


Abstract
Mer1p activates the splicing of at least three pre-mRNAs (AMA1, MER2, MER3) during meiosis in the yeast Saccharomyces cerevisiae. We demonstrate that enhancer recognition by Mer1p is separable from Mer1p splicing activation. The C-terminal KH-type RNA-binding domain of Mer1p recognizes introns that contain the Mer1p splicing enhancer, while the N-terminal domain interacts with the spliceosome and activates splicing. Prior studies have implicated the U1 snRNP and recognition of the 5′ splice site as key elements in Mer1p-activated splicing. We provide new evidence that Mer1p may also function at later steps of spliceosome assembly. First, Mer1p can activate splicing of introns that have mutated branch point sequences. Secondly, Mer1p fails to activate splicing in the absence of the non-essential U2 snRNP protein Snu17p. Thirdly, Mer1p interacts with the branch point binding proteins Mud2p and Bbp1p and the U2 snRNP protein Prp11p by two-hybrid assays. We conclude that Mer1p is a modular splicing regulator that can activate splicing at several early steps of spliceosome assembly and depends on the activities of both U1 and U2 snRNP proteins to activate splicing.
INTRODUCTION
Precursor messenger RNAs (pre-mRNAs) in eukaryotes contain intervening sequences or introns that must be removed to create a functional RNA. The process of removing introns, or splicing, occurs in the nucleus and is catalyzed by a large ribonucleoprotein complex called the spliceosome (reviewed in 1). In many eukaryotes, especially higher eukaryotes, the primary transcripts can be alternatively spliced, leading to the production of functionally diverse protein isoforms (2). During alternative splicing, splicing regulators modulate the activity of the spliceosome to alter the selection of splice sites to be used by either recruiting components of the spliceosome to the alternative splice sites or blocking the spliceosome from choosing the default splice sites (3–7).
The action of splicing regulators suggests that formation of the earliest splicing complex, the commitment complex 1 (CC1) in yeast or the E complex in metazoans, represents an important control point for splicing regulation. This complex involves base pairing between the 5′ splice site and the 5′ end of U1 snRNA (8,9) as well as a multitude of U1 snRNP protein–intron interactions (10,11). With the assistance of the accessory factors Bbp1p/SF1 and Mud2p/U2AF, commitment complex 2 (CC2) forms and initially identifies the branch point sequence (BPS) which will react with the 5′ splice site during the first chemical step of splicing (12–15). However, splice site selection is not always finalized at this stage and can be altered (16,17). Following CC2 or E complex formation, the U2 snRNP can bind to form the pre-spliceosome or A complex. Pre-spliceosome formation requires ATP, and the intron BPS is recognized by base pairing to U2 snRNA (18–20). Precisely how U1 and U2 interact with each other in these early complexes remains unclear. Subsequent spliceosome assembly events lead to replacement of U1 by U6 at the 5′ splice site and generation of the catalytic form of the spliceosome (21).
The mechanisms by which splicing regulators control splicing complex formation are understood only in the most simple terms. Both positive and negative splicing regulators seem to be modular, selecting transcripts for regulation through their RNA-binding domains, and exerting their action through their auxiliary domains (7). The nature of the function of the auxiliary domains remains mysterious. On the basis of ‘tethering’ experiments in which the activation domain of the splicing factor is fused to a heterologous RNA-binding domain and tested for the ability to control splicing of a transcript containing a heterologous RNA-binding site, it has been concluded that activation domains regulate the association of snRNPs on nearby regions of the transcript during the early steps of spliceosome assembly. By regulation of early snRNP–pre-mRNA interactions, splicing is regulated (22–26). Several regulators appear to act at later stages of the splicing pathway, including modulation of spliceosome function rather than assembly (27). Thus, the specific mechanisms by which splicing activation domains function are of continuing interest.
Mer1p is a splicing regulator that is produced during meiosis in the yeast Saccharomyces cerevisiae (28). It has a KH domain RNA-binding motif (29) and can specifically bind RNA that contains a Mer1p enhancer element (30). Several findings support a function for Mer1p at the very first stage of spliceosome assembly when the U1 snRNP binds to pre-mRNA to form CC1. First, all natural Mer1p-activated introns possess weak 5′ splice sites. Secondly, the role of Mer1p in splicing of the intron in the MER2 gene can be bypassed by a mutant U1 snRNA that can form additional base pairs to the 5′ splice site sequence (31). Thirdly, Nam8p, a non-essential U1 snRNP protein that can be cross-linked to pre-mRNA near the 5′ splice site (10,11), is required for Mer1p-activated splicing (30,32). Fourthly, Mer1p co-immunoprecipitates U1 snRNA (30). A reasonable hypothesis based on these data is that Mer1p stabilizes U1 snRNP binding to pre-mRNAs that contain the Mer1p enhancer element, thereby increasing the efficiency with which they progress to productive splicing complexes. In this paper, we provide new evidence that Mer1p can activate splicing of pre-mRNAs with weak BPSs, and that Mer1p function also relies on components of the U2 snRNP. These data suggest that Mer1p may function in the formation or stabilization of multiple splicing complexes including the pre-spliceosome.
MATERIALS AND METHODS
Strains and plasmids
The construction of many of the plasmids used for splicing analysis in this study were described before (30). The MS2 Coat gene was amplified from pCT119 (a gift from D. S. Peabody) by PCR using Vent DNA polymerase and appropriate primers containing unique restriction enzyme sites. The MS2 Coat DNA was ligated into pGAC14 plasmid [2µ, TRP1, MS2 Coat transcription under control of the GPD promoter (33)] between the BamHI and SalI sites to form pGCOAT. The MS2 Coat gene was also amplified using a primer that deleted the stop codon and added a hexaglycine linker to the C-terminus. This fragment was then ligated into pGMER1 to form pGCTMER1, producing a single ORF for an N-MS2 Coat-6gly-Mer1-C fusion protein. To construct the MS2 Coat-Mer1p fusion lacking the KH domain, first the N-terminal 180 codons of MER1 were amplified by PCR and ligated into pGAC14 to form pGAD (activation domain). The MS2 Coat-6gly PCR product was then ligated into pGAD to form pGCTAD for the expression of the MS2 Coat-6gly-Mer1p activation domain fusion protein. The resulting polypeptides coded by the above plasmids were found to be expressed and stable in yeast extracts when assayed by western analysis using a polyclonal antibody raised against Mer1p (data not shown). KH46 (Matα, ura3-52, leu2-3, 112, trp1-1, lys2, his3-1, ade2-101, cup1Δ::ura-3–52) yeast were used for these studies and for isolating total RNA for primer extension analysis with one exception: the snu17Δ strain was purchased from Invitrogen.
The Kunkel method of site-specific in vitro mutagenesis (34) was used to construct pRS316CUP-AMA1bp. This plasmid carries the Mer1p-activated AMA1/SPO70 intron fused to CUP1, but the UACUAAC intron BPS is changed to UAUUAAC. The same method was also used to construct pRS316CUP-MS2.1ACT G5A. This plasmid uses the GPD promoter for transcription of an actin pre-mRNA that has a GUAUGU to GUAUAU mutation (G5A) in the 5′ splice site sequence, an MS2 operator located 35 nt downstream from the 5′ splice site, and the CUP1 ORF fused to exon 2 of actin. A tighter binding variant of the MS2 operator (35) was utilized, and the sequence of the stem was altered to abolish a cryptic 5′ splice site that is contained within the wild-type stem sequence (CGTACaCCaucaGGGTACG, upper case letters form the stem, lower case letters in the loop or bulge). The new sequence (CCTAGaCCaucaGGCTAGG) maintained the same overall secondary structure base pairing potential as the wild-type (Fig. 1).
Figure 1.

(A) Mer1p domains and fusion constructs used in this study. Mer1p contains a C-terminal KH domain RNA-binding motif and an N-terminal domain required for activating splicing. CTMER has the MS2 Coat gene fused to the N-terminus of the entire MER1 gene, whereas CTAD has the MS2 Coat gene fused to the activation domain fragment of MER1. The MS2 Coat construct serves as a control that produces unfused MS2 Coat protein. The AD construct codes for the N-terminal activation domain from MER1, and the KH construct codes for the C-terminal fragment of Mer1p which contains the KH domain and short C-terminal peptide. (B) The sequences of the MS2 operator and MER1 splicing enhancer elements used to modify the actin intron.
Primer extension analysis was described before (30) and performed on duplicate or triplicate samples. Primer extension products representing spliced and unspliced RNAs were quantitated by phosphorimaging. The formula S/(S + U), where S is spliced product and U is unspliced pre-mRNA, was used to calculate splicing efficiency. Primer sequences were specific for plasmid-encoded mRNA and bind to the CUP1 sequences in the second exon. The CUP1 primer sequence is 5′-GGCACTCATGACCTTCATTTTGG.
Two-hybrid plasmids and assays
The construction of the U2 snRNP genes in pACT2 plasmids has been described before (36,37). The U1 snRNP genes NAM8, SNU71, SNU56, LUC7, YHC1 and the U2 snRNP gene SNU17 were cloned into pACT2 by PCR amplifying the genes from genomic DNA. The MER1 ORF or activation domain fragment was amplified by PCR and ligated in pBTM116 to form pBTM-MER1 or pBTM-AD, which fuses the lexA DNA binding domain to Mer1p. Colorimetric assays were performed (36,37) with strain L40 carrying pBTM and pACT2 derivatives after 3–4 days of growth on selective media.
RESULTS
The Mer1p activation domain stimulates splicing independently of the KH domain
Based on prior work (30), we suggested that the C-terminal KH domain of Mer1p recognizes the splicing enhancer in the intron RNA, while the N-terminal portion of Mer1p activates splicing. To test this, the entire Mer1p protein (CTMER1) or just the N-terminal domain of Mer1p (CTAD for MS2 Coat + activation domain) was fused with the MS2 bacteriophage coat protein, which binds a small, defined stem–loop structure (see Fig. 1) (35). The ability of these fusion proteins to activate splicing of modified actin pre-mRNAs that contain the Mer1p enhancer or the MS2 operator (the MS2 Coat protein binding site) was tested.
Consistent with the weak 5′ splice site requirement shown previously using the GUUCGU mutation (30), the actin 5′ splice site mutant GUAUGU to GUAUAU (G5A) pre-mRNA containing the Mer1p splicing enhancer can be activated by Mer1p (Fig. 2, lane 9). Fusion of MS2 Coat to the N-terminus of Mer1p does not prevent the MS2 Coat-Mer1p fusion from activating splicing (lane 7). However, neither MS2 Coat protein alone nor the Mer1p fusion protein lacking the KH domain is able to activate splicing, presumably because neither protein can bind the enhancer (lanes 6 and 8). If instead of the Mer1p enhancer, the pre-mRNA contains the MS2 operator, a different result is observed. With the MS2 operator in place of the Mer1p enhancer, the MS2 Coat-Mer1p fusion is able to activate splicing (lanes 2 and 5) about as well as Mer1p through its own enhancer (lane 9). If the KH domain is removed from the MS2 Coat-Mer1p fusion, the resulting polypeptide (CTAD) can still activate splicing, albeit to a lesser extent (lane 3). The CTAD protein also lacks the C-terminal 22 residues that follow the KH domain, and their loss may explain why the CTAD protein does not activate splicing to the same extent as the full-length CTMER1 protein. Neither MS2 Coat protein nor Mer1p activates splicing of the MS2 operator pre-mRNA (lanes 1 and 4), presumably because MS2 Coat has no activation domain, and Mer1p cannot bind this pre-mRNA. We conclude that the KH domain is not essential for splicing activation and serves primarily to select introns that have the Mer1p splicing enhancer. A bacteriophage RNA binding domain fused to the N-terminus of Mer1p can replace the KH domain provided that an appropriate RNA binding site is present in the intron. Thus, the N-terminal segment of Mer1p, which has been shown to associate with U1 snRNP (30), must contain protein elements necessary for splicing activation independent of intron recognition.
Figure 2.

The KH domain of Mer1p can be replaced with another RNA-binding domain. Primer extension analysis was used to measure splicing of modified actin RNAs produced in vivo. Actin RNAs were expressed from a plasmid and contained a G5A 5′ splice site mutation to reduce splicing efficiency and either the Mer1p enhancer element or the MS2 operator element near the 5′ splice site. Yeast cells also included one of the plasmids from Figure 1. Lanes 2 and 5 are replicates. The dark band between the 262 and 345 nt markers in lanes 1–5 results from a primer extension stop at the MS2 operator hairpin. Phosphorimaging quantitation of splicing efficiency (percent spliced) is calculated by the formula S/(S + U) × 100 where S = spliced and U = unspliced. The average values for duplicate samples are reported.
Over-expression of the N-terminal part of Mer1p squelches Mer1p-activated splicing
If the activation domain and RNA-binding domains of Mer1p have separable functions as suggested above, then over-expression of the N-terminal domain could have a dominant-negative effect on the activity of full-length Mer1p. A similar phenomenon, termed squelching (38), has been useful in dissecting the function of modular transcription factors. Similar to transcriptional squelching, over-expression of the Mer1p N-terminal domain squelches splicing activation by full-length Mer1p (Fig. 3, lane 3). Essentially, no splicing activation of AMA1 reporter pre-mRNA occurs if cells express both Mer1p and the activation domain polypeptide. Over-expressed KH domain fragment does not squelch splicing activation by Mer1p (lane 2). We conclude that Mer1p activation can be inhibited in a dominant-negative fashion or squelched by N-terminal segments of the Mer1p protein. This suggests that activation involves the interaction of the N-terminal domain of Mer1p with another splicing factor. This experiment, together with that shown in Figure 2, demonstrates that the N-terminal domain of Mer1p contains a splicing activation function. Western blots were performed to assess the ratios of Mer1p N-terminal domain and KH RNA-binding domain fragments to full-length Mer1p, in order to assess the amounts of protein required for squelching. Although all plasmids used have high copy (2µ) replication origins, the truncated proteins are expressed from the GPD promoter, whereas the full-length Mer1p protein is expressed from the ADH promoter. Western analysis using a polyclonal antibody raised against recombinant Mer1p shows that all three Mer1p-derived proteins are produced and stable in yeast extracts. Furthermore, constructs containing the GPD promoters produce ∼50- to 100-fold more protein than the ADH-containing constructs (data not shown). Presumably the activation domain fragment prevents full-length Mer1p from interacting with the necessary splicing factors to stimulate splicing.
Figure 3.
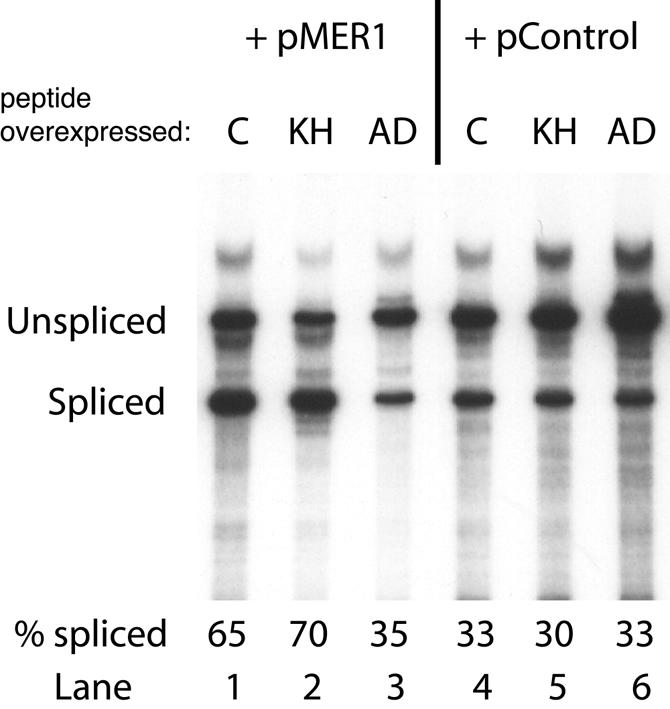
Over-expression of the activation domain of Mer1p has a dominant-negative effect on full-length Mer1p. Yeast were transformed with a MER1 expression plasmid (pMER1) or a control plasmid lacking MER1 (pControl). Additionally, the yeast contained the KH domain plasmid (KH), the activation domain plasmid (AD) or a control plasmid (C) that lacks any of the MER1 domains. Total RNA was isolated and splicing of the AMA1 pre-mRNA was measured by primer extension on duplicate samples. Splicing efficiencies for each sample are reported below each lane.
Mer1p activates the splicing of introns with mutations in the branch point sequence
Given the above results and our previous demonstration that the N-terminal segment of Mer1p binds the U1 snRNP (30), we began to explore the mechanism of Mer1p activation. Mer1p is capable of activating the splicing of introns that bear features known to reduce the efficiency of 5′ splice site recognition, including weak 5′ splice sites, large 5′ exons or an intronic splicing silencer (30,31). If Mer1p activation is specific to overcoming inefficient 5′ splice site recognition, then an intron with an efficiently recognized 5′ splice site but a weakly recognized branch point might be expected to be refractory to Mer1p activation. To test this idea, we introduced mutations to the BPS of the AMA1 intron and to a modified actin intron that contains the Mer1p enhancer element and determined whether they could be activated by Mer1p. When the BPS is altered from UACUAAC to UAUUAAC, splicing efficiency is diminished from ∼30% spliced to <1% spliced for AMA1 reporter mRNA (Fig. 4, lanes 2 and 4). When Mer1p is expressed, splicing of the AMA1 reporter pre-mRNA containing the mutant BPS is activated to 33% spliced (lane 3). Since the AMA1 intron retains its splicing silencer between the 5′ splice site and the Mer1p enhancer (30), it could be argued that Mer1p activation could still occur by increasing 5′ splice site recognition. To address this possibility, introns with efficiently utilized 5′ splice sites were also tested. The AMA1 NT7-15 mutant has a disruption to the splicing silencer element that renders the intron both efficiently spliced and insensitive to further activation by Mer1p (30) (Fig. 4, lanes 5 and 6). When the NT7-15 mutant carries the UACUAAC to UAUUAAC mutation in the BPS, its splicing is inefficient, but is activated by Mer1p (Fig. 4, lanes 7 and 8). An actin intron containing a wild-type GUAUGU 5′ splice site and the Mer1p enhancer element, but with a mutated BPS, can also be activated by Mer1p, albeit to a much lower extent (data not shown). The weaker splicing activation observed for the actin BPS mutant might be due to the much larger distance between the BPS and enhancer in the ACT1 reporter (∼240 nt) as compared with the AMA1 reporter (∼50 nt). We conclude that Mer1p activation can be imposed on pre-mRNAs with inefficient BPSs, suggesting Mer1p may act at multiple points in the splicing pathway after 5′ splice site recognition.
Figure 4.
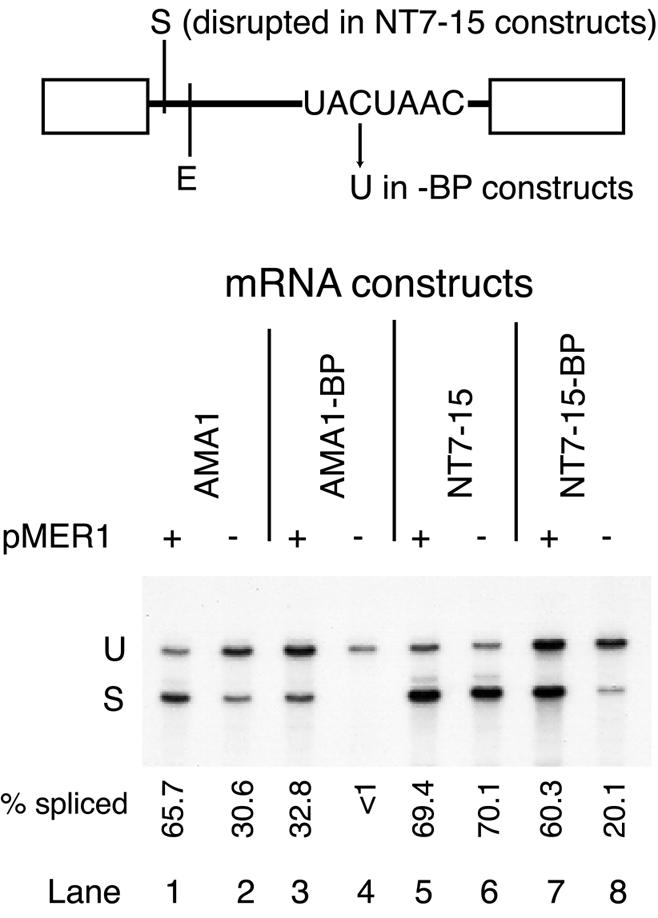
Mer1p activates splicing of introns with BPS mutations. Primer extension analysis of splicing of various AMA1 reporter mRNAs isolated from yeast containing a MER1 expression plasmid (+ pMER1) or a control plasmid (–). Five micrograms of total RNA were used in each reaction. The –BP constructs contain the C to U mutation in the third position of the BPS. The NT7-15 construct is an AMA1 intron with alterations that disrupt the splicing silencer at nucleotides 7–15 of the intron; it does not require Mer1p for efficient splicing. The relative positions of the splicing silencer (S) and enhancer (E) with respect to the 5′ splice site and BPS are indicated above the gel phosphorimage. Splicing efficiencies were averaged for several replicates and are reported below each lane. U and S refer to cDNAs from unspliced and spliced RNAs, respectively.
BPS mutations are reported to have an effect on mRNA export from the nucleus, since mutations in both the BPS and the branch point binding protein Bbp1p lead to efficient export of unspliced mRNA (39,40). It is possible that the UAUUAAC BPS mutation causes efficient export to the cytoplasm. Interestingly, RNA derived from reporters containing BPS mutations in this study are less abundant than from reporters without BPS mutations in the absence of Mer1p expression (Fig. 4, compare sum of signal in lane 2 with that in lane 4 or total signal in lane 6 with that in lane 8) suggesting that they are subject to a decay process that destroys them. In addition to activating splicing, Mer1p seems to generally increase the abundance of RNA from the mutant BPS reporters (compare lane 3 with 4, or lane 7 with 8). One consequence of Mer1p action might be to prevent export of unspliced pre-mRNAs to the cytoplasm and stabilize them in the nucleus, such that prolonged attempts at splicing can occur. This could occur indirectly by stabilizing the association of Bbp1p with the branch point as seems to occur in the wild-type transcript, or directly through Mer1p–pre-mRNA interactions. Further studies need to be performed to determine if Mer1p has a role in nuclear retention or stabilization of enhancer-containing pre-mRNAs.
The non-essential U2 snRNP protein Snu17p is required for Mer1p-activated splicing
To identify splicing factors that are involved in Mer1p-activated splicing, we have been analyzing Mer1p-activated splicing in strains of yeast deleted of non-essential genes involved in RNA processing and export. If Mer1p can activate splicing without the missing non-essential factor, then that factor is not required for Mer1p splicing activation. The only known example of a non-essential gene required for Mer1p splicing is NAM8, also known as MRE2 (30,32). Many non-essential genes involved in RNA processing have been tested for Mer1p-activated splicing. Other than Nam8p, none of the known non-essential U1 snRNP and commitment complex proteins U1A (Mud1p), Cbp20p, Cbp80p, and the accessory protein Mud2p (30 and data not shown) are required for Mer1p splicing activation. Surprisingly, the U2 snRNP protein Snu17p is required for Mer1p-activated splicing (Fig. 5). Yeast lacking Snu17p cannot activate the splicing of the AMA1, MER2 and MER3 reporter pre-mRNAs. Loss of Mer1p-activated splicing does not occur with the deletion of the other non-essential U2 snRNP proteins Cus2p and U2A′ (Lea1p) (30). Snu17p is a non-essential subunit of the multimeric U2 snRNP-associated protein complex SF3b (41). This complex contains several conserved, essential splicing proteins such as Hsh155p, Hsh49p, Cus1p and Rse1p known to be required for U2 snRNP binding to pre-mRNA in yeast and humans (42). The ability of Mer1p to activate splicing must not only involve the U1 snRNP, but the U2 snRNP as well.
Figure 5.

Mer1p-activated splicing requires the U2 snRNP protein Snu17p. Primer extension analysis of AMA1, MER2 and MER3 reporter mRNAs from SNU17 and snu17Δ yeast containing a MER1 expression plasmid (+ pMER1) or a control plasmid (–). Percent spliced is reported as the average of several replicate samples.
Mer1p interacts with U1 and U2 snRNP proteins
Functional requirements for Mer1p splicing activation identify two snRNP proteins, the U1 snRNP protein Nam8p (30) and the U2 snRNP protein Snu17p (Fig. 5). In addition, Mer1p binds the U1 snRNP in vitro (30). To test for possible protein–protein interactions between Mer1p and U1 and U2 snRNP proteins we performed two-hybrid tests (Table 1). Although Nam8p is required for Mer1p to activate splicing, there is no detectable two-hybrid interaction between Nam8p and Mer1p. Two essential U1 snRNP proteins, Snu71p and Snu56p, are destabilized from the U1 snRNP when Nam8p is absent (43). Both of these proteins give a positive signal in the two-hybrid test with Mer1p. Mer1p also weakly interacts with the U1 snRNP protein Luc7p and two accessory splicing factors that are thought to be part of CC2. These factors, Mud2p and Bbp1p, have been implicated in recognizing the BPS prior to the U2 snRNP interaction (14,15,39). Several U2 snRNP proteins were also tested, and surprisingly, Prp11p interacts strongly with Mer1p. Like Nam8p, Snu17p does not interact with Mer1p even though it is required for Mer1p function. A second U2 snRNP protein (Prp9p) that interacts with Prp11p (44), also weakly interacts with Mer1p. This weak two-hybrid signal between Mer1p and Prp9p may result from an interaction that is bridged by Prp11p as opposed to a direct interaction between Mer1p and Prp9p. The weak interactions seen between Mer1p and Bbp1p or Mud2p may also be indirectly mediated by Prp11p. Mud2p interacts with Prp11p (45) and Bbp1p (13). Strong positives from the two-hybrid analysis were repeated using a bait construct that contained the activation domain fragment of Mer1p. The results mirrored those obtained with the full-length Mer1p (Table 1). This indicates that the Mer1p activation domain is the primary mediator of the interactions observed with these splicing factors. Even for the strong interactions noted here it is difficult to know for certain whether the interactions are direct or mediated by other proteins, however, the data are consistent with previously published immunoprecipitation results (30). Thus, although two-hybrid interactions do not always reliably predict direct protein–protein interactions, these data support the interpretation that Mer1p interacts with the U1 snRNP, as well as with other components of both the commitment complexes and the pre-spliceosome.
Table 1. Two-hybrid results with Mer1p or the activation domain fragment of Mer1p, and U1 or U2 snRNP proteins.
| pACT+ | Blue intensity with pBTM-MER1 |
|---|---|
| Vector |
– |
| NAM8 |
– |
| SNU56 |
++ |
| SNU71 |
+++ |
| MER1 |
– |
| U1C |
– |
| LUC7 |
+ |
| MUD2 |
+ |
| BBP |
++ |
| CUS1 |
– |
| CUS2 |
– |
| HSH49 |
– |
| HSH155 |
– |
| SUB2 |
– |
| PRP9 |
+ |
| PRP11 |
++++ |
| prp11-1 |
– |
| PRP5 |
– |
| PRP21 |
– |
| SNU17 |
– |
| |
Blue intensity with pBTM-AD |
| SNU56 |
++ |
| SNU71 |
+++ |
| PRP11 | ++++ |
Minus signs indicate no accumulation of blue color. Plus signs indicate development of blue color with more plus signs corresponding to darker blue color development.
DISCUSSION
Our results indicate that Mer1p has a modular structure containing a C-terminal RNA-binding KH domain and an N-terminal splicing activation domain. Splicing can be activated by the N-terminal domain of Mer1p so long as it is connected to an RNA-binding domain and the pre-mRNA contains the appropriate target sequence. Moreover, over-expression of the N-terminal activation domain of Mer1p has a dominant-negative effect on full-length Mer1p, presumably by squelching the interaction between Mer1p and its target splicing factor(s). Several other splicing regulators from higher eukaryotes also have modular structures, most notably SR proteins (46). We propose that the role of the KH domain is to select introns that have the Mer1p splicing enhancer, while the role of the N-terminal domain is to recruit splicing factors to the pre-mRNA.
Our results also demonstrate that Mer1p cannot activate splicing without two non-essential proteins from U1 and U2 snRNPs, and suggest that Mer1p participates in several early splicing complexes. The observation that Mer1p can activate the splicing of introns containing a mutated BPS indicates that Mer1p functions after formation of CC1, which does not require the BPS (47). The BPS is recognized after CC1 has formed, first by two accessory proteins, Bbp1p and Mud2p to form CC2 (13,15,45), and subsequently by the U2 snRNP to form the pre-spliceosome (18,19,48). Our results cannot distinguish whether splicing activation of pre-mRNAs containing the mutated BPS occurs during CC2 formation or pre-spliceosome formation. However, the requirement for Snu17p, a component of the U2-associated factor SF3b, suggests that Mer1p may act as late as the pre-spliceosome stage, when U2 and SF3b are thought to bind the pre-mRNA. If Mer1p simply stimulated the formation of CC1 and dissociated from the complex before the pre-spliceosome has formed, then we would not expect a U2 snRNP protein to be required for Mer1p-activated splicing. Lastly, two-hybrid interactions between Mer1p and U2 snRNP proteins support the conclusion that Mer1p is a component of the pre-spliceosome. These results help build a model for Mer1p-activated splicing that is illustrated in Figure 6. In this model, Mer1p interacts with components of the U1 and U2 snRNPs to increase the rate of formation or stability of early splicing complexes and allows for an active spliceosome to form on enhancer-containing pre-mRNAs.
Figure 6.

A model summarizing interactions between Mer1p and splicing factors during the formation of early splicing complexes. In this model, Mer1p stimulates the formation of these early splicing complexes on pre-mRNAs that contain the Mer1p splicing enhancer or prevents the dissociation of these early splicing complexes such that spliceosome assembly can progress to the formation of an active spliceosome. Proteins that interact with Mer1p by two-hybrid analysis are indicated by a lightning bolt between the protein and Mer1p. Thicker bolts indicate stronger two-hybrid interactions. Proteins that cross-link to the 5′ splice site region of pre-mRNA are indicated by a red X. Proteins required for Mer1p to activate splicing, but for which no direct physical interaction with Mer1p can be detected, are connected to Mer1p with orange arrows. The branch point adenosine of the pre-mRNA is indicated by an A, and the enhancer element is colored red.
Previously, we reported that Mer1p co-immunoprecipitates U1 but not U2 snRNA (30). However, the work presented in this report implicates the U2 snRNP in Mer1p-activated splicing. The absence of any U2 snRNA in the Mer1p immunoprecipitate may reflect a loose or transient association with the U2 snRNP that does not persist after stringent washing conditions. Salt concentrations much lower than typically used in immunoprecipitation experiments are known to support the reversible dissociation of SF3a and SF3b subunits from U2 snRNA (36,49,50). In addition, accessory splicing factors that are not integral components of snRNPs, like Mud2p and Bbp1p, also fail to quantitatively co-immunoprecipitate snRNA even in the presence of pre-mRNA (13). It is also possible that the epitopes on Mer1p (mol. wt ∼33 kDa) are not accessible in pre-spliceosomes (mol. wt ∼2 MDa) or other splicing complexes. Therefore, the absence of U2 snRNA in Mer1p immunoprecipitates is not as informative as the finding that Mer1p requires a U2 snRNP protein to activate introns with weak 5′ splice sites, or that Mer1p can activate introns with weak BPSs.
Why are Nam8p and Snu17p required for Mer1p to activate splicing?
Previously, we have shown that Nam8p is required for Mer1p to activate splicing, but that Nam8p is not required for Mer1p to co-immunoprecipitate U1 snRNA (30). Hence, it seems unlikely that Nam8p provides a direct binding site to the U1 snRNP. Nam8p, like other U1 snRNP proteins, has been shown to be important for splicing introns with weak 5′ splice sites or for pre-mRNAs lacking a 5′ cap and can stabilize commitment complexes (11,43). Nam8p cross-links to nucleotides immediately downstream of the 5′ splice site sequence, close to where the enhancer element is found. Without Nam8p, two essential U1 snRNP proteins are destabilized from U1 snRNP preparations in vitro (43). These two proteins, Snu71p and Snu56p, interact with Mer1p by two-hybrid analysis. In the absence of Nam8p, Mer1p associates with the U1 snRNP and might stabilize the binding of Snu71p and Snu56p. However, Mer1p may not be able to stabilize the resulting commitment complex without the additional stability thought to be imparted by the Nam8p–intron interaction (10,11). We suggest that Nam8p helps to stabilize the proteins Mer1p must associate with to recruit U1 snRNP, or helps to stabilize the association between U1 and the pre-mRNAs selected by Mer1p.
Snu17p enters splicing complexes as part of the U2 snRNP and is thought to function in spliceosome assembly after addition of the tri-snRNP but before U1 snRNP release (41). The metazoan homolog of Snu17p has been identified as p14, which can be cross-linked to the pre-mRNA branch point nucleotide (51). When Snu17p is present, U1 and U4 are rapidly displaced from the assembling spliceosome after the binding of the tri-snRNP (41). While non-essential, the loss of Snu17p retards progression of spliceosome assembly and leads to the formation of a complex that contains all five snRNPs. Hence, Snu17p may be involved in displacing both the U1 and U4 snRNPs from assembling spliceosomes. Without Snu17p, Mer1p cannot activate splicing. This suggests that either Mer1p recognizes a structure containing or formed by Snu17p in the U2 snRNP in order to recruit or stabilize the association of U2, or is present during the progression of the assembling spliceosome when Snu17p functions.
A model for splicing activation by Mer1p is emerging. In this model, Mer1p functions at several early steps of spliceosome assembly. Based on its ability to interact with enhancer RNA and several splicing factors, we propose that Mer1p recruits the U1 and U2 snRNPs to enhancer-containing pre-mRNAs. Mer1p might accelerate the formation of these early splicing complexes or stabilize them, thus allowing subsequent spliceosomal intermediates and active spliceosomes to form on pre-mRNAs that otherwise would not be good substrates for splicing. Other non-mutually exclusive mechanisms are possible too. In addition to its direct involvement with splicing, Mer1p may also bind to unspliced pre-mRNA and help retain it in the nucleus and protect it from degradation for prolonged attempts at splicing. Nuclear retention of pre-mRNA by another splicing factor, Bbp1p/mammalian SF1, has also been described (40), as well as a competitive relationship between splicing and nuclear degradation (52).
It is becoming apparent that the mechanisms underlying splicing regulation can be quite complex (7). The timing of splicing regulation is no longer thought to be limited to prior to the first catalytic step, and the mechanisms of splicing regulation are no longer thought to be limited to influencing the formation of the initial complex in assembling spliceosomes (27,53). In studying a seemingly simple splicing activation event, Mer1p-dependent activation from an intronic splicing enhancer, we have uncovered requirements for two constitutive splicing factors associated with two different snRNPs. As with metazoan splicing activators and repressors (46,54), Mer1p fusions with MS2 Coat protein demonstrate the modular nature of Mer1p and the transportability of its splicing activation domain. The precise mechanisms by which the Mer1p and other activation domains stimulate splicing remain to be determined.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by NIHGMS grant GM40478 to M.A. M.S. was supported by an American Cancer Society postdoctoral fellowship (PF-00-329-01) and by University of Missouri-St Louis Mission Enhancement and University of Missouri Research Boards.
REFERENCES
- 1.Staley J.P. and Guthrie,C. (1998) Mechanical devices of the spliceosome: motors, clocks, springs and things. Cell, 92, 315–326. [DOI] [PubMed] [Google Scholar]
- 2.Graveley B.R. (2001) Alternative splicing: increasing diversity in the proteomic world. Trends Genet., 17, 100–107. [DOI] [PubMed] [Google Scholar]
- 3.Green M.R. (1991) Biochemical mechanisms of constitutive and regulated pre-mRNA splicing. Annu. Rev. Cell Biol., 7, 559–599. [DOI] [PubMed] [Google Scholar]
- 4.Cooper T.A. and Mattox,W. (1997) The regulation of splice-site selection and its role in human disease. Am. J. Hum. Genet., 61, 259–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chabot B. (1996) Directing alternative splicing: cast and scenarios. Trends Genet., 12, 472–478. [DOI] [PubMed] [Google Scholar]
- 6.Lopez A.J. (1998) Alternative splicing of pre-mRNA: developmental consequences and mechanisms of regulation. Annu. Rev. Genet., 32, 279–305. [DOI] [PubMed] [Google Scholar]
- 7.Black D.L. (2003) Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem., 72, 291–336. [DOI] [PubMed] [Google Scholar]
- 8.Siliciano P.G. and Guthrie,C. (1988) 5′ Splice site selection in yeast: genetic alterations in base-pairing with U1 reveal additional requirements. Genes Dev., 2, 1258–1267. [DOI] [PubMed] [Google Scholar]
- 9.Seraphin B. and Rosbash,M. (1989) Mutational analysis of the interactions between U1 small nuclear RNA and pre-mRNA of yeast. Gene, 82, 145–151. [DOI] [PubMed] [Google Scholar]
- 10.Zhang D. and Rosbash,M. (1999) Identification of eight proteins that cross-link to pre-mRNA in the yeast commitment complex. Genes Dev., 13, 581–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Puig O., Gottschalk,A., Fabrizio,P. and Seraphin,B. (1999) Interaction of the U1 snRNP with nonconserved intronic sequences affects 5′ splice site selection. Genes Dev., 13, 569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruskin B., Zamore,P.D. and Green,M.R. (1988) A factor, U2AF, is required for U2 snRNP binding and splicing complex assembly. Cell, 52, 207–219. [DOI] [PubMed] [Google Scholar]
- 13.Abovich N. and Rosbash,M. (1997) Cross-intron bridging interactions in the yeast commitment complex are conserved in mammals. Cell, 89, 403–412. [DOI] [PubMed] [Google Scholar]
- 14.Berglund J.A., Abovich,N. and Rosbash,M. (1998) A cooperative interaction between U2AF65 and mBBP/SF1 facilitates branchpoint region recognition. Genes Dev., 12, 858–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berglund J.A., Chua,K., Abovich,N., Reed,R. and Rosbash,M. (1997) The splicing factor BBP interacts specifically with the pre-mRNA branchpoint sequence UACUAAC. Cell, 89, 781–787. [DOI] [PubMed] [Google Scholar]
- 16.Seraphin B., Kretzner,L. and Rosbash,M. (1988) A U1 snRNA:pre-mRNA base pairing interaction is required early in yeast spliceosome assembly but does not uniquely define the 5′ cleavage site. EMBO J., 7, 2533–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seraphin B. and Rosbash,M. (1990) Exon mutations uncouple 5′ splice site selection from U1 snRNA pairing. Cell, 63, 619–629. [DOI] [PubMed] [Google Scholar]
- 18.Wu J. and Manley,J.L. (1989) Mammalian pre-mRNA branch site selection by U2 snRNP involves base pairing. Genes Dev., 3, 1553–1561. [DOI] [PubMed] [Google Scholar]
- 19.Parker R., Siliciano,P.G. and Guthrie,C. (1987) Recognition of the TACTAAC box during mRNA splicing in yeast involves base pairing to the U2-like snRNA. Cell, 49, 229–239. [DOI] [PubMed] [Google Scholar]
- 20.Zhuang Y. and Weiner,A.M. (1989) A compensatory base change in human U2 snRNA can suppress a branch site mutation. Genes Dev., 3, 1545–1552. [DOI] [PubMed] [Google Scholar]
- 21.Kandels-Lewis S. and Seraphin,B. (1993) Involvement of U6 snRNA in 5′ splice site selection. Science, 262, 2035–2039. [DOI] [PubMed] [Google Scholar]
- 22.Lynch K.W. and Maniatis,T. (1996) Assembly of specific SR protein complexes on distinct regulatory elements of the Drosophila doublesex splicing enhancer. Genes Dev., 10, 2089–2101. [DOI] [PubMed] [Google Scholar]
- 23.Tian M. and Maniatis,T. (1993) A splicing enhancer complex controls alternative splicing of doublesex pre-mRNA. Cell, 74, 105–114. [DOI] [PubMed] [Google Scholar]
- 24.Vilardell J. and Warner,J.R. (1994) Regulation of splicing at an intermediate step in the formation of the spliceosome. Genes Dev., 8, 211–220. [DOI] [PubMed] [Google Scholar]
- 25.Siebel C.W., Fresco,L.D. and Rio,D.C. (1992) The mechanism of somatic inhibition of Drosophila P-element pre-mRNA splicing: multiprotein complexes at an exon pseudo-5′ splice site control U1 snRNP binding. Genes Dev., 6, 1386–1401. [DOI] [PubMed] [Google Scholar]
- 26.Lam B.J., Bakshi,A., Ekinci,F.Y., Webb,J., Graveley,B.R. and Hertel,K.J. (2003) Enhancer-dependent 5′-splice site control of fruitless pre-mRNA splicing. J. Biol. Chem., 278, 22740–22747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lallena M.J., Chalmers,K.J., Llamazares,S., Lamond,A.I. and Valcarcel,J. (2002) Splicing regulation at the second catalytic step by Sex-lethal involves 3′ splice site recognition by SPF45. Cell, 109, 285–296. [DOI] [PubMed] [Google Scholar]
- 28.Engebrecht J.A., Voelkel-Meiman,K. and Roeder,G.S. (1991) Meiosis-specific RNA splicing in yeast. Cell, 66, 1257–1268. [DOI] [PubMed] [Google Scholar]
- 29.Siomi H., Matunis,M.J., Michael,W.M. and Dreyfuss,G. (1993) The pre-mRNA binding K protein contains a novel evolutionarily conserved motif. Nucleic Acids Res., 21, 1193–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spingola M. and Ares,M.,Jr (2000) A yeast intronic splicing enhancer and Nam8p are required for Mer1p-activated splicing. Mol. Cell, 6, 29–38. [DOI] [PubMed] [Google Scholar]
- 31.Nandabalan K., Price,L. and Roeder,G.S. (1993) Mutations in U1 snRNA bypass the requirement for a cell type-specific RNA splicing factor. Cell, 73, 407–415. [DOI] [PubMed] [Google Scholar]
- 32.Nakagawa T. and Ogawa,H. (1997) Involvement of the MRE2 gene of yeast in formation of meiosis-specific double-strand breaks and crossover recombination through RNA splicing. Genes Cells, 2, 65–79. [DOI] [PubMed] [Google Scholar]
- 33.Lesser C.F. and Guthrie,C. (1993) Mutational analysis of pre-mRNA splicing in Saccharomyces cerevisiae using a sensitive new reporter gene, CUP1. Genetics, 133, 851–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kunkel T.A., Bebenek,K. and McClary,J. (1991) Efficient site-directed mutagenesis using uracil-containing DNA. Methods Enzymol., 204, 125–139. [DOI] [PubMed] [Google Scholar]
- 35.Carey J., Lowary,P.T. and Uhlenbeck,O.C. (1983) Interaction of R17 coat protein with synthetic variants of its ribonucleic acid binding site. Biochemistry, 22, 4723–4730. [DOI] [PubMed] [Google Scholar]
- 36.Pauling M.H., McPheeters,D.S. and Ares,M.,Jr (2000) Functional Cus1p is found with Hsh155p in a multiprotein splicing factor associated with U2 snRNA. Mol. Cell. Biol., 20, 2176–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Igel H., Wells,S., Perriman,R. and Ares,M.,Jr (1998) Conservation of structure and subunit interactions in yeast homologues of splicing factor 3b (SF3b) subunits. RNA, 4, 1–10. [PMC free article] [PubMed] [Google Scholar]
- 38.Gill G. and Ptashne,M. (1988) Negative effect of the transcriptional activator GAL4. Nature, 334, 721–724. [DOI] [PubMed] [Google Scholar]
- 39.Rain J.C. and Legrain,P. (1997) In vivo commitment to splicing in yeast involves the nucleotide upstream from the branch site conserved sequence and the Mud2 protein. EMBO J., 16, 1759–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rutz B. and Seraphin,B. (2000) A dual role for BBP/ScSF1 in nuclear pre-mRNA retention and splicing. EMBO J., 19, 1873–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gottschalk A., Bartels,C., Neubauer,G., Luhrmann,R. and Fabrizio,P. (2001) A novel yeast U2 snRNP protein, Snu17p, is required for the first catalytic step of splicing and for progression of spliceosome assembly. Mol. Cell. Biol., 21, 3037–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jurica M.S. and Moore,M.J. (2003) Pre-mRNA splicing: awash in a sea of proteins. Mol. Cell, 12, 5–14. [DOI] [PubMed] [Google Scholar]
- 43.Gottschalk A., Tang,J., Puig,O., Salgado,J., Neubauer,G., Colot,H.V., Mann,M., Seraphin,B., Rosbash,M., Luhrmann,R. and Fabrizio,P. (1998) A comprehensive biochemical and genetic analysis of the yeast U1 snRNP reveals five novel proteins. RNA, 4, 374–393. [PMC free article] [PubMed] [Google Scholar]
- 44.Legrain P. and Chapon,C. (1993) Interaction between PRP11 and SPP91 yeast splicing factors and characterization of a PRP9–PRP11–SPP91 complex. Science, 262, 108–110. [DOI] [PubMed] [Google Scholar]
- 45.Abovich N., Liao,X.C. and Rosbash,M. (1994) The yeast MUD2 protein: an interaction with PRP11 defines a bridge between commitment complexes and U2 snRNP addition. Genes Dev., 8, 843–854. [DOI] [PubMed] [Google Scholar]
- 46.Graveley B.R. and Maniatis,T. (1998) Arginine/serine-rich domains of SR proteins can function as activators of pre-mRNA splicing. Mol. Cell, 1, 765–771. [DOI] [PubMed] [Google Scholar]
- 47.Seraphin B. and Rosbash,M. (1991) The yeast branchpoint sequence is not required for the formation of a stable U1 snRNA-pre-mRNA complex and is recognized in the absence of U2 snRNA. EMBO J., 10, 1209–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Legrain P., Seraphin,B. and Rosbash,M. (1988) Early commitment of yeast pre-mRNA to the spliceosome pathway. Mol. Cell. Biol., 8, 3755–3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brosi R., Groning,K., Behrens,S.E., Luhrmann,R. and Kramer,A. (1993) Interaction of mammalian splicing factor SF3a with U2 snRNP and relation of its 60-kD subunit to yeast PRP9. Science, 262, 102–105. [DOI] [PubMed] [Google Scholar]
- 50.Brosi R., Hauri,H.P. and Kramer,A. (1993) Separation of splicing factor SF3 into two components and purification of SF3a activity. J. Biol. Chem., 268, 17640–17646. [PubMed] [Google Scholar]
- 51.Will C.L., Schneider,C., MacMillan,A.M., Katopodis,N.F., Neubauer,G., Wilm,M., Luhrmann,R. and Query,C.C. (2001) A novel U2 and U11/U12 snRNP protein that associates with the pre-mRNA branch site. EMBO J., 20, 4536–4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bousquet-Antonelli C., Presutti,C. and Tollervey,D. (2000) Identification of a regulated pathway for nuclear pre-mRNA turnover. Cell, 102, 765–775. [DOI] [PubMed] [Google Scholar]
- 53.Graveley B.R. (2002) Sex, AGility and the regulation of alternative splicing. Cell, 109, 409–412. [DOI] [PubMed] [Google Scholar]
- 54.Del Gatto-Konczak F., Bourgeois,C.F., Le Guiner,C., Kister,L., Gesnel,M.C., Stevenin,J. and Breathnach,R. (2000) The RNA-binding protein TIA-1 is a novel mammalian splicing regulator acting through intron sequences adjacent to a 5′ splice site. Mol. Cell. Biol., 20, 6287–6299. [DOI] [PMC free article] [PubMed] [Google Scholar]