

Abstract
Background
Mitochondria are the main manufacturers of cellular ATP in eukaryotes. The plant mitochondrial genome contains large number of foreign DNA and repeated sequences undergone frequently intramolecular recombination. Upland Cotton (Gossypium hirsutum L.) is one of the main natural fiber crops and also an important oil-producing plant in the world. Sequencing of the cotton mitochondrial (mt) genome could be helpful for the evolution research of plant mt genomes.
Methodology/Principal Findings
We utilized 454 technology for sequencing and combined with Fosmid library of the Gossypium hirsutum mt genome screening and positive clones sequencing and conducted a series of evolutionary analysis on Cycas taitungensis and 24 angiosperms mt genomes. After data assembling and contigs joining, the complete mitochondrial genome sequence of G. hirsutum was obtained. The completed G.hirsutum mt genome is 621,884 bp in length, and contained 68 genes, including 35 protein genes, four rRNA genes and 29 tRNA genes. Five gene clusters are found conserved in all plant mt genomes; one and four clusters are specifically conserved in monocots and dicots, respectively. Homologous sequences are distributed along the plant mt genomes and species closely related share the most homologous sequences. For species that have both mt and chloroplast genome sequences available, we checked the location of cp-like migration and found several fragments closely linked with mitochondrial genes.
Conclusion
The G. hirsutum mt genome possesses most of the common characters of higher plant mt genomes. The existence of syntenic gene clusters, as well as the conservation of some intergenic sequences and genic content among the plant mt genomes suggest that evolution of mt genomes is consistent with plant taxonomy but independent among different species.
Introduction
Mitochondria, where the oxidative phosphorylation and the various biochemical processes take place throughout metabolism, are the main manufacturers of cellular ATP in eukaryotes. The plant mitochondrial genome contains large number of foreign DNA and repeated sequences undergone frequently intramolecular recombination, making it extraordinarily difficult to sequence plant mitochondrial (mt) genomes, particularly those of angiosperms [1], [2]. With the sequencing efforts over the past decade, the number of complete mt genomes has been greatly increased [3]–[5]. These finished mt genomes allow a deep analysis on the evolution of the higher plant mt genomes in aspects of gene orders, genome structure, and migration sequences as well as phylogenetic analysis.
Angiosperm mt genomes vary dramatically in size [6]. The size variation likely stems from their tendency to integrate DNA from other genomes [7]–[11] and the propensity for repeated sequences [12], [13]. Even so, large numbers of homologous sequences are distributed through the plant mt genome, including many noncoding sequences. Compared the mt genome of Brassica napus with that of Arabidopsis thaliana and Beta vulgaris, the values of shared sequences were in good agreement with the phylogenetic relationship among these three species [14].
Because of low rates of nucleotide substitution [15], [16], the mitochondrial genes are often used in plant evolutionary analysis, especially for construction of ancient phylogenetic relationships [5], [17], [18]. MtDNA trees are largely congruent with those constructed with chloroplast genes and nuclear genes, showing that mt genes are informative markers for evolution analysis across angiosperms. Gene orders are frequently not conserved across species [16], [19], possibly due to the mitochondrial penchant for recombination [12], [20]. Conservation of gene clusters are frequently used to infer evolution relationship among animal mt genomes [21], however, little researches have been performed in plant mt genomes [22], [23].
Here we report the first complete Gossypium mt genome derived from the widely cultivated upland species, Gossypium hirsutum. This sequence represents a major circular molecule that is 621,884 bp in length. The upland cotton mt genome possesses most of the common characters of higher plant mt genomes and maintains essential protein-coding genes and tRNA genes. Phylogenetic analyses, as well as analyses of conserved sequences, tRNAs and gene clusters among 25 mt genomes (24 angiosperms and Cycas taitungensis), indicate that (1) evolution of mt genomes is independent among different species, and (2) the evolution of the mt genomes is consistent with plant taxonomy as a whole (the upland cotton mt genome is much closer with Carica papaya than other angiosperms).
Results and Discussion
Genome assembly and features of Gossypium hirsutum mitochondrial genome
1. Genome assembly
The Gossypium hirsutum mt genome was sequenced using the Roche 454 GS FLX platform, which generated 286,792 reads with an average length of 399 bp. Cleaned reads were assembled by Newbler (Version 2.53), and contigs were subsequently joined via PCR into three scaffolds according to the from-to relationship among contigs (Table S1). Primers were designed and used to screen a Fosmid library [24] for clones to join the three scaffolds. Of the eight identified positive clones, two clones were selected for shotgun sequencing to finish the gaps, while end-sequencing of the remaining six clones were performed to verify the finished genome. Finally, the upland cotton mt genome was assembled into a single, circular molecule, with the length 621,884 bp and GC content 45.0% (Accession Number JX065074).
2. Gene annotation
68 genes were annotated in the cotton mt genome, including 35 protein-coding genes, four rRNA genes and 29 tRNA genes (Figure 1, Table S2). Among the eight multi-copy genes (i.e., nad1, rps3, rrn26, trnW, trnS(GCT), trnP, trnfM and trnM), nad1 gene contains an additional copy with exon b and exon c, and rps3 gene has an extra pseudogene-like copy which lacks 544 bp on the 3′ end of exon 2. Five genes (rps1, rps2, rps11, rps13 and rps19) are partially deleted and several remnant fragments of those deleted loci are annotated in the genome, with the largest fragment only 54 bp in length (derived from rps19). The 1.5 kb intron of rpl2 gene reported in other sequenced higher plant mt genomes is not found in the G. hirsutum mt genome.
Figure 1. Genome map of Gossypium hirsutum mitochondrial genome.

The map shows both the gene map (outer circle) and repeat map (inner map). Genes exhibited on the inside of outer circle are transcribed in a clockwise direction, while genes on the outside of outer circle are transcribed in a reverse direction. The inner circle reveals the distribution of repeats in G. hirsutum mt genome. The yellow lines represent > = 1 kb repeats, the blue lines represent <100 bp repeat and the red lines represent repeat between 100 bp and 1 kb.
The protein-coding genes in the upland cotton mt genome comprise a total length of 61,582 bp (9.9%), nearly half of which is intronic sequence (exons = 31,721 bp; introns = 29,861 bp), while tRNA genes and rRNA genes only represent 2,234 bp and 8,826 bp of the genome. The percentages of genic contents except the tRNA content differ significantly due to the variation of mt genome size in angiosperms (Figure 2A). However, the sequence length distribution is very similar to other sequenced seed plant mt genomes, with the exception of the rRNA content (Figure 2B); it is slightly elevated in the G. hirsutum mt genome due to the duplication of rrn26 (3,374 bp).
Figure 2. Gene composition of different mitochondrial genomes.

The percentage of different genic sequence (A) and the length of different genic sequence (B).
3. Gene clusters
Except the tRNA genes, ten gene clusters are annotated in the upland cotton mt genome (Table 1). Genes that comprise such clusters are usually separated by short intergenic regions or even partially overlapped in coding sequences and transcribed from the same strand. The gene orders differ markedly in higher plant mt genomes and four plant mt genomes are chosen to compare the gene orders with G. hirsutum mt genome. As showed in Figure 3, the G. hirsutum mt genome shares 10 clusters with C. papaya (Figure 3A), seven with R. communis (Figure 3B), six and four with A. thaliana (Figure 3C) and Z. mays (Figure 3D).
Table 1. Information of gene clusters in Gossypium hirsutum mt genome.
| Gene cluster | Location and Interval | Type |
| rpl16-rps3 | 593447..593881-(-28bp)-593853..597250 | III |
| cob-rps14 | 547956..549134-(1363bp)-550498..550800 | II |
| rpl2-rpl5-nad5c | 532722..533726-(497bp)-534224..534805-(1117bp)-535923..535944 | II |
| nad2abc-sdh3 | 418598..420365-(999bp)-421265..421699 | I |
| mttB-nad9 | 308702..309502-(184bp)-309687..310259 | IV |
| sdh4-cox3 | 258268..258666-(-72bp)-258594..259391 | I |
| cox1-rps10 | 260808..262400-(186bp)-262587..263768 | II |
| atp9-nad5ab | 162829..163140-(220bp)-163361..165659 | I |
| nad3-rps12 | 129383..129754-(48bp)-129803..130159 | II |
| nad1e-matR-nad1d | 81154..81412-(806bp)-82219..84186-(661bp)-84848..84905 | IV |
Boldface: Interval length between two genes.
Type I represents gene cluster composed of respiratory genes; Type II represents gene cluster composed of respiratory genes; Type III represents gene cluster composed of respiratory genes; Type IV represents gene cluster compose of respiratory genes.
Figure 3. Gene order and existed clusters between the mitochondrial gene maps of Gossypium and other four angiosperms.

Gene order of the protein-coding and rRNA-coding genes, and the former's trans-spliced exons were based on the mt genome of G. hirsutum arranging from top to bottom. Genes of other four mt genomes were indicated by the corresponding numbers given to cotton genes listed on the left margin. Duplicate genes carried the same number. From left to right for (A) C. papaya, (B) R. communis, (C) A. thaliana and (D) Z. mays.
4. Repeated sequence
343 repeat sequences larger than 20 bp were detected in G. hirsutum mt genome (Figure 1). In total, the detected repeats occupied 22.9% of the mt genome. Of the 343 repeats, most of them exist as short (20 bp to 39 bp), scattered repeats, about 10% (35 repeats) are larger than 100 bp (Table 2) and 1% (four repeats) larger than 10 kb, (R1, 27,495 bp; R2, 10,623 bp; R3 10,302 bp; and R4, 10,251 bp). Copy number for the larger repeats (100+ bp) varied narrowly from two (22 repeats) to four (two repeats) copies. The smaller repeats were also tabulated, and appeared to have distinct distributions and copy number variations (Table 3).
Table 2. Repeats (>100 bp) in Gossypium hirsutum mt genome.
| No. | Size (bp) | Identity (%) | Copy-1 | Copy-2a | Copy-3a | Copy-4a | Typeb | ||||
| start | end | start | end | start | end | start | End | ||||
| R01 | 27495 | 99.92 | 437002 | 464489 | 594397 | 621884 | DR | ||||
| R02 | 10623 | 99.81 | 224949 | 235564 | 532802 | 522190 | IR | ||||
| R03 | 10302 | 99.98 | 130185 | 140486 | 340674 | 330373 | IR | ||||
| R04 | 10251 | 99.86 | 64505 | 74747 | 247693 | 257941 | DR | ||||
| R05 | 879 | 100 | 140497 | 141375 | 330372 | 329494 | IR | ||||
| R06 | 399 | 99.5 | 225720 | 226118 | 421200 | 420802 | 532032 | 531634 | IR/DR | ||
| R07 | 349 | 99.43 | 81010 | 81358 | 226466 | 226118 | 531286 | 531634 | IR/DR | ||
| R08 | 260 | 86.54 | 519189 | 519438 | 555385 | 555140 | IR | ||||
| R09 | 259 | 98.07 | 226519 | 226775 | 273990 | 273734 | 531233 | 530978 | IR/DR | ||
| R10 | 256 | 83.2 | 56397 | 56644 | 68151 | 67904 | 251342 | 251095 | IR/DR | ||
| R11 | 229 | 99.13 | 39574 | 39802 | 119894 | 119666 | IR | ||||
| R12 | 203 | 99.51 | 260466 | 260668 | 430908 | 430706 | IR | ||||
| R13 | 194 | 100 | 70998 | 71191 | 254191 | 254384 | 427256 | 427449 | DR | ||
| R14 | 175 | 100 | 147589 | 147763 | 495709 | 495883 | DR | ||||
| R15 | 174 | 98.85 | 455621 | 455794 | 550847 | 551020 | 613016 | 613189 | DR | ||
| R16 | 168 | 91.07 | 378953 | 379120 | 536243 | 536080 | IR | ||||
| R17 | 166 | 94.58 | 162194 | 162357 | 310412 | 310248 | IR | ||||
| R18 | 162 | 91.98 | 278372 | 278532 | 506946 | 506786 | IR | ||||
| R19 | 160 | 97.5 | 225357 | 225515 | 439973 | 439816 | 532394 | 532236 | 597366 | 597208 | IR/DR |
| R20 | 159 | 99.37 | 278884 | 279042 | 455305 | 455463 | 612700 | 612858 | DR | ||
| R21 | 151 | 100 | 427472 | 427622 | 486639 | 486489 | IR | ||||
| R22 | 145 | 87.59 | 246993 | 247137 | 409294 | 409158 | IR | ||||
| R23 | 138 | 97.1 | 260510 | 260645 | 430864 | 430729 | 495627 | 495763 | IR/DR | ||
| R24 | 136 | 96.32 | 285066 | 285201 | 534095 | 533960 | IR | ||||
| R25 | 135 | 90.37 | 519189 | 519320 | 555385 | 555255 | IR | ||||
| R26 | 133 | 94.74 | 161921 | 162053 | 351301 | 351169 | IR | ||||
| R27 | 128 | 84.38 | 278561 | 278686 | 506757 | 506635 | IR | ||||
| R28 | 127 | 92.91 | 279043 | 279168 | 455473 | 455598 | 612868 | 612993 | DR | ||
| R29 | 123 | 88.62 | 260519 | 260638 | 430855 | 430736 | 495636 | 495755 | 578212 | 578092 | IR/DR |
| R30 | 118 | 99.15 | 185254 | 185371 | 309736 | 309619 | IR | ||||
| R31 | 113 | 100 | 81796 | 81908 | 438852 | 438740 | 596247 | 596135 | IR/DR | ||
| R32 | 113 | 98.23 | 86305 | 86417 | 307767 | 307655 | IR | ||||
| R33 | 107 | 100 | 70936 | 71042 | 254129 | 254235 | 502859 | 502753 | IR/DR | ||
| R34 | 101 | 85.15 | 302711 | 302803 | 513954 | 514053 | DR | ||||
| R35 | 101 | 97.03 | 425155 | 425255 | 555258 | 555158 | IR | ||||
Boldface: IR copy, compared with copy-1 as control.
DR and IR: direct and reverse repeats, respectively; IR/DR: both direct repeat and reverse repeat among multiple copies.
Table 3. Frequency distribution of repeat lengths in the mt genome of Gossypium hirsutum.
| Size, bp | 20–39 | 40–59 | 60–79 | 80–99 | 100–999 | > = 1000 |
| Number | 192 | 69 | 35 | 11 | 32 | 4 |
| Total length of repeats, bp | 10, 747 | 9, 667 | 9, 567 | 8, 365 | 18, 368 | 117, 300 |
| Coverage, % | 1.7 | 1.6 | 1.5 | 1.3 | 3.0 | 18.9 |
5. Cp-like sequences
Integration of cp-like sequences is a common occurrence in plant mitochondrial genomes, and G. hirsutum is no exception. 27 chloroplast-derived sequences (80% or higher identity to the G. hirsutum chloroplast genome) are found in the mt genome, contributing 6,833 bp (1.1% of the genome size) with segments ranging from 36 bp to 2,185 bp. 12 of the 27 chloroplast-derived sequences are tRNA related sequences, three are photosynthesis related sequences and the rest are other type of chloroplast sequences.
Migration of cpDNA in plant mt genomes
Chloroplast-derived sequences play an important role in plant mt genomes. Many researches have shown that cp-like tRNA genes are essential to maintain normal translation [18], [25]–[27] and cp-like sequences can act as functional genes and gene promoters [28], [29]. Besides, mitochondrial plastid DNA also contributes codons to mitochondrial protein-coding sequences and has a role in posttranscriptional RNA processing [10].
14 species that chloroplast genomes are available were chosen to analyze cp-like migration in plant mt genomes (Table 4). The length of individually integrated sequences varies widely, from 20 bp to 12 kb. The capacity of cpDNA in plant mt genomes also differs greatly; the total amount of cpDNA exceeds 60 kb in Vitis vinifera mt genome, whereas it represents less than 2 kb in Silene latifolia and Vigna radiate. Besides, the size of the largest integrated fragment varied from 275 bp (Silene latifolia) to 12 kb (Carica papaya). Based on the above data, the migration of cpDNA in plant mt genomes seems to be an independent and random event.
Table 4. Information of chloroplast homologous sequences in plants.
| Species | Total length of chloroplast homologous sequence in mt genome | Numbers of chloroplast homologs | Coverage of chloroplast homologous sequence |
| Arabidopsis thaliana | 4803 | 24 | 1.3% |
| Brassica napus | 8749 | 23 | 3.9% |
| Carica papaya | 21368 | 25 | 4.5% |
| Nicotiana tabacum | 11184 | 37 | 2.6% |
| Cucurbita pepo | 88208 | 204 | 9.0% |
| Gossypium hirsutum | 6833 | 27 | 1.1% |
| Vigna radiata | 2109 | 17 | 0.5% |
| Vitis vinifera | 64357 | 73 | 8.3% |
| Ricinus communis | 5649 | 26 | 1.1% |
| Silene latifolia | 1998 | 16 | 0.8% |
| Sorghum bicolor | 26357 | 45 | 5.6% |
| Triticum aestivum | 13855 | 36 | 3.1% |
| Zea mays | 23445 | 39 | 4.1% |
| Oryza sativa ssp indica | 33176 | 41 | 6.7% |
| Oryza sativa ssp japonica | 33157 | 41 | 6.7% |
Blast was performed to check the homology of cp-like migration in plant mt genomes. Five cp-derived fragments (trnH, trnM, trnN, trnP and trnW) were found conserved in all analyzed mt genomes and one (trnD) and two (trnC and trnF) cp-derived fragment were found conserved in dicots and monocots, respectively. In addition, some of these conserved cp-derived fragments maintain the same sequence arrangement relationship with mitochondrial genes (Figure 4), indicating these migration events are very ancient and occurred before the species differentiation.
Figure 4. Linkage map between cp-homologous and mitochondrial sequences in higher plant mt genomes.

Origin and distribution of tRNAs in plant mt genomes
The ancestral mt genome possesses an intact set of transfer RNAs (tRNAs), however, a large number of tRNAs undergo loss, migration and inactivation during mt genome evolution [27]. Different with the human mt genome, which keeps a minimal but complete set of tRNA genes, the number of tRNA genes in numerous plant mt genomes is insufficient for translation, although a certain number of tRNA genes have been brought in via DNA migration [30]–[32].
To evaluate the origin and distribution of tRNA genes, tRNAscan-SE [33] was adopted to predict the number and types of tRNA genes. Most of the analyzed species keep 16–21 kinds of tRNA genes and because of extensive loss of genes in Silene latifolia [18], only 10 were annotated. These results suggest that nuclear encoded tRNAs are necessary to maintain the normal translation in higher plant mt genomes.
Based on chloroplast genomes, 19 native (mitochondria-originated) tRNA genes and 19 cp-like tRNA genes are defined in higher plant mt genomes (Figure 5A). As showed in Figure 5B and 5C, although we found the uptake of four cp-like tRNAs (trnD, trnF, trnN, and trnW) and lose of four native tRNAs (trnD, trnF, trnN, and trnW) seems to occur during the same period, the uptake and lose of the rest cp-like tRNA genes (Figure 5B) and native tRNA genes (Figure 5C) are more likely to be occurred in different period of evolution. Besides, some cp-like tRNA genes have scattered distribution and some native tRNA genes are irregularly lost among higher plant mt genomes, showing the gain and lose of tRNA genes occurred independently during the evolution.
Figure 5. Distribution map of tRNA genes in 25 plants.

Figure 5A shows distribution of tRNA genes in higher plant mt genomes: the yellow boxs represented native tRNA genes, the green cells represent cp-like tRNA genes; Figure B shows uptake of cp-like tRNA genes during different evolutionary period; Figure C shows loss of native tRNA genes during different evolutionary period. The three Oryza genomes: 1, Oryza rufipogon; 2, Oryza sativa subsp indica; 3, Oryza sativa subsp japonica. The two Beta genomes are: 1, Beta vulgaris subsp maritima; 2, Beta vulgaris subsp vulgaris.
Gene orders and gene clusters in plant mt genomes
The gene orders differ tremendously among plant mt genomes. In this research, we compared the gene orders across the 25 species and counted the number of syntenic gene clusters (genes that keep the same order; Table 5). In general, the closer species in evolution share more clusters. However, there are also some inconsistent cases, C. taitungensis and C. lanatus share 13 gene clusters, much more than the number between C. lanatus and the other angiosperms; the cluster number between T. aestivum and C. lanatus is larger than that between C. lanatus and the other dicots. These exceptions probably were due to the frequently recombination during the plant mt genomes. Recombination can break the previous clusters and result in novel ones, while multiple recombination events can lead to generate the same synteny gene clusters too.
Table 5. Numbers of synteny gene clusters across 25 plant mt genomes.
| Species | Ct | At | Bvm | Bvv | Bj | Bn | Bo | Cap | Cl | Cup | Gh | Nt | Rc | Sl | Vr | Vv | Or | Ori | Orj | Sb | Td | Ta | Zl | Zm |
| A. thaliana | 8 | |||||||||||||||||||||||
| Beta1 | 8 | 6 | ||||||||||||||||||||||
| Beta2 | 7 | 6 | 39 | |||||||||||||||||||||
| B. juncea | 8 | 19 | 6 | 7 | ||||||||||||||||||||
| B. napus | 8 | 19 | 6 | 7 | 39 | |||||||||||||||||||
| B. oleracea | 8 | 19 | 6 | 7 | 39 | 39 | ||||||||||||||||||
| C. papaya | 11 | 11 | 8 | 8 | 11 | 11 | 11 | |||||||||||||||||
| C. lanatus | 13 | 9 | 10 | 10 | 10 | 10 | 11 | 16 | ||||||||||||||||
| C. pepo | 13 | 10 | 9 | 10 | 9 | 9 | 10 | 16 | 23 | |||||||||||||||
| G. hirsutum | 9 | 8 | 4 | 4 | 9 | 9 | 9 | 11 | 12 | 12 | ||||||||||||||
| N. tabacum | 10 | 8 | 9 | 9 | 8 | 8 | 8 | 14 | 14 | 16 | 11 | |||||||||||||
| R.communis | 12 | 10 | 11 | 11 | 10 | 9 | 10 | 14 | 18 | 16 | 8 | 15 | ||||||||||||
| S. latifolia | 7 | 7 | 10 | 10 | 7 | 7 | 7 | 10 | 8 | 9 | 6 | 12 | 9 | |||||||||||
| V. radiata | 10 | 10 | 6 | 6 | 11 | 10 | 11 | 11 | 11 | 11 | 10 | 12 | 13 | 8 | ||||||||||
| V. vinifera | 12 | 10 | 10 | 10 | 12 | 12 | 12 | 12 | 17 | 14 | 7 | 12 | 13 | 9 | 10 | |||||||||
| O. rufipogon | 9 | 6 | 5 | 5 | 5 | 6 | 6 | 7 | 10 | 9 | 6 | 6 | 7 | 5 | 6 | 10 | ||||||||
| Oryza1 | 9 | 5 | 5 | 5 | 5 | 6 | 6 | 7 | 10 | 9 | 6 | 5 | 7 | 5 | 6 | 10 | 40 | |||||||
| Oryza2 | 9 | 5 | 5 | 5 | 5 | 6 | 6 | 7 | 10 | 9 | 6 | 5 | 7 | 5 | 6 | 10 | 40 | |||||||
| S. bicolor | 7 | 4 | 6 | 5 | 5 | 4 | 4 | 6 | 8 | 5 | 5 | 5 | 5 | 2 | 5 | 9 | 8 | 9 | 9 | |||||
| T. dactyloides | 6 | 6 | 4 | 4 | 6 | 5 | 5 | 7 | 6 | 5 | 6 | 5 | 7 | 2 | 5 | 6 | 8 | 9 | 9 | 7 | ||||
| T. aestivum | 7 | 6 | 5 | 5 | 8 | 6 | 6 | 10 | 11 | 10 | 6 | 8 | 9 | 4 | 7 | 8 | 11 | 11 | 11 | 9 | 10 | |||
| Z. luxurians | 7 | 7 | 5 | 5 | 5 | 5 | 5 | 7 | 8 | 6 | 5 | 6 | 6 | 3 | 5 | 7 | 9 | 9 | 9 | 11 | 17 | 11 | ||
| Z. mays | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 9 | 8 | 7 | 4 | 5 | 6 | 4 | 5 | 8 | 11 | 9 | 9 | 10 | 13 | 11 | 20 | |
| Z. perennis | 7 | 8 | 6 | 6 | 6 | 7 | 7 | 7 | 9 | 7 | 4 | 8 | 6 | 3 | 5 | 8 | 10 | 11 | 11 | 11 | 15 | 11 | 30 | 17 |
Note: Numbers of synteny gene clusters differed across C. taitungensis (Ct), A. thaliana (At), Beta vulgaris subsp maritima (Bvm), Beta vulgaris subsp vulgaris (Bvv), B. juncea (Bj), B. napus (Bn), B. oleracea (Bo), C. papaya (Cap), C. lanatus (Cl), C. pepo (Cup), G. hirsutum (Gh), N. tabacum (Nt), R.communis (Rc), S. latifolia (Sl), V. radiata (Vr), V. vinifera (Vv), O. rufipogon (Or), Oryza sativa subsp indica (Ori), Oryza sativa subsp japonica (Orj), S. bicolor (Sb), T. dactyloides (Td), T. aestivum (Ta), Z. luxurians (Zl), Z. mays (Zm) and Z. perennis. The two Beta genomes in the first row were: 1, Beta vulgaris subsp maritima; 2, Beta vulgaris subsp vulgaris, and the two Oryza genomes in the first row were: 1, Oryza sativa subsp indica; 2, Oryza sativa subsp japonica. Any two genes linked were counted as one synteny gene cluster.
There are also some conserved syntenic gene clusters among higher plant mt genomes. Alverson reported that 14 syntenic gene clusters are shared between C. lanatus and C. pepo [13]. We checked gene clusters in the 25 mt genomes and found five gene clusters conserved in all the plant mt genomes (Figure 6 and Table 6). There are also four and one gene clusters that are specific conserved in dicots and monocots respectively. The genes that compose these clusters share short intergenic region or even overlap in the CDS region. The gene cluster atp4-nad4L, for example, exists in all dicots surveyed, except for Gossypium hirsutum; the cluster nad1e-matR exists in all the plant but Beta, Nicotiana and Silene, indicating a lineage specific disruption of this cluster.
Figure 6. Distribution of conserved gene clusters.
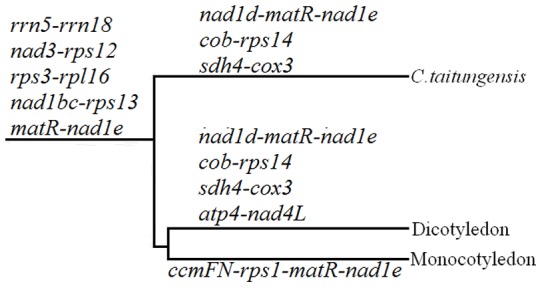
Table 6. Distribution of closely linked clusters in Gossypium hirsutum and other plant mt genomes.
| rrn5- rrn18 | nad3- rps12 | rps3- rpl16 | nad1d- matR- nad1e | nad1d- matR | matR- nad1e | sdh4- cox3 | cob- rps14 | nad1bc- rps13 | atp4- nad4L | ccmFn- rps1-matR- nad1e | ccmFN- rps1 | |
| C.taitungensis | + | + | + | + | + | + | + | + | + | − | − | + |
| A.thaliana | + | + | + | + | + | + | + | + | # | + | # | # |
| Beta1 | + | + | # | − | + | − | + | # | + | + | − | # |
| Beta2 | + | + | # | − | + | − | + | # | + | + | − | # |
| B.juncea | + | + | + | + | + | + | + | + | # | + | # | # |
| B.napus | + | + | + | + | + | + | + | + | # | + | # | # |
| B.oleracea | + | + | + | + | + | + | + | + | # | + | # | # |
| C.papaya | + | + | + | + | + | + | + | + | + | + | − | + |
| C.lanatus | + | + | + | + | + | + | + | + | + | + | − | + |
| C.pepo | + | + | + | + | + | + | + | + | + | + | − | − |
| G.hirsutum | + | + | + | + | + | + | + | + | # | − | # | # |
| N.tabacum | + | + | + | − | + | − | + | + | + | + | − | − |
| R.communis | + | + | + | + | + | + | + | + | + | + | − | − |
| S. latifolia | + | # | # | − | + | − | # | + | + | + | − | # |
| V. radiata | + | + | + | + | + | + | + | + | # | + | − | − |
| V. vinifera | + | + | + | + | + | + | + | + | + | + | − | − |
| O.rufipogon | + | + | + | − | − | + | # | − | + | − | + | + |
| Oryza1 | + | + | + | − | − | + | # | − | + | − | + | + |
| Oryza2 | + | + | + | − | − | + | # | − | + | − | + | + |
| S.bicolor | + | + | + | − | − | + | # | − | + | − | + | + |
| T.dactyloides | + | + | + | − | − | + | # | # | + | − | + | + |
| T.aestivum | + | + | + | − | − | + | # | − | + | − | + | + |
| Z.luxurians | + | + | + | − | − | + | # | # | + | − | + | + |
| Z.mays | + | + | + | − | − | + | # | # | + | − | + | + |
| Z.perennis | + | + | + | − | − | + | # | # | + | − | + | + |
Note:
+, presence of the gene cluster; −, absence of the gene cluster; #, absence for gene lose.
The two Oryza genomes are: 1, Oryza sativa Indica Group; 2, Oryza sativa Japonica Group.
The two Beta genomes are: 1, Beta vulgaris subsp. maritima; 2, Beta vulgaris subsp. vulgaris.
The origin of conserved syntenic gene clusters is still unclear. According to the Ka/Ks ratio of these gene clusters, most of them undergo purify selection and the remaining undergo neutral evolution, indicating the important role of natural selection on these gene clusters. The genes in each of gene clusters are transcribed from the same strand, implying that they may function in a co-transcription manner; the rps3-rp116-nad3-rps12 cluster in rice shares the same promoter and undergoes co-transcription [34]; three clusters (rrn5-rrn18, rps3-rpl16 and nad3-rps12) were reported co-transcribed in Phoenix dactylifera [35]. Besides, these clusters may also be helpful to predict functional coupling between genes in angiosperms [36].
Conserved sequence and phylogenetic analysis
Homologous sequences are distributed among the plant mt genomes, including a certain region of non-coding sequences. To calculate the length of shared sequences among different species, the chloroplast-derived sequences and extra copies of large repeats were removed from the analyzed mt genomes before blasting against the other mt genomes. As shown in Table S3, species closely related share the most sequences, even outside of the coding regions; species belong to different families share fewer and species belong to different groups (gymnosperm, monocots and dicots) share the fewest. These results indicate that the length of homologous sequence among plant mt genomes is consistent with taxonomy, despite the exceptional variability among these mt genomes. The Silene latifolia, member of the Caryophyllaceae family, is the least-shared species among the 24 angiosperms because of extensive loss of genomic sequence [18].
21 respiratory chain related genes that exist in all higher plants were selected for phylogenetic analysis (Table S4), including 17 respiratory complex genes and four cytochrome c biogenesis genes. These genes were first concatenated in a head-to-tail format, and phylogenetic trees were completed with both maximum likelihood method (ML; Figure 7A) and neighbor-joining (NJ; Figure 7B) method. The phylogenetic trees were congruent with the plant taxonomy and NCBI taxonomy common tree (Figure 8). To further assess the utility of the mt genes in phylogenetic reconstruction, these 21 were divided into five groups according to the function of their proteins, and genes in each group were assembled in a head-to-tail arrangement. These trees show more or less differences with the common tree. Three of the five functional groups (Complex I, V and cytochrome c biogenesis genes) reconstruct the divergence of monocots and dicots but showing slightly different evolution relationships (Figure S1 and Figure S2), the Complex III and IV gene sets fail even to reconstruct the monocot-dicot division (Figure S3).
Figure 7. Phylogenetic trees of 21 respiratory related genes.

The ML tree (A) and the NJ tree (B). Genes used were listed in Table S4, including 17 respiratory complex genes and four cytochrome c biogenesis genes.
Figure 8. NCBI common tree of 25 analyzed species.

The evolutionary rate of mitochondrial genes varies greatly among plant species [5], phylogenetic analysis of single gene differs with the plant taxonomy. In this research, we tried phylogenetic analysis of functional groups and 21 conserved genes. Compared with previous reports [5], [17], [18], phylogenetic tree of 21 conserved mitochondrial genes shows best coincidence with NCBI taxonomy common tree.
Conclusion
Plant mitochondrial genomes are fascinating molecules, whose lability and striking differences in evolutionary rates among genic and intergenic regions have generated significant interest. The G. hirsutum mt genome possesses most of the common characters of higher plant mt genomes. The comparative analysis presented here allows a more comprehensive understanding of mitochondrial genome evolution in higher plant. The existence and conservation of gene clusters, origin and distribution of tRNA genes, as well as the conservation of some intergenic sequences and genic contents suggest that evolution of mt genomes is consistent with plant taxonomy. But the highly dynamic genome structures (genome size, gene orders and gene content) reflects that recombination of higher plant mt molecular is independent and random among different species.
Materials and Methods
Plant material and mitochondrial DNA extraction
Mitochondria were obtained from 7-days-old etiolated seedlings of a variety of upland cotton (Gossypium hirsutum L.), ‘Sumian No. 20 (Xu244)’. Etiolated seedlings were ground with homogenate buffer in the proper proportion and after pulping, nuclei and debris were removed by centrifugation at 3,000 rpm for 16 min at 4°C, the supernatant was transferred to a new tube and centrifugation was carried out for 40 min at 8,500 rpm at 4°C to isolate mitochondria. Purified mitochondria were obtained by discontinuous sucrose density gradient centrifugation. After digestion of nuclear DNA with DNase I, mitochondria were lysed by CTAB at 65°C for 30 min. The lysis solution was extracted by chloroform: isoamyl alcohol for 2–3 times and then absolute ethyl alcohol was used to precipitate the mitochondrial DNA (mtDNA).
Genome sequencing and assembly
Upland cotton mtDNA were sequenced using 454 in Beijing Institute of Genomics, Chinese Academy of Sciences. Purified mtDNA was used to construct sequencing library, according to the manufacturer's manual for the 454 GS FLX Titanium. The reads were assembled into contigs by 454 GS FLX platform after removing the adaptor and contaminant sequences [37].
The relationship among contigs was acquired according to the from-to relationship. Then, primers were designed to join the contigs and fill the genomic gaps. After sequencing of PCR bands, the contig were assembled in scaffolds.
Mitochondrial genome library construction and clone sequencing
Mitochondrial genome Fosmid library for G. hirsutum was constructed following CopyControl Fosmid Library Production Kit (Epicentre, Cat. No. CCFOS110). Mitochondria genomic DNA was random mechanical sheared, size-fractioned by pulsed field gel electrophoresis, and ligated to pCC1FOS vector. The packaged phage infected the EpI300-T1R host cell and then well-separated colonies were randomly picked to accomplish the fosmid library construction [24].
The library was screened by primers designed on the conserved genes and scaffold terminals. The positive clones were chose for shotgun sequencing in Beijing Institute of Genomics, Chinese Academy of Sciences. The terminal sequencing of positive clones operated in Invitrogen Life Technologies Corporation.
Genome annotation and sequence analysis
Just like the method described in Alverson's report [13], a local database was built with mt genome sequences available in NCBI, which contained all protein and ribosomal RNA (rRNA) of previously sequenced plant mitochondrial genomes. Protein coding genes and ribosomal RNA genes were identified by performing local blast searches against the database. The tRNAscan-SE [33] was used to predict the tRNA genes. NCBI blast and local blast was used to identify putatively conserved regions among different plant mt genomes. The gene map was created by OGDraw (http://ogdraw.mpimp-golm.mpg.de/).
AB-blast was used to identify repeat sequences in G. hirsutum and other plant mt genomes (Table S5) as described previously [4], [9], [13]–[14], [18]–[19], [38]–[45]. The repeat sequence distribution map was drawn by Circos. The genome was searched against itself and local Perl scripts were adopted to run detail analysis. We used local R scripts to identify gene clusters by comparing every two mt genomes. Then we used the MEGA 5.0 to draw phylogenetic tree based on clustered genes. These 21 genes were 17 respiratory complex genes (atp1, atp4, atp6, atp8, atp9, cob, cox1, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7, nad9) and four cytochrome c biogenesis genes (ccmB, ccmC, ccmFC, ccmFN) (Table S4).
Supporting Information
Phylogenetic trees of NADH dehydrogenase genes and cytochrome c biogenesis genes. The ML tree (A) and NJ tree (B) were based on NADH dehydrogenase genes. The ML tree (C) and the NJ tree (D) were based on cytochrome c biogenesis genes.
(TIF)
Phylogenetic trees of ATPase genes. The ML tree (A) and the NJ tree (B).
(TIF)
Phylogenetic trees of apocytochrome b genes and cytochrome c oxidase genes. The ML tree (A) and NJ tree (B) were based on apocytochrome b genes. The ML tree (C) and the NJ tree (D) were based on cytochrome c oxidase genes.
(TIF)
Partial primers of PCR in genome assembling.
(DOC)
Genes annotated in the Gossypium hirsutum mt genome.
(DOC)
Size of shared sequences among 25 plant mt genomes.
(XLS)
Information of genes in phylogenetic tree.
(DOC)
Information of mitochondrial genomes involved in this study.
(DOC)
Acknowledgments
We thank Prof Shu-Miaw Chao at Biodiversity Research Center of Academia Sinica, Taipei, China, and Dr. Lida Zhang at Shanghai Jiao Tong University and Dr. Yi Huang at Chinese Academy of Agricultural Sciences for helps during data analysis, Prof. Xuequn Liu at South-Central University for Nationalities for helpful discussion. We are deeply indebted to Prof. Ying-guo Zhu and Dr. Shaoqing Li at Wuhan University for supplying the experimental platform.
Funding Statement
This work was supported by grants from the National Natural Science Foundation of China (NSFC grant number 31171591), the National High Technology Research and Development Program (grant number 2011AA10A102), Key Project of MOE (grant number 107012), and Training Program Foundation for the New Century Excellent Talents in University (NCET-06-0106) to J. Hua, and the support by the Innovation Fund for Graduate Student of China Agricultural University (KYCX2010024) to S. Li. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Palmer JD, Herbo LA (1987) Unicircular structure of the Brassica hirta mitochondrial genome. Curr genet 11: 565–570. [DOI] [PubMed] [Google Scholar]
- 2. Ward BL, Anderson RS, Bendich AJ (1981) The mitochondrial genome is large and variable in a family of plants (Cucurbitaceae). Cell 25: 793–803. [DOI] [PubMed] [Google Scholar]
- 3. Oda K, Yamato K, Ohta E, Nakamura Y, Takemura M, et al. (1992) Gene organization deduced from the complete sequence of liverwort Marchantia polymorpha mitochondrial DNA: A primitive form of plant mitochondrial genome. J Mol Biol 223: 1–7. [DOI] [PubMed] [Google Scholar]
- 4. Unseld M, Marienfeld JR, Brandt P, Brennicke A (1997) The mitochondrial genome of Arabidopsis thaliana contains 57 genes in 366,924. Nat Genet 15: 57–61. [DOI] [PubMed] [Google Scholar]
- 5. Ma PF, Guo ZH, Li DZ (2012) Rapid sequencing of the bamboo mitochondrial genome using Illumina technology and parallel episodic evolution of organelle genomes in grasses. PLoS One 7: e30297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Quetier F, Vedel F (1977) Heterogeneous population of mitochondrial DNA molecules in higher plants. Nature 268: 365–368. [Google Scholar]
- 7. Stern DB, Lonsdale DM (1982) Mitochondrial and chloroplast genomes of maize have a 12-kilobase DNA sequence in common. Nature 299: 698–702. [DOI] [PubMed] [Google Scholar]
- 8. Vaughn JC, Mason MT, Sper-Whitis GL, Kuhlman P, Palmer JD (1995) Fungal origin by horizontal transfer of a plant mitochondrial group I intron in the chimeric coxI gene of Peperomia. J Mol Evol 41: 563–572. [DOI] [PubMed] [Google Scholar]
- 9. Goremykin VV, Salamini F, Velasco R, Viola R (2009) Mitochondrial DNA of Vitis vinifera and the Issue of Rampant Horizontal Gene Transfer. Mol Biol Evol 26: 99–110. [DOI] [PubMed] [Google Scholar]
- 10. Wang D, Rousseau-Gueutin M, Timmis JN (2012) Plastid sequences contribute to some plant mitochondrial genes. Mol Biol Evol 29: 1707–1711. [DOI] [PubMed] [Google Scholar]
- 11. Alverson AJ, Rice DW, Dickinson S, Barry K, Palmer JD (2011) Origins and recombination of the bacterial-sized multichromosomal mitochondrial genome of cucumber. Plant Cell 23: 2499–2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kitazaki K, Kubo T (2010) Cost of having the largest mitochondrial genome: evolutionary mechanism of plant mitochondrial genome. J Bot 2010: 1–12. [Google Scholar]
- 13. Alverson AJ, Wei X, Rice DW, Stern DB, Barry K, et al. (2010) Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol Biol Evol 27: 1436–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Handa H (2003) The complete nucleotide sequence and RNA editing content of the mitochondrial genome of rapeseed (Brassica napus L.): comparative analysis of the mitochondrial genomes of rapeseed and Arabidopsis thaliana. Nucleic Acids Res 31: 5907–5916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wolfe KH, Li W-H, Sharp PM (1987) Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. P Natl Acad Sci USA 84: 9054–9058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Palmer JD, Herbon LA (1988) Plant mitochondrial DNA evolved rapidly in structure, but slowly in sequence. J Mol Evol 28: 87–97. [DOI] [PubMed] [Google Scholar]
- 17. Qiu YL, Li L, Wang B, Xue JY, Hendry TA, et al. (2010) Angiosperm phylogeny inferred from sequences of four mitochondrial genes. J Syst Evol 48: 391–425. [Google Scholar]
- 18. Sloan DB, Alverson AJ, Štorchová H, Palmer JD, Taylor DR (2010) Extensive loss of translational genes in the structurally dynamic mitochondrial genome of the angiosperm Silene latifolia . BMC Evol Biol 10: 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ogihara Y (2005) Structural dynamics of cereal mitochondrial genomes as revealed by complete nucleotide sequencing of the wheat mitochondrial genome. Nucleic Acids Res 33: 6235–6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Palmer JD, Shields CR (1984) Tripartite structure of the Brassica campestris mitochondrial genome. Nature 307: 437–440. [Google Scholar]
- 21. Boore JL, Brown WM (1998) Big trees from little genomes: mitochondrial gene order as a phylogenetic tool. Curr Opin Genet Dev 8: 668–674. [DOI] [PubMed] [Google Scholar]
- 22. Liu Y, Wang B, Cui P, Li L, Xue JY, et al. (2012) The mitochondrial genome of the lycophyte Huperzia squarrosa: the most archaic form in vascular plants. PLoS One 7: e35168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu Y, Xue J-Y, Wang B, Li L, Qiu Y-L (2011) The mitochondrial genomes of the early land plants Treubia lacunosa and Anomodon rugelii: dynamic and conservative evolution. PLoS One 6: e25836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li SS, Liu GZ, Chen ZW, Wang YM, Li PB, et al. (2013) Construction and initial analysis of five Fosmid libraries of mitochondrial genomes of cotton (Gossypium). Chin Sci Bull 58: 1–7. [Google Scholar]
- 25. Clifton SW (2004) Sequence and comparative analysis of the maize NB mitochondrial genome. Plant Physiol 136: 3486–3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sugiyama Y, Watase Y, Nagase M, Makita N, Yagura S, et al. (2004) The complete nucleotide sequence and multipartite organization of the tobacco mitochondrial genome: comparative analysis of mitochondrial genomes in higher plants. Mol Genet Genomics 272: 603–615. [DOI] [PubMed] [Google Scholar]
- 27. Dietrich A, Small I, Cosset A, Weil J, Marechal-Drouard L (1996) Editing and import: strategies for providing plant mitochondria with a complete set of functional transfer RNAs. Biochimie 78: 518–529. [DOI] [PubMed] [Google Scholar]
- 28. Nakazono M, Nishiwaki S, Tsutsumi N, Hirai A (1996) A chloroplast-derived sequence is utilized as a source of promoter sequences for the gene for subunit 9 of NADH dehydrogenase (nad9) in rice mitochondria. Mol Gen Genet 252: 371–378. [DOI] [PubMed] [Google Scholar]
- 29. Adams KL, Daley DO, Whelan J, Palmer JD (2002) Genes for two mitochondrial ribosomal proteins in flowering plants are derived from their chloroplast or cytosolic counterparts. Plant Cell 14: 931–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Marechal-Drouard L, Guillemaut P, Cosset A, Arbogast M, Weber F, et al. (1990) Transfer RNAs of potato (Solanum tuberosum) mitochondria have different genetic origins. Nucleic Acids Res 18: 3689–3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Maréchal-Drouard L, Weil J, Dietrich A (1993) Transfer RNAs and transfer RNA genes in plants. Annu Rev Plant Biol 44: 13–32. [DOI] [PubMed] [Google Scholar]
- 32. Salinas T, Duchêne A-M, Maréchal-Drouard L (2008) Recent advances in tRNA mitochondrial import. Trends Biochem Sci 33: 320–329. [DOI] [PubMed] [Google Scholar]
- 33. Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25: 0955–0964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nakazono M, Itadani H, Wakasugi T, Tsutsumi N, Sugiura M, et al. (1995) The rps3-rpl16-nad3-rps12 gene cluster in rice mitochondrial DNA is transcribed from alternative promoters. Curr Genet 27: 184–189. [DOI] [PubMed] [Google Scholar]
- 35. Fang Y, Wu H, Zhang T, Yang M, Yin Y, et al. (2012) A complete sequence and transcriptomic analyses of date palm (Phoenix dactylifera L.) mitochondrial genome. PLoS One 7: e37164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Overbeek R, Fonstein M, D'souza M, Pusch GD, Maltsev N (1999) The use of gene clusters to infer functional coupling. P Natl Acad Sci USA 96: 2896–2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang T, Zhang X, Hu S, Yu J (2011) An efficient procedure for plant organellar genome assembly, based on whole genome data from the 454 GS FLX sequencing platform. Plant Methods 7: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chaw S-M, Chun-Chieh Shih A, Wang D, Wu Y-W, Liu S-M (2008) The mitochondrial genome of the gymnosperm Cycas taitungensis contains a novel family of short interspersed elements, Bpu sequences, and abundant RNA editing sites. Mol Biol Evol 25: 603–615. [DOI] [PubMed] [Google Scholar]
- 39. Fujii S, Kazama T, Yamada M, Toriyama K (2010) Discovery of global genomic re-organization based on comparison of two newly sequenced rice mitochondrial genomes with cytoplasmic male sterility-related genes. BMC Genomics 11: 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tian X (2006) The rice mitochondrial genomes and their variations. Plant Physiol 140: 401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Notsu Y, Masood S, Nishikawa T, Kubo N, Akiduki G, et al. (2002) The complete sequence of the rice (Oryza sativa L.) mitochondrial genome: frequent DNA sequence acquisition and loss during the evolution of flowering plants. Mol Genet Genomics 268: 434–445. [DOI] [PubMed] [Google Scholar]
- 42. Allen JO, Fauron CM, Minx P, Roark L, Oddiraju S, et al. (2007) Comparisons among two fertile and three male-sterile mitochondrial genomes of maize. Genet 177: 1173–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kubo T, Nishizawa S, Sugawara A, Itchoda N, Estiati A, et al. (2000) The complete nucleotide sequence of the mitochondrial genome of sugar beet (Beta vulgaris L.) reveals a novel gene for tRNACys (GCA). Nucleic Acids Res 28: 2571–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rivarola M, Foster JT, Chan AP, Williams AL, Rice DW, et al. (2011) Castor bean organelle genome sequencing and worldwide genetic diversity analysis. PLoS One 6: e21743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Alverson AJ, Zhuo S, Rice DW, Sloan DB, Palmer JD (2011) The mitochondrial genome of the legume Vigna radiata and the analysis of recombination across short mitochondrial repeats. PLoS One 6: e16404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Phylogenetic trees of NADH dehydrogenase genes and cytochrome c biogenesis genes. The ML tree (A) and NJ tree (B) were based on NADH dehydrogenase genes. The ML tree (C) and the NJ tree (D) were based on cytochrome c biogenesis genes.
(TIF)
Phylogenetic trees of ATPase genes. The ML tree (A) and the NJ tree (B).
(TIF)
Phylogenetic trees of apocytochrome b genes and cytochrome c oxidase genes. The ML tree (A) and NJ tree (B) were based on apocytochrome b genes. The ML tree (C) and the NJ tree (D) were based on cytochrome c oxidase genes.
(TIF)
Partial primers of PCR in genome assembling.
(DOC)
Genes annotated in the Gossypium hirsutum mt genome.
(DOC)
Size of shared sequences among 25 plant mt genomes.
(XLS)
Information of genes in phylogenetic tree.
(DOC)
Information of mitochondrial genomes involved in this study.
(DOC)