


Abstract
Reversible protein phosphorylation provides a major regulatory mechanism in eukaryotic cells. Due to the high variability of amino acid residues flanking a relatively limited number of experimentally identified phosphorylation sites, reliable prediction of such sites still remains an important issue. Here we report the development of a new web-based tool for the prediction of protein phosphorylation sites, DISPHOS (DISorder-enhanced PHOSphorylation predictor, http://www.ist.temple.edu/DISPHOS). We observed that amino acid compositions, sequence complexity, hydrophobicity, charge and other sequence attributes of regions adjacent to phosphorylation sites are very similar to those of intrinsically disordered protein regions. Thus, DISPHOS uses position-specific amino acid frequencies and disorder information to improve the discrimination between phosphorylation and non-phosphorylation sites. Based on the estimates of phosphorylation rates in various protein categories, the outputs of DISPHOS are adjusted in order to reduce the total number of misclassified residues. When tested on an equal number of phosphorylated and non-phosphorylated residues, the accuracy of DISPHOS reaches 76% for serine, 81% for threonine and 83% for tyrosine. The significant enrichment in disorder-promoting residues surrounding phosphorylation sites together with the results obtained by applying DISPHOS to various protein functional classes and proteomes, provide strong support for the hypothesis that protein phosphorylation predominantly occurs within intrinsically disordered protein regions.
INTRODUCTION
Intrinsically unstructured proteins are frequently involved in key biological processes such as cell cycle control, transcriptional and translational regulation, membrane fusion and transport, and signal transduction (1,2). A high percentage of cell-signaling and cancer-associated proteins are predicted to have long disordered regions, suggesting the general importance of intrinsic disorder for signaling and regulation (3). An investigation of the functions performed by intrinsically disordered regions reveals that they are often involved in molecular recognition and protein modifications including phosphorylation (4).
Protein phosphorylation represents an important regulatory mechanism in eukaryotic cells. At least one-third of all eukaryotic proteins are estimated to undergo reversible phosphorylation (5). Phosphorylation modulates the activity of numerous proteins involved in signal transduction, and regulates the binding affinity of transcription factors to their coactivators and DNA thereby altering gene expression, cell growth and differentiation (6). Phosphorylation sites frequently cluster within functionally important protein domains, i.e. the majority of phosphorylation sites of Mdm2 are located in its p53- and p14-ARF-binding regions (7), and the phosphorylation of PEST motifs influences ubiquitin-mediated protein degradation (8).
The phosphorylation sites in proteins were found within intrinsically disordered regions in some cases, and within regions of well ordered structure in other instances. With regard to the structural consequences of phosphorylation, both disorder to order and order to disorder transitions have been observed to follow the phosphorylation event (9). Conformational changes upon phosphorylation often affect protein function. For example, serine phosphorylation of the peptide corresponding to the calmodulin binding domain of human protein p4.1 influences the ability of the peptide to adopt an alpha-helical conformation and thereby impairs the calmodulin-peptide interaction (10). Another example is the v-cyclin-CDK6-mediated phosphorylation of two serines in the unstructured loop of Bcl-2, which abolishes its anti-apoptotic potential (11).
Experimental difficulties in the large-scale identification of protein phosphorylation sites stimulated the development of computational approaches to predict these sites from protein sequence. Two major bioinformatics tools currently available for proteome-wide identification of phosphorylation sites are a neural-network based predictor NetPhos (12) and a motif-based service Scansite (13,14). Both NetPhos and Scansite are available via the Internet. However, due to the high degree of variability in consensus patterns around phosphorylation sites caused by the diversity and the large number of kinases (15), and the still relatively small number of non-redundant, experimentally verified sites, high-accuracy prediction of phosphorylation sites remains an open research area.
To gain further insight regarding the role of disorder in the phosphorylation process, we investigated more than 1500 experimentally determined phosphorylation sites in eukaryotic proteins and compared them with ordered and disordered protein regions. The similarity in sequence complexity, amino acid composition, flexibility parameters, and other properties between phosphorylation sites and disordered protein regions suggests that intrinsic disorder in and around the potential phosphorylation target site is an essential common feature for eukaryotic serine, threonine and tyrosine phosphorylation sites. Based on this observation and state-of-the-art machine learning principles, we constructed a new predictor of phosphorylation sites, DISPHOS (DISorder-enhanced PHOSphorylation predictor) that has an improved accuracy in comparison to the widely used NetPhos and Scansite. Explicit use of disorder prediction, expansion of the training data set, model choice, feature selection/extraction and training and testing processes all contributed to an improvement in phosphorylation site prediction accuracy, which reached values of 76.0 ± 0.3, 81.3 ± 0.3 and 83.3 ± 0.3% for serine (S), threonine (T) and tyrosine (Y), respectively. Furthermore, estimates of fractions of phosphorylated residues (class priors) in various kingdoms, proteomes and protein functional categories were used to adjust the outputs of the predictor and minimize the number of misclassified residues in proteins from these groups.
MATERIALS AND METHODS
Data sets
We created databases of positive (P) and negative (NP) sites by extracting 25-residue long sequences centered at S, T and Y sites from the eukaryotic proteins in SWISS-PROT using ‘Eukaryota’ in the organism field. To construct the positive data sets (PS, PT and PY), only the residues annotated as ‘phosphorylation’ in the ‘MOD_RES’ field were selected, and the sites annotated as ‘potential’, ‘probable’ and ‘by similarity’ were omitted. The control data sets of non-phosphorylated sites (NS, NT and NY) were extracted from the same proteins and represented all S, T and Y residues that did not have the ‘phosphorylation’ or ‘phosphorylation’ combined with ‘potential’, ‘probable’ or ‘by similarity’ annotation.
We then combined the SWISS-PROT sites with the PhosphoBase sites and removed all P- and NP-sites that had more than 30% sequence identity (excluding the middle residue) inside the combined data sets. This relatively conservative threshold used for such short fragments (16) was selected in part to prevent situations where non-redundant fragments are associated with considerably different numbers of homologs at >30% sequence identity. Additionally, a data set biased towards some non-redundant fragments would not only influence generalization of the predictor, but also produce inaccurate estimates of the predictor’s performance on non-redundant data. Thus, in effect, only seven residues out of 24 were allowed to match in the pairwise alignments in which no gaps were allowed.
The negative data set may contain numerous un-annotated positive sites, which inevitably contribute to noise in the data set. Furthermore, due to the presence of homologous proteins and as a consequence of combining two databases, the same site may be annotated as phosphorylated in one protein, but not in its close homolog or duplicate. Such disagreement in annotations may be a result of different experiments involving different kinases. Thus, the same site would be included in both positive and negative data sets. To address this problem, all NP-sites with >30% identity with any of the P-sites were discarded.
Even after the elimination of sequence redundancy, the combined data set may still be biased with respect to the kinases involved in phosphorylation of the remaining P-sites. To investigate this issue further, we extracted detailed annotations (including kinase names) for all phosphorylation sites from our positive data sets. This allowed us to calculate that at least one phosphorylation site for over 60 different kinases is included in our positive data sets even after redundancy elimination, and that on average ∼40% of the P-sites are phosphorylated by the unknown/unannotated kinases. Moreover, none of the well studied kinases is associated with >10% of the P-sites in our positive data sets. Thus, we consider our data sets to be reasonably diverse and not severely biased both in terms of sequence redundancy and kinase representation.
Other data sets used in this study were: (i) all disorder—disordered regions characterized by X-ray diffraction (extracted from PDB-Select-25), NMR and CD (extracted from the literature); (ii) Globular-3D—the ordered protein regions extracted from Protein Data Bank (PDB): fibrous sequences such as coiled coils, collagen and silk fibroins were removed from this data set (17); (iii) PDB order—ordered protein regions from PDB-Select-25, a non redundant set of PDB structures; (iv) 12 protein functional categories, constructed as previously described (3); (v) viral, bacterial, archaeal and eukaryotic proteins were extracted from SWISS-PROT using the keyword search in the organism field; (vi) the seven eukaryotic proteomes were downloaded from NCBI (ftp://ftp.ncbi.nlm.nih.gov/genomes/).
Feature construction and selection/extraction
The input to a predictor of phosphorylation sites is a 25 residue long sequence with S, T or Y in the center. The position-specific features were constructed using the standard orthogonal (binary) representation (18). Briefly, for each position of the input sequence we constructed a 20-dimensional vector of 0s with a 1 only for the residue observed at this position. Since the central residue is always S, T or Y, this site was not included, resulting in 24 (positions) × 20 (amino acid residues) = 480 binary features. Predictions for residues near N- or C-termini were made using half of the features.
Another 20 features represented the relative amino acid frequencies over the window of 25, and the outputs from five predictors of disorder [VLXT (17), VL2 and VLC, VLV, VLS (19)] were also employed as features to construct the phosphorylation predictor. Three more features used the information from the three secondary structure predictors: PHD (20), SSPAL (21) and NNSSP (22). The predictions of helix, sheet and loop for PHD, and helix and sheet for both SSPAL and NNSSP, totaling seven features, were utilized. Seven additional features were added: sequence complexity (23), net charge (K + R – D – E) (24), aromatic content (W + F + Y), hydrophobic moment (25), hydrophobicity (26), flexibility scale (27), and Janin’s scale for surface exposed and buried residues (28).
In total, we collected a set of 519 features (480 binary and 39 real-valued). A binary target variable (1 for P-site, 0 for NP-site) was then added to each example. Consequently, two matrices were constructed for each residue (S, T and Y): matrix P for positive sites and matrix N for the negative control set.
The small number of positive examples, high dimensionality, correlation among features, and the sparse nature of each sample required dimensionality reduction. We applied the Fisher’s permutation test (29) to the set of 480 binary features and selected only those that had significant or near-significant differences in position-specific amino acid compositions between P- and NP-sites (P < 0.1). Since the sample remained sparse and still contained correlated features, we then performed PCA (30) and utilized a smaller fixed number of principal components in predictor construction. The forward-selection algorithm (31) was used to select the best of the remaining 39 non-binary features.
Predictor training and testing
We combined each set of positive examples PS, PT and PY with their corresponding sets of negative examples NS, NT and NY to construct linear predictors based on logistic regression, a maximum-likelihood technique suited for classification problems. Linear predictors were used due to the small sample sizes, especially in the cases of threonine and tyrosine sites, and possibility of noise.
The training algorithm used to construct DISPHOS is as follows. Ten percent of both positive and negative sets were selected at random to form a test set. The remaining 90% of the positive examples and a random selection of the same size from the remaining 90% of negative examples were included in a balanced training set, and a predictor was trained. The training was repeated for I = 30 random selections of negative examples, and the prediction on the test set was made by averaging raw outputs from all I models. To avoid the dependence of prediction results on the choice of the test set, the whole procedure was repeated until the confidence intervals of the performance measures dropped below a pre-specified threshold.
Performance evaluation
To evaluate the performance of the predictors, we measured sensitivity and specificity for a given set of parameters used for dimensionality reduction and model construction. This approach is commonly used in cases of imbalanced class sizes. Sensitivity (sn) is defined as the percentage of positive examples, i.e. phosphorylation sites, correctly predicted, while specificity (sp) is the percentage of negative examples correctly predicted (32). Assuming that the class sizes are equal, the accuracy of prediction (acc) can be expressed as the arithmetic mean of sensitivity and specificity. This sets the results of a prediction at random to 50% accuracy. Since all experiments were repeated n times, together with accuracy, we also report 95% confidence intervals calculated as ±2σ/√n, where σ is the standard deviation of the estimated parameter (sn or sp).
Estimation of class distributions in functional protein categories and genome-wide predictions
Estimates of class distributions in the unlabeled sets of residues are generally obtained by simply applying predictors to the unlabeled data. However, it is often the case in bioinformatics that class distributions in the labeled (or training) data sets are significantly different from those in the unlabeled data sets. For example, PDB is biased towards crystallizable proteins as compared to SWISS-PROT, and predictors trained on the PDB data may not achieve accurate estimates on SWISS-PROT. Thus, class distributions in the unlabeled data cannot be directly calculated as fractions of residues predicted to belong to each class (P and NP). In order to estimate probabilities of each class in various protein groups, we used the approach briefly presented below.
Let us denote the fraction of residues predicted to be phosphorylated in an unlabeled set by q(1) and fraction of residues predicted not to be phosphorylated by q(0), where q(1) = 1 – q(0). In matrix formulation we can express these fractions as q = [q(0) q(1)]T. The predicted class distribution q in an unlabeled data set can be expressed as:
![]()
where p = [p(0) p(1)]T is the unknown true class distribution in the unlabeled data set and
![]()
such that sn and sp represent estimates of correctly predicted phosphorylation and non-phosphorylation sites, respectively, and they are both calculated using only labeled residues. From equation 1, an estimate of the class probabilities in an unlabeled data set can now be obtained as:
![]()
Since, in general, E[P–1] ≠ E[P]–1, Vucetic and Obradovic proposed a bootstrapping-based algorithm to estimate p (33). In this approach the predictor is iteratively retrained, and a new matrix P is calculated based on the newly estimated class fractions until convergence of p is reached. Here, we provide the convergence of p by shifting the decision threshold instead of retraining predictors. In each cycle, p was calculated using 200 bootstrap replicates of the unlabeled data set, which was created after all proteins from a group were concatenated into a single long string.
A major limitation of this approach regarding the accuracy of estimates occurs in the presence of noise, especially in relatively extreme cases, that is, when p(1) is low or (rarely) high. In the case when p(1) ≈ 0, sn approaches 0, sp approaches 1 and the condition number of P becomes progressively high (in our examples, over 10 000 for the serine data set), indicating that the estimation system becomes increasingly sensitive to very small errors in estimations of sn and sp (34). We believe that experimentally determined phosphorylation sites are correctly labeled with high accuracy, and thus the estimates of sn were considered accurate. In contrast, the confidence in negative examples is significantly lower because of the possibility that certain residues were still not experimentally confirmed to be phosphorylated. Consequently, parameter sp was estimated using the negative data set and not the data set of true negatives; so it is likely underestimated proportionally to the noise level.
To obtain a more accurate estimate of sp, we used the following reasoning. The level of noise in all three data sets was estimated using Tomek links (35). Tomek links have effectively been used for noise reduction (36), but they cannot distinguish between borderline examples (examples near class boundaries) and true noise. However, in the case when confidence in the labels of the positive class is high, the amount of borderline examples can be detected using the positive set, and the approximation of noise in the negative class can be simply obtained as a difference between fractions of ‘noisy’ examples from both classes (under reasonable assumptions that the noise is uniform and that the similar fractions of borderline examples are in each class). Fractions of noisy examples were found by using balanced data sets with PCA-reduced dimensionality to 15 (after feature selection). This procedure was repeated multiple times with different random selections of negative examples, while all positive examples were used in each run. The experiments on various artificial data of similar size showed that one-sided noise could be accurately estimated by Tomek links and K-nearest neighbors if it is not too high (>30–40%). Once the noise was estimated, the accuracy on negative data was recalculated in each iteration of the bootstrap-based procedure. Specifically, the corrected specificity spnew is calculated from sp = (1 – sn)·p(1) + spnew·p(0), and then substituted in P for the old parameter sp. Parameter p(1) = 1 – p(0) represents the estimated noise level in the negative data set.
Adjusting predictor outputs according to estimated class priors
Predictors trained using a specific class distribution are known not to be optimal when applied to unlabeled data with different class priors (37). Here, we use estimated class priors given a group of proteins (specific kingdom, organism or functional category) to adjust the predictor outputs such that the total number of misclassified residues is minimized. Given the class distribution of the training set pT = [pT(0) pT(1)]T and the estimated distribution of class priors for a particular group of unlabeled residues p, the adjusted a posteriori probabilities of both classes are calculated as:
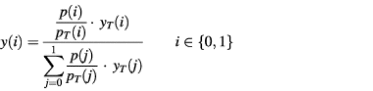
where yT(i) is the a posteriori probability outputted by a predictor trained using class distribution pT, which is in our case [0.5 0.5]T. The output of DISPHOS is y(1), i.e. the probability that the residue is phosphorylated.
RESULTS
In order to construct an improved predictor, we created a new database of P- and NP-sites by extracting annotated and unannotated S, T and Y sites from SWISS-PROT and combining them with the sites from PhosphoBase, the database used to train NetPhos (12). We will use the term ‘site’ to indicate the phosphorylation site itself and the 24 flanking residues, 12 residues upstream and 12 residues downstream from the actual phosphorylation site. Our choice of the 25 amino acid window was based on experimental data for kinases that are known to contact 7–12 residues adjacent to the site of modification (38).
First, we evaluated the accuracy of the NetPhos predictor using the P- and NP-sites derived from SWISS-PROT, excluding the ones that had >30% identity with the sites from PhosphoBase. The prediction accuracy of NetPhos (as defined in Materials and Methods) on this out-of-sample set of 107 serine, 36 threonine and 43 tyrosine positive sites combined with 500 negative sites for each residue, reached 69.1% for S (sensitivity 81.3 ± 3.7%, specificity 56.8 ± 2.2%), 71.9% for T (sensitivity 66.7 ± 7.8%, specificity 77.2 ± 1.9%) and 68.9% for Y (sensitivity 69.8 ± 7.0%, specificity 68.0 ± 2.1%). Note that the accuracy of NetPhos on PhosphoBase sites shown in the original paper (12) was calculated under the assumption of 100% specificity, which is clearly not the case.
Next, we estimated the accuracy of Scansite (14). Since the profiles for Scansite are not publicly available, we tested its accuracy using 100 positive and 100 negative examples for each S, T and Y residue randomly selected from our data sets. We used medium stringency for the evaluation as a reasonable balance between the sensitivity and specificity of Scansite. The prediction accuracy estimated for Scansite was 64.0% for S (sensitivity 38.0 ± 4.9%, specificity 90.0 ± 3.0%), 66.5% for T (sensitivity 43.0 ± 5.0%, specificity 90.0 ± 3.0%) and 68.5% for Y (sensitivity 49.0 ± 5.0%, specificity 88.0 ± 3.2%).
All subsequent experiments were performed on the combined data sets consisting of the sites from both SWISS-PROT and PhosphoBase (Table 1). A detailed description of the data set construction is given in Materials and Methods.
Table 1. Phosphorylation and non-phosphorylation sites used in the current study.
| |
P-sitesa | NP-sites | ||
|---|---|---|---|---|
| No. of initial sites | No. of final sitesb | No. of initial sites | No. of final sitesb | |
| S |
1135 |
613 |
29 425 |
10 798 |
| T |
265 |
140 |
22 243 |
9051 |
| Y | 301 | 136 | 13 035 | 5103 |
aCombined sites from SWISS-PROT and PhosphoBase.
bSequences with >30% identity were removed from the initial set.
Analysis of amino acid properties surrounding phosphorylation sites
We analyzed the properties of amino acids surrounding each site and determined which residues were enriched or depleted at specific positions. Figure 1 shows the residues for which the observed differences in relative frequencies between P- and NP-sites are statistically significant (P < 0.05). Statistical significance was calculated using Fisher’s permutation test (29). For the S, T and Y sites we observed 164, 56 and 84 compositional differences, respectively, between P- and NP-sites with P-values <0.05. A positive difference signifies that the P-sites are enriched in the corresponding residue, while a negative difference signifies depletion. Each residue was assigned a property: surface or buried (28) (Fig. 1A), charged or neutral (Fig. 1B), hydrophobic or hydrophilic (39) (Fig. 1C), high or low flexibility parameters (HFP and LFP, respectively) (27) (Fig. 1D). The percentages of residues enriched and depleted in each category are shown in Table 2.
Figure 1.

The amino acid residues significantly enriched and depleted around phosphorylation sites. Each residue is assigned a property: surface (red) or buried (black) according to Janin’s scale (28) (A), charged (black) or neutral (green) (B), hydrophobic (black) or hydrophilic (blue) according to Eisenberg’s scale (39) (C), high (pink) or low (blue) flexibility index according to flexibility scale (27) (D). Following the definition of Vihinen et al. (27) alanine and threonine were considered to have high flexibility if flanked by residues with HFP, and they were considered to have low flexibility if surrounded by residues with LFP.
Table 2. The percentages of amino acid residues that are significantly (P < 0.05) enriched and depleted around phosphorylation sites.
| Surface (%) | Buried (%) | Charged (%) | Neutral (%) | Hydrophobic (%) | Hydrophilic (%) | HFPa (%) | LFPb (%) | |
|---|---|---|---|---|---|---|---|---|
| S enriched |
95 |
5 |
57 |
43 |
13 |
87 |
100 |
0 |
| S depleted |
25 |
75 |
6 |
94 |
68 |
32 |
11 |
89 |
| T enriched |
79 |
21 |
54 |
46 |
25 |
75 |
93 |
7 |
| T depleted |
29 |
71 |
7 |
93 |
71 |
29 |
25 |
75 |
| Y enriched |
80 |
20 |
36 |
64 |
53 |
47 |
84 |
16 |
| Y depleted | 31 | 69 | 13 | 87 | 80 | 20 | 31 | 69 |
aResidues with high flexibility parameters by Vihinen et al. (27).
bResidues with low flexibility parameters.
All three types of phosphorylation sites are clearly enriched in surface exposed residues and depleted in buried residues (Fig. 1A and Table 2). Interestingly, the most prevalent amongst the surface residues surrounding serine are serine, lysine, arginine and glutamic acid, with glutamic acid found predominantly downstream from the phosphorylation site. The enrichment in serine agrees with the results of previous analysis (12) and confirms the observation that serines tend to cluster. Another distinctive characteristic of the serine sites is the depletion of cysteine, leucine, isoleucine and the aromatic residues. The decreased frequency of leucine and isoleucine is also observed around T and Y sites. The abundance of aspartic and glutamic acids in close proximity to the Y site, and proline at the positions distant to Y, is a signature of tyrosine phosphorylation sites.
The analysis of the charged versus neutral residues surrounding phosphorylation sites suggests that all three sites are strongly depleted in the neutral residues (Fig. 1B and Table 2), and S and T are slightly enriched in the charged ones. Among the residues that are under-represented around S, T and Y sites, neutral residues constitute 94, 93 and 87%, respectively (Table 2). Interestingly, the reciprocal distribution of charged versus neutral amino acids does not hold for Y: this site is both enriched and depleted in the neutral residues. The high frequency of uncharged proline around Y sites might explain this result.
The partition of the residues into hydrophobic and hydrophilic classes shows that all three sites are depleted in the hydrophobics (Fig. 1C and Table 2), with S (87%) and T (75%) being especially enriched in hydrophilics. The under-representation of hydrophobics is mainly due to the depletion of leucine, isoleucine and aromatic residues around all three sites.
We used a flexibility scale (27) that is based on B-factor values to evaluate the distribution of amino acid residues with HFP and LFP (Fig. 1D and Table 2). Following the definition of Vihinen et al. (27), alanine and threonine were considered to have HFP if flanked by residues with HFP, and they were considered to have LFP if surrounded by residues with LFP. The prevalence of HFP residues and the depletion of LFP residues around all sites are observed; 100, 93 and 84% of residues enriched around S, T and Y sites, respectively, belong to the HFP category (Table 2). The prevalence of HFP residues around potential phosphorylation sites may facilitate accessibility of the site to the kinase.
The distribution of the disorder-promoting (R, K, E, P and S) and order-promoting (C, W, Y, I and V) residues (17) around phosphorylation sites suggests that all sites have amino acid distributions characteristic for intrinsically disordered protein regions. The disorder-promoting amino acids are significantly enriched, and order-promoting amino acids are significantly depleted around all P-sites in our data sets. This observation indicates that the addition of the disorder feature may be important for phosphorylation site prediction.
In addition to the overall comparisons (Table 2), we examined amino acids and positions that show the largest signals (or lowest P-values) in Fisher’s permutation test. The top 10 enrichments and the top 10 depletions of such residues are shown in Table 3. For serine, the P-values for the six most enriched and the two most depleted amino acids exhibit the greatest difference between P- and NP-sites with P < 0.0001, while for T the P-values for all depleted amino acids show smaller significant differences with P > 0.001. As for Y, the five most enriched amino acids also show the greatest differences between P- and NP-sites with P < 0.0001. Thus, the presence of particular amino acids at certain positions appears to be more important than the opposite, although the absence of particular amino acids is still statistically significant in all cases.
Table 3. The top 10 amino acids enriched and depleted around known phosphorylation sites as determined by Fisher’s permutation test (29).
| Enriched | Depleted | ||||
|---|---|---|---|---|---|
| Position | Residue | P-value | Position | Residue | P-value |
| Serine sites | |||||
| –3 |
R |
<0.0001 |
–11 |
L |
<0.0001 |
| –2 |
R |
<0.0001 |
–5 |
C |
<0.0001 |
| +1 |
P |
<0.0001 |
–3 |
C |
0.0002 |
| +2 |
E |
<0.0001 |
+2 |
L |
0.0002 |
| +3 |
E |
<0.0001 |
+2 |
N |
0.0002 |
| +4 |
S |
<0.0001 |
+3 |
L |
0.0002 |
| –6 |
K |
0.0001 |
–6 |
L |
0.0003 |
| –5 |
R |
0.0001 |
–3 |
F |
0.0003 |
| –4 |
S |
0.0001 |
–2 |
N |
0.0003 |
| +2 |
S |
0.0001 |
–4 |
V |
0.0004 |
| Threonine sites | |||||
| –3 |
R |
<0.0001 |
–4 |
L |
0.0019 |
| –2 |
R |
<0.0001 |
+7 |
L |
0.0032 |
| +1 |
P |
<0.0001 |
+1 |
N |
0.0063 |
| –12 |
K |
0.0001 |
–2 |
N |
0.0064 |
| +12 |
E |
0.0006 |
+1 |
F |
0.0106 |
| +2 |
R |
0.0007 |
–3 |
L |
0.0141 |
| –9 |
S |
0.0015 |
+4 |
I |
0.0152 |
| +2 |
D |
0.0028 |
+2 |
L |
0.0162 |
| +6 |
K |
0.0054 |
+1 |
A |
0.0165 |
| –6 |
G |
0.0060 |
–4 |
A |
0.0178 |
| Tyrosine sites | |||||
| –4 |
E |
<0.0001 |
–4 |
L |
0.0001 |
| –1 |
E |
<0.0001 |
+2 |
L |
0.0005 |
| +3 |
M |
<0.0001 |
–4 |
I |
0.0009 |
| +5 |
P |
<0.0001 |
+8 |
I |
0.0009 |
| +9 |
P |
<0.0001 |
+6 |
F |
0.0019 |
| –3 |
E |
0.0001 |
–3 |
L |
0.0021 |
| +2 |
N |
0.0001 |
–2 |
L |
0.0023 |
| –2 |
P |
0.0003 |
–10 |
N |
0.0045 |
| –7 |
S |
0.0004 |
+11 |
I |
0.0054 |
| –3 | N | 0.0007 | –10 | V | 0.0065 |
Interestingly, several positions that are distant from the potential phosphorylation site also have significant differences between P and NP-sites, for example, position –11 for S, position –12 for T and position +9 for Y (Table 2). This result suggests that although the recognition patterns for the currently known kinases usually involve only positions in close proximity to the potential phosphorylation site (usually within –5 to +5), the more distant positions may also be important. Thus, the kinase recognition patterns may be extended in the future as new kinases are discovered.
Sequence complexity of P-sites versus disordered protein regions
One feature that distinguishes disordered protein regions from ordered is sequence complexity K2 measured by Shannon’s entropy and applied to amino acid sequences by Wootton (23). We discovered previously that the SWISS-PROT database is characterized by higher amounts of both low-complexity segments and predicted disordered regions in comparison to PDB (17,40). Here we determined the sequence complexity K2 for the P- and NP-sites, and compared them to the K2 of ordered and disordered protein regions (Fig. 2). The K2 distribution for the phosphorylation sites is very similar to the K2 distribution for the disordered segments, whereas the K2 for the NP-sites is very close to the K2 for ordered globular segments. Moreover, the differences in cumulative percentages for P- and NP-sites (Fig. 2, inset) show that from 1.4 to 5 times more P-sites have K2 values between 2.9 and 3.8, indicating the enrichment of P-sites in low complexity segments. The correlation of low K2 with phosphorylation is an interesting and important result of this analysis.
Figure 2.

Sequence complexity distributions. Sequence complexity K2 was calculated for the sliding window of 45 residues. The data set ‘all disorder’ consisted of disordered regions characterized by X-ray diffraction (extracted from PDB-Select-25), NMR and CD (extracted from the literature). Globular-3D data set consisted of the ordered protein regions extracted from PDB and fibrous sequences such as coiled coils, collagen and silk fibroins were removed from this data set. A lower sequence complexity is observed for P-sites as compared with NP-sites (inset).
Amino acid compositions of P- and NP-sites and disordered protein regions
Disordered and ordered protein regions differ in amino acid composition, with W, C, F, I, Y, V, L and N being significantly depleted and A, R, G, Q, S, P, E and K being significantly enriched in regions of disorder (17). We compared amino acid compositions of P- and NP-sites and disordered protein regions (Fig. 3). The results are presented as the difference between the composition of each data set and the composition of ordered globular protein regions: (Cdata set – CGlobular-3D)/CGlobular-3D.
Figure 3.

Comparison of amino acid compositions between disordered protein regions, P- and NP-sites. The composition for each data set is shown in comparison with the ordered Globular-3D data set. The results are presented as the difference between the composition of each data set and the composition of ordered globular protein regions: (Cdata set – CGlobular-3D) / CGlobular-3D. A negative bar indicates that the data set is depleted in the corresponding amino acid, and the positive bar indicates enrichment. Amino acid residues on the X-axes are arranged according to the flexibility scale (27). The middle residues for P- and NP-sites representing actual phosphorylation sites were excluded from the calculations. The error bars correspond to 1 SD.
Overall, the amino acid compositions of P-sites and disordered regions are very similar. In particular, they are both significantly depleted in rigid, buried, neutral amino acids (W, C, F, I, Y, V and L), and are significantly enriched in flexible, surface-exposed serine, proline, glutamic acid and lysine (Fig. 3). Interestingly, we observed >3-fold enrichment in serine for phosphorylation sites as compared with both non-phosphorylation sites and disordered protein regions. The extreme positive S peak for P-sites yet again suggests the tendency of serines to cluster and the possibility of sequential phosphorylation at multiple sites.
Predictor construction and accuracy estimation
We first used only position-specific amino acid frequencies (Materials and Methods) to construct a predictor for each site and to estimate its accuracy. The purpose of this experiment was to compare the accuracy achieved by DISPHOS with the accuracy of NetPhos (12) when the same types of features over the same window size were used. The major differences between these two predictors originate from the data set construction, model choice, feature selection/extraction, and training and testing processes, as discussed below. We also used the initial predictor developed on the balanced training set to iteratively estimate the class priors in various groups of proteins. These estimates, in turn, enabled us to adjust the outputs of the initial predictor thus minimizing the total number of misclassified residues in each protein group.
We expanded the positive and especially negative data sets (Table 1) by combining phosphorylation sites derived from two databases, PhosphoBase and SWISS-PROT. Three new data sets (samples) corresponding to S, T and Y sites, respectively, were then constructed (Materials and Methods). After the feature construction process, all data sets were high-dimensional and sparse (due to data representation), noisy (due to possible mislabeling of positive and especially negative sites), and highly imbalanced (with a large number of negative versus small number of positive sites). The ratios of positive versus negative sites were 1:18, 1:65 and 1:38 for S, T and Y, respectively. Additionally, only small sets of confirmed positive sites were available for T and Y after removing similar sequences (Table 1).
Before model training we performed dimensionality reduction using a feature selection process (Materials and Methods) and principal component analysis (PCA). Then, following the approach of Radivojac et al. (41), we applied a bagging-like combination of logistic regression models (Fig. 4) to construct a predictor. Therefore, we carefully approached the problem of class imbalance that usually requires special treatment since predictor performance may be highly degraded (42). Neural-network based predictors did not surpass the accuracy of the bagged linear models.
Figure 4.

The process of model building and testing.
When only significant position-specific features were used, the best accuracy for S (74.9 ± 0.3%) was achieved using a dimensionality of 40 and a P-value of 0.1 in the feature selection process (Table 4). This accuracy exceeded that of NetPhos on the out-of-sample data set (69.2%) indicating significantly better performance of DISPHOS when the larger data set and the refined set of position-specific features were used. Similar results were observed for predictors using 30 dimensions and P-values of 0.1 for T and Y: 78.9 ± 0.6% versus 71.9% for T, and 81.3 ± 0.5% versus 60.6% for Y. Without ensembles of classifiers, i.e. for I = 1, the accuracy drops by two to three percentage points in each case.
Table 4. Comparison of the accuracies (% ± 95% confidence intervals) between the NetPhos and DISPHOS.
| Site |
NetPhos |
DISPHOS position-specific |
Final DISPHOS | |||
|---|---|---|---|---|---|---|
| Round 1a | Round 2 | Round 3 | Round 4 | |||
| S |
69.2 |
74.9 ± 0.3 |
75.6 ± 0.3 |
75.8 ± 0.4 |
76.0 ± 0.3 |
76.0 ± 0.3 |
| |
|
|
VL2 disorder |
VLXT disorder |
Relative frequency G |
Relative frequency I |
| T |
71.9 |
78.9 ± 0.6 |
80.5 ± 0.3 |
81.0 ± 0.3 |
81.1 ± 0.3 |
81.3 ± 0.3 |
| |
|
|
VL2 disorder |
Relative frequency S |
NNSSP helix |
Relative frequency F |
| Y |
60.6 |
81.3 ± 0.5 |
82.0 ± 0.3 |
83.3 ± 0.3 |
83.3 ± 0.3 |
83.0 ± 0.3 |
| Relative frequency Q | Relative frequency T | Relative frequency K | Surface exposure | |||
aThe achieved accuracy of predictor together with the best feature for each selection round is shown. The accuracies of S, T and Y phosphorylation sites predictors increased by 6.8, 9.4 and 22.7%, respectively, in comparison with NetPhos.
The addition of non-binary features resulted in a further increase in the accuracy of DISPHOS. Using the forward-selection algorithm (31), we iteratively added new real-valued features to the sample until no increase in predictor accuracy was observed. Four rounds of feature selection were experimentally determined to be sufficient for each model. In every round only the feature that increased accuracy the most was chosen, and only the remaining features were used as candidates in the next round. The best features with their accuracies are summarized in Table 4.
As expected, the disorder feature increased the prediction accuracy for serine and threonine (Table 4). In a few instances, the relative frequency of an amino acid was also selected as the best feature (Table 4). Relative frequency features reflect the presence or absence of a particular amino acid anywhere in the window of 25, and not only at a specific position. Interestingly, none of the non-binary features from Figure 1 were selected as the best for increasing predictor accuracy, most likely because they were already implicitly included in the predictor through disorder and position-specific features.
We report sensitivity and specificity of DISPHOS to be in the range from 76% for serine to 83% for tyrosine. However, the positive and negative examples were not labeled with the same confidence. Some of the negative examples were likely mislabeled due to the lack of experimental verification. This noise may have lead to underestimated predictor specificities. Using the approach described in Materials and Methods, we estimated the amount of noise in the negative data sets to be ∼14% for serine, ∼5% for threonine and ∼8% for tyrosine. Subsequently, the correction formula provided us with more accurate specificities. We believe that the true specificities for the S, T, and Y predictors are ∼85, ∼84 and ∼88%, respectively, pushing the overall prediction accuracy to 80% for serine, 82% for threonine and 86% for tyrosine. Similar corrections applied to NetPhos and Scansite would increase the accuracy of these methods as well. Hence, an immediate step towards improving the results of this study should be acquiring (or selecting) better quality data that will enable us to obtain (i) improved phosphorylation predictors and (ii) more accurate estimates of sn and sp, both leading to higher quality estimates of phosphorylation in nature.
Prediction of phosphorylation sites in protein functional categories from SWISS-PROT
We previously predicted disorder in various protein functional categories extracted from the SWISS-PROT database (3). Here, we predicted phosphorylation sites in the same protein data sets using DISPHOS and a new estimation method (Materials and Methods). The estimated percentages of predicted S, T and Y phosphorylation sites for each data set and for disordered and ordered protein regions are shown in Figure 5.
Figure 5.

Estimated percentages of S, T and Y phosphorylation sites in 12 functional protein categories from SWISS-PROT and in disordered and ordered data sets. The y-axis indicates the estimated percentage of phosphorylated S, T or Y residues in each data set. The data set ‘all disorder’ consisted of disordered protein regions characterized by X-ray diffraction (extracted from PDB-Select-25), NMR and CD (extracted from the literature). The data set ‘PDB order’ consisted of the ordered protein regions with known 3D structure extracted from PDB. The error bars correspond to 1 SD.
The most interesting result is that we predict over 10 times more phosphorylation sites in completely disordered proteins than in ordered proteins from PDB, which strongly supports the idea of a tight interconnection between protein phosphorylation and disorder. Moreover, the number of predicted phosphorylation sites in Figure 5 correlates highly with the amount of predicted disorder (3) in all protein categories.
Previously we showed that regulatory, cancer-associated and cytoskeletal proteins have about twice as much predicted disorder than proteins involved in degradation, biosynthesis and metabolism (3). Here, we observed very similar but even more profound differences in the percentages of predicted serine phosphorylation sites for the same protein data sets: 57.4 ± 0.7, 42.2 ± 1.1 and 40.6 ± 0.9% for regulatory, cancer-associated and cytoskeletal proteins versus 10.8 ± 1.7, 8.7 ± 0.6 and 5.9 ± 1.1% for degradation, biosynthesis and metabolism data sets, respectively (Fig. 5). These differences support the hypothesis that proteins involved in regulatory and signaling cellular functions undergo more frequent phosphorylation/dephosphorylation than proteins involved predominantly in catalysis.
Prediction of phosphorylation sites in various kingdoms and proteomes
It is well known that regulation in bacteria occurs via histidine or aspartic acid phosphorylation, involved in two-component signaling pathways (43). However, the putative homologs of eukaryotic protein serine/threonine kinases and phosphatases, found in bacteria and archaea, suggest the possibility of alternative eukaryotic-like signal transduction pathways in these species (44). Therefore, we were interested in comparing the DISPHOS predictions on viral, archaeal and bacterial proteins with predictions on proteins from seven complete or almost complete eukaryotic genomes (Table 5).
Table 5. Prediction of phosphorylation sites on proteins from SWISS-PROT and on seven entire proteomes.
| Databases |
Estimated %a of phosphorylation sites | ||
|---|---|---|---|
| S | T | Y | |
| SWISS-PROT | |||
| Eukaryotes |
17.5 ± 2.0 |
4.7 ± 0.9 |
6.8 ± 1.0 |
| Bacteria |
2.0 ± 0.8 |
1.5 ± 0.8 |
4.5 ± 0.7 |
| Archaea |
2.5 ± 0.8 |
1.6 ± 0.7 |
5.0 ± 0.7 |
| Viruses |
7.0 ± 0.8 |
3.0 ± 0.8 |
5.1 ± 1.0 |
| Proteomes | |||
| Arabidopsis thaliana |
18.9 ± 1.0 |
5.4 ± 1.0 |
8.2 ± 1.0 |
| Caenorhabditis elegans |
18.6 ± 1.5 |
5.5 ± 1.0 |
6.3 ± 1.0 |
| Drosophila melanogaster |
32.0 ± 2.2 |
8.9 ± 1.3 |
7.9 ± 0.8 |
| Homo sapiens |
26.9 ± 2.0 |
9.5 ± 1.7 |
8.5 ± 1.2 |
| Mus musculus |
21.4 ± 1.0 |
7.0 ± 0.9 |
7.5 ± 0.9 |
| Rattus norvegicus |
20.9 ± 0.9 |
7.0 ± 0.9 |
7.5 ± 1.0 |
| Saccharomyces cerevisiae | 19.2 ± 1.2 | 3.0 ± 0.8 | 6.7 ± 0.9 |
a±1 SE (correct under the assumption that the noise level in the negative set is accurately estimated).
The percentages of predicted S, T and Y phosphorylation sites in each data set are shown in Table 5. We predict that on average from 18 (Caenorhabditis elegans) to 32% (Drosophila melanogaster) of all serine residues in eukaryotic proteomes may become phosphorylated, whereas this number ranges from only 2% in bacteria to 7% in viruses. Likewise, the percentage of predicted threonine phosphorylation sites in most eukaryotic proteomes (5–9%) exceeds those in bacteria (2%) and archaea (2%). We estimate that viruses and yeast have equal percentages (3%) of threonine phosphorylation sites. Interestingly, the estimated percentage of threonine phosphorylation sites in higher eukaryotes exceeds those in yeast, worm and plant proteomes. The percentage of tyrosine phosphorylation sites in all data sets is very similar and ranges from 4 to 5% in prokaryotes, viruses and archaea, and from 6 to 8% in eukaryotes (Table 5).
DISCUSSION
Predictor optimization and application
Although we developed a relatively high-accuracy predictor, there are several limitations that may still greatly impact the proteome-wide estimates. The most obvious source of predictor inaccuracy is the unreliably labeled, negative training set. Numerous un-annotated or not-yet-discovered phosphorylation sites are likely to be present in the NP training set, even though we removed all sites >30% identical to P-sites. In addition, estimates of the noise level are influenced by the assumptions made in our approach (Materials and Methods). Another evident source of inaccuracy is the small positive training set.
As mass spectrometry-based technologies are becoming available for high-throughput determination of phosphorylation sites (45), it will be possible to expand the positive data set and also to clean the negative data set, subsequently designing a predictor with even higher accuracy. Meanwhile, the current predictor can be utilized to gain insight into large-scale phosphorylation patterns of entire proteomes. In addition, it can be used to predict new phosphorylation sites in proteins involved in various cell-signaling pathways that might improve our understanding of their biologically relevant functions and regulation.
Implications for substrate recognition
Kinase substrates typically bind to the enzyme with weak affinity, and yet phosphorylation by each kinase is specific (46). High specificity coupled with low affinity is ideal for signaling. One way that such a combination of characteristics can be achieved is via coupled binding and folding (47). The low net affinity arises because the positive free energy associated with the disorder-to-order transition reduces the magnitude of the negative free energy arising from the interactions within the contact surface. The usefulness of protein disorder for such high specificity/low affinity signaling interactions was pointed out almost 25 years ago (47).
While our approach does not yield sequence patterns for the substrates of specific kinases, there are clear relationships between our results and those in the PROSITE database (48) for several kinases. The motif [RK]-(2)x-[ST], where S or T is the phosphorylation site and x can be any residue, is preferentially phosphorylated by cAMP- and cGMP-dependent protein kinases and exactly corresponds to the enrichment pattern reported here for both S and T (Fig. 1). Moreover, the known substrates and inhibitors of cAMP-dependent protein kinase as well as the optimal sequence derived from the peptide library for this enzyme both have arginine at positions –2 and –3, and this signature is considered to be very important for phosphorylation (38). The phosphorylation sites [ST]-x-[RK] and [ST]-x(2)-[DE] for protein kinase C and casein kinase II, respectively, are also identical to the sites observed here (Fig. 1). Another example that supports the validity of our analysis is a very strong preference for proline at position +1 for the serine phosphorylation site by the cyclin-dependent serine/threonine protein kinases (49). Using a peptide library of substrates, Songyang et al. (38) found one more position +3 where arginine or lysine is highly preferred by cyclin-dependent kinases. This finding agrees with the known phosphorylation sites for these enzymes and with the significant position-specific residues for S and T discovered here using a bioinformatics approach (Fig. 1).
There is one site for tyrosine protein kinases ([RK]-x(2)-[DE]-x(3)-Y or [RK]-x(3)-[DE]-x(2)-Y) in PROSITE that only partially corresponds to the pattern discovered by our analysis. We did not observe either R or K at position –7 upstream from Y, but we found both D and E at positions –3 and –4. The abundance of the acidic residues upstream from the Y site is also a signature for phosphorylation by Src kinase (50). Interestingly, the substrate specificities of SH2 domains from other tyrosine protein kinases (Fyn, Lck, Fgr and Abl) corresponding to the consensus sequence Y(Ph)EE(I/V), where Y(Ph) means phosphotyrosine (51), exactly match the one found here (Fig. 1). Although there are a number of exceptions to all listed consensus patterns, our results are in good agreement with the experimentally determined phosphorylation motifs.
The pattern-based methods such as those using PROSITE patterns emphasize amino acids that occur at particular positions and overlook amino acids that are excluded from the same or other positions in the pattern (this, however, is not true for PROSITE profiles or Pfam hidden Markov models). As shown in Table 3, the absence of a particular amino acid from a motif can be as important as the presence of an amino acid within a motif. If viewed from the protein folding perspective, avoiding steric clashes and the placement of hydrophobic amino acids on the protein surface can be as essential as forming well packed interfaces. Thus, modifying pattern-matching methods to explicitly include the absence of residues from particular positions in the pattern should lead to improved performance of these methods.
Protein phosphorylation from structural biology perspective
Several observations support our results indicating that phosphorylation commonly occurs within intrinsically disordered protein regions.
Relatively few regions of disorder have been structurally characterized, yet a significant fraction of them contain phosphorylation sites (4). Overall, disordered regions have a much higher frequency of known phosphorylation sites than ordered regions, suggesting a strong preference for locating phosphorylation sites in the regions of intrinsic disorder. Disordered regions also have significantly larger fractions of predicted phosphorylation sites than do ordered regions (Fig. 5). Our previous observations show that ∼12 or ∼7% of ordered serines have high B-factors, i.e. 2 or 3 SD above protein means, respectively (52). Yet, we predict that only 1% of serines in the ordered regions could be phosphorylated (Fig. 5). Similarly, albeit to a lesser extent, 9% (5%) of ordered threonines have high B-factors, while only 1% of ordered threonines are predicted to be phosphorylated. These data raise the possibility that protein phosphorylation of serines and threonines predominantly occur within intrinsically disordered regions and not merely on surface residues. The analysis of tyrosine sites shows that 4% (2%) of residues have high B-factors, while we predict 6% of ordered tyrosines to be phosphorylated. Therefore, tyrosines appear to be phosphorylatable both in intrinsically disordered and surface exposed ordered states.
Three biologically important protein kinase inhibitors, PKIα (53), p27 (54) and p21 (55), are polypeptides that bind to their respective kinases via very well characterized disorder-to-order transitions. Similar disorder-to-order transitions, although less extensively characterized, exist for a number of actual kinase peptide substrates. The part of the PKIα inhibitor that covers the active site (56,57) and nine additional bound peptide substrates found in PDB all have extended, irregular conformations that are consistent with disordered structure (see the following structures in PDB: 1ATP, 1IR3, 1O6I, 1QMZ, 1PHK, 1O6K, 1GY3, 1CDK and 1JBP).
The bound substrate or inhibitor peptides have essentially no intra-chain backbone hydrogen bonding while having extensive hydrogen bonding between their backbones and the backbones or side chains of their kinase partners (56–60). The formation of these hydrogen bonds would not be possible if the sites of phosphorylation were located within ordered regions. That is, this hydrogen bond formation requires that the peptide substrates have available backbone hydrogen bonding potential just prior to association with kinase, and such availability is simply incommensurate with ordered protein structure (61). Thus, these data strongly suggest that the peptide substrates of kinases must be disordered. While the extensive hydrogen bonding between peptide substrates and their kinase partners has been noted by many researchers, the implications for the order–disorder status of phosphorylation sites was previously overlooked.
As shown above, the available structural data are consistent with a strong preference for phosphorylation to occur in regions of intrinsic disorder. There are, however, very few counter examples, in which the 3D structures of phosphorylation sites have been observed in the absence of phosphorylation (9). One such example is sigma-factor SpoIIAA from Bacillus subtilis, whose structure was solved in both the phosphorylated (1H4X) and unphosphorylated (1H4Z) forms. The main differences between these two forms are found within segment 83–98, with residues 93–95 being disordered in the 1H4Z structure. Although the actual phosphorylation site (Ser57) is ordered in both structures, the exceptionally slow rate of SpoIIAA phosphorylation by SpoIIAB does not exclude the possibility of local unfolding of the region surrounding Ser57 just prior to phosphate attachment. Another example is isocitrate dehydrogenase (IDH) from Escherichia coli. Both unphosphorylated and Ser113-phosphorylated forms of IDH are ordered in crystal structures, and no large-scale conformational change is observed in the unliganded enzyme on in vitro phosphorylation by IDH kinase/phosphatase. Interestingly, IDH kinase/phosphatase is an unusual protein that does not exhibit the extensive sequence homology to other protein kinases. Therefore, the mechanism of phosphorylation by IDH kinase/phosphatase and other eukaryotic kinases may differ significantly in terms of requirement for a region to be unfolded prior to phosphorylation.
Parallel studies on the structural requirements for protease digestion sites can provide further insight on the phosphorylation process. While regions of intrinsic disorder are clearly and strongly favored over regions of order as sites of protease digestion (62,63), a few trypsin-sensitive sites were observed to be located within protein structured domains. These regions, however, inevitably require local unfolding prior to protease digestion in order to become accessible to the protease. It was shown that the folded forms could not fit into the enzyme active site without disordering at least 12 residues surrounding the sites of trypsin digestion (64).
In view of the structural aspects of protease digestion, we suggest several possible scenarios for the phosphorylation counter examples occurring in ordered protein regions. (i) Intrinsic disorder may not be universally required for all kinase substrates. From the data presented herein, phosphorylation of a site within an ordered protein region would require a kinase–substrate interaction that is markedly different from those characterized to date. (ii) The region to be phosphorylated undergoes an order to disorder transition just prior to association with the kinase, thereby exposing its backbone hydrogen bonding potential. (iii) The observed structures are crystallization artifacts, with intrinsic disorder prevailing in the regions surrounding the sites of phosphorylation. Further studies on the counter-example proteins are needed to decide among the three alternatives given above. Such studies could provide important new understanding of protein phosphorylation.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank Chad Haynes from the Rockefeller University for expert programming support and for making the predictor available via the Internet. We also thank Allen Nicholson from Temple University and the anonymous reviewers for their helpful comments and suggestions. This study was supported by NIH grant 1R01 LM07688 awarded to A.K.D and Z.O., and NSF grants NSF-CSE-IIS-9711532 and NSF-IIS-0196237 to Z.O. and A.K.D.
REFERENCES
- 1.Wright P.E. and Dyson,H.J. (1999) Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J. Mol. Biol., 293, 321–331. [DOI] [PubMed] [Google Scholar]
- 2.Dyson H.J. and Wright,P.E. (2002) Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol., 12, 54–60. [DOI] [PubMed] [Google Scholar]
- 3.Iakoucheva L.M., Brown,C.J., Lawson,J.D., Obradovic,Z. and Dunker,A.K. (2002) Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol., 323, 573–584. [DOI] [PubMed] [Google Scholar]
- 4.Dunker A.K., Brown,C.J., Lawson,J.D., Iakoucheva,L.M. and Obradovic,Z. (2002) Intrinsic disorder and protein function. Biochemistry, 41, 6573–6582. [DOI] [PubMed] [Google Scholar]
- 5.Marks F. (1996) Protein Phosphorylation. VCH Weinheim, New York, Basel, Cambridge, Tokyo. [Google Scholar]
- 6.Zor T., Mayr,B.M., Dyson,H.J., Montminy,M.R. and Wright,P.E. (2002) Roles of phosphorylation and helix propensity in the binding of the KIX domain of CREB-binding protein by constitutive (c-Myb) and inducible (CREB) activators. J. Biol. Chem., 277, 42241–42248. [DOI] [PubMed] [Google Scholar]
- 7.Hay T.J. and Meek,D.W. (2000) Multiple sites of in vivo phosphorylation in the MDM2 oncoprotein cluster within two important functional domains. FEBS Lett., 478, 183–186. [DOI] [PubMed] [Google Scholar]
- 8.Rechsteiner M. and Rogers,S.W. (1996) PEST sequences and regulation by proteolysis. Trends Biochem. Sci., 21, 267–271. [PubMed] [Google Scholar]
- 9.Johnson L.N. and Lewis,R.J. (2001) Structural basis for control by phosphorylation. Chem. Rev., 101, 2209–2242. [DOI] [PubMed] [Google Scholar]
- 10.Vetter S.W. and Leclerc,E. (2001) Phosphorylation of serine residues affects the conformation of the calmodulin binding domain of human protein 4.1. Eur. J. Biochem., 268, 4292–4299. [DOI] [PubMed] [Google Scholar]
- 11.Ojala P.M., Yamamoto,K., Castanos-Velez,E., Biberfeld,P., Korsmeyer,S.J. and Makela,T.P. (2000) The apoptotic v-cyclin-CDK6 complex phosphorylates and inactivates Bcl-2. Nature Cell Biol., 2, 819–825. [DOI] [PubMed] [Google Scholar]
- 12.Blom N., Gammeltoft,S. and Brunak,S. (1999) Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol., 294, 1351–1362. [DOI] [PubMed] [Google Scholar]
- 13.Yaffe M.B., Leparc,G.G., Lai,J., Obata,T., Volinia,S. and Cantley,L.C. (2001) A motif-based profile scanning approach for genome-wide prediction of signaling pathways. Nat. Biotechnol., 19, 348–353. [DOI] [PubMed] [Google Scholar]
- 14.Obenauer J.C., Cantley,L.C. and Yaffe,M.B. (2003) Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res., 31, 3635–3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manning G., Whyte,D.B., Martinez,R., Hunter,T. and Sudarsanam,S. (2002) The protein kinase complement of the human genome. Science, 298, 1912–1934. [DOI] [PubMed] [Google Scholar]
- 16.Rost B. (1999) Twilight zone of protein sequence alignments. Protein Eng., 12, 85–94. [DOI] [PubMed] [Google Scholar]
- 17.Romero P., Obradovic,Z., Li,X., Garner,E.C., Brown,C.J. and Dunker,A.K. (2001) Sequence complexity of disordered protein. Proteins, 42, 38–48. [DOI] [PubMed] [Google Scholar]
- 18.Qian N. and Sejnowski,T.J. (1988) Predicting the secondary structure of globular proteins using neural network models. J. Mol. Biol., 202, 865–884. [DOI] [PubMed] [Google Scholar]
- 19.Vucetic S., Brown,C.J., Dunker,A.K. and Obradovic,Z. (2003) Flavors of protein disorder. Proteins, 52, 573–584. [DOI] [PubMed] [Google Scholar]
- 20.Rost B., Sander,C. and Schneider,R. (1994) PHD—an automatic mail server for protein secondary structure prediction. Comput. Appl. Biosci., 10, 53–60. [DOI] [PubMed] [Google Scholar]
- 21.Salamov A.A. and Solovyev,V.V. (1997) Protein secondary structure prediction using local alignments. J. Mol. Biol., 268, 31–36. [DOI] [PubMed] [Google Scholar]
- 22.Salamov A.A. and Solovyev,V.V. (1995) Prediction of protein secondary structure by combining nearest-neighbor algorithms and multiple sequence alignments. J. Mol. Biol., 247, 11–15. [DOI] [PubMed] [Google Scholar]
- 23.Wootton J.C. (1993) Statistic of local complexity in amino acid sequences and sequence databases. Comput. Chem., 17, 149–163. [Google Scholar]
- 24.Xie Q., Arnold,G.E., Romero,P., Obradovic,Z., Garner,E. and Dunker,A.K. (1998) The sequence attribute method for determining relationships between sequence and protein disorder. Genome Inform. Ser. Workshop Genome Inform., 9, 193–200. [PubMed] [Google Scholar]
- 25.Eisenberg D., Weiss,R.M. and Terwilliger,T.C. (1984) The hydrophobic moment detects periodicity in protein hydrophobicity. Proc. Natl Acad. Sci. USA, 81, 140–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sweet R.M. and Eisenberg,D. (1983) Correlation of sequence hydrophobicities measures similarity in three-dimensional protein structure. J. Mol. Biol., 171, 479–488. [DOI] [PubMed] [Google Scholar]
- 27.Vihinen M., Torkkila,E. and Riikonen,P. (1994) Accuracy of protein flexibility predictions. Proteins, 19, 141–149. [DOI] [PubMed] [Google Scholar]
- 28.Janin J. (1979) Surface and inside volumes in globular proteins. Nature, 277, 491–492. [DOI] [PubMed] [Google Scholar]
- 29.Efron B. and Tibshirani,R.J. (1993) An Introduction to the Bootstrap. Chapman and Hall, New York. [Google Scholar]
- 30.Haykin S. (1999) Neural Networks: A Comprehensive Foundation, 2nd edn. Prentice Hall, Upper Saddle River, NJ. [Google Scholar]
- 31.Bishop C.M. (1995) Neural Networks for Pattern Recognition. Oxford University Press, Oxford, UK. [Google Scholar]
- 32.Hastie T., Tibshirani,R. and Friedman,J.H. (2001) The Elements of Statistical Learning: Data Mining, Inference and Prediction. Springer Verlag, New York. [Google Scholar]
- 33.Vucetic S. and Obradovic,Z. (2001) Classification on data with biased class distribution. Proceedings of the 12th European Conference of Machine Learning, Freiburg, Germany, September 5–7, 2001. Lecture Notes in Computer Science Volume 2167. Springer, Verlag, Heidelberg, pp. 527–538.
- 34.Belsley D.A., Kuh,E. and Welsch,R.E. (1980) Regression Diagnostics. John Wiley and Sons, New York. [Google Scholar]
- 35.Tomek I. (1976) Two modifications of CNN. IEEE Trans. Syst. Man Cybern., 6, 769–772. [Google Scholar]
- 36.Kubat M. and Matwin,S. (1997) Addressing the curse of imbalanced training sets: one-sided selection. In Fisher,D.H.,Jr (ed.), Proceedings of the 14th International Conference on Machine Learning (ICML-97), Nashville, Tennessee, July 8–12, 1997. Morgan Kaufmann, San Francisco, CA, pp. 179–186. [Google Scholar]
- 37.Saerens M., Latinne,P. and Decaestecker,C. (2002) Adjusting the outputs of a classifier to new a priori probabilities: a simple procedure. Neural Comput., 14, 21–41. [DOI] [PubMed] [Google Scholar]
- 38.Songyang Z., Blechner,S., Hoagland,N., Hoekstra,M.F., Piwnica-Worms,H. and Cantley,L.C. (1994) Use of an oriented peptide library to determine the optimal substrates of protein kinases. Curr. Biol., 4, 973–982. [DOI] [PubMed] [Google Scholar]
- 39.Eisenberg D. (1984) Three-dimensional structure of membrane and surface proteins. Annu. Rev. Biochem., 53, 595–623. [DOI] [PubMed] [Google Scholar]
- 40.Romero P., Obradovic,Z. and Dunker,A.K. (1999) Folding minimal sequences: the lower bound for sequence complexity of globular proteins. FEBS Lett., 462, 363–367. [DOI] [PubMed] [Google Scholar]
- 41.Radivojac P., Obradovic,Z., Brown,C.J. and Dunker,A.K. (2003) Prediction of boundaries between intrinsically ordered and disordered protein regions. Pac. Symp. Biocomput., 8, 216–227. [PubMed] [Google Scholar]
- 42.Kubat M., Holte,R.C. and Matwin,S. (1998) Detection of oil spills in satellite radar images of sea surface. Mach. Learn., 30, 195–215. [Google Scholar]
- 43.Alex L.A. and Simon,M.I. (1994) Protein histidine kinases and signal transduction in prokaryotes and eukaryotes. Trends Genet., 10, 133–138. [DOI] [PubMed] [Google Scholar]
- 44.Leonard C.J., Aravind,L. and Koonin,E.V. (1998) Novel families of putative protein kinases in bacteria and archaea: evolution of the ‘eukaryotic’ protein kinase superfamily. Genome Res., 8, 1038–1047. [DOI] [PubMed] [Google Scholar]
- 45.Zhou H., Watts,J.D. and Aebersold,R. (2001) A systematic approach to the analysis of protein phosphorylation. Nat. Biotechnol., 19, 375–378. [DOI] [PubMed] [Google Scholar]
- 46.Gould C. and Wong,C.F. (2002) Designing specific protein kinase inhibitors: insights from computer simulations and comparative sequence/structure analysis. Pharmacol. Ther., 93, 169–178. [DOI] [PubMed] [Google Scholar]
- 47.Schulz G.E. (1979) Nucleotide binding proteins. In Balaban,M. (ed.), Molecular Mechanism of Biological Recognition. Elsevier/North-Holland Biomedical Press, New York, pp. 79–94.
- 48.Falquet L., Pagni,M., Bucher,P., Hulo,N., Sigrist,C.J., Hofmann,K. and Bairoch,A. (2002) The PROSITE database, its status in 2002. Nucleic Acids Res., 30, 235–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nigg E.A. (1991) The substrates of the cdc2 kinase. Semin. Cell Biol., 2, 261–270. [PubMed] [Google Scholar]
- 50.Hunter T. and Cooper,J.A. (1984) Tyrosine protein kinases and their substrates: an overview. Adv. Cyclic Nucleotide Protein Phosphorylation Res., 17, 443–455. [PubMed] [Google Scholar]
- 51.Zhou S. and Cantley,L.C. (1995) Recognition and specificity in protein tyrosine kinase-mediated signalling. Trends Biochem. Sci., 20, 470–475. [DOI] [PubMed] [Google Scholar]
- 52.Radivojac P., Obradovic,Z., Smith,D.K., Zhu,G., Vucetic,S., Brown,C.J., Lawson,J.D. and Dunker,A.K. (2004) Protein flexibility and intrinsic disorder. Protein Sci., 13, 71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hauer J.A., Johnson,D.A. and Taylor,S.S. (1999) Binding-dependent disorder-order transition in PKI alpha: a fluorescence anisotropy study. Biochemistry, 38, 6774–6780. [DOI] [PubMed] [Google Scholar]
- 54.Bienkiewicz E.A., Adkins,J.N. and Lumb,K.J. (2002) Functional consequences of preorganized helical structure in the intrinsically disordered cell-cycle inhibitor p27(Kip1). Biochemistry, 41, 752–759. [DOI] [PubMed] [Google Scholar]
- 55.Kriwacki R.W., Hengst,L., Tennant,L., Reed,S.I. and Wright,P.E. (1996) Structural studies of p21Waf1/Cip1/Sdi1 in the free and Cdk2-bound state: conformational disorder mediates binding diversity. Proc. Natl Acad. Sci. USA, 93, 11504–11509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bossemeyer D., Engh,R.A., Kinzel,V., Ponstingl,H. and Huber,R. (1993) Phosphotransferase and substrate binding mechanism of the cAMP-dependent protein kinase catalytic subunit from porcine heart as deduced from the 2.0 Å structure of the complex with Mn2+ adenylyl imidodiphosphate and inhibitor peptide PKI(5–24). EMBO J., 12, 849–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Narayana N., Cox,S., Shaltiel,S., Taylor,S.S. and Xuong,N. (1997) Crystal structure of a polyhistidine-tagged recombinant catalytic subunit of cAMP-dependent protein kinase complexed with the peptide inhibitor PKI(5–24) and adenosine. Biochemistry, 36, 4438–4448. [DOI] [PubMed] [Google Scholar]
- 58.Lowe E.D., Noble,M.E., Skamnaki,V.T., Oikonomakos,N.G., Owen,D.J. and Johnson,L.N. (1997) The crystal structure of a phosphorylase kinase peptide substrate complex: kinase substrate recognition. EMBO J., 16, 6646–6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.ter Haar E., Coll,J.T., Austen,D.A., Hsiao,H.M., Swenson,L. and Jain,J. (2001) Structure of GSK3beta reveals a primed phosphorylation mechanism. Nature Struct. Biol., 8, 593–596. [DOI] [PubMed] [Google Scholar]
- 60.Hubbard S.R. (1997) Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J., 16, 5572–5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McDonald I.K. and Thornton,J.M. (1994) Satisfying hydrogen bonding potential in proteins. J. Mol. Biol., 238, 777–793. [DOI] [PubMed] [Google Scholar]
- 62.Fontana A., Polverino de Laureto,P. and De Filippis,V. (1993) Molecular aspects of proteolysis of globular proteins. In van den Tweel,W., Harder,A. and Buitelear,M. (eds), Protein Stability and Stabilization. Elsevier Science, Amsterdam, pp. 101–110. [Google Scholar]
- 63.Fontana A., Zambonin,M., Polverino de Laureto,P., De Filippis,V., Clementi,A. and Scaramella,E. (1997) Probing the conformational state of apomyoglobin by limited proteolysis. J. Mol. Biol., 266, 223–230. [DOI] [PubMed] [Google Scholar]
- 64.Hubbard S.J., Eisenmenger,F. and Thornton,J.M. (1994) Modeling studies of the change in conformation required for cleavage of limited proteolytic sites. Protein Sci., 3, 757–768. [DOI] [PMC free article] [PubMed] [Google Scholar]