

FRET-based sensors for cAMP and PKA activity reveal that mitochondrial subcompartments host segregated cAMP cascades with distinct functional and kinetic signatures.
Abstract
Cyclic AMP (cAMP)-dependent phosphorylation has been reported to exert biological effects in both the mitochondrial matrix and outer mitochondrial membrane (OMM). However, the kinetics, targets, and effectors of the cAMP cascade in these organellar domains remain largely undefined. Here we used sensitive FRET-based sensors to monitor cAMP and protein kinase A (PKA) activity in different mitochondrial compartments in real time. We found that cytosolic cAMP did not enter the matrix, except during mitochondrial permeability transition. Bicarbonate treatment (expected to activate matrix-bound soluble adenylyl cyclase) increased intramitochondrial cAMP, but along with membrane-permeant cAMP analogues, failed to induce measureable matrix PKA activity. In contrast, the OMM proved to be a domain of exceptionally persistent cAMP-dependent PKA activity. Although cAMP signaling events measured on the OMM mirrored those of the cytosol, PKA phosphorylation at the OMM endured longer as a consequence of diminished control by local phosphatases. Our findings demonstrate that mitochondria host segregated cAMP cascades with distinct functional and kinetic signatures.
Introduction
The classical second messenger, cAMP, has recently gained attention as a player in the control of mitochondrial function. Typically, cAMP relies on its main effector, protein kinase A (PKA). This enzyme can be free in the cytosol or confined to precise subcellular locations thanks to a family of proteins called A kinase anchoring proteins (AKAPs; Feliciello et al., 2005; Pagliarini and Dixon, 2006; O’Rourke et al., 2011). Specific AKAPs such as AKAP 121 and SPHKAP/SKIP are known to tether PKA to the outer mitochondrial membrane (OMM) and intermembrane space in proximity of local targets (Lieberman et al., 1988; Feliciello et al., 2005; Kovanich et al., 2010; Means et al., 2011). This spatial organization allows timely phosphorylation of mitochondrial proteins that regulate apoptosis (BAD; Harada et al., 1999), mitochondrial shape (Drp1; Cribbs and Strack, 2007; Chang and Blackstone, 2007), and cristae maintenance (ChChd3; Darshi et al., 2011; Means et al., 2011).
Recently, the existence of an independent intramitochondrial cAMP signaling circuit was also reported. This cascade consists of a matrix-localized cAMP source, the bicarbonate-activated soluble adenylyl cyclase (sAC; Wuttke et al., 2001; Zippin et al., 2003), PKA holoenzyme (Sardanelli et al., 2006; Acin-Perez et al., 2009a, 2011a), and the cAMP-degrading enzyme PDE2A (Acin-Perez et al., 2011b). Matrix-confined PKA was proposed to phosphorylate cytochrome c oxidase (COXIV-I) to enhance oxidative phosphorylation (OXPHOS; Acin-Perez et al., 2011a). However, prior data suggesting that PKA activation inhibits OXPHOS (Bender and Kadenbach, 2000; Robin et al., 2003; Helling et al., 2008) appear to be in striking conflict with this model.
Although the presence of a complete cAMP signaling machinery in the matrix suggests that mitochondria might be able to use a local cAMP messenger system in situ, the dynamic nature of these putative cAMP signals and how they impact downstream effectors is largely unknown. Additionally, the extent to which cAMP produced in the cytoplasm reaches different mitochondrial compartments and the question of how PKA tethered to mitochondria responds to cAMP compared with the enzyme in the bulk cytoplasm have only been partially addressed (DiPilato et al., 2004; Allen and Zhang, 2006; Acin-Perez et al., 2009b; Agnes et al., 2010).
Here we targeted fluorescence resonance energy transfer (FRET)-based cAMP and PKA activity sensors to the OMM and matrix to visualize changes in mitochondrial cAMP and PKA activity in real time in live cells. Our data demonstrate that mitochondria harbor at least two distinct cAMP microdomains (matrix and OMM) with unique signaling characteristics that separate them functionally from the cytosolic cAMP pathways.
Results & discussion
Among the most important information-bearing molecules, cAMP is known to influence mitochondrial function; however, whether cAMP penetrates the mitochondrial matrix is less clear (DiPilato et al., 2004; Acin-Perez et al., 2009b). To address this issue, we targeted the pH-insensitive FRET- and Epac-based cAMP sensor EpacH90 (van der Krogt et al., 2008) to the mitochondrial matrix. This construct (“mito-EpacH90”) colocalized with the marker MitoTracker red, which is consistent with mitochondrial localization (Fig. 1 A).
Figure 1.

Cyclic AMP produced in the cytosol does not reach the mitochondrial matrix. (A) Confocal images of HeLa cells expressing mito-EpacH90 and loaded with MitoTracker red suggesting proper localization of mito-EpacH90. (B) cAMP measurements in intact cells expressing cytosolic EpacH90 (black trace) or mito-EpacH90 (red trace; mean of five cells); typical of n = 9 experiments; 34 mito-H90, 19 Epac-H90 cells. (C) Digitonin-permeabilized HeLa cells expressing mito-D3cpv (black traces) or mito-EpacH90 (red trace; n = 6 experiments; 10 mito-D3cpv cells, 13 mito-EpacH90 cells; red trace indicates the mean of three cells). (D) Mixed populations of permeabilized HeLa cells expressing mito-D3cpv (black trace) or mito-EpacH90 (red traces; n = 3 experiments; 5 mito-D3cpv cells, 8 mito-EpacH90 cells). After Ca2+ pulses, mitochondria changed morphology and failed to retain Ca2+, (hallmarks of MPT), coincident with responses of mito-EpacH90 to exogenous cAMP. (E) Permeabilized HeLa cells expressing mito-EpacH90 (n = 5 experiments; 20 cells) subjected to increasing [Ca2+] in the presence of cAMP (10 µM). 100 µM Ca2+ induced a dramatic increase in cAMP measured by mito-EpacH90. (F) Cells expressing mito-EpacH90 bathed in Hepes-buffered normal Ringer’s solution (continuous perfusion) then switched to CO2/HCO3−-buffered Krebs-Ringer’s, inducing an apparent cAMP rise (presumably due to matrix sAC activation). Data from two representative cells are shown (n = 6 repeats; 16 mito-EpacH90 cells). Error bars indicate mean ± SD.
HeLa cells expressing mito-EpacH90 or EpacH90 (cytosolic) were mixed and seeded onto glass coverslips so that cells harboring both types of sensor could be visualized in the same microscope field. Cells expressing EpacH90 responded to the cAMP-generating agonist forskolin (FSK) in the presence of the phosphodiesterase (PDE) inhibitor 3-isobutyl-methylxanthine (IBMX) and also to the Epac-specific membrane-permeant cAMP analogue 8-(4-chloro-phenylthio)-2′-O-methyladenosine-3′-5′-cyclic monophosphate (8CPT-OMe-cAMP). However, although neighboring cells expressing mito-EpacH90 readily detected 8CPT-OMe-cAMP, they were insensitive to FSK and IBMX, which suggests that the messenger generated in the cytosol did not reach the matrix (Fig. 1 B).
We next used mixed populations of digitonin-permeabilized HeLa cells expressing either mito-EpacH90 or the matrix-targeted Ca2+ sensor mito-D3cpv (Filippin et al., 2005; Palmer and Tsien, 2006) together with the marker mCherry (Shaner et al., 2004). Mitochondria displayed the expected Ca2+ uptake (Baughman et al., 2011; De Stefani et al., 2011), indicating successful permeabilization. Meanwhile nearby cells expressing mito-EpacH90 did not respond to exogenous Ca2+ or cAMP (10 µM) but only to the membrane-permeable cAMP analogue 8-bromo-adenosine 3′-5′-cyclic monophosphate (8Br-cAMP; Fig. 1 C; see also Fig. S1 A).
Collectively, these data demonstrate that cAMP cannot permeate the inner mitochondrial membrane. This is in agreement with prior studies using indirect biochemical approaches in isolated mitochondria (Acin-Perez et al., 2009b), but contrasts previous cAMP FRET imaging measurements by DiPilato et al. (2004). This discrepancy most likely lies in the localization signal used for our cAMP sensor, which minimized the fraction of reporter mislocalized to the cytosol.
Next, we investigated whether conditions exist that permit cytosol and matrix to exchange information via cAMP. We reasoned that this communication might be most prominent during the mitochondrial permeability transition (MPT; Newmeyer and Ferguson-Miller, 2003; Hajnóczky et al., 2006). We induced MPT in digitonin-permeabilized HeLa cells using Ca2+ overload (Bopassa et al., 2005) while monitoring mitochondrial Ca2+ and cAMP contemporaneously. Upon MPT, exogenous cAMP rapidly entered the matrix (Fig. 1 D). Treatment with 10 µM Ca2+ in the presence of 10 µM cAMP did not alter the matrix cAMP content; however, addition of 100 µM Ca2+ induced massive cAMP entry after roughly 60 s (Fig. 1 E). Our findings suggest that during MPT, cAMP is free to diffuse into the matrix, intermingling these two otherwise independent cascades.
An intramitochondrial sAC has been proposed to generate cAMP in response to metabolically produced HCO3/CO2 (Acin-Perez et al., 2009b). As shown in Fig. 1 F, mito-EpacH90 responded with an increase of the FRET ratio when we switched acutely from Hepes-buffered media to HCO3−/CO2-buffered solutions. These data are consistent with the idea that bicarbonate anion is capable of eliciting cAMP signals in the matrix by allowing sAC to independently generate cAMP (Acin-Perez et al., 2009b, 2011a).
We next asked whether cAMP produced via HCO3−/CO2 would stimulate the previously reported matrix-resident PKA (Sardanelli et al., 2006; Acin-Perez et al., 2009b; Agnes et al., 2010). PKA activity was monitored using matrix-targeted versions of the FRET-based sensors AKAR3 and AKAR4 (Allen and Zhang, 2006; Depry et al., 2011; “mito-AKAR3; mito-AKAR4”; Fig. 2 A). HCO3−/CO2 caused the predicted increase in matrix cAMP content; however, contrary to our expectations, there was no increase in PKA activity in neighboring cells expressing mito-AKAR4 (identified by the marker mCherry; Fig. 2 B).
Figure 2.

PKA activity in the mitochondrial matrix measured using targeted FRET-based reporters. (A) Confocal images of HeLa cells expressing mito-AKAR4 loaded with MitoTracker red. (B) Mixed populations of HeLa cells transfected with mito-EpacH90 (black trace; mean of three cells) or mito-AKAR4 (red trace; mean of four cells) were initially bathed in Hepes-buffered solution and then switched to CO2/HCO3−-buffered solution (n = 5 experiments; 10 mito-AKAR4 cells, 12 mito-EpacH90 cells). (C) HeLa expressing mito-AKAR3 (two representative cells; red traces) or OMM-AKAR3 (two representative cells; black traces; n = 8; 20 OMM-AKAR3 cells, 20 mito-AKAR3 cells). (D) HeLa cells expressing OMM-AKAR3 (two representative cells; black traces) compared with neighboring mito-AKAR3 positive cells (two representative cells; red traces; n = 10 experiments; 24 OMM-AKAR3 cells, 28 mito-AKAR3 cells). (E) Cells expressing mito-AKAR3 (red trace; mean of three cells) or mito-EpacH90 plus mCherry (black traces; mean of six cells; typical of n = 4 experiments; 10 mito-AKAR3 cells, 16 mito-EpacH90 cells). (F) HEK cells transfected with mito-AKAR4 (red trace) or OMM-AKAR4 together with mCherry (black trace; mean of four cells); typical of n = 3 experiments; 6 OMM-AKAR4 cells, 8 mito-AKAR4 cells). Error bars indicate mean ± SD.
As controls we generated sensors targeted to the OMM (“OMM-AKAR3” and “OMM-AKAR4”). As shown in Fig. 2 C, in mixed populations of HeLa cells expressing mito-AKAR3 or OMM-AKAR3, mito-AKAR3 (red traces) did not respond to cytosolic cAMP generated by FSK/IBMX, whereas OMM-AKAR3 in neighboring cells (black traces) did. This result was expected based on the finding that cytosolic cAMP does not reach the matrix. However, 8-(4-chloro-phenylthio)-adenosine-3′-5′-cyclic monophosphate (8CPT-cAMP; a potent activator of PKA; see Fig. S1B) also failed to elicit PKA activity in the matrix as measured by mito-AKAR3 (Fig. 2 D; red traces; see also Fig. S1 C). 8CPT-cAMP rapidly reached the mitochondrial matrix (Fig. 2 E), but failed to elicit detectable PKA activity. In HEK cells, OMM-AKAR4 responded to cytosolic cAMP elevations induced by prostaglandin E2 (PGE2) and FSK/IBMX. Addition of 8CPT-cAMP did not elicit additional elevation of the ratio (likely because of saturation of the probe), whereas the PKA inhibitor N-[2-(p-Bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H89) reversed the FRET signal. However, neighboring cells expressing mito-AKAR4 again showed no ratio changes during any of these treatments (Fig. 2 F).
Although the preceding findings would imply that the matrix does not contain an endogenous PKA holoenzyme capable of responding to intramitochondrial cAMP signals, we also considered the trivial possibility that the AKAR sensors simply do not perform properly in the milieu of the matrix. As shown in Fig. S1 D, reduction of the bulky targeting signal (4Cox8) on the AKAR reporter did not alter our results, nor did we see FRET changes after delivery of exogenous cAMP to the matrix during MPT (Fig. S1 E). We also confirmed that the AKAR sensors were functional in the relatively alkaline environment of the matrix (Fig. S1 F).
As a further control, we prepared matrix-targeted versions of mCherry-tagged constitutively active PKA “mito-PKACat-mCherry” (based on the catalytic subunit α; Day et al., 2011) and the potent protein kinase inhibitor PKI (isoform α), also tagged with mCherry (“mito-PKI-mCherry”). The mitochondrial localization of these constructs was ascertained by confocal microscopy (Fig. S2, A and B).
As detailed in Fig. S2 (C and D), the cytosolic parent constructs were extremely effective in activating (cyto-PKACat-mCherry) or inhibiting (cyto-PKI-mCherry) PKA. In addition, the fact that cyto-PKACat-mCherry did not affect the FRET signal of mito-AKAR4 and that PKI targeted to the matrix did not influence cytosolic or OMM PKA activity (Fig. S2, E–H) attests to the fidelity of our targeting strategy and demonstrates that we can independently manipulate PKA activity in these different compartments.
As summarized in Fig. 3 A, mito-AKAR4 consistently reported significantly higher starting FRET ratios when coexpressed with mito-PKACat-mCherry compared with neighboring controls. This effect was completely reversed by mito-PKI-mCherry, and remained unaltered in the presence of cytosolic PKI. Importantly, matrix-targeted PKI had no effect on the initial mito-AKAR4 ratio, which suggests a lack of basal phosphorylation of the matrix-targeted sensor under resting conditions.
Figure 3.
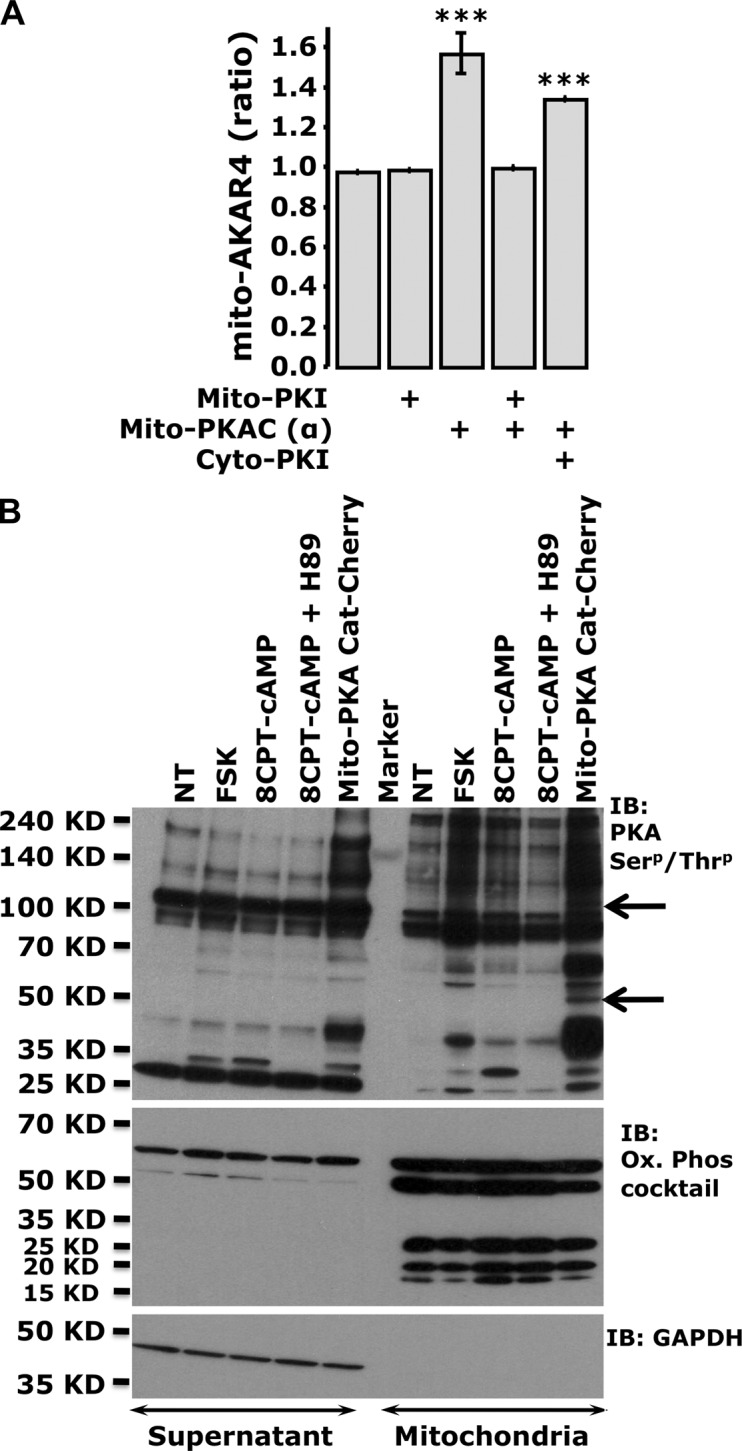
Overexpression of PKA in the matrix is detected by AKAR sensors, and induces specific phosphorylation patterns. (A) Summary of the starting mito-AKAR4 ratios in the presence of mito-PKACat-mCherry, mito-PKI-mCherry, and cyto-PKI-mCherry (***, P < 0.0002 with respect to control). Error bars indicate mean ± SD. (B) Phosphorylation status of cytosolic and mitochondria-enriched fractions of HEK cells treated with cAMP-generating agonists or cell-permeant cAMP analogues, or transfected with mito-PKACat-mCherry. A phospho-(Ser/Thr) PKA substrate antibody unveiled mito-PKACat-mCherry–dependent mitochondria-specific phosphorylation bands (arrows). The purity of mitochondria was tested using an antibody cocktail against the human OXPHOS subunits, whereas GAPDH was used to detect any cytosolic contamination (typical of n = 3 independent experiments).
Collectively, these data indicate that mito-AKAR4 is able to report PKA activity, but the endogenous PKA is either not present in the matrix of HeLa and HEK cells (contrasting previous conclusions; Acin-Perez et al., 2009b; Agnes et al., 2010; Lu et al., 2013), or its activity is so low that it cannot be detected. These two cell types are predominantly glycolytic, therefore it will be interesting to determine whether these properties differ in metabolically active cells (e.g., heart, brain) that depend principally on OXPHOS for their energy needs.
Interestingly, when we expressed mito-PKACat-mCherry and examined PKA-dependent phosphorylation patterns by Western blotting, at least two mitochondrial-specific bands were observed (Fig. 3 B). Meanwhile, activation of endogenous PKA through FSK (which generates extra-mitochondrial cAMP only) or 8CPT-cAMP (which potentially influences both the matrix and cytosol) did not yield this same mitochondrial-specific phosphorylation pattern. It is perhaps noteworthy that our measurements using a matrix-localized ATP sensor, mito-GO-ATeam2 (Nakano et al., 2011), were not consistent with an effect of matrix-specific cAMP signals on ATP production (Fig. S3, A–C). Meanwhile, expression of mito-PKACat-mCherry did alter the resting mitochondrial ATP content as measured by mito-GO-ATeam2 (Fig. S3 D). In a recent paper, Lu et al. (2013) found that the mitochondrial transcription factor TFAM can be phosphorylated by overexpression of catalytically active PKA in the matrix of HEK cells (Lu et al., 2013). However, we interpret our data above and those of Lu et al. (2013) cautiously; PKA is a very strong kinase, and its expression in restricted domains such as the matrix might result in promiscuous phosphorylation of proteins with less than optimal PKA consensus sites (Ubersax and Ferrell, 2007).
During the course of our studies, we consistently observed that OMM-AKAR4 responses were very robust compared with the cytosol, which was also noted by Allen and Zhang (2006) using AKAR3 targeted to the OMM via a different targeting strategy. As shown in Fig. 4 A, in HEK cells challenged with the cAMP-generating agonist isoproterenol (ISO; 10 nM), both OMM-AKAR4 and AKAR4 responded strongly. However upon ISO removal, OMM-AKAR4 exhibited strikingly slower termination kinetics. In HEK expressing either OMM-EpacH90 or EpacH90, all cells responded similarly to ISO, suggesting that cAMP rises and is degraded similarly in cytosol and OMM (Fig. 4 B). In HeLa cells, treatment with a low dose of FSK (2 µM) remained undetected both in cytosol and OMM because of the strong PDE activity in that cell type. However, both domains responded equally upon treatment with IBMX, unveiling the underlying cAMP production (Fig. 4 C). When we measured PKA activity using the same protocol in cells expressing OMM-AKAR4 or AKAR4 (Fig. 4 D), the termination of the signal at the OMM was again dramatically slower.
Figure 4.

Comparison of cAMP and PKA signals at the OMM and cytosol. (A) Mixed populations of HEK cells expressing OMM-AKAR4 (two representative cells; red traces) or soluble AKAR4 (two representative cells; black traces). Both sensors responded to 10 nM isoproterenol (ISO); however, the OMM-AKAR4 signal reversed with significant delay compared with AKAR4 upon ISO removal (n = 4 experiments; 14 AKAR4 cells, 16 OMM-AKAR4 cells). (B) cAMP responses to ISO measured by OMM-EpacH90 (red trace; mean of four cells) or cytosolic EpacH90 (black trace) in HEK 293 cells (n = 3 experiments; 9 EpacH90 cells, 12 OMM-EpacH90 cells). (C) cAMP kinetics measured by OMM-EpacH90 (red trace; mean of three representative cells) or cytosolic EpacH90 (black trace; mean of four representative cells) in HeLa cells (n = 3 experiments; 10 EpacH90 cells, 19 OMM-EpacH90 cells). (D) Comparison of AKAR4 and OMM-AKAR4. Termination of PKA activity by agonist removal or H89 resulted in slower FRET reversal specifically at the OMM; two representative cells are depicted (n = 3 repeats; 12 AKAR4 cells, 14 OMM-AKAR4 cells).
PDEs are major players in the generation of cAMP microdomains (Zaccolo, 2011). However, their role in the generation of persistent PKA activity at the OMM appeared to be limited. We therefore considered the role of local Ser/Thr protein phosphatases, known constituents of macromolecular complexes coordinated by AKAPs (Feliciello et al., 2005; Carlucci et al., 2008), in this process. As shown in Fig. 5 A, the potent Ser/Thr phosphatase inhibitor calyculin A attenuated the H89-induced recovery of both AKAR4 and OMM-AKAR4 in HeLa cells stimulated with FSK/IBMX. These data suggest that the termination of PKA activity at the OMM depends largely on calyculin A–sensitive phosphatases.
Figure 5.

Phosphatase-dependent termination of PKA signals at the OMM and cytosol. (A) Acute addition of 20 nM calyculin A to mixed populations of cells expressing OMM-AKAR4 (one representative cell; red trace) or AKAR4 (one representative cell; black trace; n = 6 experiments; 14 AKAR4 cells, 14 OMM-AKAR4 cells). (B) HEK cells expressing OMM-AKAR4 or AKAR4. (B, right) Mean of the slope of the responses to ISO (an estimate of PKA activity; n = 9 experiments; 40 AKAR4 cells, 22 OMM-AKAR4 cells) or 10 µM H89 on top of FSK/IBMX (an estimate of phosphatase activity). AKAR4, 39 cells; OMM-AKAR4, 25 cells; n = 12 experiments (***, P < 0.00015). (C) Mixed populations of HeLa cells expressing cyto-PKACat-mCherry together with OMM-AKAR4 (one representative cell; red trace) or with nontargeted AKAR4 (one representative cell; black trace; n = 11 repeats; 9 AKAR4 cells; 16 OMM-AKAR4 cells). The high starting ratios reversed with different kinetics upon addition of 10 µM H89 (P < 0.0002). (C, right) Mean slope of the responses to H89 across all experiments (***, P < 0.0002). Error bars indicate mean ± SD.
In HEK cells challenged with 10 nM ISO, the onset of PKA activation was similar in cytosol and OMM (Fig. 5 B, right). However, the termination of the response after PKA inhibition by H89, which reflects the local phosphatase activity, was again significantly slower at the OMM (P < 0.0002; Fig. 5 B, right). Finally AKAR4 and OMM-AKAR4 responded differently to H89 in cells expressing the catalytically active PKACat-mCherry. As expected, both sensors displayed a much higher starting ratio in the presence of PKACat-mCherry (Fig. 5 C; see also Fig. S2 C), but the H89-induced decline in the OMM-AKAR4 ratio was still significantly slower compared with that of AKAR4 (Fig. 5 C, right), which indicates that these differences did not depend entirely on local PKA activity.
Collectively, these data indicate that the OMM is a privileged cAMP signaling microdomain with high PKA activity. This microenvironment promotes cAMP-triggered activity not only by concentrating PKA in situ via specific AKAPs (Feliciello et al., 2005), but also, as demonstrated by our data, by hosting a less active Ser/Thr phosphatase population. We propose that this pro-PKA OMM maintains the phosphorylation state of local targets such Drp1, BAD, and Ca2+ release channels at ER–OMM junctions (Rizzuto et al., 1998; Harada et al., 1999; Cribbs and Strack, 2007; Chang and Blackstone, 2007; Csordás et al., 2010; Merrill et al., 2011) even in the face of waning cytosolic PKA activity.
In this work we found that the mitochondrial matrix of HEK and HeLa cells contains low or null PKA activity. However, this raises the question of how mitochondrial proteins that contain PKA consensus can be phosphorylated in the matrix (Covian and Balaban, 2012). Our finding that the OMM is rich in PKA activity together with the observation that the intermembrane space contains a functional AKAP (Means et al., 2011) raises the possibility that PKA targets might be phosphorylated during their transition into the matrix.
Materials and methods
Reagents
8Br-cAMP, 8CPT-cAMP, 8CPT-OMe-cAMP, and N6-Benzoyladenosine-3′, 5′-cyclic monophosphate (6Bnz-cAMP-AM) were purchased from Biolog Inc. and EMD Millipore. H89 was purchased from Enzo Life Sciences. All restriction enzymes were obtained from New England Biolabs, Inc. Custom-made primers and MitoTracker red/green were purchased from Invitrogen. Unless otherwise noted, all other reagents were obtained from Sigma-Aldrich.
Generation of mitochondrial-targeted FRET-based sensors
The FRET-based cAMP sensor EpacH90 was a generous gift of Kees Jalink (Netherlands Cancer Institute, Amsterdam, Netherlands). AKAR3 and AKAR4 were provided by Jin Zhang (John Hopkins University School of Medicine, Baltimore, MD). Hiromi Imamura (The Hakubi Project, Kyoto University, Kyoto, Japan) provided us with the mitochondrial ATP sensors, mito-GO-ATeam2/3. The matrix targeting signal 4Cox8 was extracted from mito-D3cpv (a gift of Giulietta Di Benedetto and Tullio Pozzan, University of Padova, and Venetian Institute of Molecular Medicine, Padua, Italy) using HindIII and introduced to the N terminus of EpacH90 (pcDNA3), AKAR3, and AKAR4 (pcDNA3′). The OMM targeting peptide yTom70 (derived from yTOMmCherry; provided by Gyorgy Hajnoczky, Thomas Jefferson University, Philadelphia, PA) was introduced into these plasmids using custom-made primers encoding this fragment and pre-designed restriction enzyme sites for HindIII. The protein kinase A inhibitor isoform alpha (purchased from Origene Technologies, Rockville, MD) was amplified with primers bearing BamHI sites and subcloned in-frame with mCherry in pcDNA3. PKACat-mCherry (PKA catalytic subunit alpha from mouse) was a gift from Susan S. Taylor (University of California at San Diego, La Jolla, CA) and Roger Y. Tsien (University of California at San Diego and Howard Hughes Medical Institute, La Jolla, CA). Matrix targeting for PKI-mCherry and PKACat-mCherry was achieved using the 4cox8 sequence. All constructs were tested functionally and verified by sequencing (Dana Farber DNA Resource Core, Boston MA).
Cell culture and transfection
HeLa and HEK293 cells were obtained from the American Type Culture Collection and were grown in DMEM + 10% FBS. Both cell lines were maintained in a humidified CO2 (5%) incubator at 37°C and were split every 2–3 d after reaching 80% confluence. All plasmids were transfected using Effectene transfection reagent (QIAGEN).
Confocal imaging
HeLa cells were seeded onto glass coverslips and transfected with sensors the next day. 24 h after transfection, the cells were loaded with 10 nM MitoTracker red or 25 nM MitoTracker green for 15 min at 37°C. Subsequently, cells were rinsed four times with Hepes-buffered Ringer’s solution containing (in mM): 125 NaCl, 25 Hepes, 10 glucose, 5 K2HPO4, 1 MgSO4, and 1 CaCl2, pH 7.40. Coverslips were mounted onto a home-built flow-through perfusion chamber, and cells were bathed in Hepes-buffered Ringer’s solution at room temperature and imaged under a 60× Plan-Apochromat (NA 1.40) oil immersion objective lens using a confocal microscope (Eclipse C1si; Nikon). Images were collected on live cells with Kalman filtering (n = 6) using the EZ-C1 software (Nikon).
Cell permeabilization
Cells were briefly rinsed with an intracellular-like buffer (125 mM KCl, 25 mM NaCl, 10 mM Hepes, 0.5 mM MgCl2, 1 mM ATP, 500 µM EGTA, and 211 µM CaCl2, pH 7.25) complemented with 5 mM succinate and 5 mM glutamate. Consequently, cells were treated with intracellular buffer containing digitonin (5–10 µg/ml). Cellular permeabilization was determined on stage by visual evaluation of the plasma membrane morphology under bright-field illumination.
Western blotting
HEK cells were transfected with mito-PKAC (a) mCherry or left untransfected. 24 h later, proteins were extracted from the transfected populations, while the nontransfected cells were treated with cAMP analogues (8CPT-cAMP, 8Br-cAMP, or 6Bnz-cAMP) or FSK in the presence or absence of the PKA inhibitor H89 for 1 h at 37°C. Subsequently, cells were lysed using RIPA buffer (Sigma-Aldrich) complemented with a protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific). Cell lysates were sonicated and insoluble material was removed by centrifugation at 14,000 g for 10 min at 4°C. Mitochondria were extracted using a commercial mitochondria isolation kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Total cell lysates or mitochondria-enriched fractions were resolved on 5–20% Tris-glycine SDS-PAGE gels (Wako Chemicals, Inc.) and electroblotted onto polyvinylidene fluoride (PVDF) membranes (Hybond-P; GE Healthcare). After transfer, PVDF membranes were blocked for 3 h at room temperature in 5% BSA–Tris-buffered saline/Tween 20 (TBST; 10 mM Tris HCl, pH 8.0/150 mM NaCl/0.1% Tween 20). Next, the membranes were incubated overnight at 4°C with a phospho-(Ser/Thr) PKA substrate antibody diluted 1:700 in 5% BSA-TBST. The day after, the membranes were washed four times with TBST and incubated at room temperature for 1 h with a peroxidase-conjugated secondary antibody (1:2,000; Santa Cruz Biotechnology, Inc.). Peroxidase activity was detected with enhanced chemiluminescence (ECL Advance Western blotting detection kit; GE Healthcare). GAPDH antibody (1:4,000; Santa Cruz Biotechnology, Inc.) was used as a loading control for the total cell lysates and to detect cytosolic protein contamination in the mitochondrial enriched samples, whereas a total OXPHOS Human WB antibody cocktail against complexes I–V (1:700; Abcam) was used to determine mitochondrial enrichment.
Ratio imaging
Cells were mounted onto a home-built perfusion chamber and bathed in Hepes-buffered Ringer’s solution containing (in mM): 125 NaCl, 25 Hepes, 10 glucose, 5 K2HPO4, 1 MgSO4, and 1 CaCl2, pH 7.40. For the experiments of sAC activation, we acutely switched the superfusion from Hepes Ringer’s to Krebs-Henseleit solution containing (in mM): 120 NaCl, 2.09 K2HPO4, 0.34 KH2PO4, 24 NaHCO3, 1 MgSO4, 1 CaCl2, and 10 d-glucose. Krebs-Henseleit solution was gassed continuously with 95% O2/5% CO2 to maintain a pH of 7.4. Real-time FRET imaging experiments were performed at room temperature using fluorescence ratio imaging systems built around TE200 and TE2000-U inverted fluorescence microscopes (both from Nikon) equipped with a QuantEM 512 camera (Photometrics) or an ORCA ER camera (Hamamatsu Photonics), respectively. MetaFluor software (Molecular Devices) was used to control filter wheels (Sutter Instrument) placed in the excitation and emission path, and to acquire ratio data. Coverslips were mounted in a home-built flow-through perfusion chamber, and cells were imaged using 40× Plan Fluor (NA 1.30) or 60× Plan-Apochromat total internal reflection fluorescence (NA 1.45) oil immersion objective lenses. FRET emission ratios for the following reporters were acquired every 5–10 s: 485nm/535 nm for EpacH90 (440 nm excitation), 535 nm/485 nm for AKAR-based and mito-D3cpv sensors (440 nm excitation), and 560 nm/510 nm for mito-GOATeam2&3 (470 nm excitation). The fluorescence of mCherry (excitation 585 nm, emission 610 nm) did not interfere with any of these measurements (Lefkimmiatis et al., 2009). Figures depict typical traces chosen from a single representative experiment that was repeated multiple times; “n” refers to the number of repeats for a given experiment.
Statistics
Data are presented as mean ± SD. Student’s t tests for unpaired data and one-way analysis of variance (Anova) with Bonferroni post hoc tests were used to determine the significance between treatments. The number of replicates and samples are indicated in figure legends (***, P < 0.0002).
Online supplemental material
Fig. S1 shows validation of mito-EpacH90 and functional validation of the mitochondrial matrix PKA activity sensors. Fig. S2 shows the mitochondrial localization and functional validation of the OMM-AKAR4 construct, constitutively active PKA, and PKA inhibitors. Fig. S3 depicts measurements of intramitochondrial free ATP using mito-GO-ATeam2 in response to FSK/IBMX (affects cytosolic cAMP) versus membrane permeant analogues (affects both cytosolic and matrix), and the effects of PKA and PKI on mitochondrial ATP content. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201303159/DC1.
Supplementary Material
Acknowledgments
We are very grateful to Giulietta Di Benedetto, Tullio Pozzan, Gyorgy Hajnoczky, and Silvana Curci for critical discussions. We thank Jon Nichols and K.-T. Chuang for reading the manuscript, and Jon Nichols and Christina Dragon for technical assistance.
This paper was supported by grants to A.M. Hofer from the VA Office of Research and Development (1 I01 BX000968-01) and from the NIH/NIDDK (1R21DK088197-0).
Footnotes
Abbreviations used in this paper:
- 8Br-cAMP
- 8-bromo-adenosine 3′-5′-cyclic monophosphate
- 8CPT-cAMP
- 8-(4-chloro-phenylthio)-adenosine-3′-5′-cyclic monophosphate
- 8CPT-OMe-cAMP
- 8-(4-chloro-phenylthio)-2′-O-methyladenosine-3′-5′-cyclic monophosphate
- AKAP
- A kinase anchoring protein
- H89
- N-[2-(p-Bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide
- IBMX
- 3-isobutyl-methylxanthine
- ISO
- isoproterenol
- MPT
- mitochondrial permeability transition
- OMM
- outer mitochondrial membrane
- OXPHOS
- oxidative phosphorylation
- PDE
- phosphodiesterase
- sAC
- soluble adenylyl cyclase
References
- Acin-Perez R., Salazar E., Brosel S., Yang H., Schon E.A., Manfredi G. 2009a. Modulation of mitochondrial protein phosphorylation by soluble adenylyl cyclase ameliorates cytochrome oxidase defects. EMBO Mol Med. 1:392–406 10.1002/emmm.200900046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acin-Perez R., Salazar E., Kamenetsky M., Buck J., Levin L.R., Manfredi G. 2009b. Cyclic AMP produced inside mitochondria regulates oxidative phosphorylation. Cell Metab. 9:265–276 10.1016/j.cmet.2009.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acin-Perez R., Gatti D.L., Bai Y., Manfredi G. 2011a. Protein phosphorylation and prevention of cytochrome oxidase inhibition by ATP: coupled mechanisms of energy metabolism regulation. Cell Metab. 13:712–719 10.1016/j.cmet.2011.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acin-Perez R., Russwurm M., Günnewig K., Gertz M., Zoidl G., Ramos L., Buck J., Levin L.R., Rassow J., Manfredi G., Steegborn C. 2011b. A phosphodiesterase 2A isoform localized to mitochondria regulates respiration. J. Biol. Chem. 286:30423–30432 10.1074/jbc.M111.266379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agnes R.S., Jernigan F., Shell J.R., Sharma V., Lawrence D.S. 2010. Suborganelle sensing of mitochondrial cAMP-dependent protein kinase activity. J. Am. Chem. Soc. 132:6075–6080 10.1021/ja909652q [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen M.D., Zhang J. 2006. Subcellular dynamics of protein kinase A activity visualized by FRET-based reporters. Biochem. Biophys. Res. Commun. 348:716–721 10.1016/j.bbrc.2006.07.136 [DOI] [PubMed] [Google Scholar]
- Baughman J.M., Perocchi F., Girgis H.S., Plovanich M., Belcher-Timme C.A., Sancak Y., Bao X.R., Strittmatter L., Goldberger O., Bogorad R.L., et al. 2011. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 476:341–345 10.1038/nature10234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender E., Kadenbach B. 2000. The allosteric ATP-inhibition of cytochrome c oxidase activity is reversibly switched on by cAMP-dependent phosphorylation. FEBS Lett. 466:130–134 10.1016/S0014-5793(99)01773-1 [DOI] [PubMed] [Google Scholar]
- Bopassa J.C., Michel P., Gateau-Roesch O., Ovize M., Ferrera R. 2005. Low-pressure reperfusion alters mitochondrial permeability transition. Am. J. Physiol. Heart Circ. Physiol. 288:H2750–H2755 10.1152/ajpheart.01081.2004 [DOI] [PubMed] [Google Scholar]
- Carlucci A., Lignitto L., Feliciello A. 2008. Control of mitochondria dynamics and oxidative metabolism by cAMP, AKAPs and the proteasome. Trends Cell Biol. 18:604–613 10.1016/j.tcb.2008.09.006 [DOI] [PubMed] [Google Scholar]
- Chang C.-R., Blackstone C. 2007. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 282:21583–21587 10.1074/jbc.C700083200 [DOI] [PubMed] [Google Scholar]
- Covian R., Balaban R.S. 2012. Cardiac mitochondrial matrix and respiratory complex protein phosphorylation. Am. J. Physiol. Heart Circ. Physiol. 303:H940–H966 10.1152/ajpheart.00077.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cribbs J.T., Strack S. 2007. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 8:939–944 10.1038/sj.embor.7401062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordás G., Várnai P., Golenár T., Roy S., Purkins G., Schneider T.G., Balla T., Hajnóczky G. 2010. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol. Cell. 39:121–132 10.1016/j.molcel.2010.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darshi M., Mendiola V.L., Mackey M.R., Murphy A.N., Koller A., Perkins G.A., Ellisman M.H., Taylor S.S. 2011. ChChd3, an inner mitochondrial membrane protein, is essential for maintaining crista integrity and mitochondrial function. J. Biol. Chem. 286:2918–2932 10.1074/jbc.M110.171975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day M.E., Gaietta G.M., Sastri M., Koller A., Mackey M.R., Scott J.D., Perkins G.A., Ellisman M.H., Taylor S.S. 2011. Isoform-specific targeting of PKA to multivesicular bodies. J. Cell Biol. 193:347–363 10.1083/jcb.201010034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depry C., Allen M.D., Zhang J. 2011. Visualization of PKA activity in plasma membrane microdomains. Mol. Biosyst. 7:52–58 10.1039/c0mb00079e [DOI] [PubMed] [Google Scholar]
- De Stefani D., Raffaello A., Teardo E., Szabò I., Rizzuto R. 2011. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 476:336–340 10.1038/nature10230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiPilato L.M., Cheng X., Zhang J. 2004. Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signaling within discrete subcellular compartments. Proc. Natl. Acad. Sci. USA. 101:16513–16518 10.1073/pnas.0405973101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feliciello A., Gottesman M.E., Avvedimento E.V. 2005. cAMP-PKA signaling to the mitochondria: protein scaffolds, mRNA and phosphatases. Cell. Signal. 17:279–287 10.1016/j.cellsig.2004.09.009 [DOI] [PubMed] [Google Scholar]
- Filippin L., Abad M.C., Gastaldello S., Magalhães P.J., Sandonà D., Pozzan T. 2005. Improved strategies for the delivery of GFP-based Ca2+ sensors into the mitochondrial matrix. Cell Calcium. 37:129–136 10.1016/j.ceca.2004.08.002 [DOI] [PubMed] [Google Scholar]
- Hajnóczky G., Csordás G., Das S., Garcia-Perez C., Saotome M., Sinha Roy S., Yi M. 2006. Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium. 40:553–560 10.1016/j.ceca.2006.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada H., Becknell B., Wilm M., Mann M., Huang L.J., Taylor S.S., Scott J.D., Korsmeyer S.J. 1999. Phosphorylation and inactivation of BAD by mitochondria-anchored protein kinase A. Mol. Cell. 3:413–422 10.1016/S1097-2765(00)80469-4 [DOI] [PubMed] [Google Scholar]
- Helling S., Vogt S., Rhiel A., Ramzan R., Wen L., Marcus K., Kadenbach B. 2008. Phosphorylation and kinetics of mammalian cytochrome c oxidase. Mol. Cell. Proteomics. 7:1714–1724 10.1074/mcp.M800137-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovanich D., van der Heyden M.A., Aye T.T., van Veen T.A., Heck A.J., Scholten A. 2010. Sphingosine kinase interacting protein is an A-kinase anchoring protein specific for type I cAMP-dependent protein kinase. ChemBioChem. 11:963–971 10.1002/cbic.201000058 [DOI] [PubMed] [Google Scholar]
- Lefkimmiatis K., Srikanthan M., Maiellaro I., Moyer M.P., Curci S., Hofer A.M. 2009. Store-operated cyclic AMP signalling mediated by STIM1. Nat. Cell Biol. 11:433–442 10.1038/ncb1850 [DOI] [PubMed] [Google Scholar]
- Lieberman S.J., Wasco W., MacLeod J., Satir P., Orr G.A. 1988. Immunogold localization of the regulatory subunit of a type II cAMP-dependent protein kinase tightly associated with mammalian sperm flagella. J. Cell Biol. 107:1809–1816 10.1083/jcb.107.5.1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B., Lee J., Nie X., Li M., Morozov Y.I., Venkatesh S., Bogenhagen D.F., Temiakov D., Suzuki C.K. 2013. Phosphorylation of human TFAM in mitochondria impairs DNA binding and promotes degradation by the AAA+ Lon protease. Mol. Cell. 49:121–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Means C.K., Lygren B., Langeberg L.K., Jain A., Dixon R.E., Vega A.L., Gold M.G., Petrosyan S., Taylor S.S., Murphy A.N., et al. 2011. An entirely specific type I A-kinase anchoring protein that can sequester two molecules of protein kinase A at mitochondria. Proc. Natl. Acad. Sci. USA. 108:E1227–E1235 10.1073/pnas.1107182108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill R.A., Dagda R.K., Dickey A.S., Cribbs J.T., Green S.H., Usachev Y.M., Strack S. 2011. Mechanism of neuroprotective mitochondrial remodeling by PKA/AKAP1. PLoS Biol. 9:e1000612 10.1371/journal.pbio.1000612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano M., Imamura H., Nagai T., Noji H. 2011. Ca2+ regulation of mitochondrial ATP synthesis visualized at the single cell level. ACS Chem. Biol. 6:709–715 10.1021/cb100313n [DOI] [PubMed] [Google Scholar]
- Newmeyer D.D., Ferguson-Miller S. 2003. Mitochondria: releasing power for life and unleashing the machineries of death. Cell. 112:481–490 10.1016/S0092-8674(03)00116-8 [DOI] [PubMed] [Google Scholar]
- O’Rourke B., Van Eyk J.E., Foster D.B. 2011. Mitochondrial protein phosphorylation as a regulatory modality: implications for mitochondrial dysfunction in heart failure. Congest. Heart Fail. 17:269–282 10.1111/j.1751-7133.2011.00266.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarini D.J., Dixon J.E. 2006. Mitochondrial modulation: reversible phosphorylation takes center stage? Trends Biochem. Sci. 31:26–34 10.1016/j.tibs.2005.11.005 [DOI] [PubMed] [Google Scholar]
- Palmer A.E., Tsien R.Y. 2006. Measuring calcium signaling using genetically targetable fluorescent indicators. Nat. Protoc. 1:1057–1065 10.1038/nprot.2006.172 [DOI] [PubMed] [Google Scholar]
- Rizzuto R., Pinton P., Carrington W., Fay F.S., Fogarty K.E., Lifshitz L.M., Tuft R.A., Pozzan T. 1998. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 280:1763–1766 10.1126/science.280.5370.1763 [DOI] [PubMed] [Google Scholar]
- Robin M.-A., Prabu S.K., Raza H., Anandatheerthavarada H.K., Avadhani N.G. 2003. Phosphorylation enhances mitochondrial targeting of GSTA4-4 through increased affinity for binding to cytoplasmic Hsp70. J. Biol. Chem. 278:18960–18970 10.1074/jbc.M301807200 [DOI] [PubMed] [Google Scholar]
- Sardanelli A.M., Signorile A., Nuzzi R., Rasmo D.D., Technikova-Dobrova Z., Drahota Z., Occhiello A., Pica A., Papa S. 2006. Occurrence of A-kinase anchor protein and associated cAMP-dependent protein kinase in the inner compartment of mammalian mitochondria. FEBS Lett. 580:5690–5696 10.1016/j.febslet.2006.09.020 [DOI] [PubMed] [Google Scholar]
- Shaner N.C., Campbell R.E., Steinbach P.A., Giepmans B.N.G., Palmer A.E., Tsien R.Y. 2004. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22:1567–1572 10.1038/nbt1037 [DOI] [PubMed] [Google Scholar]
- Ubersax J.A., Ferrell J.E., Jr 2007. Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 8:530–541 10.1038/nrm2203 [DOI] [PubMed] [Google Scholar]
- van der Krogt G.N.M., Ogink J., Ponsioen B., Jalink K. 2008. A comparison of donor-acceptor pairs for genetically encoded FRET sensors: application to the Epac cAMP sensor as an example. PLoS ONE. 3:e1916 10.1371/journal.pone.0001916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuttke M.S., Buck J., Levin L.R. 2001. Bicarbonate-regulated soluble adenylyl cyclase. JOP. 2(4, Suppl):154–158 [PubMed] [Google Scholar]
- Zaccolo M. 2011. Spatial control of cAMP signalling in health and disease. Curr. Opin. Pharmacol. 11:649–655 10.1016/j.coph.2011.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zippin J.H., Chen Y., Nahirney P., Kamenetsky M., Wuttke M.S., Fischman D.A., Levin L.R., Buck J. 2003. Compartmentalization of bicarbonate-sensitive adenylyl cyclase in distinct signaling microdomains. FASEB J. 17:82–84 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.