

Abstract
Purpose
Congenital hereditary endothelial dystrophy 2 (CHED2) is an autosomal recessive disorder caused by mutations in the solute carrier family 4, sodium borate transporter, member 11 (SLC4A11) gene. The purpose of this study was to identify the genetic cause of CHED2 in six Indian families and catalog all known mutations in the SLC4A11 gene.
Methods
Peripheral blood samples were collected from individuals of the families with CHED2 and used in genomic DNA isolation. PCR primers were used to amplify the entire coding region including intron-exon junctions of SLC4A11. Amplicons were subsequently sequenced to identify the mutations.
Results
DNA sequence analysis of the six families identified four novel (viz., p.Thr262Ile, p.Gly417Arg, p.Cys611Arg, and p.His724Asp) mutations and one known p.Arg869His homozygous mutation in the SLC4A11 gene. The mutation p.Gly417Arg was identified in two families.
Conclusions
This study increases the mutation spectrum of the SLC4A11 gene. A review of the literature showed that the total number of mutations in the SLC4A11 gene described to date is 78. Most of the mutations are missense, followed by insertions-deletions. The present study will be helpful in genetic diagnosis of the families reported here.
Introduction
Congenital hereditary endothelial dystrophy (CHED) is a rare inherited disorder of the corneal endothelium, characterized by corneal opacification and nystagmus. CHED is usually evident at the time of birth or in the early years of life. This disorder is due to the malfunction and degeneration of the corneal endothelium that lead to corneal edema, especially of the stroma, and give the cornea the appearance of ground glass [1]. The condition is known to occur in two genetic forms: autosomal dominant (CHED1, MIM# 121700) and autosomal recessive (CHED2, MIM# 217700), the latter more severe and usually more common. CHED1 and CHED2 have been mapped to chromosome 20 at two distinct loci [2,3]. Vithana et al. [4] identified the CHED2 gene solute carrier family 4, sodium borate transporter, member 11 (SLC4A11) through a positional candidate gene approach. SLC4A11, also known as bicarbonate transporter-related protein-1 (BTR1), is a member of the SLC4 family of bicarbonate transporters. In humans, the SLC4A and SLC26 families are the main bicarbonate transporters [5]. The SLC4A11 gene consists of 18 coding exons and is expressed in several organs and tissues, including the eye, blood, lung, ovary, colon, mouth, embryonic tissue, pancreas, kidney, skin, cranial nerve, ascites, prostate, and brain. The gene encodes an 891-amino-acid-long protein with a calculated molecular mass of 100 kDa, which contains 14 transmembrane domains along with multiple intracellular phosphorylation sites and two extra cellular N-glycosylation sites [6]. Homozygous mutations in SLC4A11 have also been shown to cause another rare autosomal recessive disorder, corneal dystrophy and perceptive deafness (CDPD, MIM# 217400) or Harboyan syndrome [7]. Interestingly, heterozygous mutations in SLC4A11 cause Fuchs endothelial corneal dystrophy-4 (FECD4, MIM# 613268) [8-10].
Mutations in SLC4A11 have been reported in families with CHED2 from different populations, including India [4,11-19]. We reported earlier two homozygous mutations in the SLC4A11 gene in two families with CHED2 ascertained at the Minto Eye Hospital, Bangalore. Here, we report on the mutation analysis of this gene in six additional families with CHED2 identified at the same hospital. We have also reviewed the literature on the contribution of this gene in CHED2, FECD4, and CDPD.
Methods
Families
We recruited seven patients from six consanguineous families (Figure 1), three boys and four girls, aged 3-6 years, at the Minto Eye Hospital, Bangalore, Karnataka. Consanguinity was due to maternal uncle and niece marriage in all the families. All family members were examined in detail by D. Ravi Prakash and S. Yathish. Their state of health at the time of recruitment was good with the exception that all affected individuals from six families had congenital bilateral cloudy cornea (Figure 2). None of the parents had cloudy cornea or any other systemic involvement.
Figure 1.

Deoxyribonucleic acid sequence analysis of individuals. Sequencing chromatograms of the heterozygous parents and affected homozygous individuals from family 3, 4, 5, 6, 7, and 8 are shown. Arrows mark the nucleotide change in a heterozygous state in parents and in a homozygous state in affected individuals. + and m denote the wild type and the mutant alleles.
Figure 2.
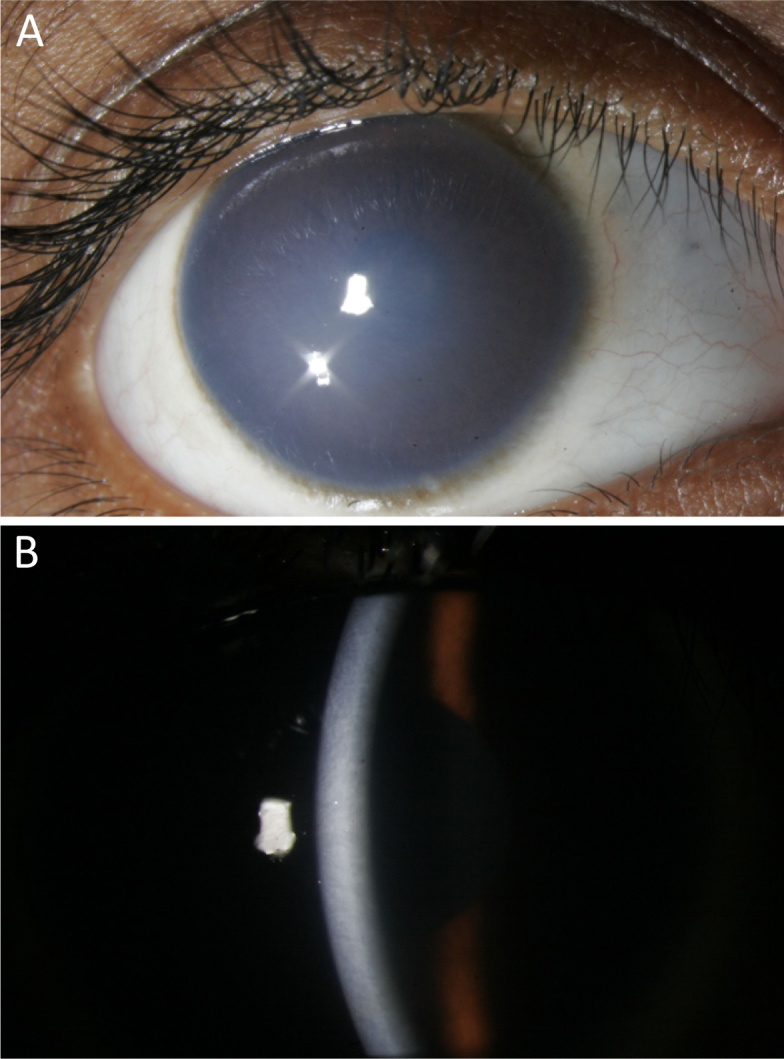
Clinical features of affected individual II-1 from family 7. A: Cornea showing opacification. B: Slit-lamp examination of the cornea showing thickening and opacification.
Genetic analysis
For genetic analysis, 3–5 ml of peripheral blood sample was drawn from each individual in a Vacutainer EDTA tube (Becton Dickinson, Franklin Lakes, NJ) and used for genomic DNA isolation using a Wizard genomic DNA extraction kit (Promega, Madison, WI). This research followed the tenets of the Declaration of Helsinki and the guidelines of the Indian Council of Medical Research, New Delhi. To determine if CHED2 in these families is due to mutations in the SLC4A11 gene, the entire coding region of the SLC4A11 gene was amplified using primers that amplify all coding exons and their intron-exon junctions [12]. Mutations were identified by sequencing the PCR products from one affected individual from each family on an ABIprism A310-automated sequencer (Life Technologies, Carlsbad, CA). PCR was performed in a total volume of 25 µl containing 50 ng of genomic DNA, 1.5 mM MgCl2, 200 µM of each deoxynucleotide triphosphate, 1X buffer, and 1 unit of Taq DNA polymerase (Sigma-Aldrich, Bangalore, India) using a PTC-100 thermocycler (MJ Research, Waltham, MA). Before sequencing, the PCR products were purified using a GenElute gel extraction kit (Sigma-Aldrich, St. Louis, MO). Once a mutation was identified, all members of the family were sequenced to identify the mutation. Allele-specific PCR was performed to determine if a specific mutation was present in 50 ethnically matched normal controls (Table 1).
Table 1. Primers used for mutation analysis in normal controls by allele-specific PCR.
| Mutation | Primer sequence (5′-3′) | Tm (°C) | Amplicon size (bp) |
|---|---|---|---|
| c.1831T>C |
F:GCGTGCGAGAGATCCTGTCCGACC
14R*: AGTAGGGGACAGGCTACTGCTATGCC |
70 |
168 |
| c.1249G>A |
F:GACCATAGCCGGGCAGAGCATCA
11R*:GGGCTGAACCAGATCCCAAGCCTTGA |
66 |
379 |
| c.2170C>G |
F:CTTGGATCCATGCCGCCTACCCCGA
16R*:GGCCAGAGGCTCCCCACTCCTCAG |
61 |
149 |
| c.785C>T | 8F*:CCCGGGCAGGGCCTCCTCTGTTTC R:GCGCGCCACCTCCATCGCAGTCTTAA | 72 | 86 |
Abbreviations: F, forward primer; R, reverse primer, Tm, annealing temperature; and, bp, base pairs. *Primers are described in Kumar et al. [12].
To find the functional significance of the mutated amino acid residues, SLC4A11 protein sequences from different species were aligned by the ClustalW2 program. To predict the effect of mutations on SLC4A11 function, we used two bioinformatics programs: PolyPhen-2 and MutationTaster. The output score from the PolyPhen-2 program ranges from 0 to a positive number, where 0 is neutral, and a high positive number is damaging to protein function. The output from the MutationTaster program is a p (probability) value. A p value close to 1 indicates the high “security” of the prediction that the mutation is damaging to protein function.
Results and Discussion
DNA sequence analysis of the SLC4A11 gene showed four novel mutations, c.1831T>C (p.Cys611Arg), c.1249G>A (p.Gly417Arg), c.2170C>G (p.His724Asp), and c.785C>T (p.Thr262Ile) in families 3, 4, 5, and 6, respectively, and a known mutation, c.2606G>A (p.Arg869His), in family 8 in a homozygous state (Figure 1; Table 2). Interestingly, the c.1249G>A (p.Gly417Arg) mutation was also observed in family 7 (Figure 1).
Table 2. Effect of novel mutations on SLC4A11 function by the in silico analysis.
| Sl.# | Family | Mutation | PolyPhen-2 score | Mutation Taster score |
|---|---|---|---|---|
| 1 |
Family 3 |
c.1831T>C |
Probably damaging with |
Disease causing with |
| (p.Cys611Arg) |
a score of 0.99 |
a p value of 0.99 |
||
| 2 |
Family 4 and |
c.1249 G>A |
Probably damaging with |
Disease causing with |
| Family 7 |
(p.Gly417Arg) |
a score of 1 |
a p value of 0.99 |
|
| 3 |
Family 5 |
c.2170 C>G |
Probably damaging with |
Disease causing with |
| (p.His724Asp) |
a score of 1 |
a p value of 0.99 |
||
| 4 |
Family 6 |
c.785C>T |
Probably damaging |
Disease causing with |
| (p.Thr262Ile) | with a score of 1 | a p value of 0.99 |
Based on the following criteria, we considered the four novel changes mutations. 1) The changes were segregating in the family (Figure 1). 2) The changed amino acids were highly conserved across species (Figure 3). 3) The changes were not observed in 50 normal controls (data not shown). 4) The PolyPhen-2 program predicted all four changes would probably be damaging (Table 2). 5) The MutationTaster program predicted the four changes would be disease causing (Table 2).
Figure 3.

Conservation of the amino acid residues across different species. Arrows mark the conservation of mutated amino acid residues Cys611, Gly417, His724, and Thr262 across different species in SLC4A11. The number refers to the position of the amino acid residue.
We performed a literature review to catalog all the mutations described to date in the SLC4A11 gene. With the four novel mutations described in the present study, the total number of mutations in this gene reaches 78 (Table 3). These include 42 missense, nine nonsense, four splice site, and 23 insertion-deletion mutations (Table 3). The mutations are scattered across the gene (Table 3), suggesting that its entire coding region needs to be sequenced in an affected individual to identify the mutation.
Table 3. Known mutations in the SLC4A11 gene.
| Sl. no. | Mutation | Exon/ intron (IVS) | Nature of mutation | State of zygosity | Effect on protein | Phenotype | Number and ethnic origin of family | Reference |
|---|---|---|---|---|---|---|---|---|
| 1 |
c.99_100delTC (p.S33SfsX18) |
2 |
Deletion |
Heterozygous |
Truncation of protein and addition of novel amino acids |
FECD4 |
1 Chinese |
[8] |
| 2 |
c.140delA(p.Y47SfsX69) |
2 |
Deletion |
Homozygous |
Truncation of protein and addition of novel amino acids |
CHED2 |
1 Indian |
[11] |
| 3 |
c.246_247delTTinsA (p.R82RfsX33) |
2 |
Indel |
Homozygous |
Truncation of protein and addition of novel amino acids |
CHED2 |
1 Indian |
[14] |
| 4 |
c.306delC
(p.G103VfsX13) |
3 |
Deletion |
Compound heterozygous with an unknown second mutation |
Truncation of protein and addition of novel amino acids |
CHED2 |
1 Indian |
[11] |
| 5 |
c.334C>T (p.R112X) |
3 |
Nonsense |
Homozygous and compound heterozygous with c.2318C>T (p.773L) and c.1751C>A (p.T773K) |
Truncation of protein |
CHED2 |
3 Indian |
[11] |
| 6 |
c.353_356delAGAA
(p.K118TfsX11) |
4 |
Deletion |
Homozygous |
Truncation of protein and addition of novel amino acids |
CHED2 |
2 Indian |
[4] |
| 7 |
c.374G>A
(p.R125H) |
4 |
Missense |
Homozygous |
May have an effect on
N-terminal cytoplasmic
domain |
CHED2 |
1 Indian |
[16] |
| 8 |
c.427G>A
(p.E143K) |
4 |
Missense |
Homozygous |
May have an effect on
N-terminal cytoplasmic
domain |
CHED2 |
1 Indian |
[13] |
| 9 |
c.520delGCTTCGCC
(p.R158fs) |
4 |
Out-of-frame deletion |
Homozygous |
Truncation of
protein |
CHED2 |
1 Saudi Arabian |
[18] |
| 10 |
c.473_481delGCTTCGCCAinsC
(p.R158PfsX3) |
4 |
Indel |
Homozygous |
Truncation of protein and
addition of novel amino acids,
absence of all TMD |
CHED2 |
1 Indian |
[16] |
| 11 |
c.473_480del8 bp
(p.R158QfsX4) |
4 |
Deletion |
Homozygous |
Truncation of protein and
addition of novel amino acids |
CHED2
and CDPD |
2 Indian,
1 Gipsy (Eastern European) |
[7,11] |
| 12 |
c.478G> A (p.A160T) |
4 |
Missense |
Homozygous |
May have an effect on
N-terminal cytoplasmic
domain |
CHED2 |
2 Indian |
[14,16] |
| 13 |
c.501G>C
(p.E167D) |
4 |
Missense |
Heterozygous |
Reduction in the mature 120 kDa form, with addition of 100 kDa species |
FECD4 |
Northern European
(No. of families not mentioned) |
[10] |
| 14 |
c.618_619delAG
(p.V208AfsX38) |
5 |
Deletion |
Homozygous |
Truncation of protein and
addition of novel amino acids |
CHED2 |
2 Indian |
[11] |
| 15 |
c.625C>T
(p.R209W) |
5 |
Missense |
Homozygous |
May have an effect on N-terminal cytoplasmic domain |
CHED2 |
2 Indian |
[11] |
| 16 |
c.637T>C
(p.S213P) |
5 |
Missense |
Compound heterozygous with
c.2566A>G (p.M856V) |
May have an effect on N-terminal cytoplasmic domain |
CDPD |
1 Sephardi Jewish |
[7] |
| 17 |
c.638C>T
(p.S213L) |
5 |
Missense |
Homozygous |
May have an effect on N-terminal cytoplasmic domain |
CHED2 |
1 Indian |
[11] |
| 18 |
c.654 (−97)_c.778 (−1488)del698
(p.C218KfsX49) |
5–6 |
Deletion |
Homozygous |
Truncation of protein and
addition of novel amino acids,
absence of all TMDs |
CHED2 |
1 Indian |
[16] |
| 19 |
c.743G>A (p.S232N) |
6 |
Missense |
Compound heterozygous with
c.1033A>T (p.Arg329X) |
Loss of function or membrane localization |
CHED2 |
1 US family of
Chinese ancestry |
[15] |
| 20 |
c.697C>T (p.R233C) |
6 |
Missense |
Homozygous |
May have an effect on
N-terminal cytoplasmic
domain |
CHED2 |
1 Indian |
[11] |
| 21 |
c.720G>A (p.W240X) |
6 |
Nonsense |
Homozygous |
Truncation of protein |
CHED2 |
1 British |
[13] |
| 22 |
c.785C>T (p.T262I) |
6 |
Missense |
Homozygous |
Damaging to protein function |
CHED 2 |
1 Indian |
Present study |
| 23 |
c.806C>T
(p.A269V) |
7 |
Missense |
Homozygous |
May have an effect on
N-terminal cytoplasmic
domain |
CHED2 |
2 Indian |
[16] |
| 24 |
c.812C>T
(p.T271M) |
7 |
Missense |
Homozygous |
May have an effect on
N-terminal cytoplasmic
domain |
CHED2 |
1 Saudi Arabian |
[17] |
| 25 |
c.845G>C
(p.R282P) |
7 |
Missense |
Heterozygous |
Immature protein |
FECD4 |
Northern European
(No. of families not mentioned) |
[10] |
| 26 |
c.859_862delGAGA
insCCT
(p.E287PfsX21) |
7 |
Indel |
Homozygous |
Truncation of protein and
addition of novel amino acids,
absence of all TMDs |
CHED2 |
1 Indian |
[12] |
| 27 |
c.878_889del12 p.E293_E296del |
7 |
Deletion |
Homozygous |
May have an effect on
N-terminal cytoplasmic
domain |
CHED2 |
1 Indian |
[11] |
| 28 |
c.1033A>T (p.R329X) |
7 |
Nonsense |
Compound heterozygous with
c.743G>A (p.Ser232Asn) |
Premature truncation of the
transcript |
CHED2 |
1 US family of
Chinese ancestry |
[15] |
| 29 |
c.996+26C_+44Cdel19 |
IVS-7 |
Deletion |
Homozygous |
Not Known |
CHED2 |
2 Indian |
[11] |
| 30 |
c.1044+25del19nt |
IVS-7 |
Deletion |
Homozygous |
Not known |
CHED2 |
1 Saudi Arabian |
[18] |
| 31 |
c.1091–1G>C |
IVS-8 |
Splice site |
Homozygous |
Not known |
CHED2 |
1 Indian |
[11] |
| 32 |
c.1156T>C (p.C386R) |
9 |
Missense |
Homozygous |
Disruption of TMD 1 |
CHED2 |
4 Indian |
[13,16,19] |
| 33 |
c.1228G>C
(p.G394R) |
9 |
Missense |
Homozygous |
Disruption of TMD1 |
CHED2 |
1 Saudi Arabian |
[18] |
| 34 |
c.1195G>A (p.E399K) |
9 |
Missense |
Heterozygous |
Aberrant glycosylation and cellular localization |
FECD4 |
1 Indian |
[8] |
| 35 |
c.1202C>A (p.T401L) |
9 |
Missense |
Compound heterozygous with c.1418T>G
(p.L473R) |
Not known |
CHED2 |
1 Indian |
[11] |
| 36 |
c.1249 G>A (p.G417R) |
10 |
Missense |
Homozygous |
Damaging to protein function |
CHED2 |
2 Indian |
Present study |
| 37 |
c.1253G>A (p.G418D) |
10 |
Missense |
Homozygous |
Disruption of TMD 2 |
CHED2 |
1 Indian,
1 Saudi Arabian |
[11,18] |
| 38 |
c.1317_1322del6ins8
(p.L440VfsX6) |
10 |
Indel |
Homozygous |
Truncation of protein and
addition of novel amino acids |
CHED2 |
1 Indian |
[11] |
| 39 |
c.1378_1381delTACGinsA
(p.Y460_A461 delinsT) |
11 |
Indel |
Homozygous |
Not known |
CDPD |
1 Dominican Republican |
[7] |
| 40 |
c.1391G>A (p.G464D) |
11 |
Missense |
Homozygous |
Conformation change |
CHED2 |
3 Pakistani |
[4] |
| 41 |
c.1463G>A
(p.R488K) |
11 |
Missense |
Homozygous |
Not known |
CDPD |
1 Moroccan |
[7] |
| 42 |
c.1466C>T (p.S489L) |
12 |
Missense |
Homozygous |
Conformation change |
CHED2 |
1 Pakistani, 1 Indian |
[4,11] |
| 43 |
c.1577A>G
(p.Y526C) |
12 |
Missense |
Heterozygous |
Partial loss of localization at the membrane |
FECD4 |
Northern European
(No. of families not mentioned) |
[10] |
| 44 |
c.1704_1705delCT (p.H568HfsX177) |
13 |
Deletion |
Homozygous |
Truncation of protein and
addition of novel amino acids |
CHED2 |
1 Indian |
[14] |
| 45 |
c.1723G>A
(p.V575M) |
13 |
Missense |
Heterozygous |
Partial loss of localization at the membrane |
FECD4 |
Northern European
(No. of families not mentioned) |
[10] |
| 46 |
c.1748G>A
(p.G583D) |
13 |
Missense |
Heterozygous |
Immature protein |
FECD4 |
Northern European
(No. of families not mentioned) |
[10] |
| 47 |
c.1751C>A (p.T584K) |
13 |
Missense |
Homozygous and compound heterozygous with c.334C>T (p.Arg112X) |
Disruption of TMD 6 |
CHED2 |
2 Indian |
[11] |
| 48 |
c.1813C>T (p.R605X) |
14 |
Nonsense |
Homozygous and compound heterozygous with an unknown second mutation |
Truncation of protein |
CHED2 |
6 Indian |
[4,11,14] |
| 49 |
c.1831T>C
(p.C611R) |
14 |
Missense |
Homozygous |
Damaging to protein function |
CHED2 |
1 Indian |
Present study |
| 50 |
c.1894G>T (p.E632X) |
14 |
Nonsense |
Homozygous |
Truncation of protein |
CHED2 |
2 Indian |
[11,14] |
| 51 |
IVS15 −6 _ −16
delins GGCCGGCCGG |
IVS-15 |
Indel |
Homozygous |
Inactivation of
splice
acceptor site |
CHED2 |
1 Indian |
[4] |
| 52 |
c.2014_2016delTTC
(p.F672del) |
15 |
In-frame deletion |
Homozygous |
Disruption of TMD8 |
CHED2 |
1 Indian |
[12] |
| 53 |
c.2067–6_-16delinsGGCCGGCCGG |
IVS-15 |
Splice site |
Homozygous |
Inactivation of an acceptor
splice site |
CHED2 |
1 Indian |
Cited in [16] |
| 54 |
c.2114+1G>A |
IVS-15 |
Donor Splice site |
Homozygous |
Inclusion of
intron 15 |
CHED2 |
1 Saudi Arabian |
[18] |
| 55 |
c.2126G>A (p.G709E) |
15 |
Missense |
Heterozygous |
Aberrant glycosylation and cellular localization |
FECD |
1 Chinese |
[8] |
| 56 |
c.2170 C>G
(p.His724Asp) |
15 |
Missense |
Homozygous |
Damaging to protein structure |
CHED2 |
1 Indian |
Present study |
| 57 |
c.2224G>A
(p.G742R) |
16 |
Missense |
Heterozygous |
Reduction in the mature 120-kDa form, with addition of 100-kDa species |
FECD |
Northern European
(No. of families not mentioned) |
[10] |
| 58 |
c.2233_2240dup
TATGACAC
(p.T747TfsX6) |
16 |
Duplication |
Compound heterozygous with c.2528T>C
(p.L843P) |
Aberrantly truncated protein of 916 residues |
CDPD |
1 South American Indian |
[7] |
| 59 |
c.2236C>T
(p.R757X) |
16 |
Nonsense |
Homozygous |
Protein truncation |
CHED2 |
2 Saudi Arabian |
[18] |
| 60 |
c.2240 +1G>A |
IVS-16 |
Splice site |
Homozygous and compound heterozygous with an unknown second mutation |
Inactivation of splice donor site |
CHED2 |
1 British,
1 Indian |
[13,19] |
| 61 |
c.2261C>T (p.T754M) |
17 |
Missense |
Heterozygous |
Aberrant glycosylation and cellular localization |
FECD4 |
1 Chinese |
[8] |
| 62 |
c.2263C>T
(p.R755W) |
17 |
Missense |
Homozygous |
Disruption of TMD 11 |
CHED2 |
3 Indian |
[11,13,16] |
| 63 |
c.2264G>A (p.R755Q) |
17 |
Missense |
Homozygous
and compound heterozygous with c.2623C>T (p.Arg875X) |
Conformation change |
CHED2 |
4 Indian,
1 Myanmar |
[4,11,13,14] |
| 64 |
c.2318C>T (p.P773L) |
17 |
Missense |
Homozygous
and compound heterozygous with c.334C>T (p.R112X) |
Disruption of TMD 11 |
CHED2 |
3 Indian |
[11,16] |
| 65 |
c.2389_2391delGAT
(p.D797del) |
17 |
Deletion |
Homozygous |
Disruption of TMD 12 |
CHED2 |
1 Indian |
[11] |
| 66 |
c.2398C>T
(p.Q800X) |
17 |
Nonsense |
Compound heterozygous with
c.2437–1G>A |
Truncation of protein |
CHED2 |
1 British |
[13] |
| 67 |
c.2407C>T
(p.Gln803X) |
17 |
Nonsense |
Homozygous |
Truncation of protein |
CHED2 |
1 Indian |
[11] |
| 68 |
c.2411G>A (p.R804H) |
18 |
Missense |
Homozygous |
Conformation change |
CHED2 |
1 Indian family |
[14] |
| 69 |
c.2420delTinsGG (p.L807RfsX71) |
18 |
Missense |
Homozygous |
Truncation of protein and
addition of novel amino acids |
CHED2 |
1 Indian family |
[14] |
| 70 |
c.2423_2454del 32nt
(p.Leu808ArgfsX110) |
17 |
Deletion |
Compound heterozygous with
c.2528T>C (p.Leu843Pro) |
Aberrantly truncated protein of 916 residues |
CDPD |
1 Dutch |
[7] |
| 71 |
c.2470G>A (p.V824M) |
18 |
Missense |
Homozygous |
Not known |
CHED2 |
6 Indian |
[7,11,19] |
| 72 |
c.2498C>T (p.T833M) |
18 |
Missense |
Homozygous |
Conformation change |
CHED2 |
2 Indian |
[14] |
| 73 |
c.2500G>A
(p.G834S) |
18 |
Missense |
Heterozygous |
Immature protein |
FECD |
Northern European
(No. of families not mentioned) |
[10] |
| 74 |
c.2506 C>T
(p.Q836X) |
18 |
Nonsense |
Compound heterozygous with c.2318C>T (p.P773L) |
Truncation of protein |
CHED2 |
1 Indian |
[16] |
| 75 |
c.2518–2520 delCTG
(p.L840del) |
18 |
In-frame deletion |
Homozygous |
Disrupts the appropriate assembly or localization of protein in the membrane |
CHED2 |
1 Indian |
[19] |
| 76 |
c.2605C>T (p.R869C) |
18 |
Missense |
Homozygous |
Conformation change |
CHED2 |
3 Indian,
1 Middle
Eastern |
[4,11,13] |
| 77 |
c.2606G>A (p.R869H) |
18 |
Missense |
Homozygous |
Damaging to protein structure |
CHED2 |
3 Indian |
[14],
Present study |
| 78 | c.2618T>C (p.L873P) | 19 | Missense | Homozygous | Disruption of TMD 14 | CHED2 | 1 Indian | [16] |
CDPD is a degenerative corneal disorder characterized by the association of congenital hereditary endothelial dystrophy with progressive sensorineural hearing loss. The ocular manifestations in CDPD include diffuse bilateral corneal edema occurring with severe corneal clouding, blurred vision, visual loss, and nystagmus, which are usually present at birth or within the neonatal period and are indistinguishable from CHED2. The sensorineural hearing loss is slowly progressive and can be identified only during the second decade of life [20]. As stated, homozygous mutations in SLC4A11 cause not only CHED2 but also CDPD. One of the mutations, c.473_480del8bp (p.R158QfsX4), causes CHED2 and CDPD (Table 3).
Why some individuals also develop perceptive deafness along with corneal dystrophy due to mutations in SLC4A11 is unclear. However, it could be due to an additional environmental effect and/or genetic modifiers. Morris et al. [21] showed differential expression of SLC4A11 in the inner ear of mice specifically in the region of the stria vascularis. Taking this fact into account, Desir et al. [7] postulated that corneal dystrophy and perceptive deafness might have a common origin in the neural crest cells from which the stria vascularis and the corneal endothelium develop. Further, four mutations (p.S213P, p.Y460_A461 delinsT, p.R488K, and p.Leu808ArgfsX110) are specific to only CDPD (Table 3), and none of the 11 heterozygous mutations causing FECD (FECD4) are found in patients with CHED2 and CDPD (Table 3). FECD is a progressive degeneration of the corneal endothelium leading to thickened Descemet’s membrane, a collagen-rich basal lamina secreted by the endothelium, and reduced vision. In patients with FECD, corneal endothelial cells die, as a result of which bumps called guttae form on the back of the cornea. This causes the cornea to swell and distort vision, resulting in pain and severe visual impairment [8,22].
Why some heterozygous mutations in SLC4A11 cause FECD4 is also not clear. However, it could be speculated on. The involvement of SLC4A11 in various corneal dystrophies suggests a significant genetic overlap occurs across several corneal dystrophies and they might share a common pathomechanism [10]. Moreover, the characteristic abnormal posterior non-banded zone of the Descemet’s membrane, which represents an abnormal function of the corneal endothelium in CHED2 and FECD4, underlies the importance of the SLC4A11 protein for the proper development and differentiation of the corneal endothelium and may explain how the same gene can be involved in the pathogenesis of CHED2 and FECD4 [8,22]. In addition, a combination of mechanisms may be at play, with partial loss of function and gradual accumulation of the aberrant misfolded protein having a role in FECD4 pathology [8].
It is not surprising to find mutations in SLC4A11 causing three different disorders. Similar to SLC4A11, mutations in the same gene are known to cause different disorders. For example, null mutations in CEP290 (NPHP6) cause Meckel syndrome (MKS4, MIM# 611134) [23], Bardet-Biedl syndrome (BBS14, MIM# 209900) [24], and Joubert syndrome (JBTS5, MIM# 610188) [25,26], while hypomorphic mutations in the same gene lead to Leber congenital amaurosis (LCA10, MIM# 611755) [27].
In summary, we have identified four novel mutations in the SLC4A11 gene in the present study. With the four novel mutations reported here, the total number of mutations described to date in SLC4A11 reaches 78. Further, this information will be useful for providing rapid prenatal diagnosis and genetic counseling to families and their relatives.
Acknowledgments
We thank the patients and their family members for their participation in this study. We also thank an anonymous reviewer for their comments to improve the manuscript. This work was funded by the University Grants Commission (No. UGC-SAP-22-0307-0013-03-469), New Delhi.
References
- 1.Ehlers N, Módis L, Møller-Pedersen T. A morphological and functional study of congenital hereditary endothelial dystrophy. Acta Ophthalmol Scand. 1998;76:314–8. doi: 10.1034/j.1600-0420.1998.760312.x. [DOI] [PubMed] [Google Scholar]
- 2.Toma NM, Ebenezer ND, Inglehearn CF, Plant C, Ficker LA, Bhattacharya SS. Linkage of congenital hereditary endothelial dystrophy to chromosome 20. Hum Mol Genet. 1995;4:2395–8. doi: 10.1093/hmg/4.12.2395. [DOI] [PubMed] [Google Scholar]
- 3.Hand CK, Harmon DL, Kennedy SM, FitzSimon JS, Collum LMT, Parfey NA. Localization of the gene for autosomal recessive congenital hereditary endothelial dystrophy (CHED2) to chromosome 20 by homozygosity mapping. Genomics. 1999;61:1–4. doi: 10.1006/geno.1999.5920. [DOI] [PubMed] [Google Scholar]
- 4.Vithana EN, Morgan P, Sundaresan P, Ebenezer ND, Tan DT, Mohamed MD, Anand S, Khine KO, Venkataraman D, Yong VH, Salto-Tellez M, Venkatraman A, Guo K, Hemadevi B, Srinivasan M, Prajna V, Khine M, Casey JR, Inglehearn CF, Aung T. Mutations in sodium-borate cotransporter SLC4A11 cause recessive congenital hereditary endothelial dystrophy (CHED2). Nat Genet. 2006;38:755–7. doi: 10.1038/ng1824. [DOI] [PubMed] [Google Scholar]
- 5.Romero MF. Molecular pathophysiology of SLC4 bicarbonate transporters. Curr Opin Nephrol Hypertens. 2005;14:495–501. doi: 10.1097/01.mnh.0000168333.01831.2c. [DOI] [PubMed] [Google Scholar]
- 6.Parker MD, Ourmozdi EP, Tanner MJ. Human BTR1, a new bicarbonate transporter superfamily member and human AE4 from kidney. Biochem Biophys Res Commun. 2001;282:1103–9. doi: 10.1006/bbrc.2001.4692. [DOI] [PubMed] [Google Scholar]
- 7.Desir J, Moya G, Reish O, Van Regemorter N, Deconinck H, David KL, Meire FM, Abramowicz MJ. Borate transporter SLC4A11 mutations cause both Harboyan syndrome and non-syndromic corneal endothelial dystrophy. J Med Genet. 2007;44:322–6. doi: 10.1136/jmg.2006.046904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vithana EN, Morgan PE, Ramprasad V, Tan DT, Yong VH, Venkataraman D, Venkatraman A, Yam GH, Nagasamy S, Law RW, Rajagopal R, Pang CP, Kumaramanickevel G, Casey JR, Aung T. SLC4A11 mutations in Fuchs endothelial corneal dystrophy. Hum Mol Genet. 2008;17:656–66. doi: 10.1093/hmg/ddm337. [DOI] [PubMed] [Google Scholar]
- 9.Hemadevi B, Srinivasan M, Arunkumar J, Prajna NV, Sundaresan P. Genetic analysis of patients with Fuchs endothelial corneal dystrophy in India. BMC Ophthalmol. 2010;10:3. doi: 10.1186/1471-2415-10-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riazuddin SA, Vithana EN, Seet LF, Liu Y, Al-Saif A, Koh LW, Heng YM, Aung T, Meadows DN, Eghrari AO, Gottsch JD, Katsanis N. Riazuddin. Missense mutations in the sodium borate cotransporter SLC4A11 cause late-onset Fuchs corneal dystrophy. Hum Mutat. 2010;31:1261–8. doi: 10.1002/humu.21356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sultana A, Garg P, Ramamurthy B, Vemuganti GK, Kannabiran C. Mutational spectrum of the SLC4A11 gene in autosomal recessive congenital hereditary endothelial dystrophy. Mol Vis. 2007;13:1327–32. [PubMed] [Google Scholar]
- 12.Kumar A, Bhattacharjee S, Prakash DR, Sadanand CS. Genetic analysis of two Indian families affected with congenital hereditary endothelial dystrophy: two novel mutations in SLC4A11. Mol Vis. 2007;13:39–46. [PMC free article] [PubMed] [Google Scholar]
- 13.Ramprasad VL, Ebenezer ND, Aung T, Rajagopal R, Yong VH, Tuft SJ, Viswanathan D, El-Ashry MF, Liskova P, Tan DT, Bhattacharya SS, Kumaramanickavel G, Vithana EN. Novel SLC4A11 mutations in patients with recessive congenital hereditary endothelial dystrophy (CHED2). Hum Mutat. 2007;28:522–3. doi: 10.1002/humu.9487. [DOI] [PubMed] [Google Scholar]
- 14.Jiao X, Sultana A, Garg P, Ramamurthy B, Vemuganti GK, Gangopadhyay N, Hejtmancik JF, Kannabiran C. Autosomal recessive corneal endothelial dystrophy (CHED2) is associated with mutations in SLC4A11. J Med Genet. 2007;44:64–8. doi: 10.1136/jmg.2006.044644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aldave AJ, Yellore VS, Bourla N, Momi RS, Khan MA, Salem AK, Rayner SA, Glasgow BJ, Kurtz I. Autosomal recessive CHED associated with novel compound heterozygous mutations in SLC4A11. Cornea. 2007;26:896–900. doi: 10.1097/ICO.0b013e318074bb01. [DOI] [PubMed] [Google Scholar]
- 16.Hemadevi B, Veitia RA, Srinivasan M, Arunkumar J, Prajna NV, Lesaffre C, Sundaresan P. Identification of mutations in the SLC4A11 gene in patients with recessive congenital hereditary endothelial dystrophy. Arch Ophthalmol. 2008;126:700–8. doi: 10.1001/archopht.126.5.700. [DOI] [PubMed] [Google Scholar]
- 17.Shah SS, Al-Rajhi A, Brandt JD, Mannis MJ, Roos B, Sheffield VC, Syed NA, Stone EM, Fingert JH. Mutation in the SLC4A11 gene associated with autosomal recessive congenital endothelial dystrophy in a large Saudi family. Ophthalmic Genet. 2008;29:41–5. doi: 10.1080/13816810701850033. [DOI] [PubMed] [Google Scholar]
- 18.Aldahmesh MA, Khan AO, Meyer BF, Alkuraya FS. Mutational spectrum of SLC4A11 in autosomal recessive CHED in Saudi Arabia. Invest Ophthalmol Vis Sci. 2009;50:4142–5. doi: 10.1167/iovs.08-3006. [DOI] [PubMed] [Google Scholar]
- 19.Paliwal P, Sharma A, Tandon R, Sharma N, Titiyal JS, Sen S, Nag TC, Vajpayee RB. Congenital hereditary endothelial dystrophy mutation analysis of SLC4A11 and genotype-phenotype correlation in a North Indian patient cohort. Mol Vis. 2010;16:2955–63. [PMC free article] [PubMed] [Google Scholar]
- 20.Harboyan G, Mamo J, Der Kaloustian V, Karam F. Congenital corneal dystrophy: progressive sensorineural deafness in a family. Arch Ophthalmol. 1971;85:27–32. doi: 10.1001/archopht.1971.00990050029005. [DOI] [PubMed] [Google Scholar]
- 21.Morris KA, Snir E, Pompeia C, Koroleva IV, Kachar B, Hayashizaki Y, Carninci P, Soares MB, Beisel KW. Differential expression of genes within the cochlea as defined by a custom mouse inner ear microarray. J Assoc Res Otolaryngol. 2005;6:75–89. doi: 10.1007/s10162-004-5046-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klintworth GK. Corneal dystrophies. Orphanet J Rare Dis. 2009;4:7–45. doi: 10.1186/1750-1172-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baala L, Audollent S, Martinovic J, Ozilou C, Babron M-C, Sivanandamoorthy S, Saunier S, Salomon R, Gonzales M, Rattenberry E, Esculpavit C, Toutain A, Moraine C, Parent P, Marcorelles P, Dauge M-C, Roume J, Le Merrer M, Meiner V, Meir K, Menez F, Beaufrère A-M, Francannet C, Tantau J, Sinico M, Dumez Y, MacDonald F, Munnich A, Lyonnet S, Gubler M-C, Génin E, Johnson CA, Vekemans M, Encha-Razavi F, Attié-Bitach T. Pleiotropic effects of CEP290 (NPHP6) mutations extend to Meckel syndrome. Am J Hum Genet. 2007;81:170–9. doi: 10.1086/519494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leitch CC, Zaghloul NA, Davis EE, Stoetzel C, Diaz-Font A, Rix S, Alfadhel M, Lewis RA, Eyaid W, Banin E, Dollfus H, Beales PL, Badano JL, Katsanis N. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat Genet. 2008;40:443–8. doi: 10.1038/ng.97. [DOI] [PubMed] [Google Scholar]
- 25.Sayer JA, Otto EA, O'Toole JF, Nurnberg G, Kennedy MA, Becker C, Hennies HC, Helou J, Attanasio M, Fausett BV, Utsch B, Khanna H, Liu Y, Drummond I, Kawakami I, Kusakabe T, Tsuda M, Ma L, Lee H, Larson RG, Allen SJ, Wilkinson CJ, Nigg EA, Shou C, Lillo C, Williams DS, Hoppe B, Kemper MJ, Neuhaus T, Parisi MA, Glass IA, Petry M, Kispert A, Gloy J, Ganner A, Walz G, Zhu X, Goldman D, Nurnberg P, Swaroop A, Leroux MR, Hildebrandt F. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet. 2006;38:674–81. doi: 10.1038/ng1786. [DOI] [PubMed] [Google Scholar]
- 26.Valente EM, Silhavy JL, Brancati F, Barrano G, Krishnaswami SR, Castori M, Lancaster MA, Boltshauser E, Boccone L, Al-Gazali L, Fazzi E, Signorini S, Louie CM, Bellacchio E, International Joubert Syndrome Related Disorders Study Group Bertini E, Dallapiccola B, Gleeson JG. Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat Genet. 2006;38:623–5. doi: 10.1038/ng1805. [DOI] [PubMed] [Google Scholar]
- 27.den Hollander AI, Koenekoop RK, Yzer S, Lopez I, Arends ML, Voesenek KE, Zonneveld MN, Strom TM, Meitinger T, Brunner HG, Hoyng CB, van den Born LI, Rohrschneider K, Cremers FP. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am J Hum Genet. 2006;79:556–61. doi: 10.1086/507318. [DOI] [PMC free article] [PubMed] [Google Scholar]