

Abstract
Background
Despite rapid progress in understanding the mechanisms that shape the evolution of proteins, the relative importance of various factors remain to be elucidated. In this study, we have assessed the effects of 16 different biological features on the evolutionary rates (ERs) of protein-coding sequences in bacterial genomes.
Results
Our analysis of 18 bacterial species revealed new correlations between ERs and constraining factors. Previous studies have suggested that transcriptional abundance overwhelmingly constrains the evolution of yeast protein sequences. This transcriptional abundance leads to selection against misfolding or misinteractions. In this study we found that there was no single factor in determining the evolution of bacterial proteins. Not only transcriptional abundance (codon adaptation index and expression level), but also protein-protein associations (PPAs), essentiality (ESS), subcellular localization of cytoplasmic membrane (SLM), transmembrane helices (TMH) and hydropathicity score (HS) independently and significantly affected the ERs of bacterial proteins. In some species, PPA and ESS demonstrate higher correlations with ER than transcriptional abundance.
Conclusions
Different forces drive the evolution of protein sequences in yeast and bacteria. In bacteria, the constraints are involved in avoiding a build-up of toxic molecules caused by misfolding/misinteraction (transcriptional abundance), while retaining important functions (ESS, PPA) and maintaining the cell membrane (SLM, TMH and HS). Each of these independently contributes to the variation in protein evolution.
Keywords: Evolutionary rates, Bacteria, Multiple features, Transcriptional abundance
Background
Amino acid substitution rates vary considerably among different proteins. Although rapid progress has been made in determining the most important factors that shape protein evolution, the challenge remains to assess the relative importance of various variables, such as gene expression level, essentiality (ESS) and protein interactions [1-10]. One early study [11] proposed a negative correlation between the severity of gene knockout effects and coding sequence evolution, which was dependent upon the notion that purifying selection should be more efficient for essential genes than those that are non-essential. A link has been discovered between protein expression levels and evolutionary rates (ERs) in both unicellular and multicellular organisms [7,12-19].
In general, genes that are highly expressed preferentially use optimal codons to improve translational efficiency. The codon adaptation index (CAI), a measure of synonymous codon usage bias, has been widely used as a proxy for gene expression levels [20]. When CAI values were used as a substitute for actual expression levels in yeast [2] and bacteria [12], only a small proportion of rate variation in protein evolution can be explained by ESS. After replacing CAI values with experimental data and controlling for gene expression levels, ESS still had significant effects on protein ERs, but did not appear to be a major determinant of protein evolution [21,22]. CAI, expression level, and protein abundance can account for most of the variation in yeast protein ERs [13]. Keeping proteins from misfolding or misinteraction result in the slow evolution of highly expressed genes, and impose a general constraint on coding sequence evolution [7,23]. However, by using noiseless variables, protein interactions have explained more ER variation than transcriptional abundance [24]. Results from another study suggest that the molecular evolution of protein-coding genes is affected by both the context of extrinsic translational expression rates and intrinsic structural-functional constraints [25].
Despite the large number of studies evaluating the effects of various mechanisms during protein evolution, the relative importance of these factors compared with transcriptional abundance remains to be elucidated. In this study, we investigated the effects of various biological features on protein-coding sequence ERs for 18 bacterial species. The following genomic variables: CAI; experiment-based expression level (EL); ESS; number of protein-protein associations (PPA); mRNA folding strength (MFS); hydropathicity score (HS); aromaticity score (AS); protein length (LEN); replication strand bias (RSB); number of transmembrane helices (TMH); and subcellular localization [cytoplasm (SLC), cytoplasmic membrane (SLM), periplasm (SLP), outer membrane (SLO), extracellular (SLE), and cell wall (SLW)] have been summarized (Table 1).
Table 1.
Bacterial features examined in this study
| Feature | Abbreviation | Type | Source |
|---|---|---|---|
| Essentiality |
ESS |
binary |
DEG |
| Evolutionary rate (Ka) |
ER |
real |
PAML |
| Codon adaption index |
CAI |
real |
Python script |
| Expression level |
EL |
real |
GEO |
| mRNA folding strength |
MFS |
real |
ViennaRNA |
| Number of protein–protein associations |
PPA |
integer |
STRING |
| Subcellular localization: cytoplasm |
SLC |
real |
PSORTb |
| Subcellular localization: cytoplasmic membrane |
SLM |
real |
|
| Subcellular localization: periplasm |
SLP |
real |
|
| Subcellular localization: outer membrane |
SLO |
real |
|
| Subcellular localization: extracellular |
SLE |
real |
|
| Subcellular localization: cell wall |
SLW |
real |
|
| Number of transmembrane helices |
TMH |
integer |
TMHMM |
| Hydropathicity score |
HS |
real |
CodonW |
| Aromaticity score |
AS |
real |
|
| Length of protein in amino acids |
LEN |
integer |
|
| Replication strand bias | RSB | binary | DoriC |
Results and discussion
Genomic feature correlates of protein ER
In this study, potential correlation between ER and 16 features (Table 1) for 18 bacterial species (Table 2) was investigated. We observed strong correlations among CAI, EL, PPA, SLC, and ER; and less strong but significant correlations among ESS, SLM, TMH, AS and ER. However, weak relationships were found among MFS, SLP, SLO, SLE, SLW, HS, LEN, RSB and ER.
Table 2.
Bacterial species investigated in this study
| Organism | Abbreviation | Codon usage separation (CUS) | GC content | Generation times [29] |
|---|---|---|---|---|
|
Acinetobacter ADP1 |
Abay |
0.845 |
40.4 |
0.5 |
|
Bacillus subtilis 168 |
Bsub |
0.869 |
43.5 |
0.43 |
|
Bacteroides thetaiotaomicron VPI-5482 |
Bthe |
0.291 |
42.8 |
1.47 |
|
Caulobacter crescentus NA1000 |
Ccre |
0.754 |
67.2 |
1.5 |
|
Escherichia coli K-12 |
Ecol |
0.879 |
50.8 |
0.35 |
|
Francisella novicida U112 |
Fnov |
0.339 |
32.5 |
3 |
|
Haemophilus influenzae Rd KW20 |
Hinf |
0.877 |
38.2 |
0.5 |
|
Helicobacter pylori 26695 |
Hpyl |
0.148 |
38.9 |
2.4 |
|
Mycoplasma genitalium G37 |
Mgen |
0.529 |
31.7 |
12 |
|
Mycoplasma pulmonis UAB CTIP |
Mpul |
0.032 |
26.6 |
1.5 |
|
Mycobacterium tuberculosis H37Rv |
Mtub |
0.246 |
65.6 |
19 |
|
Porphyromonas gingivalis ATCC 33277 |
Pgin |
0.800 |
48.4 |
2.7 |
|
Pseudomonas aeruginosa UCBPP-PA14 |
Paer |
0.915 |
66.3 |
0.5 |
|
Staphylococcus aureus NCTC 8325 |
Saur |
0.807 |
32.8 |
0.4 |
|
Streptococcus pneumoniae TIGR4 |
Spne |
0.893 |
39.7 |
0.5 |
|
Streptococcus sanguinis SK36 |
Ssan |
0.962 |
43.4 |
- |
|
Salmonella typhimurium LT2 |
Styp |
0.915 |
52.2 |
0.4 |
| Vibrio cholera O1 biovar El Tor N16961 | Vcho | 0.897 | 47.4 | 0.2 |
Transcriptional abundance
A single variable linked to transcriptional abundance (CAI, EL and protein abundance) was found to explain the dominance of observed variation in yeast ERs [13]. CAI and EL are related to transcriptional abundance, while protein abundance is a result of the combined consequences of transcription and translation. Recent studies observed that MFS was strong for more abundant proteins, resulting in stronger evolutionary constraints of more highly expressed proteins [26,27]. We used three variables (CAI, EL and MFS) to highlight the impact of transcriptional abundance on ER.
We used RNAfold (http://www.tbi.univie.ac.at/~ronny/RNA/) to predict the secondary structure of RNA and to compute the strength of mRNA folding. A recent study reported that RNAfold predicted MFS is moderately correlated with experimentally determined MFS [27]. Furthermore, it was also shown that the correlation of the computationally predicted MFS and ER was much weaker than that of the experimentally determined MFS and ER. Similarly, we also observed a low correlation between MFS predicted by RNAfold and ER in bacteria. However, in our study we found that CAI and EL were significantly linked to ERs for most bacteria, depending on rank correlation coefficients (Figure 1). The top absolute coefficients were dominated (12/18 bacterial species) by CAI-ER or EL-ER correlations. The CAI-ER coefficient in Escherichia coli was -0.464 (p = 5.45 × 10-107), which is greater than other coefficients. Of these bacteria, Helicobacter pylori showed no CAI-ER correlation (rho = -0.039, p > 0.05); this species was not subject to periods of competitive exponential growth [28]. As a result, there was a lack of translational selection related to codon usage. In an early study, codon selection for translation was observed to strongly correlate with growth rates [29]. We investigated the effects of growth on CAI-ER correlations, and found that weak correlations could be partially attributed to long-term bacterial generation times (Table 2).
Figure 1.
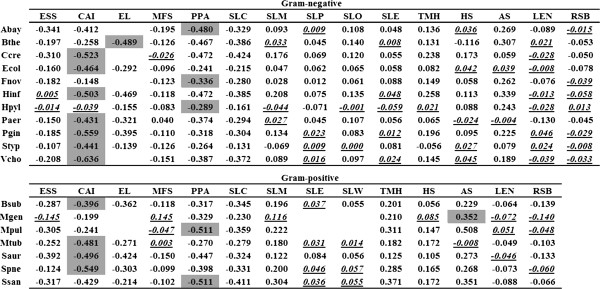
Rank correlation coefficient of variables and expression rate (ER). Gray cells indicate the highest absolute coefficients obtained for each bacterium. Blanks indicate that variables are non-existent or defective. Italicized and underlined values are non-significant at p > 0.05.
To further investigate the impact of translational selection on CAI-ER correlation, codon usage separation (CUS) was used to measure the strength of codon bias. A greater CUS value indicated a stronger codon bias mediated by translational selection. We confirmed a correlation between CUS and the CAI-ER coefficient (Pearson’s r = -0.730, p = 5.83 × 10-4). Significantly different codon usage (CUS = 0.879) was found between ribosomal proteins and other proteins in the E. coli genome (Figure 2A); however, this was not observed in the H. pylori genome (CUS = 0.148; Figure 2B). The degree to which transcriptional abundance influences ERs correlated with the strength of translational selection.
Figure 2.

Plots of correspondence analysis of relative synonymous codon usage (RSCU-CA). PC1 is the first principal component and PC2 is the second one. Ribosomal and non-ribosomal genes are represented by red and blue dots, respectively. Significantly different codon usages were observed between ribosomal proteins and other proteins in the genome of (A)E. coli, but were not observed in the genome of (B)H. pylori.
It has been previously demonstrated that translational selection across species is also strongly affected by genomic GC content [30]. We found that CAI-ER coefficients significantly correlated with GC content (Pearson’s r = -0.473; p = 0.045). CAI-ER coefficients of GC-rich bacteria are significantly greater than those for AT-rich bacteria, as translational selection is often absent in AT-rich organisms [31]. It is also known that mRNAs have a stronger secondary structure if there are more GC-rich codons [32,33]. Moreover, there is stronger selection to improve translation efficiency for weak folding at translation-initiation sites of a gene in GC-rich hosts [34]. These GC-rich organisms preferentially use GC-rich optimal codons [35]. GC-rich genomes therefore show stronger translational selection compared with AT-rich genomes. Accordingly, we found that transcriptional abundance does not always influence ERs as GC content varies across species.
A recent study found that CAI, microarray-based EL or sequencing-based EL approaches for measuring transcriptional abundance affected the assessment of the importance of transcriptional abundance to ER [9]. We found that bacteria, whose EL-ER correlation was weaker than the CAI-ER correlation, demonstrated greater CUS (0.816 vs. 0.220). For those species strongly mediated by translational selection, CAI as opposed to EL likely better explains the variation of ER. Although RNA sequencing (RNA-seq) data could be more accurate than microarray data, there is currently little RNA-seq data available for most bacterial species. In this study, we derived EL from RNA-seq data for E. coli, and used microarray data for other bacterial species. Although the sequencing-based EL-ER correlation is weaker than the CAI-ER correlation in E. coli, it is stronger than other correlations. With the development of RNA-seq experiments, we believe that the assessment of EL-ER correlations could be more accurate, and the impact of EL on ER could be stronger in certain bacterial species. To compensate for the inadequacy of each single variable to represent expression levels, we used CAI, EL and MFS to describe the impact of transcriptional abundance on ERs.
Functional importance
In an earlier study, it was proposed that ESS and protein interactions were negatively correlated with coding sequence ERs because of the constraints of important physical functions [3,6,11]. We used many types of protein associations (PPA), not only physical protein interactions (PPI), which were directly extracted from the STRING database. As expected, significant correlations between PPA/ESS and ERs were found for almost all the bacteria we investigated in our study. The strength of PPA-ER correlations was even greater than that of CAI-ER/EL-ER correlations in six organisms: Acinetobacter ADP1; Francisella novicida; H. pylori; Mycoplasma genitalium; Mycoplasma pulmonis; and Streptococcus sanguinis. In F. novicida, the ESS-ER correlation was also larger than that for CAI/EL-ER. The function of a gene is indeed an important driving force in bacterial protein evolution.
Variation in subcellular localization
Most cellular activities, including many metabolic pathways and processes, occur within the SLC. In this study, we observed significant negative correlations between SLC and ER. For example, the correlation coefficient for Caulobacter crescentus was -0.424 (p = 5.18 × 10-118). The SLM surrounds the cytoplasm of living cells, and positive correlations between SLM and ER were also observed in our study. The cell membrane functions as a selective filter, allowing molecules either to be pumped across the membrane by transmembrane transporters, or to be diffused through protein channels. These transmembrane proteins are usually specific; as a consequence, SLM proteins are fast-evolving and well adapted. We also found, as expected, that TMH positively correlated with bacterial protein ERs. The positive correlations are relatively weak between other subcellular localizations (SLP, SLO, SLE, and SLW) and the ERs of proteins. Secreted proteins located in SLO/SLE for Proteobacteria and SLW/SLE for Firmicutes were found to rapidly evolve [10]. This could be a potential explanation of why SLW, SLO and SLE rapidly evolve.
Limitations of aromatic amino acids
To manufacture proteins, microorganisms must synthesize their aromatic amino acids via the shikimate pathway. These amino acids have a limited source that impacts upon the rate at which translation errors can be corrected, and the maintenance of translation efficiency and accuracy. Therefore, the adoption of aromatic amino acids in functional or abundant proteins is not encouraged. In this study, we found that slowly evolving proteins tend to avoid adopting aromatic amino acids. In most of the investigated bacteria, AS positively and significantly correlated with ER (Figure 1).
Head-on conflict
In many bacteria, genes tend to be encoded on the leading strand. The likelihood of a gene being found on the leading strand was weakly, but significantly, associated with ER in most of the studied bacteria. As an example, RSB of Bacillus subtilis, whose genome contains over 70% leading proteins, was significantly and positively correlated with ER (Pearson’s r = -0.139; p = 7.55 × 10-11). Transcription and replication occur simultaneously in bacterial cells [36-38]. Replication progresses much faster than transcription, and inevitable conflicts occur between DNA and RNA polymerases when they bind to the same template. Co-directional collisions occur when the leading strand is the template for transcription, resulting in head-on collisions taking place when the lagging strand is the template. Head-on collisions have particularly deleterious effects, as replication forks may be arrested and transcription slowed. Over the course of evolution, transcripts are more likely to be retained if they are on the leading strand, which explains why bacterial genes on the leading strand evolve more slowly than those on the lagging strand.
Multiple factors cooperatively dominate ER
In both E. coli and B. subtilis, CAI has been identified as the most important driving force constraining ERs, through the use of partial correlation and multivariate regression analyses [12]. Drummond et al. first used principal component regression (PCR) analysis, and explained the dominant proportion of variation in yeast protein ERs by transcriptional abundance [13]. This analysis circumvents the problems of partial correlation and multivariate regression, as all principal components are orthogonal and independent. Therefore, it was useful to determine the independent contributions (R2) of biological features to yeast ERs [13]. In contrast to the situation reported for yeast, E. coli and B. subtilis[12,13], our PCR results suggest that the contributions of multiple factors are comparable for the determination of bacterial protein evolution (Figure 3). We found that Staphylococcus aureus was strongly influenced by codon bias (CUS = 0.915), with ESS, CAI, EL, PPA, SLM, TMH and HS representing 13.1, 8.5, 13.5, 9.7, 12.0, 11.3 and 11.1% of the total rate variation, respectively. These variables are comparable and account for 79.13% of the total variation. In other words, CAI and EL lose their dominance in explaining bacterial protein evolution, even in bacterial genomes with strong codon bias. Of these investigated factors, ESS, CAI, EL, PPA, SLM, THM and HS each represent over 8% of the total variation in 78 (14/18), 67 (12/18), 67 (7/12), 83 (15/18), 72 (13/18), 78 (14/18) and 67 (12/18), respectively, of bacterial genomes.
Figure 3.
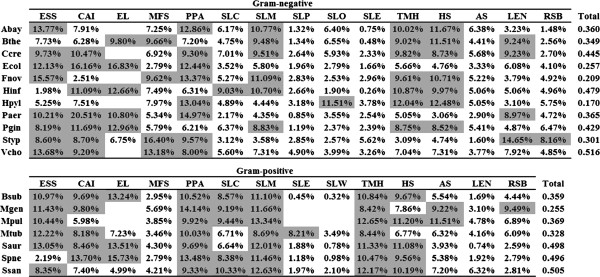
Proportion of each independent contribution (R2) to total contributions. Cells highlighted in gray indicate variables that contributed at least 8% of the total contributions to the indicated species. Blanks indicate that variables are non-existent or defective.
Based on Correspondence Analysis (CA) results, we observed the universal rule that functional factors (ESS and PPA) and transcriptional abundance (CAI and EL) were roughly grouped together, opposing the ERs in the second principal component (PC2, see Methods) (Additional file 1: Figure S1, Figure 4). Evolutionary constraints on highly transcribed proteins might prevent misfolding [7] or misinteraction [23]. This can hamper functionality and even potentially produce a large quantity of toxic proteins. In contrast, constraints on essential or high connectivity genes possibly operate to avoid the abrogation of important physiological functions. The need for translational accuracy and robustness can help explain the selection exerted on ESS, PPA, CAI, and EL. In a principal component plot obtained for yeast, ESS and PPI were distant from CAI and EL [39]. This suggests a close link between functional factors and transcriptional abundance in some bacteria that is probably dependent on ER in some way.
Figure 4.

Principal component plot of S. aureus. Functional factors (ESS and PPA) and transcriptional abundance (CAI and EL) grouped together and strongly contributed to the PC2 that is opposite to that of the ER. Similarity, SLM, TMH and HS cooperatively and negatively affected the ER at the PC2.
In all the bacterial species we investigated, SLM, TMH and HS were found to cooperatively affect ER (Additional file 1: Figure S1). These three factors have been grouped in principal component plots (Figure 4). Membrane protein transport takes place via helix-dependent protein channels embedded in cell membranes, because of their hydrophobic structure. The need to maintain transmembrane protein function may help explain the relationship among SLM, TMH, and HS.
Different forces drive ER in different species
According to PCR analysis, factors associated with transcriptional abundance (CAI, EL), important functionality (ESS, PPA) and transmembrane protein function (SLM, TMH, and HS) were the main contributors (8%) to protein ER variation in over 50% of bacterial species we studied. Transcriptional abundance is the most dominant factor in yeast [13], but not in mice [40] or bacteria (this study). The extent to which transcriptional abundance affects ERs correlates with the strength of codon bias. Our PCR analysis indicated multiple factors contribute to the rate of protein evolution in bacteria. We also found that PPA was a common important contributor to bacterial evolution, with greater effects than CAI/EL. Our results were basically identical to those presented by Plotkin and Fraser [24]-PPI appears to be responsible for most of the ER variation in yeast. The deleterious effects of protein misinteractions can affect the optimal protein concentrations and shape functional interaction networks [41]. Therefore there is a need to maintain proper interactions among high connectivity proteins as it constrains their evolution. Although ESS does not contribute strongly to yeast ERs, it is still an important factor in determining bacterial protein evolution. Our findings suggest that various forces drive protein sequence evolution in different species.
Conclusions
We have uncovered new relationships among ERs in bacterial genomes related to protein subcellular localization, transmembrane helices, hydropathicity, aromaticity, and replication strand localization. ER had a significant negative correlation with SLC, but a significant positive correlation with SLM. Because of the effects of TMH and HS on SLM, these two variables were also found to positively, although relatively weakly, correlate with bacterial protein ERs. The impact of bacterial SLM/TMH/HS and SLC on ER is independent of functional importance and transcriptional abundance. This is consistent with results from a recent study in mammalian proteins [8]. We also found that proteins that evolved slowly in bacterial genomes tended to avoid adopting aromatic amino acids. Additionally, bacterial genes on the leading strand evolved more slowly than those with genes on the lagging strand. We investigated the independent contributions of biological features to ER, and found that the dominant effect of transcriptional abundance on ER is absent in bacteria. Factors that retain important functionality (ESS, PPA), maintain cell membrane function (SLM, TMH, and HS) and avoid a build-up of toxic molecules caused by misfolding or misinteraction (CAI, EL) influence the ERs of bacterial proteins. If more RNA-Seq data are available in the future, the correlation of EL-ER could be found to be stronger in certain bacterial species than reported here. However, the influences of PPA, ESS, SLM, TMH, and HS on ER are comparable with the impact of transcriptional abundance on ER in most bacteria.
Methods
Genomic features
Essentiality
We investigated 18 bacterial species (Table 2) in the current version (7.0) of the Database of Essential Genes (DEG; http://tubic.tju.edu.cn/deg/), which hosts records of available essential genes identified by well-known genome-wide experimental techniques from a range of organisms [42]. In each of these experiments, almost all genes were investigated for their ESS scores; therefore datasets were not biased or partial. Complete coding sequences of these bacteria and their gene ESS annotations were obtained from GenBank and DEG databases, respectively.
Evolutionary rates
Orthologous gene pairs between each genome pair were identified based on reciprocal best hits using the Blastp program with criteria of E <10-5, 80% minimum residues that could be aligned, and 30% identity. Protein sequences encoded by identified orthologous gene pairs were aligned with ClustalW [43], and then back-translated into nucleotide sequences based on their original sequences. Numbers of substitutions per non-synonymous site (Ka) were calculated following Yang’s definition using the PAML package with default parameters [44]. We retained all ortholog assignments coding for more than 30 amino acids, which were not acquired by horizontal transfer, as determined by the Horizontal Gene Transfer [45] (HGT-DB; http://genomes.urv.cat/HGT-DB/) and DarkHorse [46] (http://darkhorse.ucsd.edu/) databases. Values for ERs were log-transformed after addition of a small constant (0.001).
CAI, expression level and mRNA folding strength
Transcriptional abundance was predicted from CAI, expression levels and mRNA folding strength. CAI is a species-dependent codon bias measurement that has been widely used as an empirical approach for gene expressivity, especially in microbial genomes [20]. With this methodology, dozens of ribosomal protein genes were chosen as a reference set of highly expressed genes for each genome. Our mRNA levels, derived from RNA-seq data for E. coli and microarray data for other species, under favorable environmental conditions were extracted from the Gene Expression Omnibus [47] (GEO; http://www.ncbi.nlm.nih.gov/geo/) database. Data were obtained for the following bacteria: B. subtilis (GEO Sample Accession Numbers GSM177105–GSM177118); Bacteroides thetaiotaomicron (GSM40897–GSM40906); E. coli (GSM99211–GSM99216); Haemophilus influenzae (GSM114031–GSM114033); H. pylori (GSM623401–GSM623404); Mycobacterium tuberculosis (GSM71958, GSM71988–GSM71990); Porphyromonas gingivalis (GSM590017); Pseudomonas aeruginosa (GSM462061–GSM462064, GSM462352–GSM462355); S. aureus (GSM724739–GSM724741), Streptococcus pneumonia (GSM673840); Streptococcus sanguinis (GSM908371–GSM908373); and Salmonella typhimurium (GSM874413–GSM874415). Expression level values were scaled using a logarithmic function.
The secondary structures of mRNAs, for a folding temperature under 30°C, were predicted by RNAfold within the ViennaRNA package [48]. Windows comprising 150 nucleotides were slid in 10 nucleotide steps during analysis [26]. At each nucleotide, the probability that it paired was estimated by the number of sliding windows with which it paired, divided by the number of sliding windows that include the nucleotide. We then used the average pairing possibility for an mRNA to estimate its folding strength.
Number of protein–protein associations
Protein-protein association data were obtained from the STRING database [49] (http://string-db.org/). These association data included physical PPIs and other links such as co-expression data. From the original data, we computed the number of associations for each gene using a default confidence score cutoff of 0.4.
Subcellular localization and number of transmembrane helices
We used PSORTb v3.0 [50] (http://www.psort.org/psortb/) to predict subcellular localization of proteins. Four subcellular localization types can be predicted for Gram-positive bacteria and five types can be predicted for Gram-negative bacteria. For a certain localization type, genes were assigned PSORTb prediction scores if they belong to this type, and 0 if they did not. The number of transmembrane helices was predicted from bacterial proteomes using the TMHMM Server v2.0 (http://www.cbs.dtu.dk/services/TMHMM/).
Protein hydropathicity, aromaticity, and length
We used CodonW (http://codonw.sourceforge.net/) to determine hydropathicity, aromaticity and protein length. The general average hydropathicity score for each gene product was obtained by calculating the arithmetic mean of the sum of the hydropathic indices for each amino acid. Aromaticity scores are indices for indicating frequency of aromatic amino acids.
Replication strand bias
Replication origin and terminus positions for each bacterial species were annotated using the DoriC database [51] (http://tubic.tju.edu.cn/doric/index.html). Genes were assigned a value of 1 if these positions were located on the leading strand, and 0 if otherwise.
Statistical analysis
Spearman rank correlation and PCR
Spearman’s rank correlation test was used to investigate expected direct correlations between each variable and ER. To further determine the independent contribution (R2) of each biological feature to ER, we used PCR.
CUS
To assess the impact of translation selection on codon usage, we investigated differences in relative synonymous codon usage (RSCU) between ribosomal proteins and non-ribosomal proteins using correspondence analysis (RSCU-CA). Correspondence analysis is a classical technique to reduce the dimensionality of a dataset by transforming it into its principal components. The first principal component (PC1) maximizes the standard deviation of the derived variable, while the second principal component (PC2) maximizes the standard deviation among axes uncorrelated with the first. The PC1 and PC2 could effectively explain the original 64-D codon datasets. We observed from the PC1-PC2 plot the differential codon usage pattern between ribosomal proteins and non-ribosomal proteins. Then we defined the CUS of ribosomal proteins as the percentage of ribosomal proteins falling outside the non-ribosomal protein cluster on the PC1-PC2 plot. The reference range (90%) of non-ribosomal proteins on the plot was defined as:
| (1) |
where , , and S denote the average PC1 and PC2 values of non-ribosomal proteins, and the standard deviation of the principal component value, respectively. A greater CUS indicates a greater difference in codon usage between ribosomal proteins and non-ribosomal proteins. All statistical analyses were conducted and plots generated using the R package (http://www.r-project.org/).
Abbreviations
PCR: Principal component regression; RSCU: Relative synonymous codon usage; CA: Correspondence analysis; PC: Principal component; CUS: Codon usage separation; GEO: Gene expression omnibus; DEG: Database of essential genes.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
WW participated in the design of the study, compilation of the python script, collection and preparation of data, performed the analyses, and drafted the majority of the manuscript. TZ assisted in writing the python code to implement Blast and PAML. DL double-checked the results. ZJY took part in drafting and editing the manuscript. FBG conceived and guided the study, and also assisted in writing the manuscript. All authors read and approved the final version of the manuscript.
Supplementary Material
Principal component plots of other 17 bacteria.
Contributor Information
Wen Wei, Email: doramosquito@163.com.
Tao Zhang, Email: forrest_zhang@163.com.
Dan Lin, Email: axiandan@163.com.
Zu-Jun Yang, Email: yangzujun@uestc.edu.cn.
Feng-Biao Guo, Email: fbguo@uestc.edu.cn.
Acknowledgments
This work was supported by the Program for New Century Excellent Talents in University (NCET-11-0059), the National Natural Science Foundation of China (Grant 31,071,109), and the special fund of the China Postdoctoral Science Foundation (Grant 201,104,687).
References
- Hirsh AE, Fraser HB. Protein dispensability and rate of evolution. Nature. 2001;411(6841):1046–1049. doi: 10.1038/35082561. [DOI] [PubMed] [Google Scholar]
- Pal C, Papp B, Hurst LD. Genomic function: rate of evolution and gene dispensability. Nature. 2003;421(6922):496–497. doi: 10.1038/421496b. discussion 497–498. [DOI] [PubMed] [Google Scholar]
- Fraser HB, Hirsh AE, Steinmetz LM, Scharfe C, Feldman MW. Evolutionary rate in the protein interaction network. Science. 2002;296(5568):750–752. doi: 10.1126/science.1068696. [DOI] [PubMed] [Google Scholar]
- Jordan IK, Rogozin IB, Wolf YI, Koonin EV. Essential genes are more evolutionarily conserved than are nonessential genes in bacteria. Genome Res. 2002;12(6):962–968. doi: 10.1101/gr.87702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Gu Z, Li WH. Rate of protein evolution versus fitness effect of gene deletion. Mol Biol Evol. 2003;20(5):772–774. doi: 10.1093/molbev/msg078. [DOI] [PubMed] [Google Scholar]
- Hahn MW, Kern AD. Comparative genomics of centrality and essentiality in three eukaryotic protein-interaction networks. Mol Biol Evol. 2005;22(4):803–806. doi: 10.1093/molbev/msi072. [DOI] [PubMed] [Google Scholar]
- Drummond DA, Wilke CO. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell. 2008;134(2):341–352. doi: 10.1016/j.cell.2008.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao BY, Weng MP, Zhang J. Impact of extracellularity on the evolutionary rate of mammalian proteins. Genome Biol Evol. 2010;2:39–43. doi: 10.1093/gbe/evp058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TY, Liao BY. Flagellated algae protein evolution suggests the prevalence of lineage-specific rules governing evolutionary rates of eukaryotic proteins. Genome Biol Evol. 2013;5(5):913–922. doi: 10.1093/gbe/evt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogueira T, Touchon M, Rocha EP. Rapid evolution of the sequences and gene repertoires of secreted proteins in bacteria. PLoS One. 2012;7(11):e49403. doi: 10.1371/journal.pone.0049403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson AC, Carlson SS, White TJ. Biochemical evolution. Annu Rev Biochem. 1977;46:573–639. doi: 10.1146/annurev.bi.46.070177.003041. [DOI] [PubMed] [Google Scholar]
- Rocha EP, Danchin A. An analysis of determinants of amino acids substitution rates in bacterial proteins. Mol Biol Evol. 2004;21(1):108–116. doi: 10.1093/molbev/msh004. [DOI] [PubMed] [Google Scholar]
- Drummond DA, Raval A, Wilke CO. A single determinant dominates the rate of yeast protein evolution. Mol Biol Evol. 2006;23(2):327–337. doi: 10.1093/molbev/msj038. [DOI] [PubMed] [Google Scholar]
- Pal C, Papp B, Hurst LD. Highly expressed genes in yeast evolve slowly. Genetics. 2001;158(2):927–931. doi: 10.1093/genetics/158.2.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krylov DM, Wolf YI, Rogozin IB, Koonin EV. Gene loss, protein sequence divergence, gene dispensability, expression level, and interactivity are correlated in eukaryotic evolution. Genome Res. 2003;13(10):2229–2235. doi: 10.1101/gr.1589103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian S, Kumar S. Gene expression intensity shapes evolutionary rates of the proteins encoded by the vertebrate genome. Genetics. 2004;168(1):373–381. doi: 10.1534/genetics.104.028944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos B, Bettencourt BR, Meiklejohn CD, Hartl DL. Evolution of proteins and gene expression levels are coupled in Drosophila and are independently associated with mRNA abundance, protein length, and number of protein-protein interactions. Mol Biol Evol. 2005;22(5):1345–1354. doi: 10.1093/molbev/msi122. [DOI] [PubMed] [Google Scholar]
- Popescu CE, Borza T, Bielawski JP, Lee RW. Evolutionary rates and expression level in Chlamydomonas. Genetics. 2006;172(3):1567–1576. doi: 10.1534/genetics.105.047399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingvarsson PK. Gene expression and protein length influence codon usage and rates of sequence evolution in Populus tremula. Mol Biol Evol. 2007;24(3):836–844. doi: 10.1093/molbev/msl212. [DOI] [PubMed] [Google Scholar]
- Sharp PM, Li WH. The codon Adaptation Index–a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15(3):1281–1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall DP, Hirsh AE, Fraser HB, Kumm J, Giaever G, Eisen MB, Feldman MW. Functional genomic analysis of the rates of protein evolution. Proc Natl Acad Sci U S A. 2005;102(15):5483–5488. doi: 10.1073/pnas.0501761102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, He X. Significant impact of protein dispensability on the instantaneous rate of protein evolution. Mol Biol Evol. 2005;22(4):1147–1155. doi: 10.1093/molbev/msi101. [DOI] [PubMed] [Google Scholar]
- Yang JR, Liao BY, Zhuang SM, Zhang J. Protein misinteraction avoidance causes highly expressed proteins to evolve slowly. Proc Natl Acad Sci U S A. 2012;109(14):E831–E840. doi: 10.1073/pnas.1117408109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin JB, Fraser HB. Assessing the determinants of evolutionary rates in the presence of noise. Mol Biol Evol. 2007;24(5):1113–1121. doi: 10.1093/molbev/msm044. [DOI] [PubMed] [Google Scholar]
- Wolf MY, Wolf YI, Koonin EV. Comparable contributions of structural-functional constraints and expression level to the rate of protein sequence evolution. Biol Direct. 2008;3:40. doi: 10.1186/1745-6150-3-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C, Chen X, Yang JR, Zhang J. Differential requirements for mRNA folding partially explain why highly expressed proteins evolve slowly. Proc Natl Acad Sci U S A. 2013;110(8):E678–E686. doi: 10.1073/pnas.1218066110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zur H, Tuller T. Strong association between mRNA folding strength and protein abundance in S. cerevisiae. EMBO Rep. 2012;13(3):272–277. doi: 10.1038/embor.2011.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafay B, Atherton JC, Sharp PM. Absence of translationally selected synonymous codon usage bias in Helicobacter pylori. Microbiology. 2000;146(Pt 4):851–860. doi: 10.1099/00221287-146-4-851. [DOI] [PubMed] [Google Scholar]
- Vieira-Silva S, Rocha EP. The systemic imprint of growth and its uses in ecological (meta) genomics. PLoS Genet. 2010;6(1):e1000808. doi: 10.1371/journal.pgen.1000808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin JB, Kudla G. Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet. 2011;12(1):32–42. doi: 10.1038/nrg2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naya H, Romero H, Carels N, Zavala A, Musto H. Translational selection shapes codon usage in the GC-rich genome of Chlamydomonas reinhardtii. FEBS Lett. 2001;501(2–3):127–130. doi: 10.1016/s0014-5793(01)02644-8. [DOI] [PubMed] [Google Scholar]
- Voges D, Watzele M, Nemetz C, Wizemann S, Buchberger B. Analyzing and enhancing mRNA translational efficiency in an Escherichia coli in vitro expression system. Biochem Biophys Res Commun. 2004;318(2):601–614. doi: 10.1016/j.bbrc.2004.04.064. [DOI] [PubMed] [Google Scholar]
- Kudla G, Murray AW, Tollervey D, Plotkin JB. Coding-sequence determinants of gene expression in Escherichia coli. Science. 2009;324(5924):255–258. doi: 10.1126/science.1170160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Zhou T, Wilke CO. A universal trend of reduced mRNA stability near the translation-initiation site in prokaryotes and eukaryotes. PLoS Comput Biol. 2010;6(2):e1000664. doi: 10.1371/journal.pcbi.1000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberg R, Petrov DA. General rules for optimal codon choice. PLoS Genet. 2009;5(7):e1000556. doi: 10.1371/journal.pgen.1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirkin EV, Mirkin SM. Mechanisms of transcription-replication collisions in bacteria. Mol Cell Biol. 2005;25(3):888–895. doi: 10.1128/MCB.25.3.888-895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz RT, O’Donnell M. What happens when replication and transcription complexes collide? Cell Cycle. 2010;9(13):2537–2543. doi: 10.4161/cc.9.13.12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Jinks-Robertson S. Transcription as a source of genome instability. Nat Rev Genet. 2012;13(3):204–214. doi: 10.1038/nrg3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theis FJ, Latif N, Wong P, Frishman D. Complex principal component and correlation structure of 16 yeast genomic variables. Mol Biol Evol. 2011;28(9):2501–2512. doi: 10.1093/molbev/msr077. [DOI] [PubMed] [Google Scholar]
- Liao BY, Scott NM, Zhang J. Impacts of gene essentiality, expression pattern, and gene compactness on the evolutionary rate of mammalian proteins. Mol Biol Evol. 2006;23(11):2072–2080. doi: 10.1093/molbev/msl076. [DOI] [PubMed] [Google Scholar]
- Zhang J, Maslov S, Shakhnovich EI. Constraints imposed by non-functional protein-protein interactions on gene expression and proteome size. Mol Syst Biol. 2008;4:210. doi: 10.1038/msb.2008.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Lin Y. DEG 5.0, a database of essential genes in both prokaryotes and eukaryotes. Nucleic Acids Res. 2009;37(Database issue):D455–D458. doi: 10.1093/nar/gkn858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24(8):1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Garcia-Vallve S, Guzman E, Montero MA, Romeu A. HGT-DB: a database of putative horizontally transferred genes in prokaryotic complete genomes. Nucleic Acids Res. 2003;31(1):187–189. doi: 10.1093/nar/gkg004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podell S, Gaasterland T. DarkHorse: a method for genome-wide prediction of horizontal gene transfer. Genome Biol. 2007;8(2):R16. doi: 10.1186/gb-2007-8-2-r16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M. et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013;41(D1):D991–D995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Honer Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL. ViennaRNA Package 2.0. Algorithms Mol Biol. 2011;6:26. doi: 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P. et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39(Database issue):D561–D568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, Dao P, Sahinalp SC, Ester M, Foster LJ. et al. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics. 2010;26(13):1608–1615. doi: 10.1093/bioinformatics/btq249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F, Luo H, Zhang CT. DoriC 5.0: an updated database of oriC regions in both bacterial and archaeal genomes. Nucleic Acids Res. 2013;41(Database issue):D90–D93. doi: 10.1093/nar/gks990. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Principal component plots of other 17 bacteria.