

The exocyst is recruited to secretory vesicles by the combinatorial signals of Sec4-GTP and the Snc proteins to confer both specificity and directionality to vesicular traffic.
Abstract
A screen for mutations that affect the recruitment of the exocyst to secretory vesicles identified genes encoding clathrin and proteins that associate or colocalize with clathrin at sites of endocytosis. However, no significant colocalization of the exocyst with clathrin was seen, arguing against a direct role in exocyst recruitment. Rather, these components are needed to recycle the exocytic vesicle SNAREs Snc1p and Snc2p from the plasma membrane into new secretory vesicles where they act to recruit the exocyst. We observe a direct interaction between the exocyst subunit Sec6p and the latter half of the SNARE motif of Snc2p. An snc2 mutation that specifically disrupts this interaction led to exocyst mislocalization and a block in exocytosis in vivo without affecting liposome fusion in vitro. Overexpression of Sec4p partially suppressed the exocyst localization defects of mutations in clathrin and clathrin-associated components. We propose that the exocyst is recruited to secretory vesicles by the combinatorial signals of Sec4-GTP and the Snc proteins. This could help to confer both specificity and directionality to vesicular traffic.
Introduction
Vesicular traffic between membrane-bound organelles relies on a variety of components that together ensure the selection of appropriate cargo from the donor compartment and faithful transfer of that cargo to the correct target compartment. Nearly all of these components are conserved among eukaryotic organisms (Ferro-Novick and Jahn, 1994). Most are also members of protein families in which different members play analogous roles at different stages of membrane traffic. These include rab GTPases that act as master regulators of membrane traffic (Gurkan et al., 2007; Mizuno-Yamasaki et al., 2012), SNAREs that mediate membrane fusion (Kloepper et al., 2008), and Sec1-munc18 (SM) proteins implicated in both SNARE complex assembly and in coupling SNARE assembly to efficient membrane fusion (Carr and Rizo, 2010). In addition to these components, it has recently become clear that four of the vesicle tethers that mediate the initial recognition of the target membrane by vesicles at different stages of membrane traffic are structurally related to one another, despite very low levels of sequence similarity. This collection of proteins has been alternatively termed the quatrefoil (Whyte and Munro, 2001) and CATCHR family (Yu and Hughson, 2010) and it includes the exocyst, COG, GARP, and Dsl tethering complexes. These tethers all consist of multiple subunits, each containing several helical bundles, strung in series to form rod-shaped elements (Dong et al., 2005). Although their functions remain incompletely understood, interactions with coat proteins, SNAREs, SNARE regulators, lipids, and GTPases of the rab, rho, ral, and arf families have been documented (Novick and Guo, 2002; Munson and Novick, 2006). One interpretation of these many interactions is that the tethers function as scaffolds, holding together, in the optimal orientation, all of the different components needed for efficient docking and fusion of vesicles with the appropriate target membrane (Yu and Hughson, 2010).
The exocyst was the first member of this family to be identified and characterized. This complex consists of one copy of eight different subunits. Six were identified through genetic analysis of the yeast secretory pathway (Novick et al., 1980). These are Sec3p, Sec5p, Sec6p, Sec8p, Sec10p, and Sec15p. The two remaining subunits, Exo70p and Exo84p, were identified through biochemical analysis of the purified complex (TerBush et al., 1996; Guo et al., 1999a). Each of these components is required for the final stage of the yeast secretory pathway in which secretory vesicles are directed to fuse with the plasma membrane at sites defined by the cell polarity establishment machinery. At the start of the cell cycle, vesicles are directed to the prebud site and then, as the cycle progresses, to the tip of the emerging bud. Late in the cell cycle, polarity shifts and vesicles are directed to the neck between the bud and mother cell, facilitating cytokinesis and cell separation (Novick and Brennwald, 1993). While all eight exocyst subunits concentrate at these sites, six of them, Sec5p, Sec6p, Sec8p, Sec10p, Sec15p, and Exo84p, achieve this localization exclusively by riding on secretory vesicles as they are actively transported by the type V myosin, Myo2p, along polarized actin cables (Boyd et al., 2004). The other two subunits, Sec3p and Exo70p, can localize to exocytic sites independent of actin function by directly binding to polarity determinants at the cell cortex (Guo et al., 2001). Actin-independent localization requires the N-terminal domain of Sec3p (Guo et al., 2001) and the third helical bundle of Exo70p (Hutagalung et al., 2009). Although neither of these two domains alone is needed for growth, secretion, or exocyst localization, concurrent loss of both domains is lethal even in the presence of multi-copy suppressors that can bypass a complete deletion of either gene (Hutagalung et al., 2009). Thus, it appears to be absolutely essential that at least one of these two subunits interact with polarity determinants on the target membrane.
To function as a vesicle tether, the exocyst must recognize not only the target membrane, but the vesicle membrane as well. Early evidence indicated that Sec15p functions in response to the vesicle-associated rab, Sec4p, implying an interaction with the vesicle surface (Salminen and Novick, 1989). Expression of Sec15p from the strong, inducible GAL1 promoter leads to the formation of a cluster of secretory vesicles carrying both Sec4p and the overexpressed Sec15p. By fluorescence microscopy, this is manifest as a bright patch of Sec15p that colocalizes with a patch of Sec4p. Patch formation upon Sec15p overexpression is blocked in a sec4-8 mutant strain or in a sec2-41 strain defective in the Sec4p guanine nucleotide exchange factor, Sec2p. These findings prompted our proposal that Sec15p acts in response to the activated form of Sec4p (Salminen and Novick, 1989) to tether vesicles. In the context of Sec15p overexpression, this response leads to the formation of a cluster of secretory vesicles, whereas in the context of normal Sec15p expression the response to activated Sec4 is to tether the vesicle to the plasma membrane. Two-hybrid analysis, chemical cross-linking, and pull-down studies all support a direct interaction between Sec15p and Sec4-GTP (Guo et al., 1999b). Although the Sec4p–Sec15p interaction certainly contributes to the recruitment of the exocyst to the surface of secretory vesicles, it is unlikely to be the only interaction involved as, in a sec4-8 mutant strain, Sec15p still associates with secretory vesicles, even though those vesicles fail to cluster into a patch (Salminen and Novick, 1989). In the current study, we sought to identify additional interactions between the exocyst and components on the surface of secretory vesicles. Our analysis points to a direct interaction between the exocyst component Sec6p and the redundant exocytic v-SNAREs, Snc1p and Snc2p.
Results
Visual screen for exocyst localization mutants
To identify components required for recruitment of the exocyst to secretory vesicles we performed a directed visual screen based on Sec15p localization. We introduced a plasmid expressing Sec15-GFP from the inducible GAL1 promoter into 73 strains picked from the yeast deletion library. Based on the presumption that a protein that recruits the exocyst to vesicles must itself be at least partially or transiently associated with those vesicles, we chose strains deleted for genes encoding proteins possibly associated with secretory vesicles, such as coat components, or that have been localized to the bud tip or neck where secretory vesicles concentrate before fusion with the plasma membrane.
After induction of Sec15-GFP expression by growth on galactose medium for 5–8 h, wild-type cells uniformly exhibited a bright fluorescent patch of Sec15-GFP (Fig. 1 A, a) similar to that previously seen by immunofluorescence in cells expressing Sec15p from the GAL1 promoter (Salminen and Novick, 1989). This patch was typically seen at the tip of small buds or at the neck of large budded cells. In wild-type cells and in most of the mutant strains a patch of Sec15-GFP was observed; however, in eight strains we saw a significant reduction in the fraction of cells exhibiting a fluorescent Sec15-GFP patch (Fig. 1 B). These mutants predominantly exhibited diffuse fluorescence throughout the cytoplasm such as is shown for chc1Δ cells (Fig. 1 A, b).
Figure 1.

Visual screening of Sec15p-GFP localization in deletion strains. Strains were selected from the yeast deletion library and transformed with a GAL-SEC15-GFP construct. After 5 h of galactose induction, cells were collected and imaged by fluorescence microscopy. (A) Sec15p-GFP was concentrated at exocytic sites including bud tips and bud necks in WT cells (a), yet appeared diffuse in chc1Δ cells (b). The corresponding DIC images are shown in c and d. The appearance of Sec15p-GFP patches was quantified by scoring ∼200 cells for each strain (B). Values indicate the percentage of cells showing GFP-labeled patches at the bud tip or bud neck. Bar, 10 µm.
All but one of the affected strains were deleted for either a component of the clathrin coat itself, a component known to associate with a specific class of clathrin-coated vesicles, or a component that colocalizes with clathrin by fluorescence microscopy. These include both chc1Δ and clc1Δ, the structural genes encoding clathrin heavy chain and light chain, respectively. Clathrin-coated vesicles mediate several different transport events (McNiven and Thompson, 2006). Although clathrin is perhaps best known for its role in the formation of endocytic vesicles at the plasma membrane, the same clathrin subunits also act at the Golgi and endosome to direct traffic of specific cargo proteins between these compartments. Different adaptor complexes allow the same clathrin components to be used in different cellular locations. The AP-1 adaptor complex localizes to the trans-Golgi network and recruits clathrin to nascent vesicles destined for the endosome, whereas the AP-2 and AP180/CALM adaptors localize to the plasma membrane and recruit clathrin to nascent endocytic vesicles, and AP-3 localizes to endosomes. Of all the genes encoding subunits of one of the clathrin adaptor complexes, only deletion of yap1802, encoding a homologue of the AP-180/CALM adaptor, and apm4, encoding the µ subunit of the AP-2 adaptor complex, showed significant defects in the formation of Sec15-GFP patches (Fig. 1 B). Because we observed a defect with one of the deletions of the four subunits of the AP-2 adaptor complex, but not the other three, we backcrossed the apm4 mutant and tested three different mutant strains. They exhibited on average only a 12% decrease in Sec15-GFP localization. We therefore did not pursue further analysis of the apm4 mutant.
In addition to clathrin and clathrin adaptor proteins, several other clathrin-associated proteins were implicated through our screen, including Ldb17p and Swa2p (Fig. 1 B). Ldb17p was recently shown to colocalize with clathrin at sites of endocytosis on the plasma membrane and to play a role in the selection of specific cargo for internalization (Burston et al., 2009). Swa2p, the yeast homologue of auxillin, acts in conjunction with a chaperone to disassemble clathrin coats for reutilization (Gall et al., 2000; Pishvaee et al., 2000). We also identified the uncharacterized open reading frame YHL017W in our screen. Although no function has been assigned to this gene product to date, it has been shown to colocalize with clathrin-coated vesicles by fluorescence microscopy (Huh et al., 2003). Based on this observation, we have named the gene CCP1, for clathrin co-localizing protein 1. In addition to deletions of clathrin and clathrin-associated components, we also identified the avl9Δ mutant in our screen. Avl9p is thought to play a role in the formation of a sub-class of secretory vesicles, although its molecular mechanism is not well understood (Harsay and Schekman, 2007).
Because our visual screen used a construct in which Sec15-GFP expression is driven from the strong GAL1 promoter, a condition that leads to the formation of aberrant vesicle clusters and impaired cell growth, we reexamined the mutants identified in the screen using a Sec15-3xGFP fusion construct, integrated into the SEC15 locus as the sole copy of the gene, under normal expression from its own promoter. In general, the mutants identified in our screen exhibited a similar defect in Sec15-3xGFP localization when it was expressed at normal levels (Fig. 2, A and B), although the ldb17Δ mutant was not as dramatically affected under these conditions. Importantly, both chc1Δ and clc1Δ mutants continued to exhibit a profound defect in Sec15-3xGFP localization. Because Yap1802 is redundant in function with Yap1081, we examined a yap1801Δ yap1802Δ double mutant, yet found Sec15-3xGFP mislocalization to be similar to that of the yap1802Δ single mutant.
Figure 2.

Localization of Sec15-3xGFP in selected strains. Sec15p-3xGFP was expressed at normal levels from the endogenous SEC15 promoter in the seven strains selected from the screen and a yap1801 yap1802 double mutant. The localization of Sec15p-3xGFP in wild type (wt) or cells carrying the indicated deletions is shown (A). Bar, 10 µm. The localization of Sec15p-3xGFP was quantified (B). Data represent mean ± SD from at least three independent experiments.
Two models for the role of clathrin in exocyst recruitment
We consider below two models to explain the roles of clathrin and clathrin-associated components in Sec15-GFP localization. The simplest model assumes that clathrin is used to form the secretory vesicles that then rely on the exocyst for tethering to the cell cortex and fusion with the plasma membrane. In this model, the exocyst could be recruited to secretory vesicles through a physical interaction with clathrin itself or with one of the clathrin-associated components identified in our screen. Several lines of evidence are broadly consistent with a role for clathrin in the formation of a subset of exocytic vesicles (Gall et al., 2002; Harsay and Schekman, 2007). Furthermore, other tethering complexes have been shown to bind to their respective vesicle coats.
To address this model we assessed the degree of colocalization of Chc1-RFP with Sec15-3xGFP or Sec5-3xGFP (Fig. 3). Chc1-RFP localizes to a collection of cortical puncta, representing sites of endocytosis at the cell surface and to internal puncta, representing Golgi or endosomes. In contrast, Sec15-3xGFP and Sec5-3xGFP localize to the tips of small buds and the necks of large budded cells. Very little colocalization of Chc1-RFP with either exocyst subunit was observed (Fig. 3, A and B). We also tested for colocalization of Chc1-RFP with Sec15-GFP overexpressed from the GAL1 promoter. Again, very little colocalization was seen.
Figure 3.

Localization of Chc1p and exocyst subunits. Cells coexpressing Chc1p-RFP and (A) Sec5p-3xGFP (NY2983) or (B) Sec15p-3xGFP (NY2984) were grown in selection medium to OD600 0.5 and imaged. Bar, 10 µm.
The lack of colocalization of Sec5p or Sec15p with clathrin led us to consider a second model in which clathrin and its associated components are not required directly for exocyst recruitment, but rather play an indirect role in which they serve to recycle some key secretory vesicle component from the plasma membrane after exocytic fusion. The recycled component would recruit the exocyst only after it had been repackaged into a new secretory vesicle. This model is consistent with the identification of yap1802Δ in our screen. Yap1802p is an AP-180/CALM homologue known to play a role in the initial stage of endocytosis at the plasma membrane (Maldonado-Báez et al., 2008).
To explore this model, we considered possible candidates known to be associated with secretory vesicles and internalized from the plasma membrane in a clathrin-dependent fashion. Snc1p and Snc2p (collectively termed Snc1/2p) are redundant synaptobrevin-related v-SNAREs required for the fusion of secretory vesicles with the plasma membrane (Protopopov et al., 1993). Prior studies have established that, after exocytic fusion, they are internalized from the plasma membrane by clathrin-coated vesicles and then recycled through endosomes and the Golgi into new secretory vesicles (Lewis et al., 2000; Burston et al., 2009). Internalization of Snc1/2p from the cell surface requires Yap1801/1802p and Ldb17p in addition to clathrin, consistent with the results of our screen (Burston et al., 2009). Because GFP-Snc1p is normally internalized by endocytosis before it has a chance to diffuse far from sites of exocytosis, it exhibits cortical localization that is strongly polarized to the bud relative to the mother cell. Defects in Snc1p internalization lead to depolarization of the cortical pool (Lewis et al., 2000), whereas defects in recycling back to the cell surface enhance localization to internal puncta, representing endosomes and Golgi. We examined GFP-Snc1p localization in the seven mutants identified in our screen (Fig. S1). Both clc1Δ and chc1Δ exhibited complete loss of GFP-Snc1p polarization and yap1802Δ, ldb17Δ and ccp1Δ exhibited partial loss of polarization. A yap1801Δ yap1802Δ double mutant exhibited more complete mislocalization, as previously reported (Burston et al., 2009). In the swa2Δ mutant GFP-Snc1p was predominantly in internal puncta, consistent with its known defect in the uncoating of clathrin-coated vesicles (Gall et al., 2000; Pishvaee et al., 2000). Only avl9Δ showed relatively normal GFP-Snc1p localization, suggesting that this mutant may act by an unrelated mechanism.
Snc1/2p function in exocyst localization
One prediction of this model is that loss of Snc1/2p should lead to a loss of Sec15-GFP patch formation. In our screen of the deletion library, we did observe a modest defect in Sec15-GFP localization in both the snc1Δ (25% mislocalization) and snc2Δ (35% mislocalization) strains (Fig. 1 B). As these genes are functionally redundant, and either one is sufficient for secretory function, the mild loss of localization observed in the single mutants is consistent with this model, but a more stringent test is needed. Because strains deleted for both SNC1 and SNC2 are nearly inviable, we used a strain in which the endogenous SNC1/2 genes are deleted yet growth is maintained by a copy of SNC1 expressed from the inducible GAL1 promoter (Protopopov et al., 1993). This strain grows normally in medium containing galactose, but growth begins to slow 4 h after a shift to glucose, a repressing carbon source. The level of Snc proteins is higher than normal during growth on galactose, but falls below normal after 8 h in glucose medium. We introduced fluorescent protein–tagged exocyst alleles into this strain and assessed their localization at 4 and 8 h after the shift. Sec6-2xmCH, Sec8-2xmCH, and Sec15-3xGFP all showed a dramatic loss in polarized localization by 8 h growth in glucose medium (Fig. 4, A and B).
Figure 4.

Snc depletion or blocking its internalization leads to exocyst mislocalization. (A and B) WT (NY2977, NY2978, and NY2979) and snc1Δ snc2Δ Gal-SNC1 cells (NY2980, NY2981, and NY2982) expressing Sec6p-2xmCherry, Sec8p-2xmCherry, or Sec15p-3xGFP, respectively, were grown in selection medium containing 4% galactose overnight at 25°C and then switched to selection medium containing 2% glucose at 30°C to block Snc1p expression. The localization of Sec6-2xmCherry, Sec8-2xmCherry, and Sec15-3xGFP was examined (A) and quantified (B) after 4 h and 8 h in glucose medium. Values represent the percentage of cells showing a fluorescent patch at the bud tip or bud neck. At least 100 cells were scored for each condition. Data represent mean ± SD from at least three independent experiments. Bar, 5 µm. (C and D) snc1Δ SNC2 (NY2986, NY2987) and snc1Δ snc2-V39A, M42A (NY2991, NY2992) mutant cells expressing Sec6-GFP (C) or Sec15-3xGFP (D) were grown to early log phase in SD medium at 25°C, and the cells were collected and directly examined by fluorescence microscopy (left). The percentage of cells with fluorescent protein concentrated at prebud site and small bud tip, mother-bud neck, spread over the entire medium bud cortex, or mislocalized (defused over the whole cell cytosol) was quantified (right) for wild type (WT) or the snc1Δ snc2-V39A, M42A double mutant (DM). Error bars represent SD. For three separate experiments, n ≈ 400 cells per count for both wild-type and mutant cells. (C) *, P < 0.005; **, P < 0.01. (D) *, P < 0.001; **, P < 0.02, Student’s t test.
Internalization of Snc1p from the plasma membrane can be specifically blocked by the combination of two mutations: V40A and M43A (corresponding to V39 and M42 in Snc2p; Grote et al., 2000b; Lewis et al., 2000). As a direct test of our proposed requirement for Snc1/2p recycling in exocyst recruitment, we examined Sec15-GFP and Sec6-GFP localization in strains deleted for SNC1 and expressing either wild-type Snc2p or Snc2-V39A, M42A (Fig. 4, C and D). Localization of both exocyst components was strongly affected in the snc1Δ snc2-V39M42A mutant. Broadly dispersed cortical localization was seen rather than the highly polarized pattern typical of wild-type cells. Thus, with respect to exocyst localization, specifically blocking Snc2p internalization largely recapitulates the loss of clathrin function.
Interaction of the exocyst with Snc proteins
The postulated recruitment of the exocyst by the Snc1/2 proteins predicts a physical interaction, either direct or indirect, between Snc1/2p and the exocyst. To test this prediction we fused the cytosolic domain of Snc2p to GST and used the purified and immobilized protein to test for binding of the exocyst complex from yeast lysates. We prepared lysates from eight different yeast strains, each expressing a different 13xmyc-tagged exocyst subunit from its own promoter as the sole copy of the gene. As controls we used beads carrying GST, GST-Sec22p (a v-SNARE that acts in ER–Golgi transport), and GST-Sso2p (a t-SNARE acting in exocytosis). With six of the lysates we observed significantly more binding of the tagged exocyst subunit to GST-Snc2p than to GST, GST-Sec22p, or GST-Sso2p (Fig. 5 A), suggesting that the largely assembled exocyst complex can specifically bind to Snc2p. Lower specific binding of Sec10p-13xmyc or Sec15p-13xmyc was observed, suggesting that either these tags interfere with binding to Snc2p or that a sub-complex lacking these subunits preferentially binds.
Figure 5.
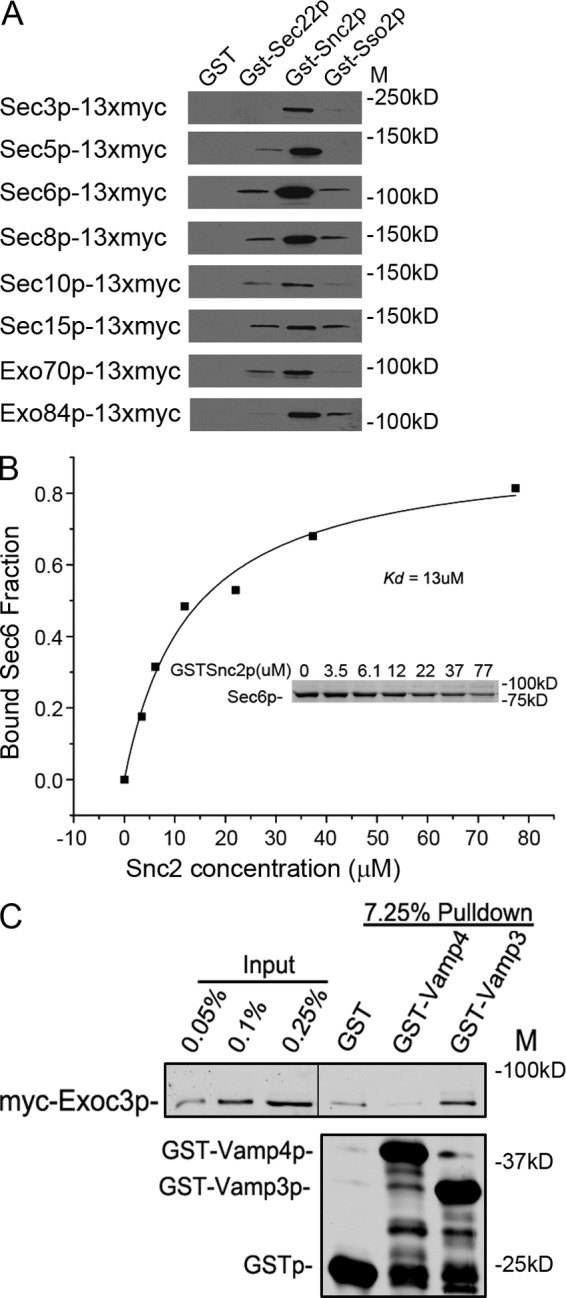
The exocyst binds to Snc2p. (A) Lysates were prepared from eight different yeast strains, each expressing, as the sole copy of the gene, a different exocyst subunit fused at the C terminus to a 13xmyc tag. The lysates were incubated with recombinant GST or GST fused to Sec22p, Snc2p, or Sso2p. The amount of GST-tagged protein was assessed by SDS-PAGE and Coomassie staining, using BSA as a standard. Based on this analysis, ∼3 µg was added to each binding reaction. Glutathione beads were added and washed and the bound proteins analyzed by immunoblot using anti-myc antibody. (B) The Kd of the Snc2p–Sec6p interaction was determined to be 13 ± 6 µM by a quantitative GST pull-down assay. 0–77 µM GST-Snc2p bound to glutathione beads was incubated with 0.4 µM Sec6p. After pelleting the beads, the unbound fraction of Sec6p was determined by scanning a Coomassie-stained gel of the supernatant (inset). The data shown are from a representative experiment of five independent repetitions. The Kd was calculated using Origin software. (C) Lysates of bacteria expressing GST, GST-Vamp4p, and GST-Vamp3p (0.2 µM final concentration) were mixed with 1 ml lysates of HeLa cells (transfected with plasmid pNB1462) expressing myc-Exoc3 from a 100-mm dish culture, and incubated at 4°C for 2 h. Glutathione beads (10 µl) were added and incubated for another hour. The beads were pelleted and washed three times with binding buffer before SDS-PAGE and Western blot analysis with anti-myc antibody. The black line in the top panel indicates where intervening lanes have been removed for presentation purposes.
To determine if the observed binding of the exocyst to GST-Snc2p is direct or indirect and to identify the subunit responsible for the interaction we tested individual recombinant exocyst subunits purified from Escherichia coli for binding to GST-Snc2p. Of the six exocyst subunits observed to bind from yeast lysates, three can be readily produced in bacteria and purified in large amounts, Sec6p, Exo70p, and Exo84p. Of these, only Sec6p showed efficient binding to GST-Snc2p (Fig. S2).
SNAREs can be characterized by the presence of either a glutamine (Q-SNARE) or arginine (R-SNARE) at the “zero” position at the center of the SNARE four-helix bundle (Fasshauer et al., 1998; Sutton et al., 1998). Assembled SNARE complexes typically contain one R-SNARE and three Q SNARE domains, Qa, Qb, and Qc. By this characterization, Snc1p and Snc2p are R-SNAREs. We tested for binding of Sec6p to all of the other R-SNAREs of yeast, Sec22p, Ykt6p, and Nyv1p, as well as to Sso1p and Sso2p, the redundant syntaxin homologues that serve as Qa-SNARES in exocytic fusion. No interaction was seen with any of these constructs except for very weak binding to GST-Nyv1p (Fig. S2). Using a range of GST-Snc2p concentrations in equilibrium binding reactions we determined that the binding of Sec6p is saturable with a Kd of ∼13 µM (Fig. 5 B). Because Snc1p is very similar in sequence and is functionally redundant with Snc2p in vivo, we also tested the interaction of Snc1p with Sec6p. GST-Snc1p also bound Sec6p, although the interaction appeared to be of somewhat lower affinity than the Snc2p–Sec6p interaction (Fig. S3).
To explore the evolutionary conservation of the Sec6p–Snc2p interaction we expressed the human Sec6 homologue, Exoc3, in Hela cells and mixed a cell lysate with lysates of bacteria expressing GST, GST-VAMP3, or GST-VAMP4. After pull-down of the GST constructs, we observed binding of Exoc3 to VAMP3, the vSNARE of the constitutive secretory pathway, but not to its close relative, VAMP4, or to GST (Fig. 5 C).
Defining the binding sites
To define the region of Sec6p that binds to Snc2p we expressed and purified several truncated constructs. Full-length Sec6p bound to Snc2p, yet deletion of 100, 291, or 410 residues from the N terminus prevented binding (Fig. 6 A). This result demonstrates that the N-terminal 1–100 region of Sec6p is necessary for the interaction with Snc2p. The N-terminal half of Sec6p cannot be produced alone as a soluble protein in E. coli; however, when coexpressed with a portion (aa 1–236) of its nearest neighbor in the exocyst complex, Sec8p, a soluble complex containing either Sec6p (1–292) or Sec6p (1–411) can be expressed and purified. The complex containing Sec6p (1–411) and Sec8p (1–236) bound to GST-Snc2p as efficiently as full-length Sec6p, whereas the complex containing Sec6p (1–292) and Sec8p (1–236) showed very little binding (Fig. 6 A). The Sec8p fragment was also detected in the precipitate with Sec6p (1–411). Taken together, these results imply that Snc2p binding requires both the region from aa 1–100 as well as the region from aa 292–411 of Sec6p. Furthermore, our results indicate that the interaction is not blocked by the interaction of Sec8p with Sec6p, a conclusion that is also consistent with the precipitation of at least six of the eight exocyst subunits, including Sec8p, from yeast lysates by GST-Snc2p (Fig. 5 A).
Figure 6.

The N terminus of Sec6p binds to the end of the Snc2p SNARE domain. (A) Full-length or truncated regions of Sec6p were expressed in bacteria as 6xHis-tagged constructs. The 1–411 and 1–292 fragments (indicated by asterisks) of Sec6p were coexpressed with a fragment of Sec8p (1–236) that binds to them and helps to maintain their solubility. The purified Sec6p fragments or Sec6p–Sec8p complexes (50 nM final concentration) were mixed with Snc2p-GST or Sec22p-GST (50 nM final concentration) in a 1-ml binding assay. The bound proteins were recovered on glutathione beads and analyzed by immunoblot analysis. The region from 1–411 of Sec6p is sufficient for Snc2p binding, whereas both the regions from 1–100 and 292–411 are required. (B) Fragments of Snc2p fused to GST were expressed in bacteria, purified, and mixed at 80 nM (final concentration) with 50 nM (final concentration) His-tagged Sec6p in a 1-ml binding assay. After precipitation of the GST-tagged constructs with glutathione beads (shown in the bottom panel by Coomassie staining of a gel in which intervening lanes have been removed for presentation purposes), the recovery of Sec6p was determined by immunoblot. The region from 53–92 of Snc2p is both necessary and sufficient for Sec6p binding, whereas the N-terminal 1–52 fragment shows no detectable binding. Relevant lanes from the corresponding Coomassie gel are shown in the bottom panel.
To define the region of Snc2p that is involved in binding to Sec6p, we expressed various Snc2 truncations fused to GST and tested for Sec6p binding. Upstream of the SNARE motif is a short region (aa 1–28) that proved to be dispensable for binding to Sec6p (Fig. 6 B). The region upstream of the arginine (R52) at the 0 layer at the center of the SNARE motif was also entirely dispensable for Sec6p binding and, on its own, exhibited no detectable affinity for Sec6p, whereas the region between the 0 layer and the transmembrane domain (aa 53–92) binds to Sec6p with an affinity (Kd = 18 µM) comparable to that of full-length Snc2p (Kd = 13 µM; Fig. 6 B). Thus, Sec6p binds to the second half of the SNARE motif.
To further define the binding site within this region we mutated three different charged clusters to either alanines or to residues of the opposite charge. We selected residues that are not directly involved in interactions with the other components of the exocytic SNARE complex (Fig. 7 A). The E51A E54A (M5), E51R E54R (M6), E60A D61A D64A (M3), and E60K D61R D64R (M4) mutations did not significantly affect binding to Sec6. However, both R75A R79A K82A (M1) and R75E R79E K82E (M2) strongly inhibited binding to Sec6p (Fig. 7 B). These results suggest that Sec6p binds to a fairly small region close to the end of the SNARE motif of Snc2p, adjacent to its transmembrane domain. We next made six different mutations in which each of these three charged residues (R75, R79, and K82) were changed singly to either alanine or to glutamate, a residue of the opposite charge (Fig. S4). R75A reduced binding, whereas R75E blocked binding more effectively. Both R79A and R79E were effective in blocking binding. K82A had little effect on binding, whereas K82E strongly blocked binding. Thus, each of these three residues is important for binding, with R79 being somewhat more sensitive to alteration than the other two.
Figure 7.

A positively charged patch at the end of the Snc2p SNARE domain is needed for Sec6p binding, but not liposome fusion. (A) The structure of the Snc2p–Ssop–Sec9p SNARE complex is shown with the Snc2p backbone highlighted in yellow and the three charged patches on the surface of Snc2p expanded in insets with side chains shown. Images were generated by PyMol using coordinates of 3B5N from the PDB provided by Strop et al. (2008). Snc1p in the original structure was replaced with Snc2p. (B) Binding of Sec6p to Snc2p mutants. His-tagged Sec6p (80 nM final concentration) was mixed with 50 nM (final concentration) of the various Snc2p mutants or with GST-Sec22p as a control. Both M1 (R75A R79A K82A) and M2 (R75E R79E K82E) show a loss of Sec6p binding activity, whereas M3 (E60A D61A D64A), M4 (E60K D61R D64R), M5 (E51A E54A), and M6 (E51R E54R) bind normally. (C) Kinetic fusion assay comparing different donor v-SNARE liposomes containing either wild-type Snc2p or Snc2p-M2 mutant. t-SNARE liposomes containing GST-Sec9c and His6-Sso1p (45 µl) were mixed with 5 µl v-SNARE liposomes and NBD fluorescence was monitored in a fluorescent plate reader for 2 h. Fusion with t-SNAREs (∼460 pmol of t-SNARE complex proteins, ∼55.8 nmol lipid) and wild-type Snc2p (high, ∼136 pmol protein, 4.5 nmol lipid) are shown in black circles, whereas fusion with Snc2p-M2 (high, ∼192 pmol protein, 4.6 nmol lipid) are shown in dark blue. Similarly, t-SNARE liposomes with less total protein (∼200 pmol of t-SNARE complex proteins, ∼28.3 nmol lipid) were fused with wild-type Snc2 liposomes (low, 21 pmol protein, 2.2 nmol lipid, gray circles) or Snc2p-M2 liposomes (low, ∼15 pmol protein, 1.3 nmol lipid, light blue circles). Representative traces are shown. (D) Average endpoint fusion. The black and dark blue bar graphs represent fusion with high concentrations of the v-SNARE protein (wild-type and mutant) in the liposomes, whereas gray and light blue bar graphs represent fusion with low concentrations of the v-SNARE proteins (wild type and mutant) in the donor liposomes. The histograms show mean fusion at 120 min and the error bars represent SEM. The number of replicates (n) is shown at the base of the histogram. (E) Coomassie blue–stained gel of proteoliposomes on a 10% Bis-Tris NuPAGE Novex gel (Invitrogen). Lanes 1–4 contain 5 µl of proteoliposomes, whereas lanes 5 and 6 (low concentration v-SNAREs) contain 10 µl of proteoliposomes. (F) Sec6p binds to Snc2p reconstituted into liposomes. Empty liposome (0.01 mM lipid) or liposomes containing 0.4 µM Snc2p or Snc2p-M2 were incubated with 0.025 µM His-Sec6 (final concentration) at 4°C for 2 h in 1 ml binding buffer (25 mM Hepes-KOH and 200 mM KCl, pH 7.4, with 0.5 µg/µl BSA). Liposomes were pelleted at 300,000 g for 20 min in an ultracentrifuge (Optima TLX; Beckman Coulter) with a TLA 120.2 rotor. The pellet was washed with 1 ml binding buffer and re-pelleted. The pellet was resuspended in 80 µl sample buffer. 10 µl of each sample was resolved by SDS-PAGE and analyzed by Western blot using anti-His (top) and anti-Snc (bottom) antibody. (G) Quantitation of Sec6p binding.
Although the structure of the assembled SNARE complex predicts that the M2 mutations would not affect SNARE assembly, we tested the Snc2-M2 mutant in a liposome fusion assay (Scott et al., 2000) to confirm that its critical fusion functions, which would include its ability to assemble into a SNARE complex, had not been compromised. Liposomes containing either Snc2p or the Snc2-M2p mutant were mixed with liposomes containing the tSNAREs, GST-Sec9c and His6-Sso1p. Lipid mixing, indicative of liposome fusion, was assayed by an NBD fluorescence-dequenching assay. At low protein levels there was no significant difference in the rate of fusion of Snc2-M2 liposomes relative to the wild-type control, and only a small decrease in the rate of fusion was observed at high protein levels (Fig. 7, C and D).
Because the Sec6p binding site is very close to the transmembrane domain of Snc2p, we tested the ability of Sec6p to bind to Snc2p reconstituted into liposomes. Using liposomes containing no Snc2p, full-length Snc2p, or the Snc2-M2p mutant we found that Sec6p can bind specifically to Snc2p liposomes, with at least a twofold reduction in binding to the Snc2-M2p mutant (Fig. 7, F and G). Therefore, the N-terminal region of Sec6p can bind to the membrane-proximal portion of the Snc2p SNARE motif even when Snc2p is anchored in a membrane.
Phenotype of the M2 mutant
The snc2-M2 mutations were introduced into the genome at the SNC2 locus of a snc1Δ yeast strain and the mutant protein was found to be expressed at a level similar to that of wild-type Snc2p (Fig. 8 D). The snc1Δ snc2-M2 strain was viable, but exhibited growth defects at both high and low temperatures in comparison to the snc1Δ SNC2 control (Fig. 8 A). Sec6-GFP and Sec15-3xGFP constructs were introduced into these strains. Although both tagged subunits continued to display a polarized localization in the snc1Δ snc2-M2 strain, a higher fraction of the cells showed punctate labeling of the entire bud cortex rather than the highly focused localization to the bud tip or mother-bud neck typical of the snc1Δ SNC2 control (Fig. 8, B and C). The snc1Δ snc2-M2 mutant cells were also larger than the control cells and were often misshapen, consistent with a defect in the spatial regulation of cell surface assembly.
Figure 8.
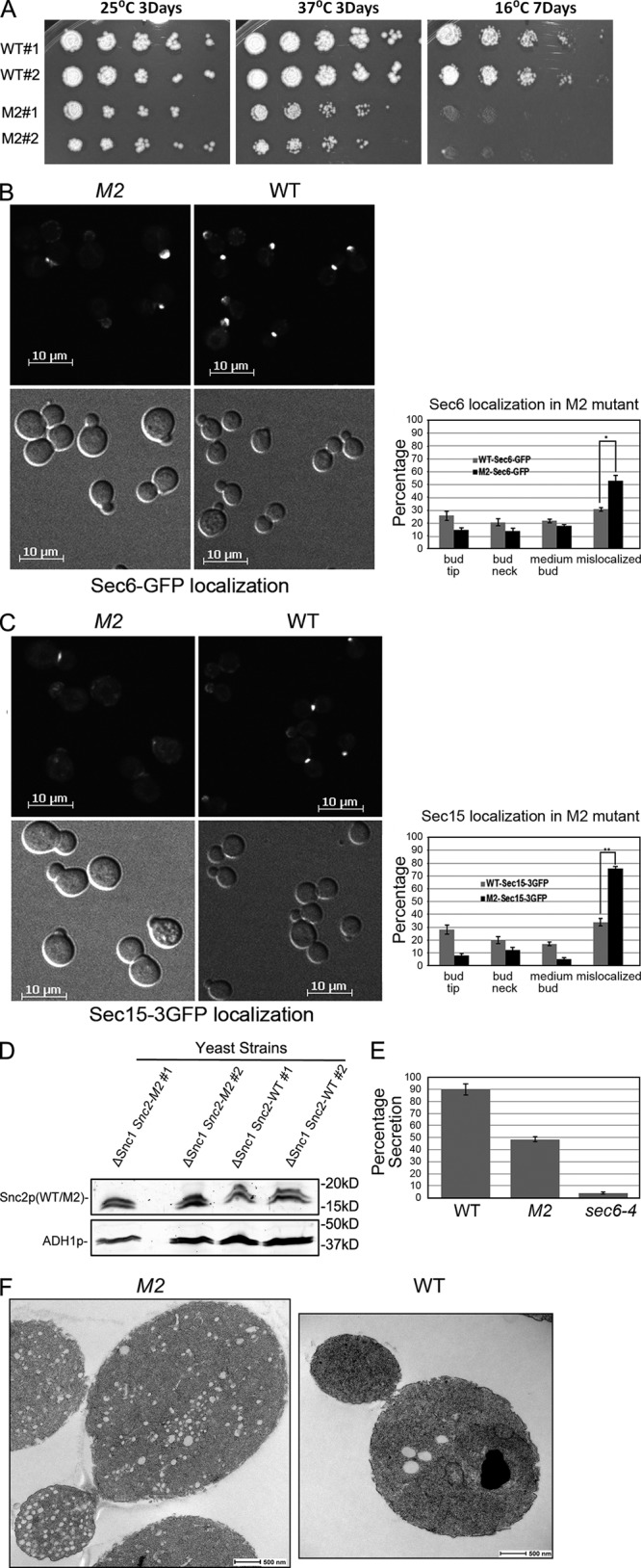
The snc2-M2 mutation inhibits growth, localization, and secretion. (A) Two independent snc1Δ SNC2 (WT, NY2985) and snc1Δ snc2-M2 (M2, NY2988) yeast strains were serially diluted by fivefold increments, plated and incubated as indicated. The snc2-M2 mutant shows growth defects at both high and low temperatures. (B) snc1Δ SNC2 (WT, NY2986) and snc1Δ snc2-M2 (M2, NY2989) yeast expressing Sec6p-GFP were grown to early log phase in SD medium at 25°C. The cells were collected and directly examined by fluorescence microscopy (left panels). The percentage of cells with Sec6p-GFP at prebud sites and small bud tips, mother-bud necks, spread over the entire medium bud cortex, or mislocalized (defused over the whole cell cytosol) was quantified (right). Error bars represent SD for two independent experiments, n = 709 for wild type and n = 792 for the snc2-M2 mutant. *, <0.02, Student’s t test. (C) snc1Δ SNC2 (WT, NY2987) and snc1Δ snc2-M2 (M2, NY2990) yeast expressing Sec15p-3xGFP were grown to early log phase in SD medium at 25°C. The cells were collected and directly examined by fluorescence microscopy (left panels). The percentage of cells with Sec15p-3xGFP at prebud sites and small bud tips, mother-bud necks, spread over the entire bud cortex, or mislocalized (defused over the whole cell cytosol) was quantified (right). Error bars represent SD for two independent experiments, n = 709 for wild type and n = 792 for the snc2-M2 mutant. **, <0.005, Student’s t test. (D) Expression of Snc2-M2p is similar to that of Snc2p. (E) Invertase assay. snc1Δ SNC2 (WT, 2985), snc1Δ snc2-M2 (M2, NY2988), and sec6-4 (NY17) yeast were grown overnight at 25°C in YP medium containing 5% glucose then transferred to YP medium containing 0.1% glucose and incubated at 37°C for 1 h. The bar graph displays the percentage of invertase synthesized during the 1-h incubation that was delivered to the cell surface. (F) snc1Δ SNC2 (NY2985, right) and snc1Δ snc2-M2 (NY2988, left) cells were grown at 25°C in YPD medium and processed for thin section electron microscopy.
The function of the secretory machinery can be assessed by measuring the efficiency of invertase secretion after a shift from repressing medium to derepressing medium. The snc1Δ SNC2 control strain secretes more than 90% of the invertase synthesized during a 1-h period of invertase derepression, whereas the snc1Δ snc2-M2 mutant secretes only ∼50%, indicating a substantial defect in the secretory machinery (Fig. 8 E).
Secretory vesicles normally fuse with the plasma membrane within a minute of their formation. Even a partial block in fusion therefore leads to a rapid accumulation of vesicles. Thin-section electron microscopy demonstrated an obvious accumulation of secretory vesicles in the snc1Δ snc2-M2 mutant cells relative to the control (Fig. 8 F).
Interaction of Sec6p with Snc2p and Sec9p
A prior study found that Sec6p interacts with the Qbc exocytic t-SNARE, Sec9p, and that this interaction requires the region of Sec6 from aa 1–300 (Sivaram et al., 2005). This interaction has a significantly higher affinity (Kd = 0.5–1 µM) than the interaction of Snc2p with Sec6p (Kd = 13 µM). Because both Snc2p and Sec9p require the N-terminal region of Sec6p for binding, we sought to determine if Sec6p can bind to both proteins at the same time or if the binding is competitive. We mixed 65 nM His-tagged Sec6p with 2.5 µM GST-Snc2p and 0–1 µM of His-tagged Sec9p. After recovery of GST-Snc2p on glutathione beads, we found no significant effect of up to 1 µM Sec9p on the binding of Sec6p to Snc2p (Fig. 9). Furthermore, Sec9p was found to associate with GST-Snc2p in the presence of Sec6p. This suggests that Sec9p and Snc2p have nonoverlapping binding sites on Sec6p and that the three proteins can form a ternary complex.
Figure 9.

Sec9p does not interfere with the Snc2p–Sec6p interaction. GST-Snc2p (2.5 µM final concentration) was incubated with 70 nM (final concentration) His-Sec6p and increasing amounts of His-MBP-Sec9c, as indicated, and then precipitated with glutathione beads. The bead-associated protein was analyzed by SDS PAGE and Coomassie staining. Black lines indicate where lanes have been rearranged for presentation purposes.
Snc and Sec4, parallel signals for exocyst recruitment
Our prior studies had suggested a role for Sec4-GTP in exocyst recruitment. Activation of Sec4p is required for patch formation upon Sec15p overexpression (Salminen and Novick, 1989) and Sec15p binds to Sec4p in its GTP-bound conformation (Guo et al., 1999b). Therefore, we considered the possibility that the Snc–Sec6p interaction acts in parallel with the Sec4–Sec15 interaction to promote exocyst recruitment. The exocyst could thereby act as a coincidence detector, requiring the combined vSNARE and rab signals for effective recruitment. A prediction of this proposal is that increasing one signal might compensate for partial loss of the other. We therefore introduced SEC4 on a high copy number plasmid into the various deletion mutants that affect exocyst localization and observed the effect on Sec15-GFP localization. Overexpression of Sec4p led to a substantial restoration of Sec15-GFP localization in all of these mutants, with the sole exception of swa2Δ (Fig. 10). For unknown reasons, overexpression of Sec4p in the swa2Δ mutant caused a severe growth defect, which may interfere with any potential restoration of Sec15-GFP localization.
Figure 10.

Overexpression of Sec4p partially restores Sec15p localization. WT or the indicated deletion mutant strains expressing Sec15-3xGFP were transformed with a 2µ circle-based plasmid overexpressing Sec4p (pNB1457) and examined by fluorescence microscopy. The percentage of cells exhibiting a Sec15p-3xGFP patch at the bud tip or mother-bud neck is shown. Data represent mean ± SD from at least three different experiments.
Discussion
We have established here a direct interaction between the Sec6p subunit of the exocyst complex and the secretory vesicle v-SNARE, Snc2p, and have shown that this interaction is critical in vivo for recruitment of the exocyst and for exocytosis. We previously showed that another exocyst subunit, Sec15p, binds to the activated form of the vesicle-associated rab, Sec4p, and proposed that this interaction is needed for recruitment of the exocyst to secretory vesicles (Salminen and Novick, 1989; Guo et al., 1999b). Results we present here suggest that the Snc proteins work in parallel with Sec4-GTP to recruit the exocyst. By binding to both Snc1/2p and Sec4p, each with relatively modest affinity, the exocyst could act as a coincidence detector, associating only with membranes carrying both key ligands. Although Snc1/2p, as a rapidly recycling, integral membrane protein, is necessarily present on both exocytic and endocytic vesicles, Sec4p is only found on the exocytic leg of this circular route and hence the exocyst preferentially associates with secretory vesicles. This asymmetry may help to establish the directionality of vesicle transport. Prior studies have demonstrated interactions of several exocyst subunits with components of the plasma membrane (Guo et al., 2001; He et al., 2007). Together, these findings help to define the molecular links by which the exocyst tethers secretory vesicles to the plasma membrane in preparation for exocytic membrane fusion.
The need to efficiently recycle Snc proteins from the plasma membrane into new secretory vesicles explains the observed requirement for clathrin in exocyst localization. Our screen identified a number of components, in addition to clathrin, that are important for Sec15-GFP localization. Yap1802p is a clathrin adaptor, homologous to the AP180/CALM adaptor proteins of mammalian cells, and has been shown to act at the plasma membrane to internalize Snc1p (Maldonado-Báez et al., 2008; Burston et al., 2009). Aux1–Swa2p is the yeast homologue of auxillin, a protein needed for disassembly of clathrin coats and therefore required for ongoing cycles of clathrin function (Gall et al., 2000; Pishvaee et al., 2000). We also identified Ldb17p, and although this protein is not as well studied, it too colocalizes with clathrin at sites of endocytosis and has been shown to be required for endocytosis of Snc1p (Burston et al., 2009). YHL017W encodes a protein we have named Ccp1 that has been shown to colocalize with clathrin in yeast (Huh et al., 2003), and although nothing more is known regarding its function, we observe a depolarization of cortical GFP-Snc1 in the ccp1 mutant that suggests a defect in internalization. The one component we identified that may act at a different site is Avl9p. This protein is required for the formation of a subset of secretory vesicles (Harsay and Schekman, 2007). The failure to form these vesicles may result in a substantial reduction in the pool of vesicle-associated exocyst components and hence a loss of Sec15-GFP localization.
Two prior studies in animal cells have noted functional connections between the exocyst and clathrin or its associated adaptors. Drosophila oocytes take up large amounts of yolk proteins through clathrin-mediated endocytosis of the yolk receptor, Yolkless. The exocyst subunit Sec5 was found to localize to clathrin-coated pits and vesicles at the plasma membrane of the oocyte (Sommer et al., 2005). Nonetheless, a sec5 mutation did not disrupt internalization of Yolkless. Rather, in a sec5 mutant, the receptor was found to accumulate in secretory granules that were blocked in exocytic fusion with the plasma membrane. No physical interaction between the exocyst and clathrin or the AP-2 adaptor was observed. These results, like those we report here, are consistent with a model in which the exocyst is recruited by a component that is concentrated in clathrin-coated pits on the plasma membrane and internalized by a clathrin-dependent mechanism. As in yeast, the relevant cargo of the clathrin-coated vesicles may be the v-SNARE required for exocytic fusion. The greatly enhanced level of endocytosis and membrane recycling in oocytes may exaggerate the pool of exocytic v-SNAREs associated with clathrin-coated pits and vesicles. The resulting high concentration of the v-SNARE might bypass the requirement for a coincident rab signal in exocyst recruitment. In this way, the exocyst would be recruited onto endocytic compartments, but still wouldn’t function until it recycles with the v-SNARE to an exocytic compartment. Although, in this situation, the exocyst might be recruited to membranes lacking exocytic rabs, activation by an exocytic rab would still be needed to initiate its tethering function.
Polarized epithelial cells rely on the exocyst for export to their basolateral surface. Sorting of cargo by the AP-1 clathrin adaptor occurs in the trans-Golgi network (TGN) and recycling endosomes. The AP-1 complex has two isoforms that differ in their µ subunit: AP-1A, which acts at the TGN, and AP-1B, which acts at the recycling endosome. LLC-PK1 cells, which lack the µ1B subunit, fail to recruit the exocyst to the recycling endosome and are defective in sorting to the basolateral surface (Fölsch et al., 2003). Although expression of µ1B was found to restore exocyst localization in LLC-PK1 cells, no direct interaction of the exocyst with clathrin or the AP-1B adaptor was detected in this study. These results are consistent with a role for clathrin and the AP-1B adaptor in sorting a key component, possibly the exocytic v-SNARE, into vesicles destined for the basolateral surface. The v-SNARE would then serve to recruit the exocyst and thereby prepare the vesicles for tethering and fusion to the basolateral plasma membrane.
Sec6p interacts with a number of different components including itself, other members of the exocyst complex, such as Sec8p, Sec10p, and Exo70p, and other components of the exocytic machinery such as Snc1/2p, Sec9p, and Sec1p (Dong et al., 2005; Sivaram et al., 2005; Munson and Novick, 2006; Morgera et al., 2012). It will be important to define when in the exocytic reaction and where in the cell each of these interactions occurs, as well as which interactions are mutually compatible and which are not. We have shown here that the interaction of Sec6p and Snc2p is compatible with the interaction of Sec6p and the Qbc-SNARE Sec9p, suggesting a possible role in SNARE complex assembly. Such a role would be fully consistent with the rapid drop in SNARE complex levels after a shift of sec6-4 cells to their restrictive temperature (Grote et al., 2000a). Nonetheless, it has been reported that addition of the pure Sec6p homodimer actually slows the formation in vitro of the Sso1p–Sec9p binary tSNARE complex that serves as the rate-limiting intermediate in assembly of the ternary Snc1p–Sso1p–Sec9p complex (Sivaram et al., 2005). We show here that the interaction of Sec6p with Snc2p can occur within the context of a largely assembled exocyst complex. Clearly, more studies will be needed to understand the roles of the other components of the exocyst and other elements of the exocytic machinery in the SNARE assembly reaction. Other, structurally related, tethering complexes have been shown to bind to both the v-SNARE and t-SNAREs that act at the same stage of membrane traffic (Sivaram et al., 2005; Yu and Hughson, 2010). These interactions may serve an important role in linking the transport vesicle, initially through the tether, to the target compartment and in catalyzing the subsequent assembly of the SNARE complex.
Materials and methods
Yeast strains, construction, and media
The strains and plasmids used in this study are listed in Tables 1–5. NY2977 and NY2978 were obtained by transforming plasmids pNB1459 and pNB1460, respectively, into NY438, whereas NY2979 was made by transforming pNB1458 into NY1745. NY2980 and NY2981 were constructed by transforming pNB1459 (pRS305-Sec6-2xmCherry) and pNB1460 (pRS305-Sec8-2xmCherry), respectively, into NY1127. NY2982 was obtained by crossing NY2979 with NY1127. The strains were grown in selection media containing 4% galactose at 25°C. To turn off the inducible GAL1 promoter, cells were diluted in selection medium containing 2% glucose starting at an OD600 of 0.1. At 2-h intervals the growth rate was determined by measuring the OD600 and cells were collected for images and immunoblot analysis. Wild-type strains were analyzed under the same conditions. NY2983 and NY2984 were obtained by transforming pNB1304 and pNB1308, respectively into strain EY1669 (Huh et al., 2003). pJM997 (Snc2-M2-His6) was generated by PCR using oligos 93 (NcoI-Snc2 5′-GGCCATGGGTTCATCATCAGTGCCATACG-3′) and 94 (Snc2-XhoI 5′-GAGATATCCTCGAGGCTGAAATGGACGACGATAGG-3′) with PNB1461 (Snc2-M2) as template. The Snc2-M2 PCR product was digested with NcoI and XhoI and ligated into pET28a-based vector (pJM235) digested with the same enzymes. Deletion of both yap1801 and 1802 was performed by disrupting the YAP1801 gene in the yap1802 deletion mutant using a PCR product to replace the ORF of YAP1801.
Table 1.
Deletion strains carrying GAL1-Sec15-GFP
| Strain | Deleted gene | Strain | Deleted gene | Strain | Deleted gene |
| HY1 | apl1 | HY29 | kin2 | HY57 | myo5 |
| HY2 | apl2 | HY30 | laa1 | HY58 | cap2 |
| HY3 | apl3 | HY31 | scd6 | HY59 | twf1 |
| HY4 | apl4 | HY32 | skt5 | HY60 | sac6 |
| HY5 | apl5 | HY33 | sro7 | HY61 | syp1 |
| HY6 | apm1 | HY34 | swa2 | HY62 | bbc1 |
| HY7 | apm2 | HY35 | YML037C | HY63 | end1 |
| HY8 | apm3 | HY36 | ccp1 | HY64 | snx4 |
| HY9 | apm4 | HY37 | YBL010C | HY65 | msh4 |
| HY10 | aps1 | HY38 | sla1 | HY66 | aat2 |
| HY11 | aps2 | HY39 | ede1 | HY67 | aat1 |
| HY12 | aps3 | HY40 | inp5 | HY68 | vac8 |
| HY13 | avl9 | HY41 | aim21 | HY69 | prb1 |
| HY14 | chc1 | HY42 | yap1801 | – | – |
| HY15 | chs1 | HY43 | yap1802 | – | – |
| HY16 | chs3 | HY44 | ldb17 | – | – |
| HY17 | chs5 | HY45 | tpm1 | – | – |
| HY18 | chs7 | HY46 | cap1 | – | – |
| HY19 | clc1 | HY47 | lsb3 | – | – |
| HY20 | drs2 | HY48 | crn1 | – | – |
| HY21 | dss4 | HY49 | aip1 | – | – |
| HY22 | ent1 | HY50 | bzz1 | – | – |
| HY23 | ent2 | HY51 | pkh2 | – | – |
| HY24 | ent3 | HY52 | rvs161 | HY80 | ptm1 |
| HY25 | ent5 | HY53 | vrp1 | HY81 | snc1 |
| HY26 | gga1 | HY54 | rvs167 | HY82 | snc2 |
| HY27 | gga2 | HY55 | arc18 | – | – |
| HY28 | kin1 | HY56 | abp1 | – | – |
Table 5.
Bacterial strains used
| Strain | Plasmid | Host Strain |
| NRB1462 | pAT110-snc2-R75A,R79A,K82A(M1); inserted into BamHI–XhoI sites of pAT110;kanr | Rosetta |
| NRB1463 | pAT110-snc2-R75E,R79E,K82E(M2); inserted into BamHI–XhoI sites of pAT110;kanr | Rosetta |
| NRB1464 | pAT110-snc2-E60A,D61A,D64A(M3); inserted into BamHI–XhoI sites of pAT110;kanr | Rosetta |
| NRB1465 | pAT110-snc2-E60K,D61R,D64R(M4); inserted into BamHI–XhoI sites of pAT110;kanr | Rosetta |
| NRB1466 | pAT110-snc2-E51A,E54A(M5); inserted into BamHI–XhoI sites of pAT110;kanr | Rosetta |
| NRB1467 | pAT110-snc2-E51R,E54R(M6); inserted into BamHI–XhoI sites of pAT110;kanr | Rosetta |
| NRB1468 | pAT110-snc2-R75A; inserted into BamHI–XhoI sites of pAT110;kanr | Rosetta |
| NRB1469 | pAT110-snc2-R75E; inserted into BamHI–XhoI sites of pAT110;kanr | Rosetta |
| NRB1470 | pAT110-snc2-R79A; inserted into BamHI–XhoI sites of pAT110;kanr | Rosetta |
| NRB1471 | pAT110-snc2-R79E; inserted into BamHI–XhoI sites of pAT110;kanr | Rosetta |
| NRB1472 | pAT110-snc2-K82A; inserted into BamHI–XhoI sites of pAT110;kanr | Rosetta |
| NRB1473 | pAT110-snc2-K82E; inserted into BamHI–XhoI sites of pAT110;kanr | Rosetta |
| NRB1474 | pQlink-His-TEV-Sec6 (1–805 aa); Ampr | Rosetta |
| NRB1475 | pQlink-His-TEV-Sec6 (292–805 aa); Ampr | Rosetta |
| NRB1476 | pQlink-His-TEV-Sec6 (411–805 aa); Ampr | Rosetta |
| NRB1477 | pQlink-His-TEV-Sec6 (1–411 aa); pCDF-GST-PreScission-Sec8(1–236 aa); Strepr,Ampr | Rosetta |
| NRB1478 | pQlink-His-TEV-Sec6 (1–292 aa); pCDF-GST-PreScission-Sec8(1–236 aa); Strepr,Ampr | Rosetta |
| NRB1480 | pCDF-GST-PreScission-Sec6(100–805 aa); Strepr | Rosetta |
| NRB1481 | pAT110-SNC2 (1–92 aa); inserted into BamHI–XhoI sites of pAT110;kanr | Rosetta |
| NRB1482 | pAT110-SNC2 (28–92 aa); inserted into BamHI–XhoI sites of pAT110;kanr | Rosetta |
| NRB1483 | pAT110-SNC2 (1–52 aa); inserted into BamHI–EcoRI sites of pAT110;kanr | Rosetta |
| NRB1484 | pAT110-SNC2 (53–92 aa); inserted into BamHI–XhoI sites of pAT110;kanr | Rosetta |
| NRB1485 | pET46-Sec6;Ampr | Rosetta |
| NRB1486 | pSV282-Sec9CT (416–651 aa); inserted into BamHI–EcoRI sites of pSV282;kanr | Rosetta |
| NRB1487 | pAT110;kanr | Rosetta |
| NRB1488 | pAT110-Vamp3; human Vamp3 cDNA inserted into BamH1–EcoR1 sites of pAT110 kanr | Rosetta |
| NRB1489 | pAT110-Vamp4; human Vamp4 cDNA inserted into the BamH1–Xho1 sites of pAT110 kanr | Rosetta |
Table 2.
The selected deletion strains expressing Sec15-3xGFP or co-expressing Sec4
| Strains | ORF |
| NY3002 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 Sec15-3xGFP::URA3 |
| NY3003 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 apm4Δ::kanMX4 Sec15-3xGFP::URA3 |
| NY3004 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 avl9Δ::kanMX4 Sec15-3xGFP::URA3 |
| NY3005 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 chc1Δ::kanMX4 Sec15-3xGFP::URA3 |
| NY3006 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 clc1Δ::kanMX4 Sec15-3xGFP::URA3 |
| NY3007 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 swa2Δ::kanMX4 Sec15-3xGFP::URA3 |
| NY3008 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ccp1Δ::kanMX4 Sec15-3xGFP::URA3 |
| NY3009 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 yap1802Δ::kanMX4 Sec15-3xGFP::URA3 |
| NY3010 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ldb17Δ::kanMX4 Sec15-3xGFP::URA3 |
| NY3011 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 Sec15-3xGFP::URA3 Sec4::LEU2 |
| NY3012 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 apm4Δ::kanMX4 Sec15-3xGFP::URA3 Sec4::LEU2 |
| NY3013 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 avl9Δ::kanMX4 Sec15-3xGFP::URA3 Sec4::LEU2 |
| NY3014 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 chc1Δ::kanMX4 Sec15-3xGFP::URA3 Sec4::LEU2 |
| NY3015 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 clc1Δ::kanMX4 Sec15-3xGFP::URA3 Sec4::LEU2 |
| NY3016 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 swa2Δ::kanMX4 Sec15-3xGFP::URA3 Sec4::LEU2 |
| NY3017 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ccp1Δ::kanMX4 Sec15-3xGFP::URA3 Sec4::LEU2 |
| NY3018 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 yap1802Δ::kanMX4 Sec15-3xGFP::URA3 Sec4::LEU2 |
| NY3019 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ldb17Δ::kanMX4 Sec15-3xGFP::URA3 Sec4::LEU2 |
Table 3.
Yeast strains used
| Strain | Genotype |
| NY17 | Mata, ura3-52, sec6-4 |
| NY438 | MATa ura3 ade8 leu2 trp1 his3 |
| NY1745 | MATα ura3 ade8 leu2 tr1 his3 |
| NY1127 | MATa ura3 ade8 leu2 trp1 his3 snc1::URA3 snc2::ADE8 (2μ GAL1p-SNC1-HA-TRP1) |
| NY2977 | MATa ura3 ade8 leu2 trp1 his3 SEC6-2xmCherry::LEU2 |
| NY2978 | MATa ura3 ade8 leu2 trp1 his3 SEC8-2xmCherry::LEU2 |
| NY2979 | MATα ura3 ade8 leu2 tr1 his3 SEC15-3xGFP::HIS3 |
| NY2980 | MATa ura3 ade8 leu2 trp1 his3 snc1::URA3 snc2::ADE8 (2μ GAL1p- SNC1-HA-TRP1) SEC6-2xmCherry::LEU2 |
| NY2981 | MATa ura3 ade8 leu2 trp1 his3 snc1::URA3 snc2::ADE8 (2μ GAL1p- SNC1-HA-TRP1) SEC8-2xmCherry::LEU2 |
| NY2982 | MATa ura3 ade8 leu2 trp1 his3 snc1::URA3 snc2::ADE8 (2μ GAL1p- SNC1-HA-TRP1) SEC15-3xGFP::HIS3 |
| NY2983 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 CHC1-mRFP1::kanMX6 SEC5-3xGFP::URA3 |
| NY2984 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 CHC1-mRFP1::kanMX6 SEC15-3xGFP::URA3 |
| NY2501 | Mata GAL+ ura3-52 leu2-3,112 his3Δ200 SEC5::13myc(HIS3) |
| NY2502 | Mata GAL+ ura3-52 leu2-3,112 his3Δ200 SEC6::13myc(HIS3) |
| NY2503 | Mata GAL+ ura3-52 leu2-3,112 his3Δ200 SEC8::13myc(HIS3) |
| NY2504 | Mata GAL+ ura3-52 leu2-3,112 his3Δ200 SEC10::13myc(HIS3) |
| NY2505 | Mata GAL+ ura3-52 leu2-3,112 his3Δ200 SEC15::13myc(HIS3) |
| NY2506 | Mata GAL+ ura3-52 leu2-3,112 his3Δ200 EXO70::13myc(HIS3) |
| NY2507 | Mata GAL+ ura3-52 leu2-3,112 his3Δ200 EXO84::13myc(HIS3) |
| NY2985 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 snc1Δ::kanMX4 |
| NY2986 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 snc1Δ::kanMX4 SEC6-GFP::URA3 |
| NY2987 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 snc1Δ::kanMX4 SEC15-3GFP::HIS3 |
| NY2988 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 snc1Δ::kanMX4 snc2-M2(75E79E82E) |
| NY2989 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 snc1Δ::kanMX4 snc2-M2(75E79E82E) SEC6-GFP::URA3 |
| NY2990 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 snc1Δ::kanMX4 snc2-M2(75E79E82E) SEC15-3GFP::HIS3 |
| NY2991 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 snc1Δ::kanMX4 snc2-V39A,M42A SEC6-GFP::URA3 |
| NY2992 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 snc1Δ::kanMX4 snc2-V39A,M42A SEC15-3GFP::HIS3 |
| NY2993 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 yap1801::his5 yap1802::kanMX4 SEC15-3xGFP::URA3 |
| NY2994 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 avl9::kanMX4 GFP-SNC1::URA3 |
| NY2995 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 chc1::kanMX4 GFP-SNC1::URA3 |
| NY2996 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 clc1::kanMX4 GFP-SNC1::URA3 |
| NY2997 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 swa2::kanMX4 GFP-SNC1::URA3 |
| NY2998 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ccp1::kanMX4 GFP-SNC1::URA3 |
| NY2999 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 yap1802::kanMX4 GFP-SNC1::URA3 |
| NY3000 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 yap1801::HIS5 yap1802::kanMX4 GFP-SNC1::URA3 |
| NY3001 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ldb17::kanMX4 GFP-SNC1::URA3 |
Table 4.
Plasmids for yeast transformation or HeLa cell transfection
| Name | Plasmid |
| pNB300 | YCp50, GAL1-Sec15 |
| PNB882 | pRS306 SEC6-GFP |
| pNB1304 | pRS306 SEC5-3xGFP |
| pNB1308 | pRS306 SEC15-3xGFP |
| pNB1456 | YCp50 GAL1-SEC15-GFP; a fragment of Sec15-GFP into BamH I–SalI sites of pNB300 |
| pNB1457 | pRS425 Sec4, a 392-bp Sec4 fragment was ligated into BamHI–XhoI sites |
| pNB1458 | pRS303 Sec15-3xGFP; inserting Sec15-3xGFP-ADHterm fragment into pRS303 SalI–SacI sites |
| pNB1459 | pRS305 Sec6-2xmCherry; inserted 1.6 kB Sec6 C-terminal fragment into XhoI–XbaI sites of pJC1-2xmCherry |
| pNB1460 | pRS305 Sec8-2xmCherry; inserted 1 kB Sec8 C-terminal fragment into XhoI–XbaI sites of pJC1-2xmCherry |
| pNB1461 | pRS306 Snc2-M2(75E79E82E) |
| pNB 1462 | pCMV-myc-Exoc3 |
| pSFNB1223 | pRS416 GFP-SNC1 |
Screening of yeast deletion library
The Saccharomyces cerevisiae strain BY4741 (MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0) and its derivatives were used in this study (Thermo Fisher Scientific). The 73 strains selected were grown at 25°C in YPD medium supplemented with G418 (200 µg/ml) to OD600 ∼1.0 and transformed with pNB1456 (constructed by fusing GFP to Sec15p in pNB300) using the lithium-acetate protocol. After appropriate selection, cells were grown overnight in SC medium containing 2% raffinose at 25°C to reach an absorbance of approximately OD600 1.0 (Abs600 nm), then diluted to OD600 0.1 in SC medium containing 2% raffinose and 2% galactose at 25°C. After 5 h, cells were collected at OD600 0.5–0.8 and Sec15-GFP localization was examined by fluorescence microscopy. To examine the localization of Sec15 expressed at normal levels under its own promoter, the eight strains identified in the initial screen were transformed with the integrating vector pNB1308.
Live-cell fluorescence microscopy and quantitative analysis
Yeast cells were grown at 25°C to early log phase (OD600 0.3–0.6). 500 µl of cells were pelleted and resuspended in growth medium. Fluorescence images were acquired with a 63× oil immersion objective (Plan Apochromat 63×/1.4 oil DIC; Carl Zeiss) on a spinning disc confocal microscopy system (Yokagawa Corporation of America), which included a microscope (Observer Z1; Carl Zeiss) equipped with an electron multiplying CCD camera (QuantEM 512SC; Photometrics). Excitation of GFP or mCherry was achieved using 488-nm argon and 568-nm argon/krypton lasers, respectively. For each sample, a z-stack of 14–18 slices with a 300-nm slice distance was generated. Images were analyzed using AxioVision software 4.8 (Carl Zeiss). Exocyst localization was scored based on localization at the tips of small buds or the necks of large buds. 100–200 cells were examined for each condition and at least three separate experiments were used to calculate the SD.
Protein purification from E. coli
GST-Snc2p, GST-Snc2p fragments, His-Sec6p, His-Sec6p fragments, and His-MBP-Sec9ct (416–651 aa) were expressed and purified from corresponding E. coli strains. In brief, cells were grown at 37°C to OD600 0.8, then 0.2 mM IPTG (final concentration) was added to induce protein expression and the culture was incubated at 16°C for 12 h with shaking. Cells were harvested by centrifugation for 10 min at 3,500 rpm and resuspended in binding buffer (20 mM Pipes, pH 6.8, 100 mM NaCl, 1% NP-40, 1 mM DTT, and 2 mM PMSF; for His-tagged protein purification no DTT was added) at a cell density of 40 OD600 U/ml. Cells were sonicated for 20-s intervals with 20-s breaks for a total of 2 min. Lysates were cleared by centrifugation at 15,000 rpm for 30 min at 4°C. The target protein was purified with glutathione–Sepharose 4B beads (GE Healthcare) for GST fusion proteins or Ni-NTA Agarose beads (QIAGEN) for His-tagged proteins. The His-Sec6 fragments of 1–411 aa (NRB1477) and 1–292 aa (NRB1478) were coexpressed with a Sec8 (1–236 aa) fragment, purified with glutathione beads and cut with PreScission protease (GE Healthcare). The protease is GST tagged and was removed with glutathione beads. The concentration of purified protein was determined by SDS-PAGE with BSA as standard. GST-Sec9c (BB442; Katz et al., 1998), pJM88, pJM81 (McNew et al., 2000), and pJM997 were expressed and purified as described previously (Scott et al., 2003) with the following modifications: cells expressing pJM81 and pJM997 were induced overnight at 20°C with 0.2 mM IPTG, and cells expressing pJM88 were induced for 4 h at 37°C with 0.3 mM IPTG. All his-tagged proteins were purified using an ÄKTAprime low-pressure chromatography system (GE Healthcare) and 1-ml HiTrap HP metal-chelating column (GE Healthcare).
Yeast lysate preparation
50 ml yeast cultures were grown overnight to 1 OD600, harvested by centrifugation at 3,000 rpm for 5 min, and washed once with distilled water and resuspended in 1.4 ml ice-cold binding buffer (20 mM Pipes, pH 6.8, 100 mM NaCl, 1% NP-40, 1 mM DTT, and 2 mM PMSF) with protease inhibitor cocktail (Roche) in a 2-ml screw cap tube (Thermo Fisher Scientific). 2 g zirconia/silica 0.5-mm beads (Biospec Products, Inc.) were added and agitated for 3 min with a mini beadbeater (Biospec Products, Inc.) at 4°C. Lysates were cleared by centrifugation at 14,000 rpm for 20 min and protein concentrations were determined using the Bio-Rad protein assay with BSA as a standard.
GST pull-down assays
GST-Snc2p pull-down of exocyst subunits.
GST-Snc2p (purified from NRB1481, final concentration 0.2 µM) and yeast lysate (derived from NY2501, NY2502, NY2503, NY2504, NY2505, and NY2506, respectively, final concentration 2 µg/µl) were mixed in a total volume of 1 ml binding buffer (20 mM Pipes, pH 6.8, 100 mM NaCl, 1% NP-40, 1 mM DTT, and 2 mM PMSF) with protease inhibitor cocktail (Roche) and incubated on a rotator at 4°C for 2 h. Then 20 µl of a 50% suspension of glutathione beads was added and incubated at 4°C for another 2 h. The beads were washed four times with 1.4 ml binding buffer. The bound protein was analyzed by Western blot with anti-myc antibody (9E10). GST-Sec22p and GST-Sso2p were used as negative controls.
Determination of the dissociation constant (Kd) for Snc2p–Sec6p interaction.
A quantitative GST pull-down assay (Pollard, 2010) was used to determine the Kd for Snc2p–Sec6p binding. In brief, GST-Snc2p (0–77 µM final concentration, purified from NRB1481) bound to glutathione beads was mixed with His-Sec6p (0.4 µM final concentration, purified from E. coli, NRB1485) in a total of 1 ml binding buffer and incubated on a rotator at 4°C for 2 h. The beads were pelleted by centrifugation at 5,000 rpm for 1 min and the supernatant protein was separated by SDS-PAGE and stained with Coomassie brilliant blue G250 (Bio-Rad Laboratories). The amount of Sec6p was quantified with ImageJ (National Institutes of Health) after scanning the gel and the Kd was calculated using Origin software (OriginLab Corporation).
Snc2p pull-down of Sec6p fragments.
GST-Snc2p (0.05 µM final concentration, purified from NRB1481) and His-Sec6 fragments (0.05 µM final concentration, purified from NRB1474, NRB1475, NRB1476, NRB1477, NRB1478, and NRB1480) were mixed in 1 ml binding buffer and the pull-down was performed as described above. GST-Sec22p was used as a negative control.
Vamp3 pull-down of Exoc3.
C-terminal Myc-tagged human Exoc3 was overexpressed by transfecting HeLa cells with a pCMV-Myc-Exoc3 plasmid (pNB1462). 36 µl FuGENE 6 transfection reagent (Promega) and 8 µg plasmid were used to transfect HeLa cells in 100-mm culture dishes at 80% confluence. The cells were harvested after 24 h, washed twice with 5 ml ice cold PBS, and lysed with 1 ml binding buffer (25 mM Hepes/KOH, pH 7.4, 150 mM NaCl, 1% NP-40, and 1 mM DTT, with protein inhibitor cocktail [Roche]). The lysate was cleared by centrifugation at 13,000 g for 15 min, mixed with bacteria lysates expressing GST (NRB1487), GST-Vamp4p (NRB1489), and GST-Vamp3p (NRB1488; 0.2 µM final concentration) and incubated at 4°C for 2 h. Glutathione beads (10 µl) were added and incubated for another hour. The beads were pelleted and washed three times with binding buffer before SDS-PAGE and Western blot analysis with anti-myc antibody.
Pull-down of Sec6p by Snc2p fragments or mutants.
GST-Snc2p fragments (0.08 µM final concentration, purified from NRB1481, NRB1482, NRB1483, and NRB1484, respectively) or GST-Snc2p mutants (purified from NRB1462–1473, respectively) were used to perform pull-down assays with Sec6p (purified from NRB1485) as described above.
Competition assay of Snc2 and Sec9 for Sec6 binding.
GST-Snc2p (from NRB1481) was bound to glutathione beads and purified. The beads were mixed with a constant amount of purified His-Sec6 (from NRB1485) and increasing amounts of His-MBP-Sec9ct (from NRB1486) in a total of 1 ml binding buffer (20 mM Pipes, pH 6.8, 100mM NaCl, 1% NP-40, 1 mM DTT, and 2 mM PMSF). The pull-down was performed as described above. Sec6p and Sec9ct pulled down by the beads were detected by Western blot using anti-His antibody (ABGENT).
Reconstitution and fusion
Proteoliposomes were produced as described previously (Scott et al., 2003). For t-SNARE liposomes, 250 µl of His8-Sso1p (15 mg/ml) in 1% n-octyl-β-d-glucopyranoside (OG) and 250 µl GST-Sec9c (23 mg/ml) were mixed for ∼15 h to form a t-SNARE complex and used to resuspend an acceptor lipid mix (POPC/DOPS [1-palmitoyl, 2-oleoyl phosphatidylcholine]/[1,2-dioleoyl phosphatidylserine], 85:15). For v-SNARE proteoliposomes, 50 µl of Snc2p-His6 (1 mg/ml) or 100 µl of Snc2p-M2-His6 (0.7 mg/ml) in 1% OG was used to resuspend a donor lipid mix (N-(7-nitro-2,1,3-benzoxadiazole-4-yl)-1,2-dipalmitoyl phosphatidylethanolamine [NBD-DPPE] and N-(lissamine rhodamine B sulfonyl) 1,2-dipalmitoyl phosphatidylethanolamine [Rhodamine-DPPE], 82:15:1.5:1.5) Proteoliposomes were formed by dilution and detergent removal achieved by flow dialysis overnight. Proteoliposomes were isolated by flotation in a discontinuous Nycodenz gradient. Protein content of the proteoliposomes was determined using an amido black protein assay (Schaffner and Weissmann, 1973) and lipid recovery determined by scintillation counting of trace tritiated 1,2-dipalmitoyl phosphatidlycholine added to the lipid mixes.
Fusion assays were also performed as described previously (Scott et al., 2003). 5 µl of labeled v-SNARE liposomes were mixed with 45 µl of unlabeled t-SNARE liposomes directly in a microtiter plate on ice. The plate was placed directly in the 37°C fluorescence plate reader (TECAN with Magellan software) without any prior preincubation.
Invertase assays
Invertase assays were adapted from the procedure described by Cai et al. (2005). In brief, cells were grown overnight at 25°C in YPD media (5% glucose) to early log phase (0.3–0.8 OD/ml). 1 OD of cells was pelleted, washed with 2 ml distilled water, and split into two equal aliquots and pelleted again. In one tube the cells were resuspended in 1 ml 10 mM NaN3 and put on ice. In the other tube cells were resuspended in 1 ml YP 0.1% glucose and grown at 37°C with shaking for 1 h. Then the cells were pelleted, resuspended in 1 ml 10 mM NaN3, and put on ice. For each sample, the external invertase was measured directly. The internal invertase was measured after cells were converted to spheroplasts and lysed in 0.5% Triton X-100.
Electron microscopy
Yeast cells of the snc1Δ snc2-M2 mutant (NY2988) and snc1Δ SNC2 (NY2985) control strains were grown at 25°C in YPD medium and processed for electron microscopy as described by Chen et al. (2012). Cells were fixed in 0.1 M cacodylate, 4% glutaraldehyde, pH 6.8, and the cell walls were enzymatically removed. The cells were then treated with 2% OsO4 and then 2% uranyl acetate before dehydration in ethanol and embedding in Spurr resin. Sections were stained with lead citrate and UrAc. Images were acquired using a G2 Spirit transmission electron microscope (Tecnai) equipped with a CCD (charge-coupled device) camera (Ultra Scan 4000; Gatan, Inc.).
Online supplemental material
Fig. S1 shows wild-type and mutant strains expressing GFP-Snc1p, imaged by fluorescence microscopy. Fig. S2 shows binding of His-tagged Sec6p, Exo70p, and Exo84p to beads carrying GST, GST-Sec22p, GST-Ykt6p, GST-Snc2p, GST-Sso1p, GST-Sso2p, or GST-Nyv1p. Fig. S3 shows binding of His-tagged Sec6p to beads carrying GST or GST-Snc1p. Fig. S4 shows binding of His-tagged Sec6p to GST-Sec22p, GST-Snc2p wild type, or to the indicated GST-Snc2p mutants. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201211148/DC1. Additional data are available in the JCB DataViewer at http://dx.doi.org/10.1083/jcb.201211148.dv.
Supplementary Material
Acknowledgments
We thank Y. Jones in the Electron Microscopy Facility in the Department of Cellular and Molecular Medicine at the University of California at San Diego, headed by M. Farquhar, for the preparation of samples for electron microscopy. We also thank E. O’Shea for the Chc1p-RFP strain.
This work was supported by grants from the National Institutes of Health (GM35370 and GM082861 to P. Novick and GM071832 to J.A. McNew).
References
- Boyd C., Hughes T., Pypaert M., Novick P. 2004. Vesicles carry most exocyst subunits to exocytic sites marked by the remaining two subunits, Sec3p and Exo70p. J. Cell Biol. 167:889–901 10.1083/jcb.200408124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burston H.E., Maldonado-Báez L., Davey M., Montpetit B., Schluter C., Wendland B., Conibear E. 2009. Regulators of yeast endocytosis identified by systematic quantitative analysis. J. Cell Biol. 185:1097–1110 10.1083/jcb.200811116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H., Zhang Y., Pypaert M., Walker L., Ferro-Novick S. 2005. Mutants in trs120 disrupt traffic from the early endosome to the late Golgi. J. Cell Biol. 171:823–833 10.1083/jcb.200505145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr C.M., Rizo J. 2010. At the junction of SNARE and SM protein function. Curr. Opin. Cell Biol. 22:488–495 10.1016/j.ceb.2010.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Novick P., Ferro-Novick S. 2012. ER network formation requires a balance of the dynamin-like GTPase Sey1p and the Lunapark family member Lnp1p. Nat. Cell Biol. 14:707–716 10.1038/ncb2523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong G., Hutagalung A.H., Fu C., Novick P., Reinisch K.M. 2005. The structures of exocyst subunit Exo70p and the Exo84p C-terminal domains reveal a common motif. Nat. Struct. Mol. Biol. 12:1094–1100 10.1038/nsmb1017 [DOI] [PubMed] [Google Scholar]
- Fasshauer D., Sutton R.B., Brunger A.T., Jahn R. 1998. Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q- and R-SNAREs. Proc. Natl. Acad. Sci. USA. 95:15781–15786 10.1073/pnas.95.26.15781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferro-Novick S., Jahn R. 1994. Vesicle fusion from yeast to man. Nature. 370:191–193 10.1038/370191a0 [DOI] [PubMed] [Google Scholar]
- Fölsch H., Pypaert M., Maday S., Pelletier L., Mellman I. 2003. The AP-1A and AP-1B clathrin adaptor complexes define biochemically and functionally distinct membrane domains. J. Cell Biol. 163:351–362 10.1083/jcb.200309020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall W.E., Higginbotham M.A., Chen C., Ingram M.F., Cyr D.M., Graham T.R. 2000. The auxilin-like phosphoprotein Swa2p is required for clathrin function in yeast. Curr. Biol. 10:1349–1358 [DOI] [PubMed] [Google Scholar]
- Gall W.E., Geething N.C., Hua Z., Ingram M.F., Liu K., Chen S.I., Graham T.R. 2002. Drs2p-dependent formation of exocytic clathrin-coated vesicles in vivo. Curr. Biol. 12:1623–1627 [DOI] [PubMed] [Google Scholar]
- Grote E., Carr C.M., Novick P.J. 2000a. Ordering the final events in yeast exocytosis. J. Cell Biol. 151:439–452 10.1083/jcb.151.2.439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grote E., Vlacich G., Pypaert M., Novick P.J. 2000b. A snc1 endocytosis mutant: phenotypic analysis and suppression by overproduction of dihydrosphingosine phosphate lyase. Mol. Biol. Cell. 11:4051–4065 10.1091/mbc.11.12.4051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W., Grant A., Novick P. 1999a. Exo84p is an exocyst protein essential for secretion. J. Biol. Chem. 274:23558–23564 10.1074/jbc.274.33.23558 [DOI] [PubMed] [Google Scholar]
- Guo W., Roth D., Walch-Solimena C., Novick P. 1999b. The exocyst is an effector for Sec4p, targeting secretory vesicles to sites of exocytosis. EMBO J. 18:1071–1080 10.1093/emboj/18.4.1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W., Tamanoi F., Novick P. 2001. Spatial regulation of the exocyst complex by Rho1 GTPase. Nat. Cell Biol. 3:353–360 10.1038/35070029 [DOI] [PubMed] [Google Scholar]
- Gurkan C., Koulov A.V., Balch W.E. 2007. An evolutionary perspective on eukaryotic membrane trafficking. Adv. Exp. Med. Biol. 607:73–83 10.1007/978-0-387-74021-8_6 [DOI] [PubMed] [Google Scholar]
- Harsay E., Schekman R. 2007. Avl9p, a member of a novel protein superfamily, functions in the late secretory pathway. Mol. Biol. Cell. 18:1203–1219 10.1091/mbc.E06-11-1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B., Xi F., Zhang X., Zhang J., Guo W. 2007. Exo70 interacts with phospholipids and mediates the targeting of the exocyst to the plasma membrane. EMBO J. 26:4053–4065 10.1038/sj.emboj.7601834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh W.K., Falvo J.V., Gerke L.C., Carroll A.S., Howson R.W., Weissman J.S., O’Shea E.K. 2003. Global analysis of protein localization in budding yeast. Nature. 425:686–691 10.1038/nature02026 [DOI] [PubMed] [Google Scholar]
- Hutagalung A.H., Coleman J., Pypaert M., Novick P.J. 2009. An internal domain of Exo70p is required for actin-independent localization and mediates assembly of specific exocyst components. Mol. Biol. Cell. 20:153–163 10.1091/mbc.E08-02-0157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz L., Hanson P.I., Heuser J.E., Brennwald P. 1998. Genetic and morphological analyses reveal a critical interaction between the C-termini of two SNARE proteins and a parallel four helical arrangement for the exocytic SNARE complex. EMBO J. 17:6200–6209 10.1093/emboj/17.21.6200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloepper T.H., Kienle C.N., Fasshauer D. 2008. SNAREing the basis of multicellularity: consequences of protein family expansion during evolution. Mol. Biol. Evol. 25:2055–2068 10.1093/molbev/msn151 [DOI] [PubMed] [Google Scholar]
- Lewis M.J., Nichols B.J., Prescianotto-Baschong C., Riezman H., Pelham H.R. 2000. Specific retrieval of the exocytic SNARE Snc1p from early yeast endosomes. Mol. Biol. Cell. 11:23–38 10.1091/mbc.11.1.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado-Báez L., Dores M.R., Perkins E.M., Drivas T.G., Hicke L., Wendland B. 2008. Interaction between Epsin/Yap180 adaptors and the scaffolds Ede1/Pan1 is required for endocytosis. Mol. Biol. Cell. 19:2936–2948 10.1091/mbc.E07-10-1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNew J.A., Parlati F., Fukuda R., Johnston R.J., Paz K., Paumet F., Söllner T.H., Rothman J.E. 2000. Compartmental specificity of cellular membrane fusion encoded in SNARE proteins. Nature. 407:153–159 10.1038/35025000 [DOI] [PubMed] [Google Scholar]
- McNiven M.A., Thompson H.M. 2006. Vesicle formation at the plasma membrane and trans-Golgi network: the same but different. Science. 313:1591–1594 10.1126/science.1118133 [DOI] [PubMed] [Google Scholar]
- Mizuno-Yamasaki E., Rivera-Molina F., Novick P. 2012. GTPase networks in membrane traffic. Annu. Rev. Biochem. 81:637–659 10.1146/annurev-biochem-052810-093700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgera F., Sallah M.R., Dubuke M.L., Gandhi P., Brewer D.N., Carr C.M., Munson M. 2012. Regulation of exocytosis by the exocyst subunit Sec6 and the SM protein Sec1. Mol. Biol. Cell. 23:337–346 10.1091/mbc.E11-08-0670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munson M., Novick P. 2006. The exocyst defrocked, a framework of rods revealed. Nat. Struct. Mol. Biol. 13:577–581 10.1038/nsmb1097 [DOI] [PubMed] [Google Scholar]
- Novick P., Brennwald P. 1993. Friends and family: the role of the Rab GTPases in vesicular traffic. Cell. 75:597–601 10.1016/0092-8674(93)90478-9 [DOI] [PubMed] [Google Scholar]
- Novick P., Guo W. 2002. Ras family therapy: Rab, Rho and Ral talk to the exocyst. Trends Cell Biol. 12:247–249 10.1016/S0962-8924(02)02293-6 [DOI] [PubMed] [Google Scholar]
- Novick P., Field C., Schekman R. 1980. Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell. 21:205–215 10.1016/0092-8674(80)90128-2 [DOI] [PubMed] [Google Scholar]
- Pishvaee B., Costaguta G., Yeung B.G., Ryazantsev S., Greener T., Greene L.E., Eisenberg E., McCaffery J.M., Payne G.S. 2000. A yeast DNA J protein required for uncoating of clathrin-coated vesicles in vivo. Nat. Cell Biol. 2:958–963 10.1038/35046619 [DOI] [PubMed] [Google Scholar]
- Pollard T.D. 2010. A guide to simple and informative binding assays. Mol. Biol. Cell. 21:4061–4067 10.1091/mbc.E10-08-0683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protopopov V., Govindan B., Novick P., Gerst J.E. 1993. Homologs of the synaptobrevin/VAMP family of synaptic vesicle proteins function on the late secretory pathway in S. cerevisiae. Cell. 74:855–861 10.1016/0092-8674(93)90465-3 [DOI] [PubMed] [Google Scholar]
- Salminen A., Novick P.J. 1989. The Sec15 protein responds to the function of the GTP binding protein, Sec4, to control vesicular traffic in yeast. J. Cell Biol. 109:1023–1036 10.1083/jcb.109.3.1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffner W., Weissmann C. 1973. A rapid, sensitive, and specific method for the determination of protein in dilute solution. Anal. Biochem. 56:502–514 10.1016/0003-2697(73)90217-0 [DOI] [PubMed] [Google Scholar]
- Scott S.V., Nice D.C., III, Nau J.J., Weisman L.S., Kamada Y., Keizer-Gunnink I., Funakoshi T., Veenhuis M., Ohsumi Y., Klionsky D.J. 2000. Apg13p and Vac8p are part of a complex of phosphoproteins that are required for cytoplasm to vacuole targeting. J. Biol. Chem. 275:25840–25849 10.1074/jbc.M002813200 [DOI] [PubMed] [Google Scholar]
- Scott B.L., Van Komen J.S., Liu S., Weber T., Melia T.J., McNew J.A. 2003. Liposome fusion assay to monitor intracellular membrane fusion machines. Methods Enzymol. 372:274–300 10.1016/S0076-6879(03)72016-3 [DOI] [PubMed] [Google Scholar]
- Sivaram M.V., Saporita J.A., Furgason M.L., Boettcher A.J., Munson M. 2005. Dimerization of the exocyst protein Sec6p and its interaction with the t-SNARE Sec9p. Biochemistry. 44:6302–6311 10.1021/bi048008z [DOI] [PubMed] [Google Scholar]
- Sommer B., Oprins A., Rabouille C., Munro S. 2005. The exocyst component Sec5 is present on endocytic vesicles in the oocyte of Drosophila melanogaster. J. Cell Biol. 169:953–963 10.1083/jcb.200411053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strop P., Kaiser S.E., Vrljic M., Brunger A.T. 2008. The structure of the yeast plasma membrane SNARE complex reveals destabilizing water-filled cavities. J. Biol. Chem. 283:1113–1119 10.1074/jbc.M707912200 [DOI] [PubMed] [Google Scholar]
- Sutton R.B., Fasshauer D., Jahn R., Brunger A.T. 1998. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature. 395:347–353 10.1038/26412 [DOI] [PubMed] [Google Scholar]
- TerBush D.R., Maurice T., Roth D., Novick P. 1996. The Exocyst is a multiprotein complex required for exocytosis in Saccharomyces cerevisiae. EMBO J. 15:6483–6494 [PMC free article] [PubMed] [Google Scholar]
- Whyte J.R., Munro S. 2001. The Sec34/35 Golgi transport complex is related to the exocyst, defining a family of complexes involved in multiple steps of membrane traffic. Dev. Cell. 1:527–537 10.1016/S1534-5807(01)00063-6 [DOI] [PubMed] [Google Scholar]
- Yu I.M., Hughson F.M. 2010. Tethering factors as organizers of intracellular vesicular traffic. Annu. Rev. Cell Dev. Biol. 26:137–156 10.1146/annurev.cellbio.042308.113327 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.