

Abstract
Context:
The p21 activated kinases (PAKs) are a family of serine/threonine kinases that are downstream effectors of small GTPase Cdc42 and Rac. PAKs regulate cell motility, proliferation, and cytoskeletal rearrangement. PAK isoform expression and activity have been shown to be enhanced in cancer and to function as an oncogene in vivo. PAKs also have been implicated in cancer progression.
Objective:
In thyroid cancer, we have previously determined that PAK overactivation is common in the invasive fronts of aggressive tumors and that it is functionally involved in thyroid cancer cell motility using molecular inhibitors. We report the development of two new PAK-inhibiting compounds that were modified from the structure OSU-03012, a previously identified multikinase inhibitor that competitively blocks ATP binding of both phosphoinositide-dependent kinase 1 (PDK1) and PAK1.
Results:
Seventeen compounds were created by combinatorial chemistry predicted to inhibit PAK activity with reduced anti-PDK1 effect. Two lead compounds were identified based on the ability to inhibit PAK1 activity in an ATP-competitive manner without discernible in vivo PDK1 inhibitory activity in thyroid cancer cell lines. Both compounds reduced thyroid cancer cell viability. Although they are not PAK-specific on a multikinase screening assay, the antimigration activity effect of the compounds in thyroid cancer cells was rescued by overexpression of a constitutively active PAK1, suggesting this activity is involved in this biological effect.
Conclusions:
We have developed 2 new multikinase inhibitors with anti-PAK activity that may serve as scaffolds for further compound development targeting this progression-related thyroid cancer target.
The p21 activated kinases (PAKs) are a family of downstream effectors of small GTPase Cdc42 and Rac that function as central regulators of cell motility and cytoskeletal rearrangement (1). Six unique PAK isoforms have been cloned that are divided into 2 groups, PAKs 1 to 3 and PAKs 4 to 6, based on the sequence and functional characteristics (2, 3). As direct targets of Cdc42 and Rac, PAKs participate in a wide range of physiological processes beyond cell motility, including cell proliferation, apoptosis regulation, and in some systems, oncogenesis (4–6).
PAK activation and overexpression has been identified in a variety of malignancies (4–6). In thyroid cancer, we previously reported an increase in PAK1 expression, PAK phosphorylation, and PAK-mediated phosphorylation of downstream effectors in the invasive fronts in aggressive papillary cancers. In additional studies, group 1 PAKs, and PAK1 in particular, were essential for thyroid cancer cell motility in vitro (7, 8). In breast cancer, PAK activity correlates with the baseline invasiveness of human breast cancer cells and breast tumor grade (9–11). In addition, PAK1 is reported to contribute to the resistance of estrogen receptor-expressing breast cancers to tamoxifen by phosphorylating the serine 305 in estrogen receptor, thereby preventing its binding with estrogen (12, 13). A role for PAK in tumorigenesis in neurofibromatosis types 1 and 2 has also been characterized in detail (14, 15), and PAK isoforms are involved in Ras-mediated tumorigenesis in several tissue types (16, 17). Thus, it has been posited that PAK may be a relevant target for cancer therapy.
Over the past several years, there has been interest in developing compounds that inhibit the function of PAK isoforms or that down-regulate PAK isoform expression (10, 17–19). We previously reported that OSU-03012, a multikinase inhibitor that inhibits phosphoinositide-dependent kinase 1 (PDK1) now in phase 1 clinical trials, also exhibits a previously unrecognized PAK inhibitory activity at low micromolar concentrations (20). Moreover, in some cell lines, the inhibition of PAK occurred at lower concentrations than PDK1. Subsequent in vitro studies demonstrated that OSU-03012 directly targets PAK1 in an ATP-competitive manner and that the antimigratory effect, but not the cell-killing effect, was rescued by overexpression of a constitutively active PAK1. Because PAKs are independent oncogenes and play a role in cancer progression, they represent a potential therapeutic target. Moreover, although there are potential advantages to inhibiting PDK1 and PAK with 1 compound, there may be limitations in terms of side effects or limitation for combinatorial therapies. We therefore embarked on the lead optimization of OSU-03012 to develop PAK inhibitors that do not also inhibit PDK1. In the present study, we report the development of 2 such compounds.
Materials and Methods
Chemistry
OSU-03012 was used as a platform to develop PAK1 inhibitors. As depicted in Figure 1 and in detail in Supplemental Figure 1 (published on The Endocrine Society's Journals Online web site at http://jcem.endojournals.org), 3 moieties of OSU-03012 were altered by using combinatorial strategies. The 2-phenanthrene in position A was modified to 3-phenanthrene to examine the conformational geometric effect on the activity; the B position CF3 group was replaced with hydrogen-forming functional groups such as hydroxyl and carboxamide groups; and the glycine at the C position was either extended or shortened by being replaced with β-alanine or being cleaved off to examine the bulkiness effect. This combinatory strategy generated 17 OSU-03012–derived compounds, and they were subjected to biological evaluation.
Figure 1.

Panel A, Structural modifications of OSU-03012 focused on sites A, B, and C. Panel B, Structures of compounds 1–17.
Chemical reagents and organic solvents were purchased from Sigma-Aldrich (St Louis, Missouri) unless otherwise mentioned. Nuclear magnetic resonance spectra (1H) were measured on a Bruker DPX 300 model spectrometer. Chemical shifts (δ) were reported in parts per million relative to the tetramethylsaline peak. Electrospray ionization mass spectrometry analyses were performed with a Micromass Q-Tof II high-resolution electrospray mass spectrometer. The purity of all tested compounds was higher than 98% by 1H nuclear magnetic resonance spectra and elemental analyses (Atlantic Microlab, Inc, Norcross, Georgia). Flash column chromatography was performed using silica gel (230–400 mesh).
Cell lines
Human thyroid cancer BCPAP, TPC1, and FTC133 cell lines were the generous gifts of Drs Rebecca Schweppe and Bryan Haugen (University of Colorado Denver, Denver, Colorado) (21) with permission from the originating laboratories: BCPAP, Dr Fabien, Centre Hospitalier Lyon-Sud, Lyon, France (22); TPC1, Dr Sato, Kanazawa University, Kanazawa, Japan (23); and FTC133, Dr Goretzki, University of Leipzig, Leipzig, Germany (24). The specific obtained cell lines were confirmed for identity by DNA fingerprinting. BCPAP was grown in RPMI 1640 medium (Life Technologies Co, Carlsbad, California) whereas FTC133 and TPC1 were grown in DMEM (Life Technologies), both supplemented with 10% fetal bovine serum (FBS) and nonessential amino acids unless as noted for experiments.
Cell viability analysis
The effect of OSU-03012 and its derivatives on cell viability was assessed by using the 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyl-2H-tetrazolium bromide (MTT) assay. Three replicates were performed in each individual experiment, and experiments were repeated at least 3 separate times. Cells were grown in 96-well plates for 24 hours and were exposed to concentrations of drugs dissolved in dimethylsulfoxide (DMSO) (final concentration 0.1%) in cell culture medium containing 1% FBS for 72 hours. Then 10 μL of 0.5 mg/ml MTT was added to each well and cells were incubated in CO2 at 37°C for 2 hours. Supernatants were removed from the wells, the reduced MTT dye was solubilized in 100 μL solution C (4 ml 1N HCl in 100 ml isopropanol), and absorbance at 570 nm was determined on a microplate reader.
Kinase assay
Recombinant active PAK1 (EMD Millipore, Billerica, Massachusetts) was incubated with selected compounds or 0.1% DMSO in the presence or absence of myelin basic protein (Sigma) in 25 μL 1× kinase buffer (Cell Signaling Technology, Danvers, Massachusetts) as previously described (20). Briefly, the addition of [γ-32P]ATP (2.5 μCi per reaction) (PerkinElmer, Waltham, Massachusetts) started the reaction, which was incubated at 30°C for 30 minutes. The reaction was terminated by the addition of 25 μL EDTA (pH 8). Results were obtained after visualization of 32P-labeled myelin basic protein after separation on 12% polyacrylamide gels and autoradiography. For the Lineweaver-Burk plot analysis, the ratio of radioactive ATP to nonradioactive ATP was not altered. The PAK1 kinase activities are the means of 3 independent experiments quantified using P81 phosphocellulose paper by a scintillation counter after 3 washes with 0.75% phosphoric acid.
Western blot and antibodies
Protein isolation and Western blot were performed as previously described (25), and 25 μg of total cell lysates was loaded into 4% to 12% SDS-PAGE, electrophoresed, and transferred to nitrocellulose membranes for immunoblotting. Primary antibodies against Akt (no. 9272), phospho-Akt (Thr308) (no. 2965), p44/42 MAPK (Erk1/2) (no. 9107), phospho-p44/42 APK (Erk1/2) (Thr202/Tyr204) (no. 4377), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (no. 2118) were obtained from Cell Signaling. Antibody against vimentin (V6630) and phospho-Ser55 vimentin were from Sigma-Aldrich, Inc and MBL Co (Nagoya, Japan), respectively. Antibodies against total and phosphorylated S6 were from Cell Signaling.
Migration assay
Cells were grown media containing 10% FBS. After 24 hours, the medium was aspirated, and medium containing 1% FBS was added for 24 hours. The cells were trypsinized, washed, and resuspended in medium containing 1% FBS. The cell concentration was calculated by hemocytometer. Then 400 μL 1% FBS medium was added into 24-well plates, and a Boyden chamber (8 μm pore) was inserted into each well. Cells were added to each insert and were allowed to attach in an incubator at 37°C at 5% CO2 for 2 hours. The cells were then exposed to various concentrations of drugs or control diluent for 3 hours. The inserts were switched to a new well containing medium containing 10% FBS, and the chamber was incubated for 12 or 24 hours depending on the migration rate of the cell line. The cells on and under the Boyden chamber membrane were fixed with 3.7% formaldehyde containing 0.05% crystal violet for 15 minutes after washing cells with PBS. The chambers were washed with distilled water. The cells on the top (nonmigrated) and bottom (migrated) sides of the membrane were collected by scraping the top and bottom of the chamber with a Q-tip, which was subsequently placed into a 1.5-ml tube. The remainder of the cells remained in the Boyden chamber. The Q-tips containing the scraped cells and the Boyden chamber containing the nonmigrated cells were separately incubated in 80% methanol and shaken at 500g for 30 minutes, and the extracted dye was measured at 570 nm. Migration was quantified as the ratio of the migrated cells over the total cells (nonmigrated plus remaining cells). Experiments were performed in duplicate on at least 3 occasions.
Migration rescue experiments
Myc-tagged constitutively activated PAK1 cDNAs were the generous gift of Dr Jonathan Chernoff (Fox Chase Cancer Center, Philadelphia, Pennsylvania) (26, 27). pRC/CMV and pcDNA3.1(+) vector were purchased from Life Technologies. BCPAP cells were grown in RPMI containing 10% FBS until 40% to 50% confluent. They were transfected with constitutively activated PAK1 cDNA or vector control, washed with PBS, trypsinized with 0.05% Trypsin-EDTA 1× (Life Technologies) for 5 to 10 minutes, collected with 10% FBS RPMI, and centrifuged at 300g for 5 minutes. Cells were resuspended with 0% FBS RPMI and counted using Countess (Life Technologies). Then a volume of 1 × 105 cells suspended in 300 μL of 0% FBS RPMI was placed into Boyden chamber inserts (8 μm pore). The inserts were placed into wells of a 24-well plate filled with 400 μL of 0% FBS RPMI. The plate was incubated at 37°C 5% CO2 for 1 hour. Then 30 μL (1 μM) of compound 4 or 15 or OSU-03012 was added to the inserts and then incubated again for 1 hour. The inserts were transferred into different wells filled with 400 μL 10% FBS RPMI. After incubation for 24 hours, the cells on either side of the insert membrane were fixed and quantified as above. Experiments were performed in triplicate on 3 occasions.
Statistical analysis
Several of the experiments were conducted on different days; thus, linear mixed-effects models were used to model any extra correlation of results within days (day-treatment random effects). We used saturated covariance structures to ensure that variances of test statistics were not underestimated. Holm's procedure was used to adjust for multiple comparisons or endpoints to strongly control type I error to α = 0.05.
Results
Screening of OSU-03012 derivatives to identify PAK1 inhibitors
The 17 derivatives along with OSU-03012, each at 7.5 μM, were screened for their ability to inhibit PAK1 activity by in vitro PAK1 kinase assay. The dose was chosen (above the IC50 of OSU-03012) to allow OSU-03012 to serve as a robust positive control (20). The PAK1 kinase assay was performed using recombinant active PAK1 with myelin basic protein as substrate in the presence of 2.5 μM ATP and 7.5 μM of the respective inhibitors. The result of this initial screen demonstrated that among the compounds tested, 6 compounds, numbers 1, 3, 4, 10, 11, and 15, inhibited PAK1 kinase activity (Figure 2A).
Figure 2.

A, In vitro kinase assay analysis of the PAK1 inhibitory activities of 17 compounds compared with OSU-03012. B, Dose-response suppression of PAK1 activity by compounds 1, 3, 4, 10, 11, 15, and OSU-03012 at indicated concentrations. C and D, ATP competition assay for PAK1 kinase. The activity of PAK1 was determined in the presence of varying concentrations of ATP (5 μM–40 μM) at indicated concentrations of compounds 4 and 15. The plot shown is representative of 3 separate experiments performed in duplicate.
To further investigate these 6 compounds, dose-response studies on PAK1 activity were performed by using PAK1 kinase assay. All 6 compounds exhibited dose-related suppression of PAK1 activity (Figure 2B and Supplemental Figure 2), and at concentrations greater than 5 μM, PAK1 activity was almost completely inhibited. Compounds 4 and 15 were slightly more potent than the other compounds. We next questioned whether compound 4 or 15 inhibited PAK1 in an ATP competitive manner as their parent compound OSU-03012. The ATP competition assays confirmed that both compounds competed with ATP binding to PAK1 as predicted based on the activity of OSU-03012 (Figure 2, C and D).
Screening of OSU-03012 derivatives for cell viability effects
We assessed the ability of the compounds to reduce cell viability in vitro by MTT assay after 72 hours of continuous exposure. Results are shown in Table 1 and are depicted as the IC50 vs simultaneously performed DMSO control. Cell counting assays were also performed for 72 hours, and results are shown in Supplemental Figure 3.
Table 1.
Potencies of Compounds for Reducing Viability of BCPAP, FTC133, and TPC1 Cells Versus DMSO Controla
| Compound | IC50, μmol/L |
||
|---|---|---|---|
| BCPAP | FTC133 | TPC1 | |
| OSU-03012 | 2.93 ± 1.05 | 2.93 ± 1.87 | 2.80 ± 1.33 |
| 1 | 2.01 ± 0.59 | 4.78 ± 4.53 | 2.60 ± 0.81 |
| 2 | 2.67 ± 0.82 | 4.83 ± 4.47 | 1.81 ± 1.24 |
| 3 | 2.34 ± 0.27 | 5.32 ± 4.05 | 3.17 ± 1.29 |
| 4 | 2.07 ± 0.06 | 2.68 ± 2.08 | 1.68 ± 0.32 |
| 5 | >10 | 5.03 ± 0.25 | >10 |
| 6 | >10 | >10 | >10 |
| 7 | 3.71 ± 1.82 | 4.53 ± 2.57 | 3.67 ± 1.52 |
| 8 | 2.90 ± 2.35 | 3.08 ± 1.63 | 4.67 ± 4.16 |
| 9 | 4.04 ± 1.38 | 4.75 ± 2.47 | 2.42 ± 0.13 |
| 10 | 6.53 ± 2.16 | 8.95 ± 0.57 | 7.39 ± 1.90 |
| 11 | 4.50 ± 1.04 | 3.94 ± 2.29 | 4.98 ± 1.94 |
| 12 | 3.76 ± 1.20 | 7.55 ± 2.19 | 5.83 ± 1.73 |
| 13 | 2.76 ± 1.56 | 4.95 ± 3.21 | 3.62 ± 0.42 |
| 14 | 3.32 ± 1.09 | 5.42 ± 1.56 | 5.98 ± 0.28 |
| 15 | 1.49 ± 0.68 | 3.37 ± 3.31 | 2.66 ± 1.05 |
| 16 | >10 | >10 | 4.38 ± 2.18 |
| 17 | >10 | >10 | 8.96 ± 1.17 |
The reported IC50 values are concentrations at which BCPAP, FTC133, and TPC1 cell viability by MTT assays are 50% of DMSO control after 72 hours of exposure. Data are presented as mean ± SD (n = 3).
PAK and PDK1 selectivity in vivo
The ability of the compounds with in vitro anti-PAK1 activity was assessed for activity in vivo in thyroid cancer cells. In addition, because the compounds are based on the structure of a PDK1 inhibitor, PDK1 inhibitory activity was also assessed. Inhibitory activity in cells was estimated using Western blot with primary antibodies directed against kinase-specific phosphorylation sites: serine 56 of vimentin for PAK and threonine 308 of Akt for PDK1. BCPAP, TPC1, and FTC133 thyroid cancer cells were used. BCPAP and TPC1 are derived from PTC and have BRAF V600E and RET/PTC1 rearrangements, respectively. FTC133 is derived from a follicular thyroid cancer with an inactivating mutation of PTEN. Similar to the kinase screening assay, cells were treated with the 6 compounds or OSU-03012 at 7.5 μM concentration or with DMSO vehicle alone. The results are shown in Figure 3. The results were not entirely consistent across the 3 cell lines, presumably related to mutation status or other factors. OSU-03012 inhibited both PDK1- and PAK-mediated phosphorylation of targets as predicted. Only compounds 4 and 15 inhibited PAK without altering PDK1 activity in at least 2 cell lines. Compound 11 did not significantly alter PAK phosphorylation of vimentin but did markedly inhibit PDK1 phosphorylation of Akt suggesting specificity for PDK1.
Figure 3.
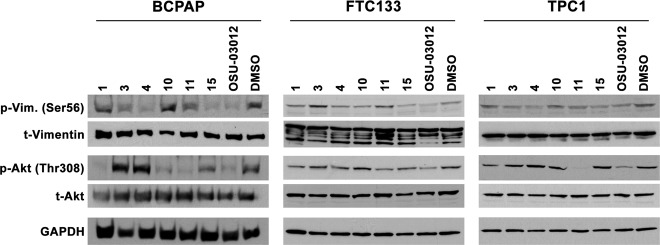
Western blot analysis of the effects of compounds with PAK inhibitory activities (1, 3, 4, 10, 11, and 15) versus OSU-03012 on PDK1 phosphorylation of Akt (Thr308) and PAK phosphorylation of vimentin (Ser56) relative to that of DMSO after 6 hours of treatment in BCPAP, FTC133, and TPC1 cells.
The dose dependency of compounds 4 and 15 was next assessed (Figure 4). Consistent with the kinase assay and initial cell line results, both compounds inhibited PAK1 in a dose-dependent manner and PDK-mediated Akt phosphorylation was not reduced (Figure 4A). Due to the important role that ERK activation plays in thyroid cancer, and its signaling activation by thyroid oncogenes, the effects of the compounds on ERK phosphorylation was also assessed. Compound 15 inhibited ERK phosphorylation at 2.5 μM in the BCPAP cells with a BRAF V600E mutation and at higher concentrations in the other 2 cell lines. In contrast, compound 4 had no demonstrable effects on ERK phosphorylation (Figure 4A). Finally, based on the multikinase assay result that demonstrated inhibition of p70S6 kinase activity (see below and Table 2), we also assessed for an effect on S6 phosphorylation and demonstrated that compound 15 inhibited S6 phosphorylation in all cell lines, whereas compound 4 inhibited this phosphorylation in BCPAP and TPC1, but not in the FTC133 cells (Figure 4B). Consistent with the signaling data, Compounds 4 and 15 were similarly effective vs OSU-03012 at inhibiting cell growth of BCPAP that has a BRAF V600E mutation but were less effective than OSU-03012 at this assay in FTC133 and TPC1 cells that have activation of PI3K and PDK1 signaling via either loss of PTEN or expression of RET/PTC, respectively (Supplemental Figure 3).
Figure 4.

A, Western blot analysis of the dose-dependent effect of compounds 4 and 15 on the phosphorylation of vimentin, Akt and ERK in BCPAP, FTC133, and TCP1 cells after 6 hours of drug treatment. B, Western blot analysis of the suppressive effects of compounds 4 and 15 on S6 phosphorylation.
Table 2.
Inhibition of Kinase in Vitro Activities by Compounds 4 and 15
| Compound 4 (% activity vs DMSO) | Compound 15 (% activity vs DMSO) | |
|---|---|---|
| CaMKI | 65 | 68 |
| CDK1/cyclinB | 83 | 95 |
| CDK5/p25 | 113 | 93 |
| CHK1 | 82 | 89 |
| C-RAF | 104 | 108 |
| EGFR | 99 | 100 |
| FAK | 87 | 96 |
| FGFR2 | 92 | 99 |
| GSK3α | 133 | 118 |
| GSK3β | 170 | 137 |
| IGF-1R, activated | 97 | 83 |
| JAK2 | 152 | 139 |
| LIMK1 | 130 | 117 |
| MEK1 | 106 | 101 |
| Met | 89 | 77 |
| p70S6K | 28 | 38 |
| PAK2 | 70 | 81 |
| PAK4 | 101 | 105 |
| PAK3 | 211 | 99 |
| PAK5 | 114 | 104 |
| PAK6 | 81 | 101 |
| PKA | 110 | 119 |
| PKBα | 103 | 99 |
| PKBβ | 76 | 84 |
| PKBγ | 33 | 56 |
| PKCα | 92 | 92 |
| PKCδ | 85 | 110 |
| Ret | 97 | 76 |
| ROCK-I | 93 | 95 |
| ROCK-II | 79 | 96 |
| SGK | 27 | 64 |
| Src(T341M) | 105 | 94 |
The 32 kinases were selected from different kinase superfamilies. Both compounds were tested using 5μM concentrations. The values are averages of triplicates with variations of less than 15%. Full names of proteins are as noted in the text or are the following in order: CaMK1, calcium/calmodulin-dependent protein kinase I; CDK1 and CDK5, cyclin-dependent kinase 1 and 5; CHK1, serine/threonine-protein kinase Chk1; EGFR, epidermal growth factor receptor; FAK, focal adhesion kinase; FGFR2, fibroblast growth factor receptor 2; GSK, glycogen synthase kinase-3; IGF-1R, insulin-like growth factor 1 receptor; JAK2, Janus kinase 2; LIMK1, LIM domain kinase 1; Met, hepatocyte growth factor receptor; PKA, protein kinase A; Ret, rearranged during transfection proto-oncogene; ROCK1 and ROCK2, Rho-associated coiled-coil-containing protein kinase 1 and 2; Src, V-Src avian sarcoma viral oncogene.
Compounds 4 and 15 are multikinase inhibitors
To further probe the selectivity of the compounds 4 and 15, they were subjected to a kinase profiling service for screening against a panel of other kinases selected based on the kinase tree at 5 μM concentration, at or above the IC50 for the 2 compounds versus PAK in both in vitro and cell-based assays. Of a panel of 32 kinases tested, 3 kinases, serum/glucocorticoid regulated kinase (SGK), ribosomal protein S6 kinase, 70 KD (p70S6K), and protein kinase Bγ (Akt 3) displayed greater than 50% inhibition by 5 μM compound 4; whereas only 1 kinase, p70S6K, was inhibited by greater than 50% by compound 15 (Table 1). The 2 compounds also did not have activity versus other PAK kinases on this assay. PAK1 was not available for testing using this system; thus, it was not possible to compare directly in this assay system.
Constitutively active PAK partially rescues migration inhibited by compounds 4 and 15
Because the compounds are multikinase inhibitors, we next sought to determine whether PAK1 inhibition was responsible for some of the biological effects of the compounds. We have previously demonstrated a critical role for PAK isoforms in thyroid cancer cell motility using several molecular inhibitors and that a constitutively active form of PAK1 rescued the effect of OSU-03012 on cell migration. Thus, we performed migration assays to assess the effects of compounds 4 and 15 on the cell motility of BCPAP, FTC133, and TPC1 cells using Boyden chambers. Preliminary experiments were performed to determine doses of each compound that did not reduce cell viability over 24 hours. As shown in Figure 5A, BCPAP cells motility was inhibited by both compounds at 2.5 μM and 5.0 μM concentrations, respectively. Both compounds also decreased cell motility in TPC1 cells, although the effect was less pronounced. In contrast, FTC133 cell motility was not inhibited by either compound. The reason for this difference is not certain. To confirm that PAK inhibition was responsible for the reduction in migration, BCPAP cells were transfected with a constitutively activated PAK1 cDNA, and migration experiments were performed in the presence or absence of compounds. Expression of the constitutively activated PAK1 rescued the antimigratory effects of compounds 4 and 15 (Figure 5B). By contrast, expression of constitutively active MAPK kinase 1 (MEK1) or p70S6 kinase did not rescue the effects of the 2 compounds on migration (Supplemental Figures 4 and 5). In comparison, the constitutively active p70S6K did have a rescue effect on OSU-03012 effects as might be predicted by its inhibition of PDK1, which regulates p70S6K (Supplemental Figure 5).
Figure 5.

Inhibition effect of compounds 4 and 15 on cell migration after 12 or 24 hours of treatment. A, Compounds 4 and 15 inhibited migration of BCPAP cells at 2.5 μM and 5.0 μM concentrations (*, P < .05 vs DMSO) after 12 hours of treatment, both compounds inhibited migration of TPC1 cells at 5 μM (*, P < .05 vs DMSO) after 24 hours of treatment, and neither compound significantly inhibited migration of FTC133 cells at either 2.5 μM or 5.0 μM concentrations after 12 hours of treatment. B, Transfection of BCPAP cells with a constitutively activated PAK1 cDNA rescues the inhibition of migration of compounds 4, 15, and OSU-03012 (*, P < .05; **, P < .01). Rescue experiments for each compound were performed on 3 separate occasions.
Discussion
PAK1 is overexpressed in the invasive fronts of thyroid cancer and is functionally important in regulating thyroid cancer cell motility (7). This study builds on our previous identification of anti-PAK kinase activity of OSU-03012, a competitive ATP-binding compound that also inhibits other kinases, including PDK1. Seventeen second-generation compounds were developed with the goal of maintaining or enhancing PAK inhibition while limiting or abolishing the anti-PDK1 effect. Six compounds were identified that inhibited PAK activity in vitro. Structure-activity relationship analysis indicates that there is a conformational geometry effect on their activity to inhibit PAK1. For example, compound 3 demonstrated slightly more potent activity based on in vitro kinase assay than OSU-03012, and in structure they are identical except for the conformation of the phenanthrene, suggesting a different binding mode of these 2 isoforms. This premise is further supported by other pairs of isoforms; for example, compound 10, which bears the same phenanthrene moiety as OSU-03012, demonstrated more potent activity in inhibiting PAK1 activity, whereas its isoform compound 9 did not show any improvement in PAK1 inhibitory activity. This may suggest that the binding mode of OSU-03012, and compound 10, favors a hydrophilic group at site B (Figure 1), whereas a hydrophobic group is able to increase the potency of the derivatives whose binding mode is similar to that of compounds 3 and 9. This hypothesis is further supported by the relative enhancement in activity of compound 15 vs compounds 2 and 7. These compounds all contain the 3-phenanthrene ring, suggesting they adopt the second conformation, paralleling the bulkiness of the groups at the B position. In summary, the preference of a functional group at the B position appears to correlate with cooperativity with phenanthrene conformation.
A bulky group at position C was observed to be associated with higher PAK1 inhibitory potency by comparing the activity of compounds with otherwise similar structures. Specifically, compounds 4, 11, 13, and 14 were more potent than compounds 9, 3, 7, and 8, respectively. In each of these compound pairs, the first compound contains a bulkier β-alanine versus a glycine. Moreover, compounds 5, 6, 16, and 17 that do not contain an amino acid residue at that site did not show any activity. Taken together, the findings support the importance of a bulky group at the C position regardless of the binding mode for anti-PAK activity.
Among all the derivatives examined, compounds 4 and 15 had comparable PAK1 inhibitory activity to OSU-03012 but did not block PDK1-mediated Akt phosphorylation on the Western blot in vivo assay. This relative selectivity may result from the larger bulkiness of compounds 4 and 15, which would be predicted to prevent binding to the ATP pocket of PDK1; on the other hand, the open ATP-binding site of PAK1 is predicted to accommodate the bulky sizes of compounds 4 and 15. This is consistent with previous reports that more selective inhibition of PAK is achieved by enlarging the size of lead compounds (28, 29).
It is interesting to note that the results of the in vitro kinase assay did not completely match the cell line Western blot data using the PAK-site–specific anti-phosphovimentin antibody. There are several potential reasons for this, including the likelihood that PAK folding and protein-protein interactions are important determinants of compound activity that are not assessed in the kinase assay. In addition, there are other mechanisms of resistance in cells such as difficulty in compounds entering cells through the cell membrane, inactivating metabolism of compounds, enhanced export of compounds, and others that may account for the differences. In vivo kinase assays would not be expected overcome much of this variability and have the additional issues related to inconsistencies with immunoprecipitation using the PAK antibodies that occur in our hands (data not shown). Nonetheless, the rescue experiments demonstrate the importance of PAK inhibition on the antimigration effect of the compounds.
The ability of the compounds to reduce the viability of BCPAP, FTC133, and TPC1 thyroid cancer cells was assessed and is shown in Table 1. Several of the compounds did not reduce cell viability in the cell lines. In contrast, compounds 4 and 15 were among the most active for each of the cell lines. Dose responses using cell counting for compounds 4, 15, and OSU-03012 are demonstrated in Supplemental Figure 3. The correlation with the anti-PAK activity was not always noted; however, PAK1 inhibition was not responsible for the cell death effect of the parent OSU-03012 compound. In light of the cell viability suppression, a dose-response study of the effect of these 2 compounds on ERK phosphorylation was also performed due its central role in mediating thyroid cancer cell proliferation and apoptosis. Interestingly, compound 15 inhibited ERK phosphorylation, whereas compound 4 did not, even at high concentrations. The mechanism for the inhibition of ERK is not certain because compound 15 did not inhibit either V-RAF-1 murine leukemia viral oncogene homolog 1 (cRAF) or MEK on the multikinase screen. Further analysis of this inhibitory effect is ongoing. In addition, based on the compound screen, we also confirmed that both compounds 4 and 15 inhibit phosphorylation of S6 consistent with the findings from the kinase profiling assay. Because of the important role of PAK on cell migration and our previous data suggesting that expression of the constitutively activated PAK rescued the effect of OSU-03012 on migration, we performed similar experiments for compounds 4 and 15. Based on initial studies, it appeared that the BCPAP cells were most sensitive to the antimigratory effects of the 2 compounds. Thus, this cell line was selected for rescue experiments, and as demonstrated in Supplemental Figure 6, there was a rescue effect on this property. As shown in Supplemental Figures 4 and 5, there was no rescue of migration using the constitutively active MEK1 or p70S6K constructs for compounds 4 and 15, although there was a rescue for OSU-03102 and the p70S6K construct. These data suggest that PAK inhibition is responsible, at least in part, for the antimigration effect of compounds 4 and 15. However, because we tested only conditions for migration in which PAK activity is known to be enhanced (ie, low confluency), these compounds should be recognized as multikinase inhibitors that included anti-PAK1 activity but are not PAK-specific. It is also important to note that migration and not invasion was evaluated in these experiments due to technical challenges in attaining consistent measurable invasion in low-confluency conditions. Thus, the other pathways may have a role in invasion that may be better tested using in vivo assay systems as are planned for the future.
In summary, we have developed several new derivatives of OSU-03012 that have maintained effects on PAK1 but do not appear to inhibit PDK1 function in vitro and in cell culture. Additional studies are required in vivo in whole animal systems to fully assess efficacy and toxicity. The compounds are multikinase inhibitors that demonstrate some degree of unique specificity. The development of compounds 4 and 15 provides a proof of concept that enlarging the size of the lead compound contributes to the development of PAK inhibitors and also demonstrates that further modification and structure-function studies are needed to devise more potent and selective inhibitors of this important kinase target for thyroid cancer and other malignancies.
Acknowledgments
This work was funded by National Institutes of Health Grants P01 CA124570 (to M.D.R.) and CA112250 (to C.-S.C.).
Current address for Y.M.: Plant Protection Institute, Henan Academy of Agricultural Sciences Zhengzhou, Henan, China.
Current address for N.P.K.: M. & N. Virani Science College, Saurashtra University Rajkot, Gujarat, India.
Disclosure Summary: M.D.R. has previously served on a medical advisory board for Veracyte and has been a member of the Board of the International Thyroid Oncology Group. The remaining authors have nothing to disclose.
Footnotes
- DMSO
- dimethylsulfoxide
- FBS
- fetal bovine serum
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyl-2H-tetrazolium bromide
- PAK
- p21 activated kinase
- PDK1
- phosphoinositide-dependent kinase 1.
References
- 1. Manser E, Leung T, Salihuddin H, Zhao ZS, Lim L. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature. 1994;367:40–46 [DOI] [PubMed] [Google Scholar]
- 2. Bokoch GM. Biology of the p21-activated kinases. Annu Rev Biochem. 2003;72:743–781 [DOI] [PubMed] [Google Scholar]
- 3. Jaffer ZM, Chernoff J. p21-activated kinases: three more join the Pak. Int J Biochem Cell Biol. 2002;34:713–717 [DOI] [PubMed] [Google Scholar]
- 4. Dummler B, Ohshiro K, Kumar R, Field J. Pak protein kinases and their role in cancer. Cancer Metastasis Rev. 2009;28:51–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ong CC, Jubb AM, Zhou W, et al. p21-activated kinase 1: PAK'ed with potential. Oncotarget. 2011;2:491–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vadlamudi RK, Kumar R. P21-activated kinases in human cancer. Cancer Metastasis Rev. 2003;22:385–393 [DOI] [PubMed] [Google Scholar]
- 7. McCarty SK, Saji M, Zhang X, et al. Group I p21-activated kinases regulate thyroid cancer cell migration and are overexpressed and activated in thyroid cancer invasion. Endocr Relat Cancer. 2010;17:989–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vasko V, Espinosa AV, Scouten W, et al. Gene expression and functional evidence of epithelial-to-mesenchymal transition in papillary thyroid carcinoma invasion. Proc Natl Acad Sci U S A. 2007;104:2803–2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Adam L, Vadlamudi R, Mandal M, Chernoff J, Kumar R. Regulation of microfilament reorganization and invasiveness of breast cancer cells by kinase dead p21-activated kinase-1. J Biol Chem. 2000;275:12041–12050 [DOI] [PubMed] [Google Scholar]
- 10. Ong CC, Jubb AM, Haverty PM, et al. Targeting p21-activated kinase 1 (PAK1) to induce apoptosis of tumor cells. Proc Natl Acad Sci U S A. 2011;108:7177–7182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vadlamudi RK, Adam L, Wang RA, et al. Regulatable expression of p21-activated kinase-1 promotes anchorage-independent growth and abnormal organization of mitotic spindles in human epithelial breast cancer cells. J Biol Chem. 2000;275:36238–36244 [DOI] [PubMed] [Google Scholar]
- 12. Holm C, Rayala S, Jirström K, Stål O, Kumar R, Landberg G. Association between Pak1 expression and subcellular localization and tamoxifen resistance in breast cancer patients. J Natl Cancer Inst. 2006;98:671–680 [DOI] [PubMed] [Google Scholar]
- 13. Rayala SK, Molli PR, Kumar R. Nuclear p21-activated kinase 1 in breast cancer packs off tamoxifen sensitivity. Cancer Res. 2006;66:5985–5988 [DOI] [PubMed] [Google Scholar]
- 14. Hirokawa Y, Nakajima H, Hanemann CO, et al. Signal therapy of NF1-deficient tumor xenograft in mice by the anti-PAK1 drug FK228. Cancer Biol Ther. 2005;4:379–381 [DOI] [PubMed] [Google Scholar]
- 15. Yi C, Wilker EW, Yaffe MB, Stemmer-Rachamimov A, Kissil JL. Validation of the p21-activated kinases as targets for inhibition in neurofibromatosis type 2. Cancer Res. 2008;68:7932–7937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chow HY, Jubb AM, Koch JN, et al. p21-Activated kinase 1 is required for efficient tumor formation and progression in a Ras-mediated skin cancer model. Cancer Res. 2012;72:5966–5975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Singhal R, Kandel ES. The response to PAK1 inhibitor IPA3 distinguishes between cancer cells with mutations in BRAF and Ras oncogenes. Oncotarget. 2012;3:700–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Murray BW, Guo C, Piraino J, et al. Small-molecule p21-activated kinase inhibitor PF-3758309 is a potent inhibitor of oncogenic signaling and tumor growth. Proc Natl Acad Sci U S A. 2010;107:9446–9451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Deacon SW, Beeser A, Fukui JA, et al. An isoform-selective, small-molecule inhibitor targets the autoregulatory mechanism of p21-activated kinase. Chem Biol. 2008;15:322–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Porchia LM, Guerra M, Wang YC, et al. 2-Amino-N-{4-[5-(2-phenanthrenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]-phe nyl} acetamide (OSU-03012), a celecoxib derivative, directly targets p21-activated kinase. Mol Pharmacol. 2007;72:1124–1131 [DOI] [PubMed] [Google Scholar]
- 21. Schweppe RE, Klopper JP, Korch C, et al. Deoxyribonucleic acid profiling analysis of 40 human thyroid cancer cell lines reveals cross-contamination resulting in cell line redundancy and misidentification. J Clin Endocrinol Metab. 2008;83:4331–4341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fabien N, Fusco A, Santoro M, Barbier Y, Dubois PM, Paulin C. Description of a human papillary thyroid carcinoma cell line. Morphologic study and expression of tumoral markers. Cancer. 1994;73:2206–2212 [DOI] [PubMed] [Google Scholar]
- 23. Tanaka J, Ogura T, Sato H, Hatano M. Establishment and biological characterization of an in vitro human cytomegalovirus latency model. Virology. 1987;161:62–72 [DOI] [PubMed] [Google Scholar]
- 24. Goretzki PE, Frilling A, Simon D, Roeher HD. Growth regulation of normal thyroids and thyroid tumors in man. Recent Results Cancer Res. 1990;118:48–63 [DOI] [PubMed] [Google Scholar]
- 25. Ringel MD, Hayre N, Saito J, et al. Overexpression and overactivation of Akt in thyroid carcinoma. Cancer Res. 2001;61:6105–6111 [PubMed] [Google Scholar]
- 26. Sells MA, Knaus UG, Bagrodia S, Ambrose DM, Bokoch GM, Chernoff J. Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr Biol. 1997;7:202–210 [DOI] [PubMed] [Google Scholar]
- 27. Sells MA, Boyd JT, Chernoff J. p21-Activated kinase 1 (Pak1) regulates cell motility in mammalian fibroblasts. J Cell Biol. 1999;145:837–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maksimoska J, Feng L, Harms K, et al. Targeting large kinase active site with rigid, bulky octahedral ruthenium complexes. J Am Chem Soc. 2008;130:15764–15765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nheu TV, He H, Hirokawa Y, et al. The K252a derivatives, inhibitors for the PAK/MLK kinase family selectively block the growth of RAS transformants. Cancer J. 2002;8:328–336 [DOI] [PubMed] [Google Scholar]