


Abstract
Post-transcriptional inhibition of HIV-1 replication can be achieved by RNA interference (RNAi). The cellular expression of short interfering RNA (siRNA) or short hairpin RNA (shRNA) homologous to regions of the HIV-1 genome decreases viral replication by the selective degradation of targeted RNA. Here, we demonstrate that another class of noncoding regulatory RNA, termed microRNA (miRNA), can be used to deliver antiviral RNAi. By incorporating sequences encoding siRNA targeting the HIV-1 transactivator protein tat into a human miR-30 pre-microRNA (pre-miRNA) backbone, we were able to express tat siRNA in cells. The tat siRNA delivered as pre-miRNA precursor was 80% more effective in reducing HIV-1 p24 antigen production than tat siRNA expressed as conventional shRNA. Our results confirm the utility of expressing HIV-1 specific siRNA through a miR-30 precursor stem–loop structure and suggest that this strategy can be used to increase the antiviral potency of RNAi.
INTRODUCTION
Post-transcriptional regulation of gene expression can be mediated by small noncoding RNAs such as short interfering RNA (siRNA) and microRNA (miRNA). siRNA are short RNA duplexes that direct the degradation of homologous mRNA through the RNA interference (RNAi) pathway (1,2). miRNA, in contrast, are single-stranded RNAs that bind to partially complementary (50–85%) 3′ untranslated regions of mRNA leading to translational repression without target degradation (3–6). Both siRNA and miRNA are ∼22 nt in length and are produced from longer double-stranded precursor RNA by the multidomain ribonuclease III enzyme Dicer (7–9). The ability of siRNA to specifically degrade selected mRNA species has rendered RNAi an indispensable tool and has allowed investigators to study the biological effects of gene disruption in vitro and in vivo. Furthermore, the ability to stably express siRNA/short hairpin RNA (shRNA) in human cells using viral vectors raises the possibility of using RNAi as a form of gene therapy to selectively inhibit the expression of disease specific genes such as oncogenes (10).
Several recent reports have demonstrated that the cellular expression of siRNA or shRNA targeting HIV-1 genes such as gag, nef, tat and rev inhibits viral replication (11–17). These studies have involved the introduction of synthetic siRNA or the genetic transfer of DNA expression cassettes capable of producing siRNA/shRNA in human cells. We hypothesized that another potential approach for delivering siRNA and inhibiting HIV-1 could involve the use of the structure of pre-microRNAs (pre-miRNAs). miRNAs have been identified in a wide range of organisms including Caenorhabditis elegans, Drosophila melanogaster and Homo sapiens. Though >100 different miRNAs have been identified, little is known regarding their function. An exception has been described in the experimental model system C.elegans where two miRNAs (lin-4 and let-7) are temporally expressed and repress the translation of mRNA encoding proteins involved in the developmental timing pathway of the worm (18,19).
A unique structural feature of miRNAs is their initial transcription as a long primary transcript (pri-miRNA) that is then processed by the nuclear enzyme Drosha into ∼70 nt precursor stem–loop hairpin RNAs (pre-miRNA) (20). Upon nuclear export, pre-miRNA is further processed by Dicer to yield the mature miRNA. One example of a miRNA is the miR-30 species that has been identified in the human HeLa cell line (5). Zeng et al. (21) have recently demonstrated that the miR-30 miRNA backbone can be altered to include sequences encoding siRNA and have used this approach to effectively degrade mRNA of endogenous human genes such as the polypyrimidine tract binding protein. McManus et al. have demonstrated that siRNA sequences incorporated into the stem of mir-26a function as effectors of RNAi with the specific degradation of homologous mRNA (4). In both reports, the nucleotide sequence introduced into the stem was 100% complementary to the targeted gene, a feature thought to be critical for commencing the RNAi cascade (22).
No reports, to our knowledge, have directly compared the efficiency of HIV-1 gene silencing that results from conventional shRNAs compared with siRNAs incorporated into a miRNA backbone. To determine whether any differences exist in these two approaches, we replaced a part of the miR-30 pre-miRNA stem sequence with a siRNA duplex sequence targeting the HIV-1 transactivator protein tat. Two miR-tat RNA expression constructs were designed that differed only with respect to the sequence of the miR-30 terminal loop (Fig. 1). The miR-tat RNA expression vectors were tested along with a previously described conventional tat shRNA expression vector in mammalian cell culture (15). We found that tat siRNA was more effectively processed from a pre-miRNA precursor as opposed to a shRNA transcript. In viral challenge experiments, we demonstrate that miR-tat RNA is 80% more potent in inhibiting HIV-1 replication than tat siRNA expressed as a conventional shRNA precursor.
Figure 1.

Schematic representation of predicted short hairpin tat and pre-micro mir-30/tat RNA sequences. Underlined arrows indicate sense and antisense tat sequences embedded in a shRNA or a miR-30 precursor RNA backbone. Bold letters in miA-tat and miB-tat represent stem–loop sequences derived from pre-miR-30. G:U wobbles are indicated with a star.
MATERIALS AND METHODS
Construction of tat shRNA and tat pre-miRNA expression cassettes
The 21 nt long HIV-1 tat sequence expressed as shRNA or pre-miRNA corresponds to nucleotide positions 5993–6013 of HIV-1NL4.3. To generate the tat-shRNA expression plasmid, two oligonucleotides, SHT-S, 5′-CTA GAG ACG AAG AGC TCA TCA GAA CAT TCA AGA GAT GTT CTG ATG AGC TCT TCG TCT TTT TC-3′, and SHT-AS, 5′-TCG AGA AAA AGA CGA AGA GCT CAT CAG AAC ATC TCT TGA ATG TTC TGA TGA GCT CTT CGT CT-3′ were annealed and introduced into XbaI/XhoI sites of plasmid pAAV-U6-MCS, as described previously (17).
The two miR-tat constructs (miA-tat and miB-tat) were generated by PCR extension of two partly complementary oligonucleotides. To create expression plasmid miA-tat, we used the oligonucleotide pair MIA-S, 5′-GTG AAC TCT AGA GCG AGA CGA AGA GCT CAT CAG AAC AGT GAA GCC ACA GAT GTG TTC TGA-3′ and MIA-AS, 5′-ATT GGA CTC GAG GCA GGA CGA AGA GCT CAT CAG AAC ACA TCT GTG GCT TCA CTG TTC TGA-3′; for miB-tat MIB-S, 5′-GTG AAC TCT AGA GCG AGA CGA AGA GCT CAT CAG AAC ACT GTG AAG CCA CAG ATG GGT GTT-3′ and MIB-AS, 5′-ATT GGA CTC GAG GCA CGA CGA AGA GCT CAT CAG AAC ACC CAT CTG TGG CTT CAC AGT GTT-3′. Oligonucleotides were annealed and primer extended with the following thermal profile, 95°C for 5 min, 50°C for 30 s, 68°C for 20 min, using Pfx-Taq polymerase (Invitrogen, Carlsbad, CA). Products were gel purified, restriction enzyme digested with XbaI/XhoI and introduced into plasmid pAAV-U6-MCS. A control vector, pAAV-U6-luc-Neo, was generated that synthesizes shRNA targeting the luciferase gene, as described previously (16).
Cell culture and transfection
293 cells were trypsinized and plated at 4 × 105 cells per well in six-well plates 24 h prior to transfection in DMEM containing 10% fetal bovine serum. One microgram of sh-tat or miA/B-tat DNA was co-transfected with 1 µg of HIV-1NL4.3 using 6 µl of Lipofectamine 2000 (Gibco-Invitrogen). Two days later, cell-free supernatant was collected and viral production was quantified by measuring HIV-1 p24 antigen levels in cellular supernatant by ELISA (Beckman-Coulter, Fullerton, CA) according to the manufacturer’s instructions.
Western blot
Supernatant was harvested from 293 cells transfected with HIV-1NL4.3 and passed through a 0.45 µm filter prior to western blot analyses. Protein extracts were prepared by washing cells with phosphate-buffered saline and lysing in TNT lysis buffer (20 mM Tris/200 mM NaCl/1% Triton X-100) containing protease inhibitors. After 20 min on ice, lysates were spun at 15 000 g for 15 min to remove insoluble material. Protein concentrations were determined by the Bradford procedure. Proteins were resolved on 12% SDS–polyacrylamide gels and transferred onto Hybond PVDF membranes (Amersham, Piscataway, NJ). For sequential probing, blots were stripped in Restore Western Blot Stripping Buffer (Pierce, Rockford, IL). A monoclonal antibody to HIV-1 p24 (183 clone H12-C) was obtained through the NIH AIDS Reagent and Reference program and used at a 1/100 dilution. The monoclonal antibody to α-tubulin (clone DM7 1A) (Sigma, St Louis, MO) was used at a 1/500 dilution. Immunodetection was performed using the peroxidase-based ECL Plus detection system (Amersham) according to the manufacturer’s instructions.
Real-time RT–PCR for intracellular HIV-1 RNA
Total cellular RNA was isolated from 293 cells with Trizol reagent (Invitrogen) and treated with RQ1 DNase I (Promega, Madison, WI) according to the manufacturer’s protocol. One microgram of DNase I-treated RNA was added to a reverse transcription (RT) reaction containing Powerscript Reverse Transcriptase (Clontech, Palo Alto, CA), 1 mM each of dNTP, 1× First-Strand buffer (Clontech), 200 ng of random hexamers (Promega), 10 U RNasin (Promega). RT was performed at 42°C for 1 h followed by heat-inactivation of the RT enzyme at 70°C for 15 min. PCR was performed with 1 µl of cDNA in a final volume of 30 µl and contained 1× Titanium Taq PCR buffer (Clontech), 20 pmol of sense primer Tat-A, 5′-ATG GAG CCA GTA GAT CCT A-3′, and antisense primer Tat-B, 5′-TGC TTT GAT AGA GAA ACT TGA TG-3′, 1 mM dNTPs, SYBR Green I (1:75 000), 10 nM fluorescein, and 1× Titanium Taq polymerase (Clontech). To normalize the samples for absolute RNA amount, a GAPDH-PCR was performed with primers G1, 5′-GAT TTC TCC CCC TTC TGC TGA TG-3′ and G2, 5′-CCT TGG CTG GGG GTG CTA A-3′. An external standard curve was created using spectrophotometrically determined copy number standards of plasmid pNL4.3. Real-time PCR was carried out in an iCycler (Bio-Rad, Hercules, CA) using the following thermal cycling profile: 95°C for 1 min, followed by 40 cycles of amplification (95°C for 15 s, 63°C for 30 s, 68°C for 30 s). All samples were run in triplicate.
Northern blot analysis of tat siRNA expression
Forty-eight hours after transfection, RNA samples were prepared from 293 cells using Trizol reagent. To determine the level of tat shRNA expression, 40 µg of total RNA was separated by electrophoresis on a 15% polyacrylamide–7 M urea gel and electroblotted for 4 h at 400 mA on a Zeta-Probe GT membrane (Bio-Rad). RNA was immobilized by UV crosslinking and baking for 1 h at 80°C. Hybridization was carried out at 42°C using Ultrahyb-Oligo Hybridization Buffer (Ambion). HIV-1 specific siRNA was probed with a 32P-labeled tat antisense oligonucleotide (5′-TGT TCT GAT GAG CTC TTC GTC-3′). Membranes were washed twice in 2× SSC, 0.1% SDS and 0.2× SSC, 0.1% SDS at 37°C.
RESULTS
Our experiments describe an alternative strategy to achieve intracellular synthesis of siRNA. By incorporating tat siRNA sequences into the stem of the human miR-30 miRNA, we were able to efficiently produce active HIV-1 specific siRNA in human cells under the control of the human U6 pol III promoter. We first followed the structural motif described by Zeng and Cullen (23) who used a loop structure (miA-tat) that differed from the endogenously produced miR-30 by 4 nt. We also created a loop structure (miB-tat) identical to that of endogenously produced miR-30 (Fig. 1). In co-transfection experiments, we observed that HIV-1 production was most effectively suppressed by miB-tat. Quantification of HIV-1 p24 antigen by immunoassay in culture supernatant revealed that miA-tat and miB-tat reduced viral activity by 25- and 45-fold, respectively (Fig. 2A and B). Most importantly, both miRNA constructs were up to 82% more effective in inhibiting HIV-1 replication than conventional tat shRNA. These results were confirmed by western blot analysis of HIV-1 p24 antigen isolated from culture supernatant and cell lysate. The production of p24 antigen was reduced in both compartments and mirrored the p24 immunoassay results. miB-tat RNA showed the most effective reduction of p24 antigen in both compartments followed by miA-tat RNA (Fig. 3).
Figure 2.


HIV-1 p24 antigen production in culture supernatant of 293 cells transfected with HIV-1NL4.3 and the indicated tat siRNA expression plasmids determined by p24 immunoassay 48 h post-transfection; luc indicates a shRNA expression plasmid targeting luciferase. (A) Absolute p24 antigen in nanograms per milliliter of culture supernatant. (B) Antiviral potency of the various tat siRNA expression plasmids relative to control plasmid luc shown as fold inhibition. Values represent averages of three independent experiments, with the range indicated.
Figure 3.
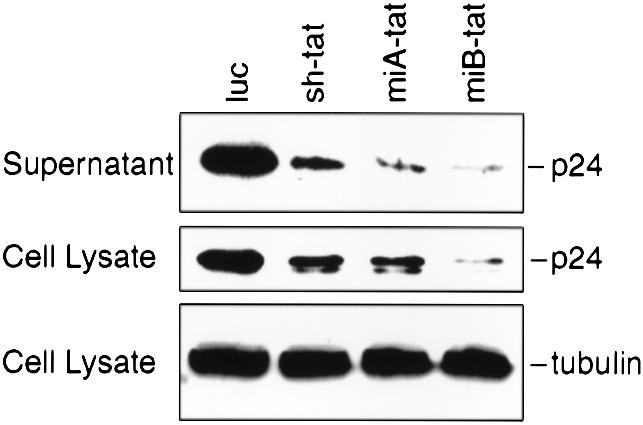
Reduction of HIV-1 p24 antigen in cell-free supernatant and cell lysate determined by western blot analysis. 293 cells were co-transfected with HIV-1NL4.3 and the indicated tat siRNA expression vectors. The control plasmid luc expresses a luciferase hairpin siRNA. Tubulin served as a loading control.
It has been demonstrated that a duplex RNA incorporated in a miRNA sequence context operates as siRNA and triggers destruction of a perfectly complementary target mRNA (4,23). To verify that the antiviral effect was indeed siRNA mediated and not due to miRNA related translational repression of protein synthesis, we further evaluated the nature of the observed reduction of viral particle production. We assessed HIV-1 RNA target degradation by quantitative real-time PCR. Once again, we observed the same pattern of antiviral activity with miB-tat being the most effective, followed by miA-tat in degrading HIV-1 RNA (Fig. 4). These results demonstrate that siRNA incorporated in a pre-miRNA backbone enters the RNAi pathway and exerts its gene silencing effect by target degradation.
Figure 4.

Reduction of total intracellular HIV-1 RNA determined by real-time PCR. Variation of RNA input was normalized by quantifying GAPDH expression by real-time PCR. To convert threshold cycles to copy number, an external standard curve was created with known copy numbers of HIV-1NL4.3. Values represent averages of three independent experiments, with the range indicated.
To examine whether the increased HIV-1 RNA degradation obtained with miB-tat was due to enhanced processing of precursor RNA to siRNA, we performed northern blot experiments to quantify the production of siRNA effector molecules. Most efficient processing of siRNA from tat short hairpin or pre-miRNAs was obtained with miB-tat RNA. Expression of sh-tat RNA resulted in the lowest level of siRNA production (Fig. 5A and B).
Figure 5.
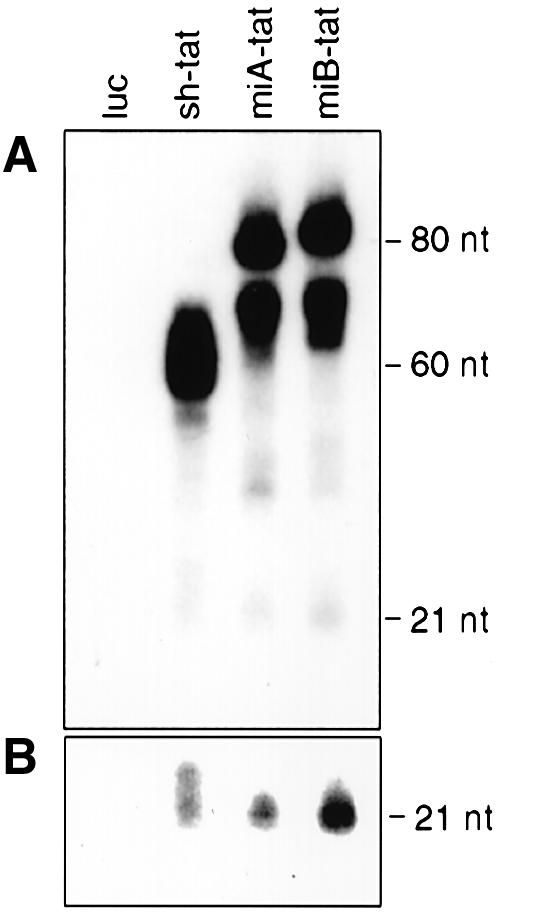
Northern blot analysis of tat shRNA, pre-miRNA and siRNA expression from 293 cells co-transfected with HIV-1NL4.3 and the indicated DNA constructs. Total cellular RNA was extracted from 293 cells 48 h after transfection and separated on a 15% polyacrylamide–7 M urea gel. Hybridization was performed using a 32P-labeled DNA oligonucleotide probe complementary to the tat sense strand. (A) Northern blot was exposed for 1 h. (B) Detection of processed tat siRNA effector molecules after 10 h of exposure.
Finally, we assessed the thermodynamic properties of all three RNA species. In modeling the secondary structure (MFold version 3.0), we found that the most potent construct (miB-tat) contained the highest local folding potential (lowest ΔG) (sh-tat ΔG, –38.5; miA-tat ΔG, –41.6; miB-tat ΔG, –45.9).
DISCUSSION
The use of small RNAs to decrease HIV-1 replication offers a new approach to antiviral gene therapy. Thus far, HIV-1 specific RNAi has been generated by the cellular introduction of synthetic 21 nt long RNA duplexes or DNA vectors harboring siRNA/shRNA expression cassettes. DNA vectors have followed a stereotypic design with the use of RNA pol III promoters to drive the expression of 19–21 nt long sense and antisense strands either separately or joined by a variably sized loop with the end result of cellular synthesis of siRNA or shRNA, respectively (12,16).
Our results confirm the utility of using the pre-miR-30 backbone as a delivery system for siRNA. The enhanced potency of gene silencing achieved with the miA/B constructs compared with conventional shRNA deserves comment. The effectiveness of RNAi mediated gene silencing is likely multifactorial and reflects the efficiency of siRNA generation, its nucleotide homology with the chosen target, the accessibility of the target RNA, the structural stability of the siRNA delivery molecule (miRNA, shRNA) and its ability to be processed by Dicer to enter the RNAi silencing complex (RISC)/miRNA ribonucleoprotein particle (miRNP). Northern blot analysis revealed that siRNA derived from miB-tat RNA was most efficiently processed in the cell. In modeling the secondary structure of the three different RNAs (MFold version 3.0), we note that the most potent construct (miB-tat) contained the highest local folding potential (lowest ΔG) (sh-tat ΔG, –38.5; miA-tat ΔG, –41.6; miB-tat ΔG, –45.9). Thus, the enhanced processing of miB-tat RNA to siRNA may be related to the higher structural stability of the stem–loop structure, which folds back more rapidly into its secondary structure that is now available for Dicer binding and cleavage. The higher rate of diced shRNA/miRNA leads to faster formation of the RISC/miRNP with the end result of more potent gene silencing. Presently, structural or sequence motifs important for Dicer recognition of its target RNA are unknown. Site-directed mutagenesis studies of pre-miR-30 demonstrated the requirement of dsRNA structure around the cleavage site of the 5′ and 3′ ends of pre-miR-30 for efficient processing into siRNA (20,23). The sequence and structural requirements of the loop structure of shRNA and miRNA precursors critical for efficient processing have not been fully investigated and remain unclear. McManus et al. found that the majority of processing within shRNA stem–loop structures occurs in the loop sequences of the hairpin (4). Thus, it may be that a yet unidentified structural motif in the pre-miRNA loop is better recognized by Dicer, or nucleases other then Dicer, and/or is more efficiently processed, resulting in increased gene silencing. In the future it will be important to determine structural elements of miRNA precursors and shRNA as well as RNase III proteins to better understand the mechanism of pre-miRNA/shRNA processing.
Our results also have a more immediate, practical application. A central challenge in using RNAi is the identification of siRNA/shRNA nucleotide sequences that lead to effective gene silencing. In our experience, nucleotide sequence selection is largely an empiric process initially guided by RNA secondary structure predictions of the target mRNA (e.g. MFold), but ultimately tested by direct experimentation. It is not uncommon for investigators to run through a panel of siRNA/shRNAs before identifying an effective species. We note that the same tat siRNA delivered by pre-miRNA is ∼80% more effective than when delivered as conventional shRNA. Thus, the use of the pre-miRNA backbone may increase the effectiveness of a moderately effective siRNA species.
Finally, a potentially important attribute of miRNAs is that a significant fraction of them occur naturally in clusters containing multiple miRNAs expressed coordinately from a single precursor RNA and processed into individual miRNAs (3,5). Thus, it may be possible to simultaneously express a series of miRNA/shRNAs targeting different regions of the viral genome, polycistronically transcribed by a single pol III promoter. Such a strategy may yield more potent inhibition of viral replication and may be a basis of effective gene therapy for HIV-1 infection.
Acknowledgments
ACKNOWLEDGEMENTS
We acknowledge the AIDS Research and Reference Reagent Program Division of AIDS, NIAID, NIH for supplying HIVNL4-3 (contributed by Dr Malcolm Martin). This work was supported by a Clinical Scientist Development Award from the Doris Duke Charitable Foundation, a Daland Fellowship in Clinical Investigation from the American Philosophical Society and a Career Development Grant from NIAID/NIH (B.R.). We thank the Lifespan/Tufts/Brown Retrovirology Core Laboratory and the Center for Cancer Research Development (NIHP20RR017695) for assay support.
REFERENCES
- 1.Tuschl T. (2002) Expanding small RNA interference. Nat. Biotechnol., 20, 446–448. [DOI] [PubMed] [Google Scholar]
- 2.Zamore P.D. (2001) RNA interference: listening to the sound of silence. Nature Struct. Biol., 8, 746–750. [DOI] [PubMed] [Google Scholar]
- 3.Lau N.C., Lim,L.P., Weinstein,E.G. and Bartel,D.P. (2001) An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science, 294, 858–862. [DOI] [PubMed] [Google Scholar]
- 4.McManus M.T., Petersen,C.P., Haines,B.B., Chen,J. and Sharp,P.A. (2002) Gene silencing using micro-RNA designed hairpins. RNA, 8, 842–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lagos-Quintana M., Rauhut,R., Lendeckel,W. and Tuschl,T. (2001) Identification of novel genes coding for small expressed RNAs. Science, 294, 853–858. [DOI] [PubMed] [Google Scholar]
- 6.Lee Y., Jeon,K., Lee,J.T., Kim,S. and Kim,V.N. (2002) MicroRNA maturation: stepwise processing and subcellular localization. EMBO J., 21, 4663–4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hammond S.M., Bernstein,E., Beach,D. and Hannon,G.J. (2000) An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature, 404, 293–296. [DOI] [PubMed] [Google Scholar]
- 8.Bernstein E., Caudy,A.A., Hammond,S.M. and Hannon,G.J. (2001) Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature, 409, 363–366. [DOI] [PubMed] [Google Scholar]
- 9.Ketting R.F., Fischer,S.E., Bernstein,E., Sijen,T., Hannon,G.J. and Plasterk,R.H. (2001) Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev., 15, 2654–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brummelkamp T.R., Bernards,R. and Agami,R. (2002) Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell, 2, 243–247. [DOI] [PubMed] [Google Scholar]
- 11.Novina C.D., Murray,M.F., Dykxhoorn,D.M., Beresford,P.J., Riess,J., Lee,S.K., Collman,R.G., Lieberman,J., Shankar,P. and Sharp,P.A. (2002) siRNA-directed inhibition of HIV-1 infection. Nature Med., 8, 681–686. [DOI] [PubMed] [Google Scholar]
- 12.Lee N.S., Dohjima,T., Bauer,G., Li,H., Li,M.J., Ehsani,A., Salvaterra,P. and Rossi,J. (2002) Expression of small interfering RNAs targeted against HIV-1 rev transcripts in human cells. Nat. Biotechnol., 20, 500–505. [DOI] [PubMed] [Google Scholar]
- 13.Jacque J.M., Triques,K. and Stevenson,M. (2002) Modulation of HIV-1 replication by RNA interference. Nature, 418, 435–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Capodici J., Kariko,K. and Weissman,D. (2002) Inhibition of HIV-1 infection by small interfering RNA-mediated RNA interference. J. Immunol., 169, 5196–5201. [DOI] [PubMed] [Google Scholar]
- 15.Boden D., Pusch,O., Lee,F., Tucker,L. and Ramratnam,B. (2003) Human immunodeficiency virus type 1 escape from RNA interference. J. Virol., 77, 11531–11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boden D., Pusch,O., Lee,F., Tucker,L., Shank,P.R. and Ramratnam,B. (2003) Promoter choice affects the potency of HIV-1 specific RNA interference. Nucleic Acids Res., 31, 5033–5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pusch O., Boden,D., Silbermann,R., Lee,F., Tucker,L. and Ramratnam,B. (2003) Nucleotide sequence homology requirements of HIV-1-specific short hairpin RNA. Nucleic Acids Res., 31, 6444–6449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee R.C., Feinbaum,R.L. and Ambros,V. (1993) The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell, 75, 843–854. [DOI] [PubMed] [Google Scholar]
- 19.Lee R.C. and Ambros,V. (2001) An extensive class of small RNAs in Caenorhabditis elegans. Science, 294, 862–864. [DOI] [PubMed] [Google Scholar]
- 20.Lee Y., Ahn,C., Han,J., Choi,H., Kim,J., Yim,J., Lee,J., Provost,P., Radmark,O., Kim,S. et al. (2003) The nuclear RNase III Drosha initiates microRNA processing. Nature, 425, 415–419. [DOI] [PubMed] [Google Scholar]
- 21.Zeng Y., Wagner,E.J. and Cullen,B.R. (2002) Both natural and designed micro RNAs can inhibit the expression of cognate mRNAs when expressed in human cells. Mol. Cell, 9, 1327–1333. [DOI] [PubMed] [Google Scholar]
- 22.Hutvagner G. and Zamore,P.D. (2002) A microRNA in a multiple-turnover RNAi enzyme complex. Science, 297, 2056–2060. [DOI] [PubMed] [Google Scholar]
- 23.Zeng Y. and Cullen,B.R. (2003) Sequence requirements for micro RNA processing and function in human cells. RNA, 9, 112–123. [DOI] [PMC free article] [PubMed] [Google Scholar]