inherited nonsyndromic deafness has been described elsewhere (16,23). Hearing impairment was symmetric and gradually progressive. Age at onset was variable, ranging from childhood to old age. Clinical data showed that these affected individuals did not have a history of exposure to aminoglycosides. Comprehensive family medical history demonstrated no muscle disease, seizures, visual problems, or other neurological disorders (23). Immortalized lymphoblastoid cell lines used for this investigation were derived from one affected matrilineal relative (III-27) of this family carrying the homoplasmic tRNASer(UCN) T7511C, ND1 T3308C and tRNAAla T5655C mutations (16) and one genetically unrelated African control individual lacking these mtDNA mutations. The sequence and subsequent RFLP analysis confirmed the presence of these homoplasmic mtDNA mutations in the affected cell line (III-27) but the absence of these mtDNA mutations in the control cell line.
As shown in Figure 1a, the T7511C transition in the tRNASer(UCN) disrupts a very conservative base-pairing, converting an A-U to a G-U base pairing on the 5′ side of the acceptor stem of this tRNA (33). This position is known to be important for tRNA identity (34) and the interaction of the tRNA with mitochondrial RNase P, which is involved in the 5′ end processing of the RNA precursors (35). The T5655C mutation, as shown in Figure 1b, lies in the 3′ end of the tRNAAla, where the position is important for tRNA identity (34). Furthermore, the T3308C mutation in the ND1 gene, as shown in Figure 1c, resulted in the replacement of the first amino acid, translation-initiating methionine with a threonine. Thus, the truncated protein was expected to be shortened by two amino acids. Interestingly, the T3308C mutation also locates in three nucleotides adjacent to the 3′ end of the tRNALeu(UUR). Unlike the T7511C mutation, the T5655C and T3308C mutations have also been found in some general populations (16,36).
Figure 1.

Sites of the mtDNA mutations in the African pedigree. (a) Location of the T7511C mutation in the secondary structure of tRNASer(UCN) (20); (b) Location of the T5655C mutation in the secondary structure of tRNAAla (33). (c) A schema of mtDNA sequence at position 3308 and adjacent sequence of ND1 and tRNALeu(UUR) from wild-type (WT) (30) and mutant (MT). Arrow indicates the position of the T3308C mutation.
The construction of cybrid cell lines
The lymphoblastoid cells derived from one affected subject (III-27) and one control individual were enucleated, and subsequently fused to a large excess of mtDNA-less human ρ°206 cells, derived from the 143B.TK– cell line (24). The cybrid clones were isolated by growing the fusion mixtures in selective DMEM medium, containing BrdU and lacking uridine (24–26). Between 25 and 45 days after fusion, 10–15 presumptive mitochondrial cybrids derived from each of the different donor cell lines were isolated, and subsequently analyzed for the presence and level of the T7511C, T5655C and T3308C mutations (16). The results confirmed the absence of the mtDNA mutations in the control clones and their presence in homoplasmy in all cybrids derived from the mutant cell line. Three cybrids derived from each donor cell line were used for the biochemical characterization described below.
Marked decrease in the levels of mitochondrial tRNASer(UCN), tRNAAla and tRNALeu(UUR)
To examine if the T7511C and T5655C mutations affect the processing of the corresponding tRNAs, the steady-state level of the tRNASer(UCN) or tRNAAla was determined by isolating total mitochondrial RNA from cybrid cell lines, separating them by a 10% polyacrylamide/7 M urea gel, electroblotting and hybridizing with a non-radioactive DIG-labeled oligodeoxynucleotide probe specific for tRNASer(UCN) or tRNAAla. After stripping the blots, the DIG-labeled oligodeoxynucleotide probes, including tRNALeu(UUR) and tRNALys as representatives of the whole H-strand transcription unit (37,38) and tRNAGlu derived from the L-strand transcription unit (37,38), were hybridized with the same blots for normalization purposes.
As shown in Figure 2, the amounts of tRNASer(UCN) and tRNAAla in mutant cells were markedly decreased, as compared to those in control cells. Surprisingly, a significant reduction in the level of tRNALeu(UUR) was observed in the mutant cell lines relative to the controls. Likely, the reduced steady-state level of tRNALeu(UUR) is due to the processing defect of this tRNA, caused by the adjacent T3308C mutation in the ND1 gene (Fig. 1c). For comparison, the average levels of tRNASer(UCN), tRNAAla and tRNALeu(UUR) in various control or mutant cell lines were normalized to the average levels in the same cell line for the tRNALys and tRNAGlu, respectively. As shown in Figure 3, the levels of tRNASer(UCN), tRNAAla and tRNALeu(UUR) in the mutant cells were significantly reduced, relative to the controls. The average levels of tRNASer(UCN) in mutant cells ranged between ∼25% of controls after normalization to tRNALys (P < 0.0028) and ∼17% of controls after normalization to tRNAGlu (P = 0.0044). Similarly, the average tRNAAla level varied from ∼52% [(after normalization to tRNALys) (P = 0.0351)] to ∼48% [(after normalization to tRNAGlu (P = 0.0432)] of controls. Furthermore, the average steady-state levels of tRNALeu(UUR) in the mutant cells was 57% of that (after normalization to tRNALys) (P = 0.0205) and 36% of that (after normalization to tRNAGlu) (P = 0.0181) in control cells.
Figure 2.
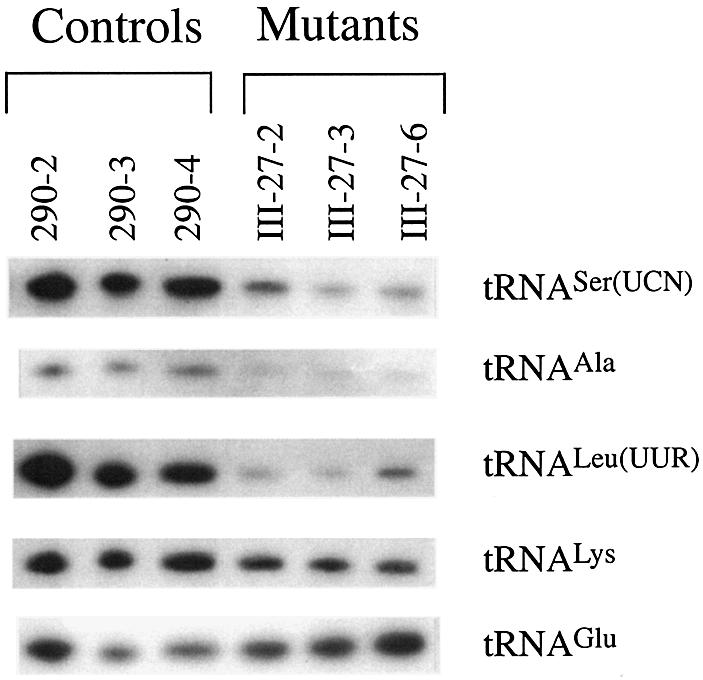
Northern blot analysis of mitochondrial tRNA. Equal amounts (5 µg) of total mitochondrial RNA from various cybrid cell lines were electrophoresed through a denaturing polyacrylamide gel, electroblotted and hybridized with DIG-labeled oligonucleotide probes specific for the mitochondrial tRNASer(UCN). The blots were then stripped and re-hybridized with DIG-labeled tRNAAla, tRNALeu(UUR), tRNALys and tRNAGlu, respectively.
Figure 3.

Quantification of mitochondrial tRNA levels. (a) Average relative tRNASer(UCN), content per cell, normalized to the average content per cell of tRNALys or tRNAGlu in three control cell lines and in three mutant cell lines. The values for the latter are expressed as percentages of the average values for the control cell lines. The calculations were based on three independent determinations of tRNASer(UCN) content in each cell line and three determinations of the content of each reference RNA marker in each cell line. (b) Average relative tRNAAla content per cell, normalized to the average content per cell of tRNALys or tRNAGlu in three control cell lines and in three mutant cell lines. Detailed calculation was the same as in (a). (c) Average relative tRNALeu(UUR) content per cell, normalized to the average content per cell of tRNALys or tRNAGlu in three control cell lines and in three mutant cell lines. Detailed calculation was the same as in (a). The error bars indicate two standard errors of the mean (S.E.M.); p indicates the significance, according to the Student’s t-test, of the difference between mutant and control values for tRNASer(UCN), tRNALeu(UUR) or tRNAAla normalized to the values for tRNALys or tRNAGlu.
An analysis of the mtDNA content of these cybrids by slot blot hybridization was performed using the DIG-labeled mt12S rRNA, and nuclear 28S rRNA probes for normalization purposes (27). The data failed to reveal any significant difference in the mtDNA/nuclear rDNA ratio between the average value for the control cells (100 ± 8%) and for the mutant cells (96 ± 22%) (P = 0.7682). Thus, the observed decrease in the tRNASer(UCN) and tRNAAla levels in the mutant relative to the control cell lines does not appear to reflect differences in the amount of mtDNA.
Significant reduction in the levels of ND1 but not ND6 mRNA
Our previous study revealed that the A7445G mutation in the precursor of tRNASer(UCN) has long-range effects on expression of the ND6 gene (20), which is co-transcribed from the mtDNA L-strand and derived from processing of large polycistronic transcripts (20). We therefore examined if the T7511C mutation in the tRNASer(UCN) affects the expression of the ND6, and whether the T3308C mutation has an effect on expression of the ND1. For this purpose, RNA transfer hybridization experiments were performed with total mitochondrial RNA from mutant and control cybrids, using the non-radioactive DIG-labeled ND6 or ND1 RNA probe, respectively. After stripping the blots, a set of DIG-labeled RNA probes: ND4, COX1 and 12S rRNA, were rehybridized with the same blots for normalization purposes.
As shown in Figure 4a, there is no obvious reduction in the amount of ND6 mRNA between the mutant and control cybrids. In contrast, the amount of ND1 mRNA was clearly lower in the mutant cell lines than in the control cell lines. To quantify the mRNA levels of ND6 and ND1, the corresponding bands in the blot were subjected to densitometric analysis. For comparison, the level of 12S rRNA, as representative of the H-strand rDNA transcription unit (37,38), and the combined levels of ND4 mRNA and COXI mRNA, as representative of the whole H-strand transcription unit (37,38), were determined on the same blots. The average levels of ND6 or ND1 mRNAs in each cell line were normalized to the average levels determined in the same cell line for each of the individual or combined reference RNAs, and expressed relative to the corresponding average values obtained in the control cell lines. The mutant cell lines exhibited slightly decreased levels of ND6 mRNA relative to the average value observed in wild-type cell lines, ranging between 88% of the average control value (after normalization to 12S rRNA) (P = 0.5438) and 95% (P = 0.8814) (after normalization to mRNAs). However, the average relative values of ND1 mRNA in the mutant cybrids, normalized with respect to those of 12S rRNA, reflected ∼43% reduction, ranging from ∼30 to 53% relative to the average control values (P = 0.0062) (Fig. 4b). Similarly, the average content values of ND1 mRNA in the mutant cybrids, after normalizing with mRNAs, exhibited an average 36% reduction, varying from ∼29 to 45% relative to the average control values (P < 0.0415) (Fig. 4c).
Figure 4.

Expression analysis of ND1 and ND6. (a) Equal amounts (2 µg) of total mitochondrial RNA from three mutant cell lines and three control cell lines were electrophoresed through a 1.8% agarose-formaldehyde gel, transferred onto a positively charged membrane and hybridized first with a DIG-labeled ND6 specific RNA probe. After stripping the blot, it was hybridized with a ND1 specific probe, and subsequently, after re-stripping of the blot, hybridized with DIG-labeled mtRNA probes ND4, COXI and 12S rRNA, respectively. (b) Average relative ND1 mRNA content per cell normalized to the average content per cell of ND4 and COXI mRNAs in three control cell lines and three mutant cell lines. The values for the latter are expressed as percentages of the average values for the control cell lines. (c) Average relative ND1 mRNA content per cell normalized to the average content per cell of 12S rRNA in three control cell lines and three mutant cell lines. Three independent determinations of ND1 mRNA content and three RNA reference markers for each cell line were used in the calculations.
Mitochondrial protein synthesis defect in the cybrid cell lines
To examine whether a defect in mitochondrial protein synthesis occurred in the cell lines carrying the mtDNA mutations, cells from each cybrid line were labeled for 30 min with [35S]methionine–[35S]cysteine in methionine-free DMEM medium in the presence of 100 µg/ml of emetine, to inhibit cytosolic protein synthesis. Figure 5a shows typical electrophoretic patterns of the mitochondrial translation products of the mutant and control cybrids. The patterns of the mtDNA-encoded polypeptides of the mutation-carrying cybrids were qualitatively identical, in terms of electrophoretic mobility of the various polypeptides, to those of the three control cybrid cell lines and of 143B.TK– (Fig. 5a).
Figure 5.

Mitochondrial protein labeling analysis. (a) Electrophoretic patterns of the mitochondrial translation products of different cybrid cell lines and of 143B.TK– cells labeled for 30 min with [35S]methionine in the presence of 100 µg of emetine per ml. Samples containing equal amounts of protein (20 µg) were run in SDS/polyacrylamide gradient gels. COI, COII and COIII, subunits I, II and III of cytochrome c oxidase; ND1, ND2, ND3, ND4, ND4L, ND5 and ND6, subunits 1, 2, 3, 4, 4L, 5 and 6 of the respiratory-chain NADH dehydrogenase; A6 and A8, subunits 6 and 8 of the H+-ATPase; CYTb, apocytochrome b. (b) Quantification of the rates of labeling of the mitochondrial translation products. The rates of mitochondrial protein labeling in three mutant cell lines and three control cell lines, determined as described elsewhere (20,31), are expressed as percentages of the value for 143B.TK– in each gel, with error bars S.E.M. Five independent labeling experiments and three to four electrophoretic analyses of each labeled preparation were carried out on each cell line. The horizontal dashed lines represent the average value for each group; p indicates the significance, according to the t-test, of the differences between mutant mean and control mean.
However, the mutant cybrids showed a clear tendency to decrease in the total rate of labeling of mitochondrial translation products relative to control cybrids. Figure 5b illustrates a quantification of the results of five labeling experiments and three to four electrophoretic runs, which was carried out by densitometric analysis of appropriate exposures of the fluorograms and normalization to the data obtained for the 143B.TK– sample included in each gel. It appears that the overall rate of labeling of the mitochondrial translation products in the mutant cell lines was decreased relative to the mean value measured in the control cell lines by ∼45–56%, with an average of 52% (P = 0.0138).
The average labeling of each polypeptide in the mutant cell lines revealed a variable decrease, ranging from 24 to 78%, relative to that in the control cell lines. As shown in Table 1, the synthesis of several high-molecular weight translation products, particularly in ND5, COI and ND4 with the highest contents of serine (UCN), leucine (UUR) and alanine codons, appeared to be more severely affected than those of the small products such as A8 and ND4L. Interestingly, the average labeling level of ND3 was decreased by ∼65%. However, the rate of synthesis of polypeptides in mutants relative to that in controls did not correlate with either the number of serine (UCN) codons or proportion of serine (UCN) residues, as well as those of leucine (UUR) or alanine codons. This observation is in contrast to previous studies with the deafness-associated A7445G mutation in the precursor of tRNASer(UCN) (20), or the MERRF A8344G mutation in the tRNALys (29).
Table 1. Usage of serine (UCN), alanine and leucine (UUR) codons in human mitochondrial genes and the average rate of labeling of the individual polypeptide in the mutant cell lines related to the average value in the control cell lines.
| Gene | Number of amino acids | Number of serine (UCN) codons | Serine (UCN) codon density (%) | Number of alanine codons | Alanine codon density (%) | Number of leucine (UUR) codons | Leucine (UUR) codon density (%) | Rate of synthesis of polypeptide in mutants (% of controls ± 2 S.E.M.) |
|---|---|---|---|---|---|---|---|---|
| ND5 |
603 |
36 |
5.9 |
44 |
7.3 |
9 |
1.5 |
22 ± 6 |
| COI |
513 |
28 |
5.5 |
40 |
7.8 |
7 |
1.4 |
33 ± 8 |
| ND4 |
459 |
33 |
7.2 |
26 |
5.7 |
9 |
2.0 |
30 ± 4 |
| Cyt b |
378 |
25 |
6.6 |
25 |
6.6 |
9 |
2.4 |
53 ± 18 |
| ND2 |
347 |
23 |
6.6 |
20 |
5.8 |
9 |
2.6 |
62 ± 12 |
| ND1 |
318 |
18 |
5.7 |
27 |
8.5 |
6 |
1.9 |
35 ± 8 |
| COIII |
260 |
16 |
6.2 |
15 |
5.7 |
3 |
1.2 |
38 ± 8 |
| COII |
227 |
9 |
4.0 |
14 |
6.1 |
5 |
2.2 |
55 ± 7 |
| A6 |
226 |
10 |
4.4 |
19 |
8.4 |
5 |
2.2 |
52 ± 12 |
| ND6 |
174 |
5 |
2.9 |
8 |
4.6 |
14 |
8.1 |
39 ± 17 |
| ND3 |
115 |
5 |
4.4 |
8 |
1.0 |
10 |
8.8 |
44 ± 18 |
| ND4L |
98 |
8 |
8.2 |
9 |
1.0 |
1 |
1.0 |
76 ± 26 |
| A8 | 68 | 4 | 5.9 | 0 | 0 | 2 | 2.9 | 67 ± 18 |
Respiration defects in the cybrid cell lines
The endogenous respiration rates of mutant and control cell lines were measured by determining the O2 consumption rate in intact cells, as described previously (24). As can be seen in Figure 6a, the rate of total O2 consumption in the mutant cell lines exhibited a variable decrease, ranging between ∼43 and 61%, relative to the mean value measured in the control cell lines, with an average reduction of ∼52% (P = 0.0021). The variations in overall respiration rate among the individual mutant cell lines, as compared to the individual control cell lines, showed a significant correlation with the corresponding variations in the rate of mitochondrial protein synthesis (r = 0.96, P < 0.001).
Figure 6.

Respiration and growth assays. (a) Average rates of total O2 consumption per cell measured in different cybrid cell lines. Four to eight determinations were carried out for each cell line. (b) Polarographic analysis of O2 consumption in digitonin-permeabilized cells of different cell lines with different substrates. The activities of the various components of the respiratory chain were determined as respiration dependent on glutamate or malate (Complex I) (solid bars; group averages indicated by dotted lines), succinate or glycerol-3-phosphate (G-3-P) (Complex III) (shaded bars; group averages indicated by short-dash lines), or ascorbate plus TMPD (Complex IV) (cross-hatched bars; group averages indicated by long-dash lines). Three to eight determinations were carried out for each cell line. (c) Ratios of DTs in galactose-containing medium to DTs in glucose- containing medium in different cell lines. Four determinations were carried out for each cell line.
In order to investigate which of the enzyme complexes of the respiratory chain was affected in the mutant cell lines, O2 consumption measurements were performed on digitonin-permeabilized cells, using different substrates and inhibitors (32). As illustrated in Figure 6b, in the mutant cell lines, the rate of malate/glutamate-driven respiration, which depends on the activities of NADH:ubiquinone oxidoreductase (Complex I), ubiquinol-cytochrome c reductase (Complex III) and cytochrome c oxidase (Complex IV), but usually reflects the rate-limiting activity of Complex I, was very significantly decreased, relative to the average rate in the control cell lines, by 43–66% (∼54% on average; P = 0.0065). Similarly, the rate of succinate/glycerol-3-phosphate (G3P)-driven respiration, which depends on the activities of Complex III and Complex IV, but usually reflects the rate-limiting activity of Complex III, was significantly affected, relative to the average rate in the control cell lines, by 32–42% (∼36% on average; P = 0.0187); furthermore, the rate of N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD)/ascorbate-driven respiration, which reflects the activity of Complex IV, exhibited a 26–40% reduction in Complex IV activity (∼33% on average; P = 0.0071). The variations in the rates of malate/glutamate-driven, succinate/G3P-driven and TMPD/ascorbate-driven respiration among the individual mutant cell lines, as compared to the individual control cell lines, showed a significant correlation with the corresponding variations in rate of mitochondrial protein synthesis (r = 0.96, P < 0.001; r = 0.98, P < 0.001; r = 0.98, P < 0.001, respectively).
Growth properties of cell lines in glucose- or galactose-containing medium
It has been shown that cell lines with impairment in their oxidative phosphorylation activity have reduced growth capacity in medium containing galactose instead of glucose (20,26,27). In order to investigate whether mtDNA mutations affect their growth capacity, the mutant and control cybrids were grown in special DMEM-galactose and in DMEM-glucose for 3 days. The DTs of the mutant cell lines in special DMEM-galactose and in DMEM-glucose were compared to those of the control cell lines in the same media. The DTs of the latter cell lines were higher in galactose medium (average 51 h) compared to the values in glucose medium (average 19 h). The DTs of the mutant cell lines in galactose medium showed a tendency to increase relative to the values in glucose medium much more than observed in the control cell lines (from an average of 27 h to an average of 128 h). As illustrated in Figure 6c, the ratios of DTs in galactose medium to those in glucose medium showed a clear increase in the mutant versus the control cell lines. Specifically, the mutant cell lines exhibited DT ratios from ∼1.6 to ∼1.9 times higher (on average 1.7 times higher, P = 0.0037) than the mean value found in control cell lines, confirming the existence of defective oxidative metabolism in the mtDNA mutation-carrying cells. The variations in DT ratio among the individual control and mutant cell lines were inversely correlated with the rate of total O2 consumption (r = 0.93; P < 0.001) and with the rate of mitochondrial protein synthesis (r = 0.90; P < 0.01).
DISCUSSION
In the present study, we have investigated the pathogenetic mechanism of the deafness-associated tRNASer(UCN) T7511C mutation, in conjunction with the ND1 T3308C and the tRNAAla T5655C mutations in a large African family. It was hypothesized that the primary defect of the T7511C mutation is a failure in the processing of L-strand RNA precursor, spanning tRNASer(UCN) as well as ND6 mRNA. In fact, ∼75–83% reductions in the level of tRNASer(UCN) were observed in the cybrid cell lines carrying the T7511C mutation. This reduced tRNA level, which is below the proposed threshold to support a normal protein synthesis rate in lymphoblastoid cells (20), led to the impairment in mitochondrial protein synthesis. However, the T7511C mutation does not significantly affect the expression level of ND6 mRNA which belongs to the same precursor as that of tRNASer(UCN) (37,38), in contrast to the fact that the A7445G mutation in the precursor of tRNASer(UCN) has long-range effects on expression of the ND6 gene (20). These results imply that the reduced levels of mitochondrial tRNASer(UCN) are likely due to an effect of the T7511C mutation on RNA processing, specifically in the 5′ end of tRNA processing defect, which is catalyzed by mitochondrial RNase P (35). In contrast, the A7445G mutation leads to the 3′ end endonucleolytic processing defect in the L-strand polycistronic RNA precursor (39). As a result, the A7445G mutation causes a significant decrease both in the amount of tRNASer(UCN) (20,21) and co-transcribed ND6 mRNA (20). Alternatively, the possible failure of aminoacylation and post-transcriptional modification of this tRNA may also contribute to the lower level of tRNASer(UCN) in the cybrids carrying the T7511C mutation, as in the case of the deafness-associated 7472insC mutation in the tRNASer(UCN) (40), the MERRF A8344G mutation in the tRNALys (29,41) and the MELAS A3243G mutation in the tRNALeu(UUR) (42,43).
The homoplasmic T5655C mutation occurs in the 3′ end (at position 73) of the tRNAAla (16). This nucleotide may act as a discriminator responsible for the identity of most tRNAs (34) and plays an important role in the recognition by their cognate aminoacyl-tRNA synthetase (44). The significant decrease in tRNAAla steady-state levels in mutant cybrids carrying the T5655C mutation may result from a failure to aminoacylate properly and of post-transcriptional modification of this tRNA. Such effects would leave the tRNA more exposed to degradation since it would not interact with protein partners effectively. Alternatively, the T5655C mutation might cause a defect in the pre-tRNA processing, thus reducing levels of the tRNAAla. However, the T5655C mutation, like other mtDNA polymorphisms, appears to be neutral as it is also present in certain human populations (16).
The homoplasmic T3308C mutation in the ND1 gene belongs to a provisional group associated with other clinical phenotypes and also occurs in some human populations, especially in the Western African haplogroup (36). The truncated ND1 polypeptide seems to retain partial function. It is possible that another methionine at position 3 of ND1 could serve as the initiation codon when the first methionine is changed by the T3308C mutation. A significant reduction in steady-state levels of both ND1 mRNA and adjacent tRNALeu(UUR) observed in the cybrids carrying this T3308C mutation is likely due to an alteration on the processing of the H-strand polycistronic RNA precursors or the destabilization of ND1 mRNA by this mutation (45).
A marked decrease in the rate of mitochondrial protein synthesis (an average decrease of ∼52%) was observed in mutant cell lines, as compared to the average rate in control cell lines. In mutant cell lines, a variable decrease in protein labeling was observed in each polypeptide. However, the rate of synthesis of polypeptides in mutants relative to that in controls did not correlate with either the number of serine (UCN) codons or proportion of serine (UCN) residues, in contrast to what was previously shown in cells carrying the A7445G mutation in the precursor of tRNASer(UCN) (20) or the MERRF A8344G mutation in tRNALys (29). Thus, the severe reduction in these high-molecular weight translation products is likely due to a combination of their high contents of serine (UCN), leucine (UUR) and alanine codons rather than serine (UCN) alone. Furthermore, ∼36–43% decrease in ND1 mRNA level contributes to greater reduction in the average labeling level of ND1 (65%) than the average reduction in overall translational products (52%) in these mutant cells. A significant correlation between the rate of mitochondrial protein synthesis and the reduced level in the tRNASer(UCN) implies that the T7511C mutation might be the primary factor responsible for the overall protein synthesis defect and subsequent respiration defect in the cybrids. In particular, the overall decrease in the rate of mitochondrial protein synthesis in the mutant cells may be mainly due to ∼75–83% reduction in the level of total tRNASer(UCN) and to the corresponding expected decrease in the amount of aminoacylated tRNASer(UCN). Indeed, >75% reduction in the level of total tRNASer(UCN) in the mutant cybrids is below a proposed threshold, which is ∼40% of the control level of the tRNASer(UCN), to support a normal rate of mitochondrial protein synthesis of the lymphoblastoid cell lines (20). On the other hand, ∼48–52% reduction in tRNAAla level caused by the T5655C mutation, and ∼43–64% reduction in tRNALeu(UUR) level resulting from the T3308C mutation, appear to contribute to the marked decrease in mitochondrial protein synthesis in the mutant cell lines carrying the T7511C mutation.
There was a very significant correlation between the rate of mitochondrial protein synthesis, and overall respiratory capacity (P < 0.001) or relative growth rate in galactose versus glucose medium (P < 0.01) in the control and mutant cell lines. This correlation is clearly consistent with the importance that the marked decrease in tRNASer(UCN) steady-state level in the mutant cell lines plays a critical role in producing their overall respiration and growth defects. On the other hand, ∼56% reduction (P < 0.0065) in glutamate/malate-dependent O2 consumption has been observed in the mutant cell lines. This reduction contrasted with a marginally significant decrease in the same cell lines in Complex III-(36%) and Complex IV-(33%) dependent O2 consumption, and indicates a specific NADH dehydrogenase deficiency. These observations indicate another factor(s) involved in the respiratory phenotype of these cell lines. Here, ∼43–53% reduction in the level of ND1 mRNA and ∼65% reduction in ND1 protein labeling, caused by the T3308C mutation, seem to be the primary contributors to the decreased Complex I activity. As a consequence, this respiratory deficiency results in a decline in ATP production in the cochlear cells (hair cells/or stria vascularis), which are essential for hearing function (3,27).
The presence of the T3308C and T5655C mutations in the African family (16) but the absences of these mtDNA mutations in the French and Japanese families (17,18) appear to account for different penetrance in these pedigrees. There is increasing evidence showing that the background sequences (haplotype) of the mtDNA modulate the severity and penetrance of the phenotypic expression of pathogenic mtDNA mutation(s) associated with some clinical abnormalities including deafness (8–11,20), blindness (46), ageing (47), Parkinson’s disease (48) and male infertility (49). Biochemical data obtained here seem to support the genetic and clinical findings that the T3308C and T5655C mutations could increase the penetrance of deafness in the African pedigree carrying the T7511C mutation.
In summary, our findings convincingly demonstrate the pathogenic mechanism leading to an impaired oxidative phosphorylation in cybrid cell lines carrying the deafness-associated tRNASer(UCN) T7511C mutation, in conjunction with the T5655C and T3308C mutations. The previous genetic and molecular studies indicate that the T7511C mutation is a primary mutation underlying the development of deafness. The fact that >75% reduction in the level of tRNASer(UCN) caused by the T7511C mutation is likely a primary contributor to ∼52% decrease in mitochondrial protein synthesis and subsequent respiratory phenotype in mutant cells appears to support genetic and molecular findings. However, the mitochondrial dysfunction caused by the tRNAAla T5655C mutation and the ND1 T3308C mutation may contribute to the phenotypic variability and penetrance of the T7511C mutation in this African family.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Li Yang and William Gibbons for their skilled technical assistance. This work was supported by National Institutes of Health (NIH) grants DC04958 and DC05230 from the National Institute on Deafness and Other Communication Disorders, NS44015 from the National Institute of Neurological Disorders and Stroke, and a Research Grant Award from the United Mitochondrial Disease Foundation to M.-X.G., and NIH grants DC01402 from the National Institute on Deafness and Other Communication Disorders to N.F.-G. and AG18412 from National Institute of Aging to F.S.
REFERENCES
- 1.Fischel-Ghodsian N. (1999) Mitochondrial deafness mutations reviewed. Hum. Mutat., 13, 261–270. [DOI] [PubMed] [Google Scholar]
- 2.Jacobs H.T. (2003) Disorders of mitochondrial protein synthesis. Hum. Mol. Genet., 15, R293–301. [DOI] [PubMed] [Google Scholar]
- 3.Prezant T.R., Agapian,J.V., Bohlman,M.C., Bu,X., Oztas,S., Qiu,W.Q., Arnos,K.S., Cortopassi,G.A., Jaber,L., Rotter,J.I., Shohat,M. and Fischel-Ghodsian,N. (1993) Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and nonsyndromic deafness. Nature Genet., 4, 289–294. [DOI] [PubMed] [Google Scholar]
- 4.Estivill X., Govea,N., Barcelo,A., Perello,E., Badenas,C., Romero,E., Moral,L., Scozzari,R., D’Urbano,L., Zeviani,M. and Torroni,A. (1998) Familial progressive sensorineural deafness is mainly due to the mtDNA A1555G mutation and is enhanced by treatment with aminoglycosides. Am. J. Hum. Genet., 62, 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matthijs G., Claes,S., Longo-Bbenza,B. and Cassiman,J.-J. (1996) Nonsyndromic deafness associated with a mutation and a polymorphism in the mitochondrial 12S ribosomal RNA gene in a large Zairean pedigree. Eur. J. Hum. Genet., 4, 46–51. [DOI] [PubMed] [Google Scholar]
- 6.Li R., Xing,G., Yan,M., Cao,X., Liu,X.Z., Bu,X. and Guan,M.X. (2004) Cosegregation of C-insertion at position 961 with A1555G mutation of mitochondrial 12S rRNA gene in a large Chinese family with maternally inherited hearing loss. Am. J. Med. Genet., 124A, 113–117. [DOI] [PubMed] [Google Scholar]
- 7.Zhao H., Li,R., Wang,Q., Yan,Q., Deng,J.H., Han,D., Bai,Y., Young,W.Y. and Guan,M.X. (2004) Maternally inherited aminoglycoside-induced and nonsyndromic deafness associated with the novel C1494T mutation in the mitochondrial 12S rRNA gene in a large Chinese family. Am. J. Hum. Genet., 74, 139–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fischel-Ghodsian N., Prezant,T.R., Fournier,P., Stewart,I.A. and Maw,M. (1995) Mitochondrial mutation associated with nonsyndromic deafness. Am. J. Otolaryngol., 16, 403–408. [DOI] [PubMed] [Google Scholar]
- 9.Sevior K.B., Hatamochi,A., Stewart,I.A., Bykhovskaya,Y., Allen-Powell,D.R., Fischel-Ghodsian,N. and Maw,M.A. (1998) Mitochondrial A7445G mutation in two pedigrees with palmoplantar keratoderma and deafness. Am. J. Med. Genet., 75, 179–185. [PubMed] [Google Scholar]
- 10.Reid F.M., Vernham,G.A. and Jacobs,H.T. (1994) A novel mitochondrial point mutation in a maternal pedigree with sensorineural deafness. Hum. Mutat., 3, 243–247. [DOI] [PubMed] [Google Scholar]
- 11.Reid F.M., Vernham,G.A. and Jacobs,H.T. (1994) Complete mtDNA sequence of a patient in a maternal pedigree with sensorineural deafness. Hum. Mol. Genet., 3, 1435–1436. [DOI] [PubMed] [Google Scholar]
- 12.Verhoeven K., Ensink,R.J., Tiranti,V., Huygen,P.L., Johnson,D.F., Schatteman,I., Van Laer,L., Verstreken,M., Van de Heyning,P., Fischel-Ghodsian,N., Zeviani,M., Cremers,C.W., Willems,P.J. and Van Camp,G. (1999) Hearing impairment and neurological dysfunction associated with a mutation in the mitochondrial tRNASer(UCN) gene. Eur. J. Hum. Genet., 7, 45–51. [DOI] [PubMed] [Google Scholar]
- 13.Tiranti V., Chariot,P., Carella,F., Toscano,A., Soliveri,P., Girlanda,P., Carrara,F., Fratta,G.M., Reid,F.M., Mariotti,C. and Zeviani,M. (1995) Maternally inherited hearing loss, ataxia and myoclonus associated with a novel point mutation in mitochondrial tRNASer(UCN) gene. Hum. Mol. Genet., 4, 1421–1427. [DOI] [PubMed] [Google Scholar]
- 14.Hutchin T.P., Parker,M.J., Young,I.D., Davis,A.C., Pulleyn,J.L., Deeble,J., Lench,N.J., Markham,A.F. and Muller,R.F. (2000) A novel mutation in the mitochondrial tRNASer(UCN) gene in a family with nonsyndromic sensorineural hearing impairment. J. Med. Genet., 37, 692–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.delCastillo F.J., Villamar,M., Moreno-Pelayo,M.A., Almela,J.J., Morera,C., Adiego,I., Moreno,F. and del Castillo,I. (2002) Maternally inherited nonsyndromic hearing impairment in a Spanish family with the 7510T>C mutation in the mitochondrial tRNASer(UCN) gene. J. Med. Genet., 39, e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sue C.M., Tanji,K., Hadjigeorgious,G., Andreu,A.L., Nishino,I., Krishna,S., Bruno,C., Hirano,M., Shanske,S., Bonilla,E., Fischel-Ghodsian,N., DiMauro,S. and Friedman,R. (1999) Maternally inherited hearing loss in a large kindred with a novel T7511C mutation in the mitochondrial DNA tRNASer(UCN) gene. Neurology, 52, 1905–1908. [DOI] [PubMed] [Google Scholar]
- 17.Chapiro E., Feldmann,D., Denoyelle,F., Sternberg,D., Jardel,C., Eliot,M.M., Bouccara,D., Weil,D., Garabedian,E.N., Coudere,R., Petit,C. and Marlin,S. (2002) Two large French pedigrees with non syndromic sensorineural deafness and the mitochondrial DNA T7511C mutation: evidence for a modulatory factor. Eur. J. Hum. Genet., 10, 851–856. [DOI] [PubMed] [Google Scholar]
- 18.Ishikawa K., Tamagawa,Y., Takahashi,K., Kimura,H., Kusakari,J., Hara,A. and Ichimura, K. (2002) Nonsyndromic hearing loss caused by a mitochondrial T7511C mutation. Laryngoscope, 112, 1494–1499. [DOI] [PubMed] [Google Scholar]
- 19.Goto Y., Noaka,L. and Horai,S. (1990) A mutation in the tRNALeu(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature, 348, 651–653. [DOI] [PubMed] [Google Scholar]
- 20.Guan M.X., Enriquez,J.A., Fischel-Ghodsian,N., Puranam,R., Lin,C.P., Marion,M.A. and Attardi,G. (1998) The deafness-associated mtDNA 7445 mutation, which affects tRNASer(UCN) precursor processing, has long-range effects on NADH dehydrogenase ND6 subunit gene expression. Mol. Cell. Biol., 18, 5868–5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reid F.M., Rovio,A., Holt,I.J. and Jacobs,H.T. (1997) Molecular phenotype of a human lymphoblastoid cell-line homoplasmic for the np-7445 deafness-associated mitochondrial mutation. Hum. Mol. Genet., 6, 443–449. [DOI] [PubMed] [Google Scholar]
- 22.Toompuu M., Tiranti,V., Zeviani,M. and Jacobs,H.T. (1999) Molecular phenotype of the np 7472 deafness-associated mitochondrial mutation in osteosarcoma cell cybrids. Hum. Mol. Genet., 8, 2275–2283. [DOI] [PubMed] [Google Scholar]
- 23.Friedman R.A., Bykhovskaya,Y., Sue,C.M., DiMauro,S., Bradley,R., Fallis-Cunningham,R., Paradies,N., Pensak,M.L., Smith,R.J., Groden,J., Li,X.C. and Fischel-Ghodsian N. (1999) Maternally inherited nonsyndromic hearing loss. Am. J. Med. Genet., 84, 369–372. [DOI] [PubMed] [Google Scholar]
- 24.King M.P. and Attardi,G. (1989) Human cells lacking mtDNA: Repopulation with exogenous mitochondria by complementation. Science, 246, 500–503. [DOI] [PubMed] [Google Scholar]
- 25.King M.P. and Attardi,G. (1996) Mitochondria-mediated transformation of human ρ° cells. Methods Enzymol., 264, 313–334. [DOI] [PubMed] [Google Scholar]
- 26.Guan M.X., Fischel-Ghodsian,N. and Attardi,G. (2001) Nuclear background determines biochemical phenotype in the deafness-associated mitochondrial 12S rRNA mutation. Hum. Mol. Genet., 10, 573–580. [DOI] [PubMed] [Google Scholar]
- 27.Guan M.X., Fischel-Ghodsian,N. and Attardi,G. (1996) Biochemical evidence for nuclear gene involvement in phenotype of nonsyndromic deafness associated with mitochondrial 12S rRNA mutation. Hum. Mol. Genet., 6, 963–971. [DOI] [PubMed] [Google Scholar]
- 28.King M.P. and Attardi,G. (1993) Post-transcriptional regulation of the steady-state levels of mitochondrial tRNAs in HeLa cells. J. Biol. Chem., 268, 10228–10237. [PubMed] [Google Scholar]
- 29.Enriquez J.A., Chomyn,A. and Attardi,G. (1995) MtDNA mutation in MERRF syndrome causes defective aminoacylation of tRNA and premature translation termination. Nature Genet., 10, 47–55. [DOI] [PubMed] [Google Scholar]
- 30.Anderson S., Bankier,A.T., Barrell,B.G., deBruijn,M.H.L., Coulson,A.R., Drouin,J., Eperon,I.C., Nierlich,D.P., Rose,B.A., Sanger,F., Schreier,P.H., Smith,A.J.H., Staden,R. and Young, I.G. (1981) Sequence and organization of the human mitochondrial genome. Nature, 290, 457–465. [DOI] [PubMed] [Google Scholar]
- 31.Chomyn A. (1996) In vivo labeling and analysis of human mitochondrial translation products. Methods Enzymol., 264, 197–211. [DOI] [PubMed] [Google Scholar]
- 32.Hofhaus G., Shakeley,R.M. and Attardi,G. (1996) Use of polarography to detect respiration defects in cell cultures. Methods Enzymol., 264, 476–483. [DOI] [PubMed] [Google Scholar]
- 33.Anderson S., Bankier,A.T., Barrell,B.G., deBruijn,M.H.L., Coulson,A.R., Drouin,J., Eperon,I.C., Nierlich,D.P., Rose,B.A., Sanger,F., Schreier,P.H., Smith,A.J.H., Staden,R. and Young,I.G. (1982) Comparison of the human and bovine mitochondrial genomes. In Slonimski,P., Borst,P. and Attardi,G. (eds), Mitochondrial Genes. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 5–43. [Google Scholar]
- 34.Normanly J. and Abelson,J. (1989) tRNA identity. Annu. Rev. Biochem., 58, 1029–1049. [DOI] [PubMed] [Google Scholar]
- 35.Puranam R.S. and Attardi,G. (2001) The RNase P associated with HeLa cell mitochondria contains an essential RNA component identical in sequence to that of the nuclear RNase P. Mol. Cell. Biol., 21, 548–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rocha H., Flores,C., Campos,Y., Arenas,J., Vilarinho,L., Santorelli,F.M. and Torroni,A. (1999) About the ‘Pathological’ role of the mtDNA T3308C mutation ellipsis. Am. J. Hum. Genet., 65, 1457–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Montoya J., Christianson,T., Levens,D., Rabinowitz,M. and Attardi,G. (1982) The pattern of transcription of the human mitochondrial rRNA genes reveals two overlapping transcription units. Proc. Natl Acad. Sci. USA, 79, 7195–7199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ojala D., Montoya,J. and Attardi,G. (1981) tRNA punctuation model of RNA processing in human mitochondria. Nature, 290, 470–474. [DOI] [PubMed] [Google Scholar]
- 39.Levinger L., Jacobs,O. and James,M. (2001) In vitro 3′-end endonucleolytic processing defect in a human mitochondrial tRNASer(UCN) precursor with the U7445C substitution, which causes nonsyndromic deafness. Nucleic Acids Res., 29, 4334–4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Toompuu M., Yasukawa,T., Suzuki,T., Hakkinen,T., Spelbrink,J.N., Watanabe,K. and Jacobs,H.T. (2002) The 7472insC mitochondrial DNA mutation impairs the synthesis and extent of aminoacylation of tRNASer(UCN) but not its structure or rate of turnover. J. Biol. Chem., 277, 22240–22250. [DOI] [PubMed] [Google Scholar]
- 41.Yasukawa T., Suzuki,T., Ishii,N., Ohta,S. and Watanabe,K. (2001) Wobble modification defect in tRNA disturbs codon–anticodon interaction in a mitochondrial disease. EMBO J., 20, 4794–4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yokogawa T., Shimada,N., Takeuchi,N., Benkowski,L., Suzuki,T., Omori,A., Ueda,T., Nishikawa,K., Spremulli,L.L. and Watanabe,K. (2000) Modification defect at anticodon wobble nucleotide of mitochondrial tRNAsLeu(UUR) with pathogenic mutations of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. J. Biol. Chem., 275, 19913–19920. [DOI] [PubMed] [Google Scholar]
- 43.Chomyn A., Enriquez,A.E., Micol,V., Fernandez-Silvas,P. and Attardi,G. (2000) The mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes syndrome-associated human mitochondrial tRNALeu(UUR) mutation causes aminoacylation deficiency and concomitant reduced association of mRNA with ribosomes. J. Biol. Chem., 275, 19198–19209. [DOI] [PubMed] [Google Scholar]
- 44.Florentz C., Sohm,B., Tryoen-Toth,P., Putz,J. and Sissler,M. (2003) Human mitochondrial tRNAs in health and disease. Cell. Mol. Life Sci., 60, 1356–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rossmanith W., Tullo,A., Potuschak,T., Karwan,R. and Sbisà,E. (1995) Human mitochondrial tRNA processing. J. Biol. Chem., 270, 12885–12891. [DOI] [PubMed] [Google Scholar]
- 46.Torroni A., Petrozzi,M., Durbano,L., Sellitto,D., Zeviani,M., Carrara,F., Carducci,C., Leuzzi,V., Carelli,V., Barboni,P., De Negri,A. and Scozzari,R. (1997) Haplotype and phylogenetic analyses suggest that one European-specific mtDNA background plays a role in the expression of Leber hereditary optic neuropathy by increasing the penetrance of the primary mutations 11778 and 14484. Am. J. Hum. Genet., 60, 1107–1121. [PMC free article] [PubMed] [Google Scholar]
- 47.Coskun P.E., Ruiz-Pesini,E. and Wallace,D.C. (2003) Control region mtDNA variants: longevity, climatic adaptation, and a forensic conundrum. Proc. Natl Acad. Sci. USA, 100, 2174–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.vanderWalt J.M., Nicodemus,K.K., Martin,E.R., Scott,W.K., Nance,M.A., Watts,R.L., Hubble,J.P., Haines,J.L., Koller,W.C., Lyons,K., Pahwa,R., Stern,M.B., Colcher,A., Hiner,B.C., Jankovic,J., Ondo,W.G., Allen,F.H., Goetz,C.G., Small,G.W., Mastaglia,F., Stajich,J.M., McLaurin,A.C., Middleton,L.T., Scott,B.L., Schmechel,D.E., Pericak-Vance,M.A. and Vance,J.M. (2003) Mitochondrial polymorphisms significantly reduce the risk of Parkinson disease. Am. J. Hum. Genet., 72, 804–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ruiz-Pesini E., Lapena,A.C., Diez-Sanchez,C., Perez-Martos,A., Montoya,J., Alvarez,E., Diaz,M., Urries,A., Montoro,L., Lopez-Perez,M.J. and Enriquez,J.A. (2000) Human mtDNA haplogroups associated with high or reduced spermatozoa motility. Am. J. Hum. Genet., 67, 682–696. [DOI] [PMC free article] [PubMed] [Google Scholar]