

Abstract
In previous work, we showed that the binding of the liver x receptor α:peroxisome proliferator-activated receptor α (LXRα:PPARα) heterodimer to the murine Cyp7a1 gene promoter antagonizes the stimulatory effect of their respective ligands. In this study, we determined if LXRα:PPARα can also regulate human CYP7A1 gene promoter activity. Co-expression of LXRα and PPARα in McArdle RH7777 hepatoma cells decreased the activity of the human CYP7A1 gene promoter in response to fibrates and 25-hydroxycholesterol. In vitro, the human CYP7A1 Site I bound LXRα:PPARα, although with substantially less affinity compared with the murine Cyp7a1 Site I. The binding of LXRα:PPARα to human CYP7A1 Site I was increased in the presence of either LXRα or PPARα ligands. In HepG2 hepatoblastoma cells, fibrates and 25-hydroxycholesterol inhibited the expression of the endogenous CYP7A1 gene as well as the human CYP7A1 gene promoter when co-transfected with plasmids encoding LXRα and PPARα. However, a derivative of the human CYP7A1 gene promoter that contains a mutant form of Site I that does not bind LXRα:PPARα was not inhibited by WY 14,643 or 25-hydroxycholesterol in both McArdle RH7777 and HepG2 cells. The ligand-dependent recruitment of LXRα:PPARα heterodimer onto the human CYP7A1 Site I can explain the inhibition of the human CYP7A1 gene promoter in response to fibrates and 25-hydroxycholesterol.
INTRODUCTION
Cholesterol 7α-hydroxylase (cyp7a) is a liver-specific enzyme that catalyzes the 7α-hydroxylation of cholesterol, the limiting step in the classical pathway responsible for the conversion of cholesterol into bile acids (1). In mice and rats the synthesis of bile acids through this pathway is under feed-forward regulation by cholesterol via a transcriptional mechanism involving the nuclear receptor known as the liver x receptor α (LXRα; NR1H3) (2–5). LXRα normally binds to a direct repeat of the hexameric hormone response element separated by four nucleotides (a DR-4 motif) as a heterodimer with retinoid x receptor (RXR; NR2B1) and is activated by oxysterols (6,7). The human CYP7A1 gene, unlike the rat and murine Cyp7a1 genes, is not stimulated by oxysterols because the human CYP7A1 gene promoter does not interact with RXR:LXRα (4,5).
Peroxisome proliferator-activated receptor α (PPARα; NR1C1) is fatty acid- and fibrate-activated nuclear receptor that is abundantly expressed in the liver (8). PPARα plays a central role in fatty acid catabolism by regulating several genes involved in this process. Ligand-bound PPARα regulates the transcription of target genes by binding as a heterodimer with RXR to its response element characterized by a DR-1 motif. Our laboratory previously reported that the human CYP7A1 and murine Cyp7a1 gene promoters are differentially regulated by fatty acids and WY 14,643 through PPARα (9). The difference is due to the existence of a PPARα:RXR binding site at the –70 nucleotide region (Site I) of the murine Cyp7a1 gene promoter. However, the exact effect of PPARα ligands on cyp7a gene expression is controversial since a number of studies employing a variety of experimental systems have reported inconsistent results (9–13).
In human clinical studies, fibric acid derivatives have been shown to increase biliary cholesterol secretion and decrease bile acid output (14,15). These effects may be explained by the repression of CYP7A1 gene expression as suggested by reduced cholesterol 7α-hydroxylation rates and decreased cyp7a activity observed in patients undergoing fibrate treatment (14,15).
We demonstrated recently that PPARα and LXRα are capable of forming an atypical heterodimer on two adjacent hexameric sequences (termed the LXRα:PPARα response element, LPRE) in the murine CYP7a1 gene promoter and repress its activity in hepatoma cells (16). The present study was undertaken to determine if the LXRα:PPARα heterodimer can also interact with the human CYP7A1 gene promoter and whether this interaction could explain the apparent reduction of cyp7a activity in response to fibrates observed in clinical studies. Here we show that the binding of LXRα:PPARα heterodimer to the human CYP7A1 gene promoter is ligand dependent and is necessary for the repression of promoter activity.
MATERIALS AND METHODS
Plasmids
The gene chimera containing the proximal promoter region of the human CYP7A1 gene (nucleotides –372 to +61) linked to the chloramphenicol acetyltransferase structural gene (hCYP7A1.pCAT), expression plasmids encoding murine PPARα, human RXRα, human LXRα and Escherichia coli β-galactosidase (pCH110) were described previously (9). The expression plasmid encoding hepatocyte nuclear factor-1 α (HNF-1α) was a gift from Dr S. Karathanasis. The mutant derivatives of the human CYP7A1 gene promoter used in this study were generated by in vitro DNA amplification using mutagenic primers and the gene chimera containing the wild-type human CYP7A1 gene promoter (9) as template. The sequence of mutagenic primers for the human CYP7A1 Site I DR-0 were: sense primer 5′-TGGCTAATTGTTTGCTTTAAAAACCAA-3′; antisense primer 5′-TAACTTGAGCTTGGTTTTTAAAGCAA-3′.
Transient transfection assays
McArdle RH7777 rat hepatoma cells (17) were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 10% newborn calf serum. The cultures were transfected at 60% confluence using the calcium phosphate–DNA co-precipitation method as described elsewhere (18). The plasmids were supplemented with sonicated salmon sperm DNA to achieve a constant mass of DNA introduced into cells in the transfection experiments. The chloramphenicol acetyltransferase activity determined for each sample was normalized to the corresponding β-galactosidase activity that was encoded by the co-transfected pCH110 plasmid. Bezafibrate, clofibrate, gemfibrozil (all from Sigma-Aldrich, Oakville, ON) and WY 14,643 (BIOMOL Research Labs, Inc., Plymouth Meeting, PA) were dissolved in dimethyl sulfoxide and added to the cell culture medium at the indicated concentrations. The oxysterol 25-hydroxycholesterol (25-HC; Sigma-Aldrich) was dissolved in ethanol. Each condition in the transfection experiments was done in triplicate and each experiment was repeated at least once.
Electrophoretic mobility shift assay
Plasmids used for production of recombinant nuclear receptors were introduced into E.coli BL21-CodonPlus™ cells (Stratagene, La Jolla, CA). The enrichment and integrity of the recombinant nuclear receptor preparations were assessed by SDS–PAGE and by immunoblotting using previously described polyclonal antibodies against PPARα and RXR (19) and a monoclonal antibody for LXRα (BD Pharmingen, Mississauga, ON, Canada). Recombinant receptors (100 ng total protein) were incubated in 40 µl for 20 min at room temperature with 4 µg of poly(dI:dC), 4 µg of bovine serum albumin (BSA), 1 µg of sonicated salmon sperm DNA, without or with varying amounts of unlabeled double stranded oligonucleotides in 1× binding buffer [40 mM Tris–HCl pH 7.9, 4 mM MgCl2, 100 mM NaCl, 2 mM EDTA, 20% glycerol, 0.2% Nonidet P-40, 2 mM dithiothreitol (DTT)]. After addition of the radiolabeled probe, the reaction was incubated for 20 min at room temperature. DNA–protein complexes were resolved by electrophoresis on a non-denaturing 4% polyacrylamide gel at 4°C. The core sequences of the probes were as follows: human CYP7A1 Site I, 5′-TTGTCAACCAAGCTCA-3′; murine Cyp7a1 Site I, 5′-TGGTCACCCAAGTTCA-3′. All oligonucleotides used in electrophoretic mobility shift assay (EMSA) were synthesized with a 4-nucleotide 5′ extension (5′-AATT-3′) to allow radiolabeling via a fill-in reaction catalyzed by the Klenow fragment of E.coli DNA polymerase I.
Measurement of cyp7a mRNA abundance and enzyme activity in HepG2 cells
Human HepG2 hepatoblastoma cells were grown in DMEM containing 5% FBS. At 70% confluence, the culture medium was replaced with fresh medium containing 5% lipoprotein-deficient medium and then treated for 24 h with either 5 µM 25-HC, 100 µM WY 14,643, 5 µM 25-HC and 100 µM WY 14,643 or vehicle [dimethyl sulfoxide and ethanol, both at 0.1% (v/v) final concentration]. Microsomes were prepared from control and treated cells that were homogenized in 0.1 M K2PO4 pH 7.4, 0.25 M sucrose, 50 mM NaF, 1 mM DTT, 1 mM EDTA buffer supplemented with a cocktail of protease inhibitors (Roche Diagnostics, Laval, QC, Canada). The total protein concentration of the microsomes was determined using the BioRad Protein Assay kit with BSA as the standard, and 100 µg were used to measure cyp7 enzyme activity according to a previously described method (20). The abundance of the cyp7a mRNA was estimated relative to the glyceraldehyde-3-phosphate dehydrogenase mRNA abundance by amplification of cDNA after reverse transcription of total RNA. The total RNA from cells treated as described above was isolated using the TRIzol™ reagent (Invitrogen Canada, Burlington, ON, Canada), and then reverse transcribed with Superscript II (Invitrogen Canada) and 18mer oligo-dT as primer following conditions recommended by the supplier. The total cDNA (2 µl) from each reaction was subsequently used as template to amplify the cyp7a and glyceraldehyde-3-phosphate dehydrogenase cDNA using Taq DNA polymerase (94°C for 1 min, annealing step at 45°C for 1 min and extension step at 72°C for 2 min; 25 cycles). The primers used to amplify human cyp7a sequences were: sense primer 5′-TAGCTCTTTACCCACAGTTAATGC-3′; antisense primer 5′-TGGACCTGGTGAATCATTTCTACC-3′. The abundance of the cyp7a mRNA was normalized to the abundance of glyceraldehyde-3-phosphate dehydrogenase mRNA, whose cDNA was amplified using the following primers: sense primer 5′-GAGCCAAACGGGTCATCATC-3′; antisense primer 5′-CATCACGCCACAGCTTTCCA-3′.
Chromatin immunoprecipitation assay
HepG2 cells (20 × 106 per assay) were incubated in medium containing 5% delipidated FBS plus vehicle (0.1% dimethyl sulfoxide for WY 14,643 and 0.1% ethanol for 25-HC), 100 µM WY 14,643, 5 µM 25-HC or 100 µM WY 14,643 + 5 µM 25-HC for 24 h. The cells were fixed by adding formaldehyde to a final concentration of 1% directly to growth medium at 37°C for 15 min. The crosslinking reaction was quenched by adding glycine to a final concentration of 0.125 M. The fixed cells were washed once in ice-cold Tris-buffered saline (20 mM Tris–HCl pH 7.6, 150 mM NaCl) containing protease inhibitors (0.1 mg/ml pepstatin, 0.1 mg/ml leupeptin, 0.1 mg/ml aprotinin, 1 mM PMSF). The cells were lysed in buffer containing 10 mM Tris–HCl pH 8, 1% Triton X-100, 0.1% SDS, 1 mM EDTA, 0.025% NaN3 for 30 min on ice. The nuclei were collected by low-speed centrifugation and then diluted to 1 ml with buffer containing 16.7 mM Tris–HCl pH 8, 16.7 mM NaCl, 0.01% SDS, 1% Triton X-100, 1.2 mM EDTA and protease inhibitors. The chromatin was sheared to ∼500-bp fragments by sonication and then centrifuged at 13 000 r.p.m. for 10 min at 4°C to clarify the sample. An aliquot of the clarified sample was taken for total DNA extraction (represents total chromatin input). The chromatin was diluted to 300 µl in elution buffer (0.1 M NaHCO3 pH 6.8, 1% SDS) prior to crosslink reversal and deproteinization. The remaining clarified sample was used for the immunoprecipitation assay. The sample was incubated with 100 µl of protein G–Sepharose beads for 1 h at 4°C with rotation to remove non-specific binding. After sedimentation, the pre-cleared supernatant was incubated with 10 µl of anti-PPARα (H-98) (sc-9000; Santa Cruz Biotechnology, Santa Cruz, CA), or 10 µl of anti-LXRα (P-20) (sc-1202; Santa Cruz Biotechnology) or 10 µl of anti-hemagglutinin (clone 3F10) (Roche) overnight at 4°C with rotation. The immune complexes were collected with 100 µl protein G–Sepharose beads for 1 h at 4°C with rotation. The beads were washed once with Wash Buffer 1 (10 mM Tris–HCl pH 8, 0.1% SDS, 1% Triton X-100, 1 mM EDTA, 0.025% NaN3) containing 500 mM NaCl, once with Wash Buffer 1 containing 140 mM NaCl, once with Wash Buffer 2 (10 mM Tris–HCl pH 8, 0.25 M LiCl, 0.5% Nonidet P-40, 0.5% sodium deoxycholate, 1 mM EDTA) then finally twice with 10 mM Tris–HCl (pH 8.0), 1 mM EDTA (TE). The washed beads were suspended in 150 µl elution buffer, rotated at room temperature for 15 min, sedimented by centrifugation, and then the eluates were recovered. The elution step was repeated and the respective eluates were combined. Crosslinks were reversed in all samples (total chromatin and immunoprecipitated chromatin) by the addition of NaCl to a final concentration of 0.3 M and then treated with 5 µg of RNAse A for 6 h at 65°C with shaking. The DNA was deproteinized by proteinase K treatment (20 µg per sample) overnight at 37°C in buffer containing 53 mM Tris–HCl pH 6.5 and 10 mM EDTA, followed by extraction with phenol/chloroform/isoamyl alcohol (25/24/1) and then precipitation in ethanol. The pellets were washed with 70% ethanol, dried and dissolved in 50 µl of TE. An aliquot (2 µl) of this was analyzed by PCR (25 cycles) using a primer pair (sense primer 5′-AAGCTTGATGAATAACTCATTCTTATC-3′; antisense primer 5′-TGCAAATCTAGGCCAAAATCTCTGAG-3′) directed to amplify the –380 to +61 nucleotide region of the human CYP7A1 gene promoter. A β-actin gene primer pair (sense primer 5′-AACACCCCAGCCATGTACG-3′; antisense primer 5′-ATGTCACGCACGATTTCCC-3′) (21) was used as the control.
Co-immunoprecipitation and immunoblotting
HepG2 cells (2 × 106 cells per assay) were removed from the dish and lysed in 250 µl of lysis buffer (50 mM Tris–HCl pH 7.4, 1% Nonidet P-40, 1 mM EDTA, 1 mM Na3VO4, 1 mM NaF and protease inhibitors) containing 150 mM NaCl. Aliquots of the lysate (250 µg total proteins) were pre-cleared with 50 µl of protein G–Sepharose beads for 1 h at 4°C with rotation and then incubated with either 10 µl of anti-PPARα, or 10 µl of anti-LXRα or 10 µl of anti-hemagglutinin overnight at 4°C with rotation. The immune complexes were collected with 100 µl of protein G–Sepharose beads for 1 h at 4°C with rotation. The beads were washed twice with lysis buffer containing 500 mM NaCl followed by two washes with the same buffer containing 150 mM NaCl. The bound proteins were eluted with 30 µl of SDS–PAGE loading buffer, boiled for 10 min and then resolved on an 8% SDS–PAGE gel. For immunoblot analysis, the proteins were probed with anti-LXRα and anti-PPARα diluted at 1:1000.
RESULTS
Co-expression of PPARα and LXRα is required for the inhibition of the human CYP7A1 gene promoter in response to WY 14,643 and 25-HC
The activity of the human CYP7A1 gene promoter in response to fibrates and 25-HC was studied in McArdle RH7777 cells co-transfected with PPARα, LXRα and RXR expression plasmids. As shown in Figure 1A, the combined transfection of LXRα and RXR did not change significantly the promoter activity in the absence or presence of 25-HC (5 µM), an LXRα ligand, in agreement with previous data (4,5). The PPARα activator WY 14,643 (100 µM) induced the human CYP7A1 gene promoter activity by ∼2.3-fold in the presence of co-expressed PPARα and RXR. The co-expression of PPARα, LXRα and RXR did not change the promoter activity as compared with the basal level in the absence of exogenous PPARα and LXRα ligands. However, the addition of WY 14,643 and 25-HC, by themselves or in combination, to transfected cells reduced promoter activity by 55%, 37% and 80%, respectively, relative to the basal promoter activity. Furthermore, the inhibition of the promoter activity by ligand-bound LXRα and PPARα was not affected by excess RXR, suggesting that the ligand-activated LXRα:PPARα heterodimer has a high affinity for Site I. These results demonstrate that the co-expression of LXRα and PPARα inhibits the CYP7A1 gene promoter in the presence of their ligands.
Figure 1.

Stimulation of the human CYP7A1 gene promoter by WY 14,643 is abolished by the co-expression of PPARα and LXRα in a ligand-dependent manner. (A) The human CYP7A1 gene promoter–reporter gene chimera (hCYP7A1.CAT) (1 µg) was introduced into McArdle RH7777 cells along with PPARα (0.5 µg), LXRα (0.5 µg) or RXR (0.5 µg) expression plasmids, as indicated. Cells were subsequently incubated for 24 h with WY 14,643, 25-HC or vehicle (0.1% dimethyl sulfoxide for WY 14,643 and 0.1% ethanol for 25-HC) in medium containing 5% lipoprotein-deficient serum. Values shown are mean ± standard deviation (SD). (B) Co-expression of PPARα and LXRα is necessary for the repression of the CYP7A1 gene promoter in response to WY 14643 and 25-HC. The cells were transfected with constant amounts of PPARα and RXR plus increasing amounts of LXRα. The amount of RXR used was equal to the total mass of PPARα and LXRα plasmids. Values shown are mean ± SD.
A titration experiment was done using a constant amount of PPARα expression plasmid (0.5 µg) and increasing amounts of LXRα expression plasmid (0.1–1.5 µg). As shown in Figure 1B, increasing amounts of LXRα had little influence on basal promoter activity in the absence of exogenous ligands. Addition of 25-HC attenuated the transactivation of the CYP7A1 gene promoter by WY 14,643-activated PPARα, and this inhibitory effect became more pronounced when higher amounts of LXRα was expressed.
Effect of different fibrates on the regulation of the human CYP7A1 promoter by PPARα and LXRα
The effect of various PPARα activators on human CYP7A1 gene promoter activity was studied in the same transfection assay system. As shown in Figure 2, bezafibrate, gemfibrozil and WY 14,643 stimulated the promoter activity by 2- to 2.5-fold, whereas clofibrate enhanced the promoter activity by only 1.5-fold. In case of the co-expression of LXRα, PPARα and RXR, the CYP7A1 gene promoter activity was also reduced by treatment of transfected cells with bezafibrate and gemfibrozil, and within a comparable range of concentrations. However, the addition of clofibrate to cells transfected with PPARα and LXRα did not diminish the promoter activity, as did the other PPARα activators used in the experiment. 25-HC also reduced the promoter activity and the combination of 25-HC and PPARα ligands resulted in far greater inhibition of CYP7A1 gene promoter activity. These data show that both PPARα and LXRα ligands can inhibit the human CYP7A1 gene promoter activity in the presence of co-expressed PPARα and LXRα.
Figure 2.

Relative potencies of various fibrates and 25-HC in the inhibition of the human CYP7A1 gene promoter in cells co-expressing LXRα and PPARα. The human CYP7A1 gene promoter–reporter gene chimera (hCYP7A1.CAT) (1 µg) was introduced into McArdle RH7777 cells along with PPARα (0.5 µg), LXRα (0.5 µg) or RXR (0.5 µg) expression plasmids, as indicated. The transfected cells were subsequently incubated for 24 h with WY 14,643, clofibrate, bezafibrate, gemfibrozil, 25-HC or vehicle (0.1% dimethyl sulfoxide for PPARα activators and 0.1% ethanol for 25-HC) in medium containing 5% lipoprotein-deficient serum.
LXRα:PPARα heterodimer binding to the human CYP7A1 Site I is potentiated by their respective ligands
We reported previously that the PPARα:RXR heterodimer can bind to Site II, but not to Site I, of the human CYP7A1 gene promoter (9). Furthermore, LXRα does not interact with the human CYP7A1 gene promoter with RXR (4,5). In our recent study we showed that a heterodimer of LXRα and PPARα can interact with the murine Cyp7a1 gene promoter at Site I in the absence of exogenous ligands (16). Although the human CYP7A1 and murine Cyp7a1 Site I sequences are polymorphic, the data presented in Figure 3 demonstrate that the human CYP7A1 Site I can nevertheless bind PPARα:RXR, albeit with less affinity as compared with the murine Cyp7a1 Site I. The interaction was decreased by an excess of unlabeled human CYP7A1 Site I. Importantly, neither PPARα nor LXRα, by themselves or with RXR, bound to the human CYP7A1 Site I.
Figure 3.
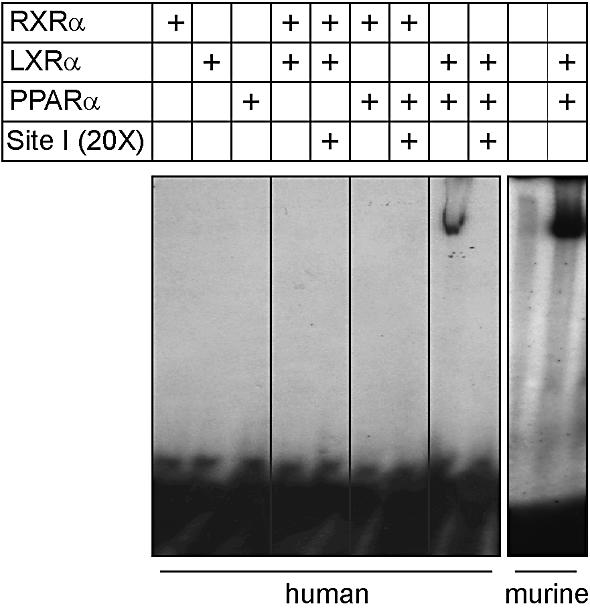
PPARα and LXRα bind to the human CYP7A1 Site I as a heterodimer. Recombinant PPARα, LXRα or RXR were incubated with radiolabeled, wild-type human CYP7A1 Site I. The sequences of the probes used in this experiment are described in Materials and Methods. The specificity of binding was assessed by competition with a 20-fold molar excess (20×) of unlabeled human CYP7A1 Site I.
The results of the transfection experiments (Fig. 1) indicated that the repression of human CYP7A1 promoter by LXRα and PPARα required the addition of their ligands. To determine if the ligand-dependent repression of CYP7A1 gene promoter activity by PPARα and LXRα is due to ligand-dependent recruitment of the LXRα:PPARα heterodimer onto the promoter, we measured the formation of the protein:probe complex in vitro in response to increasing amounts of fibrates or 25-HC added to a constant amount of LXRα and PPARα recombinant proteins. As shown in Figure 4A, the abundance of LXRα:PPARα/Site I complex formed increased with an increase in the amount of fibrates or 25-HC added to the reaction mixture. Interestingly, the induction of the LXRα:PPARα heterodimer formation by clofibrate was weak in comparison with that induced by bezafibrate, gemfibrozil or WY 14,643. It was also evident that 25-HC had a more potent effect on the heterodimer formation compared with the fibrates in this assay (Fig. 4B). Together, these data demonstrate that the binding of the LXRα:PPARα heterodimer to the human CYP7A1 Site I is stimulated by PPARα and LXRα ligands in a concentration-dependent manner.
Figure 4.

Fibrates and 25-HC increase the binding of the LXRα:PPARα heterodimer to the human CYP7A1 Site I. Radiolabeled human CYP7A1 Site I was incubated with LXRα and PPARα plus the indicated ligand or vehicle (dimethyl sulfoxide for fibrates and ethanol for 25-HC). The amount of radioactivity at the region of the gel corresponding to the probe:protein complex was quantitated using a PhosphorImager (Molecular Dynamics). The fold induction represents the ratio between the band intensity in the presence and absence of ligands. (A) Concentration-dependent effect of bezafibrate, gemfibrozil, clofibrate and WY 14,643. (B) Concentration-dependent effect of 25-HC.
LXRα:PPARα heterodimer binds to directly adjacent hexameric repeats located in the human CYP7A1 Site I
Our previous work showed that LXRα:PPARα binds to two directly adjacent hexameric elements (LPRE) in the murine Cyp7a1 Site I and to a sequence represented by an idealized DR-0 motif (16). To characterize the sequence to which the LXRα:PPARα heterodimer binds in the human CYP7A1 gene promoter, we performed EMSA using a synthetic double-stranded oligonucleotide corresponding to the first 12 residues (Site I-TR probe; Fig. 5A) that make up the CYP7A1 Site I, as well as a derivative with multibase substitutions (Site I-SU probe; Fig. 5A). The radiolabeled, intact CYP7A1 Site I bound LXRα:PPARα, and this binding was competed by an excess of the unlabeled Site I-TR probe (Fig. 5B, left). In contrast, LXRα:PPARα binding could not be displaced by increasing amounts of unlabeled Site I-SU probe (Fig. 5B, middle). Furthermore, the radiolabeled Site I-SU probe did not bind the LXRα:PPARα heterodimer. These data demonstrate that the LXRα:PPARα heterodimer interacts with the human CYP7A1 Site I at a site that is homologous to the murine Site I LPRE.
Figure 5.
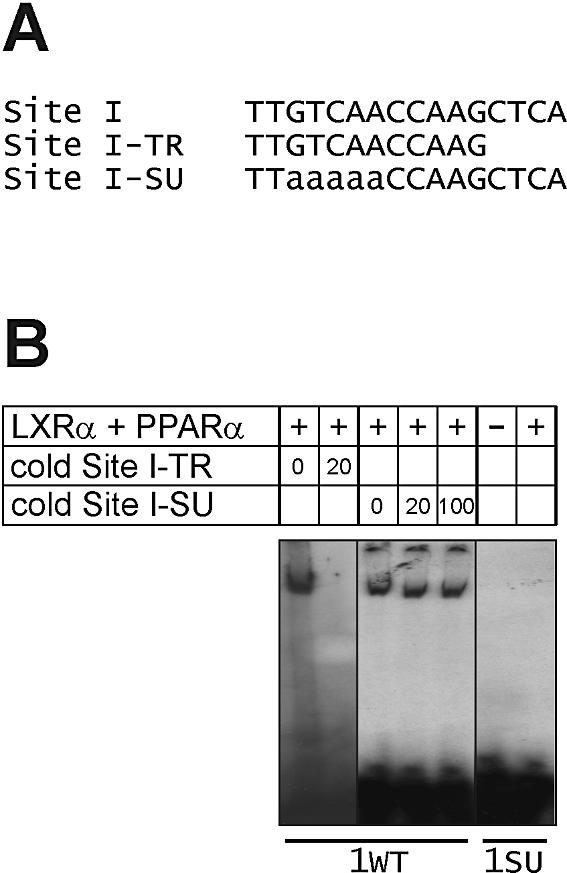
Analysis of the LXRα:PPARα heterodimer binding site in the human CYP7A1 Site I. (A) The sequences of the wild-type human CYP7A1 Site I, a derivative containing the first 12 nucleotides of Site I (designated Site I-TR), and a multibase substituted derivative (Site I-SU) are shown. (B) EMSA was performed using radiolabeled wild-type Site I (1WT) or radiolabeled Site I-SU (1SU) as probes. The 1WT probe was competed with 20-fold molar excess (20×) of unlabeled Site 1TR, or 20- and 100-fold excess of unlabeled Site 1SU.
The inhibition of the human CYP7A1 gene promoter by LXRα and PPARα requires binding of both factors to the Site I LPRE
In order to determine if the repression of the human CYP7A1 gene promoter by PPARα and LXRα is mediated by the direct binding of the LXRα:PPARα heterodimer to the human CYP7A1 Site I LPRE, we performed the transfection assays using a derivative of the human CYP7A1 gene promoter carrying the mutation in Site I that prevents the binding of the LXRα:PPARα heterodimer (i.e. the multibase substitution Site I-SU sequence shown in Fig. 5A). As shown in Figure 6, the mutant promoter had drastically reduced basal activity as compared with the wild-type promoter. The promoter activity increased upon co-expression of PPARα and RXR in the absence of WY 14,643 and was increased further upon the addition of WY 14,643. Co-expression of LXRα and RXR had no effect on promoter activity in the absence or presence of 25-HC. As predicted, the activity of the mutant promoter was not inhibited by the co-expression of PPARα, LXRα and RXR in McArdle RH7777 cells after the addition of WY 14,643 and 25-HC, either separately or in combination. Thus, the inhibition of the human CYP7A1 gene promoter in response to fibrates and 25-HC requires the formation of the LXRα:PPARα heterodimer on the CYP7A1 Site I LPRE.
Figure 6.

Binding of the LXRα:PPARα heterodimer to Site I is required to express its repressor function in McArdle RH7777 cells. A mutant derivative of the human CYP7A1 gene promoter–reporter gene chimera (mut.hCYP7A1.CAT, 1 µg) carrying the multibase substitution at Site I was transfected into hepatoma cells along with PPARα (0.5 µg), LXRα (0.5 µg) or RXR (0.5 µg) expression plasmids. The cells were treated with WY 14,643 and 25-HC, where indicated, in medium containing 5% lipoprotein-deficient serum. The promoter activities are expressed relative to the activity (100%) of the wild-type human CYP7A1 gene promoter.
Regulation of the CYP7A1 gene promoter by WY 14,643 and 25-HC in HepG2 cells
We next addressed the issue of the conflicting effects of fibrates on human CYP7A1 gene promoter activity in different cells, as reported in various studies (9–11). We have shown in a previous study (9) and in Figure 2 that the human CYP7A1 gene promoter is modestly stimulated by fibrates via PPARα and RXR in transfected cells. Although McArdle RH7777 cells are liver-derived, these cells no longer express the Cyp7a1 gene (22). In contrast, the CYP7A1 gene has remained active in HepG2 cells (23), although its activity is significantly lower compared with that in the liver. The addition of 25-HC (5 µM) and WY 14,643 (100 µM), either separately or in combination, decreased the cyp7a enzyme activity by 20, 60 and 70%, respectively (Fig. 7A, left), with corresponding reductions in cyp7a mRNA abundance (Fig. 7A, right). In transfected HepG2 cells, WY 14,643 also inhibited the human CYP7A1 gene promoter when PPARα and RXR were co-expressed (Fig. 7B, left). This effect is in contrast to that observed in McArdle RH7777 cells as shown in Figure 2. WY 14,643 treatment caused a further diminution of the human CYP7A1 gene promoter activity when LXRα was also co-expressed with PPARα and RXR. These experiments indicate that the response of the human CYP7A1 gene promoter to PPARα ligands is influenced by the cell line used in the experimental system.
Figure 7.

Regulation of CYP7A1 gene expression by fibrates and oxysterols in HepG2 cells. (A) HepG2 cells were incubated for 24 h with 5 µM 25-HC, 100 µM WY 14,643, a combination of 25-HC and WY 14,643, or vehicle (0.1% dimethyl sulfoxide for WY 14,643 and 0.1% ethanol for 25-HC), as indicated, in medium containing 5% lipoprotein-deficient serum. The cyp7a enzyme activity was measured in prepared microsomes (left) and the cyp7a mRNA abundance was estimated by reverse transcription (RT)–PCR (right). The glyceraldehyde-6-phosphate dehydrogenase mRNA abundance was used to normalize cyp7a mRNA abundance. Values are expressed relative to controls (no ligands added). (B) The wild-type human CYP7A1 promoter–reporter gene chimera (hCYP7A1.CAT, 1 µg) (left) or the mutant derivative carrying the multibase substitution at Site I (mut.hCYP7A1.CAT, 1 µg) (right) were introduced into cells along with PPARα (0.5 µg), LXRα (0.5 µg) or RXR (0.5 µg) expression plasmids, as indicated. The cells were treated with WY 14,643, where indicated, in medium containing 5% lipoprotein-deficient serum.
The response of mutant human CYP7A1 gene promoter carrying a substituted Site I (see Fig. 5) to WY 14,643 was also assessed in HepG2 cells. The activity of the mutant promoter was not altered by co-expression of PPARα and RXR, and increased when WY 14,643 was present (Fig. 7B, right). This finding is in agreement with the results obtained using McArdle RH7777 cells (Fig. 6). More importantly, the mutant promoter was still stimulated by WY 14,643 when LXRα was co-expressed along with PPARα and RXR. These results indicate that the ligand-dependent repression of the human CYP7A1 promoter mediated via binding of the LXRα:PPARα heterodimer to the Site I LPRE is reproducible in both McArdle RH7777 and HepG2 cells.
PPARα and LXRα co-expression interferes with the regulation of human CYP7A1 gene promoter by HNF-1α in transfected cells
HNF-1α binds to an element adjacent to the human CYP7A1 Site I and transactivates the human CYP7A1 gene promoter in HepG2 cells (24). We raised the question of whether the binding of LXRα:PPARα to the CYP7A1 Site I LPRE can interfere with HNF-1α action on this promoter. As shown in Figure 8, HNF-1α stimulated the CYP7A1 gene promoter by 2.2-fold in McArdle RH7777 cells. However, co-expression of PPARα and LXRα blocked the HNF-1α-mediated transactivation of the human CYP7A1 gene promoter. Furthermore, the addition of WY 14,643 or 25-HC reduced the CYP7A1 gene promoter activity to 60 and 50%, respectively.
Figure 8.

LXRα:PPARα co-expression prevents transactivation by HNF-1α in McArdle RH7777 cells. The human CYP7A1 promoter–reporter gene chimera (1 µg) was introduced into McArdle RH7777 cells, along with plasmids encoding HNF-1α (0.5 µg), PPARα (0.5 µg) LXRα (0.5 µg), or RXR (1 µg), as indicated. Cells were subsequently incubated for 24 h with WY 14,643 (100 µM) and 25-HC (5 µM) in medium containing 5% lipoprotein-deficient serum.
Formation of the LXRα:PPARα heterodimer in HepG2 cells
We carried out chromatin immunoprecipitation (ChIP) assays in untreated and ligand-treated HepG2 cells to determine if PPARα and LXRα can bind to the CYP7A1 gene promoter in intact cells. As shown in Figure 9A, a segment of the DNA spanning the Site I element of the human CYP7A1 gene promoter was co-precipitated with antibodies against PPARα or LXRα, but not with the control antibodies against hemagglutinin. Furthermore, the antibodies against PPARα or LXRα did not co-immunoprecipitate the murine actin gene, demonstrating the specificity of the ChIP assay. The co-immunoprecipitation of the CYP7A1 gene promoter with PPARα and LXRα is consistent with the idea that these receptors bind to the Site I element as a heterodimer, since neither PPARα nor LXRα bind to this site as monomers or as conventional heterodimers with RXR (5,9). In addition, it is evident from the results that binding of PPARα and LXRα to the CYP7A1 gene promoter can occur in the absence of exogenously added ligands, and that binding appears to be increased in the presence of ligands.
Figure 9.

Analysis of LXRα:PPARα heterodimer formation in untreated and ligand-treated HepG2 cells. (A) Binding of PPARα and LXRα to the human CYP7A1 gene promoter in intact cells was assessed by chromatin immunoprecipitation. The PCR amplification products (see Materials and methods for a description of the primers used in the experiment) were resolved by agarose gel electrophoresis and stained with ethidium bromide. A genomic DNA segment spanning Site I of the human CYP7A1 gene was amplified from DNA co-immunoprecipitated with antibodies against PPARα or LXRα from crosslinked HepG2 chromatin. Input DNA represents DNA purified from total chromatin. HA, hemagglutinin. (B) HepG2 cytoplasmic extracts were subjected to co- immunoprecipitation analysis using antibodies to LXRα and PPARα. The proteins immunoprecipitated by the primary antisera were separated by SDS–PAGE then analyzed by immunoblotting using the second antiserum. The antisera used in the immunoprecipitation assay (IP) are indicated at the top of the figure and the antisera used in the immunoblot (IB) analysis are indicated to the left.
We further examined the ability of PPARα and LXRα to form heterodimers in the cell cytoplasm. HepG2 cytoplasmic extracts were immunoprecipitated with anti-PPARα followed by immunoblot analysis of immunoprecipitated proteins using anti-LXRα. The experiment was also done using anti-LXRα and anti-PPARα in the immunoprecipitation and immunoblot analyses, respectively. The results shown in Figure 9B demonstrate that LXRα is co-immunoprecipitated with PPARα and vice versa, demonstrating that PPARα and LXRα can also form stable heterodimers in the cytoplasm in the absence of DNA.
DISCUSSION
We demonstrated in a recent study that LXRα and PPARα form an atypical heterodimer on the Site I regulatory region of the murine Cyp7a1 gene promoter and inhibit its activity (16). The goal of present study was to determine if LXRα and PPARα can also form a heterodimer on the human CYP7A1 gene promoter to mediate the negative regulation in response to PPARα and LXRα ligands. The sequences of the Site I regulatory region of the human CYP7A1 and murine Cyp7a1 gene promoters are divergent (9). The murine Cyp7a1 Site I can bind PPARα:RXR and RXR:LXRα (9), whereas these heterodimers do not interact with the human CYP7A1 Site I (4,5).
The present study clearly demonstrates that LXRα and PPARα also form a non-conventional heterodimer on the human CYP7A1 Site I regulatory region. This interaction was potentiated by PPARα and LXRα ligands and caused the repression of human CYP7A1 gene promoter activity. Moreover, it was apparent that LXRα:PPARα can block the stimulatory effect of HNF-1α, a transcription factor that binds to a site near Site I. The conflicting effects of PPARα ligands on human CYP7A1 gene promoter activity in cells co-expressing PPARα and RXR are dependent on the cell line that was utilized as an experimental system. The human CYP7A1 gene promoter was modestly stimulated by WY 14,643-activated PPARα in McArdle RH7777, whereas it was inhibited in HepG2 hepatoblastoma cells. Importantly, however, the inhibitory effect of LXRα and PPARα ligands on the human CYP7A1 gene promoter in cells co-expressing LXRα and PPARα was reproducible in both cell lines.
Mutagenesis of the human CYP7A1 Site I sequence to prevent the binding of the LXRα:PPARα heterodimer relieved the inhibitory effect of PPARα and LXRα ligands, and this effect was evident in both cell lines used in this study. This specific alteration of human CYP7A1 Site I structure also caused the reduction of basal promoter activity, as well as an increase in the apparent response of the mutant CYP7A1 gene promoter to WY 14,643 treatment. The decrease in basal activity was surprising since the abrogation of the LXRα:PPARα heterodimer binding was expected to relieve promoter repression. We surmise that the reduction of basal promoter activity displayed by the mutant CYP7A1 gene promoter results from the disturbance in the binding of other nuclear receptors, possibly HNF-1α (24) and/or hepatocyte nuclear factor-3 (25), near the Site I regulatory region. We demonstrated previously that WY 14,643 stimulated the human CYP7A1 gene promoter via PPARα:RXR through Site II. The stimulation of the mutant CYP7A1 gene promoter activity was most likely also mediated by PPARα:RXR through the same regulatory site. However, the apparent increase in the sensitivity of the mutant CYP7A1 gene promoter to WY 14,643 is attributable to the reduced basal activity of the mutant promoter, thereby magnifying the apparent stimulatory effect.
The ability of various fibrates to inhibit the human CYP7A1 gene promoter in transfected cells was highly correlated with the ability of these agents to stimulate the formation of the LXRα:PPARα heterodimer on the human CYP7A1 Site I in vitro. The oxysterol 25-HC also stimulated the formation of LXRα:PPARα/Site I complex in vitro, and with a potency that was comparable to WY 14,643. In transfected cells, however, 25-HC was less effective than WY 14,643 in repressing the human CYP7A1 gene promoter. The reduced efficacy of 25-HC may be due to its metabolism causing the decrease in intracellular concentration of this oxysterol and, in turn, less profound inhibition of the CYP7A1 gene promoter. Nevertheless, the repression of CYP7A1 gene expression after 25-HC treatment has been observed previously in HepG2 cells (23). In agreement with this finding, the expression of the human CYP7A1 gene expression in transgenic mice was reduced by cholesterol feeding to a level that was lower than that in chow-fed controls (5).
It was suggested previously that agonists of PPARα repress CYP7A1 gene expression in HepG2 cells by decreasing the amount of hepatocyte nuclear factor-4 α (NR2A1) that is available for binding to the CYP7A1 gene promoter (10). It appears that LXRα:PPARα inhibits the human CYP7A1 gene promoter via a different mechanism. This is based on the finding that an intact CYP7A1 Site I is necessary to achieve repression in response to LXRα and PPARα ligands and the fact that the mutant CYP7A1 gene promoter containing a mutant Site I remains responsive to stimulation by WY 14,643 in HepG2 cells.
Miyata et al. (26) documented previously the interaction of PPARα and LXRα using a yeast two-hybrid system. Although binding of the complex to DNA was not demonstrated in their study, we found that the LXRα:PPAR heterodimer is capable of binding a unique element in the murine Cyp7a1 gene promoter (16). Recently, Ide et al. (27) showed that recombinant PPARα and LXRα produced by in vitro translation can form stable interactions. In our present study, we demonstrate that PPARα and LXRα can be immunoprecipitated from HepG2 cytoplasmic extracts. The formation of PPARα and LXRα heterodimers outside the nucleus represents one way by which the activity of genes regulated by these receptors can be controlled, that is by varying the concentration of these receptors available for heterodimerization with RXR. The ability of the LXRα:PPAR heterodimer to bind DNA in a specific fashion and block transcription activation represents another level of control. In this regard, the finding that LXRα:PPARα represses the human CYP7A1 gene promoter in cell culture models is important in explaining the mechanism by which fibrates and sterols inhibit the expression of the human CYP7A1 gene.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by grants from the Canadian Institutes of Health Research. The authors thank Dr S. Jauliac for providing laboratory facilities for some of the experiments described in this study.
REFERENCES
- 1.Agellon L.B. (2002) Metabolism and function of bile acids. In Vance,D.E. and Vance,J.E. (eds), Biochemistry of Lipids, Lipoproteins and Membranes, 4th Edn. Elsevier, Amsterdam, pp. 433–448. [Google Scholar]
- 2.Lehmann J.M., Kliewer,S.A., Moore,L.B., Smith-Oliver,T.A., Oliver,B.B., Su,J.-L., Sundseth,S.S., Winegar,D.A., Blanchard,D.E., Spencer,T.A. et al. (1997) Activation of the nuclear receptor LXR by oxysterols defines a new hormone response pathway. J. Biol. Chem., 272, 3137–3140. [DOI] [PubMed] [Google Scholar]
- 3.Peet D.J., Turley,S.D., Ma,W., Janowski,B.A., Lobaccaro,J.-M., Hammer,R.E. and Mangelsdorf,D.J. (1998) Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXRα. Cell, 93, 693–704. [DOI] [PubMed] [Google Scholar]
- 4.Chiang J.Y., Kimmel,R. and Stroup,D. (2001) Regulation of cholesterol 7α-hydroxylase gene (CYP7A1) transcription by the liver orphan receptor (LXRa). Gene, 262, 257–265. [DOI] [PubMed] [Google Scholar]
- 5.Agellon L.B., Drover,V.A., Cheema,S.K., Gbaguidi,G.F. and Walsh,A. (2002) Dietary cholesterol fails to stimulate the human cholesterol 7α-hydroxylase gene (CYP7A1) in transgenic mice. J. Biol. Chem., 277, 20131–20134. [DOI] [PubMed] [Google Scholar]
- 6.Willy P.J., Umesono,K., Ong,E.S., Evans,R.M., Heyman,R.A. and Mangelsdorf,D.J. (1995) LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev., 9, 1033–1045. [DOI] [PubMed] [Google Scholar]
- 7.Janowski B.A., Grogan,M.J., Jones,S.A., Wisely,G.B., Kliewer,S.A., Corey,E.J. and Mangelsdorf,D.J. (1999) Structural requirements of ligands for the oxysterol liver X receptors LXRα and LXRâ. Proc. Natl Acad. Sci. USA, 96, 266–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Desvergne B. and Wahli,W. (1999) Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocrinol. Rev., 20, 649–688. [DOI] [PubMed] [Google Scholar]
- 9.Cheema S.K. and Agellon,L.B. (2000) The murine and human cholesterol 7α-hydroxylase gene promoters are differentially responsive to regulation by fatty acids via peroxisome proliferator-activated receptor α. J. Biol. Chem., 275, 12530–12536. [DOI] [PubMed] [Google Scholar]
- 10.Marrapodi M. and Chiang,J.Y. (2000) Peroxisome proliferator-activated receptor a (PPARα) and agonist inhibit cholesterol 7α-hydroxylase gene (CYP7A1) transcription. J. Lipid Res., 41, 514–520. [PubMed] [Google Scholar]
- 11.Patel D.D., Knight,B.L., Soutar,A.K., Gibbons,G.F. and Wade,D.P. (2000) The effect of peroxisome-proliferator-activated receptor-α on the activity of the cholesterol 7α-hydroxylase gene. Biochem. J., 351, 747–753. [PMC free article] [PubMed] [Google Scholar]
- 12.Hunt M.C., Yang,Y.Z., Eggertsen,G., Carneheim,C.M., Gafvels,M., Einarsson,C. and Alexson,S.E. (2000) The peroxisome proliferator-activated receptor α (PPARα) regulates bile acid biosynthesis. J. Biol. Chem., 275, 28947–28953. [DOI] [PubMed] [Google Scholar]
- 13.Post S.M., Duez,H., Gervois,P.P., Staels,B., Kuipers,F. and Princen,H.M. (2001) Fibrates suppress bile acid synthesis via peroxisome proliferator-activated receptor-α-mediated downregulation of cholesterol 7α-hydroxylase and sterol 27-hydroxylase expression. Arterioscler. Thromb. Vasc. Biol., 21, 1840–1845. [DOI] [PubMed] [Google Scholar]
- 14.Raedsch R., Plachky,J., Wolf,N. and Simonis,G. (1995) Biliary lipids, lithogenic index and biliary drug concentrations during etofibrate and bezafibrate treatment. Eur. J. Drug Metab. Pharmacokinet., 20, 113–118. [DOI] [PubMed] [Google Scholar]
- 15.Stahlberg D., Reihner,E., Rudling,M., Berglund,L., Einarsson,K. and Angelin,B. (1995) Influence of bezafibrate on hepatic cholesterol metabolism in gallstone patients: reduced activity of cholesterol 7α-hydroxylase. Hepatology, 21, 1025–1030. [DOI] [PubMed] [Google Scholar]
- 16.Gbaguidi G.F. and Agellon,L.B. (2002) The atypical interaction of peroxisome proliferator-activated receptor α with liver X receptor α antagonizes the stimulatory effect of their respective ligands on the murine cholesterol 7α-hydroxylase gene promoter. Biochim. Biophys. Acta, 1583, 229–236. [DOI] [PubMed] [Google Scholar]
- 17.Becker J.E., de Nechaud,B. and Potter,V.R. (1976) Two new rat hepatoma cell lines for studying the unbalanced blocked ontogeny hypothesis. In Fishman,W.H. and Sell,S. (eds), Onco-developmental Gene Expression. Academic Press, New York, NY, pp. 259–270. [Google Scholar]
- 18.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning. A Laboratory Manual. Cold Spring Harbor Press, Cold Spring Harbor, NY. [Google Scholar]
- 19.Marcus S.L., Miyata,K.S., Zhang,B., Subramani,S., Rachubinski,R.A. and Capone,J.P. (1993) Diverse peroxisome proliferator-activated receptors bind to the peroxisome proliferator-responsive elements of the rat hydratase/dehydrogenase and fatty acyl-CoA oxidase genes but differentially induce expression. Proc. Natl Acad. Sci. USA, 90, 5723–5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agellon L.B. (1997) Partial transfection of liver with a synthetic cholesterol 7α-hydroxylase transgene is sufficient to stimulate the reduction of cholesterol in the plasma of hypercholesterolemic mice. Biochem. Cell Biol., 75, 255–262. [PubMed] [Google Scholar]
- 21.Li J., Gorospe,M., Hutter,D., Barnes,J., Keyse,S.M. and Liu,Y. (2001) Transcriptional induction of MKP-1 in response to stress is associated with histone H3 phosphorylation-acetylation. Mol. Cell. Biol., 21, 8213–8224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Labonté E.D., Li,Q. and Agellon,L.B. (2000) Expression of cholesterol 7α-hydroxylase restores bile acid synthesis in McArdle RH7777 cells. Arch. Biochem. Biophys., 381, 273–277. [DOI] [PubMed] [Google Scholar]
- 23.Taniguchi T., Chen,J. and Cooper,A.D. (1994) Regulation of cholesterol 7α-hydroxylase gene expression in Hep-G2 cells. Effect of serum, bile salts, and coordinate and noncoordinate regulation with other sterol-responsive genes. J. Biol. Chem., 269, 10071–10078. [PubMed] [Google Scholar]
- 24.Chen J., Cooper,A.D. and Levy-Wilson,B. (1999) Hepatocyte nuclear factor 1 binds to and transactivates the human but not the rat CYP7A1 promoter. Biochem. Biophys. Res. Commun., 260, 829–834. [DOI] [PubMed] [Google Scholar]
- 25.Cooper A.D., Chen,J., Botelho-Yetkinler,M.J., Cao,Y., Taniguchi,T. and Levy-Wilson,B. (1997) Characterization of hepatic-specific regulatory elements in the promoter region of the human cholesterol 7α-hydroxylase gene. J. Biol. Chem., 272, 3444–3452. [PubMed] [Google Scholar]
- 26.Miyata K.S., McCaw,S.E., Patel,H.V., Rachubinski,R.A. and Capone,J.P. (1996) The orphan nuclear hormone receptor LXRα interacts with the peroxisome proliferator-activated receptor and inhibits peroxisome proliferator signaling. J. Biol. Chem., 271, 9189–9192. [DOI] [PubMed] [Google Scholar]
- 27.Ide T., Shimano,H., Yoshikawa,T., Yahagi,N., Amemiya-Kudo,M., Matsuzaka,T., Nakakuki,M., Yatoh,S., Iizuka,Y., Tomita,S. et al. (2003) Cross-talk between peroxisome proliferator-activated receptor (PPAR) α and liver X receptor (LXR) in nutritional regulation of fatty acid metabolism. II. LXRs suppress lipid degradation gene promoters through inhibition of PPAR signaling. Mol. Endocrinol., 17, 1255–1267. [DOI] [PubMed] [Google Scholar]