


Abstract
Homologous recombination in yeast can be exploited to reliably generate libraries of >107 transformants from a pool of PCR products and a linearized plasmid vector. Homology in the PCR insertion products drives shuffling of these genes in vivo by yeast homologous recombination. Two scFvs that share 89.8% homology were shuffled in vivo by homologous recombination, and chimeric genes were generated regardless of whether or not one of the scFv PCR products lacked 5′ homology to the cut vector and the second scFv PCR product lacked 3′ homology to the cut vector, or both PCR products had both 5′ and 3′ homology to the cut vector. A majority of the chimeras had single crossovers; however, double and triple crossovers were isolated. Crossover points were evenly distributed in the hybrids created and homology of as little as two nucleotides was able to produce a chimeric clone. The numbers of clones isolated with a given number of crossovers was approximated well by a Poisson distribution. Transformation efficiencies for the chimeric libraries were of the order of 104–105 transformants per microgram of insert, which is the same order of magnitude as when a single PCR product is inserted alone into the display vector by homologous recombination. This method eliminates ligation and Escherichia coli transformation steps of previous methods for generating yeast-displayed libraries, requires fewer PCR cycles than in vitro DNA shuffling and, unlike site-specific recombination methods, allows for recombination anywhere that homology exists between the genes to be recombined. This simple technique should prove useful for protein engineering in general and antibody engineering, specifically in yeast.
INTRODUCTION
Various methods have been developed for the creation of diversity within protein libraries, including random mutagenesis (1–3), in vitro DNA shuffling (4,5) and site-specific recombination (6–9). Random mutagenesis techniques utilize either a non-proof-reading DNA polymerase in the presence of MnCl2 (10), mutator Escherichia coli strains (11) or nucleotide analogs that cannot be correctly read by the DNA polymerase (12). Random mutagenesis has the advantage of allowing for the isolation of beneficial mutations anywhere within the gene that may not be obvious a priori. However, point mutation methods do not allow for a radical restructuring of the contact regions and therefore are restrictive in the sequence space that can be probed. DNA shuffling methods have the advantage of being able to generate hybrid genes that contain portions of sequence space that have already proven to be functional. DNA shuffling consists of four steps: DNase I digestion of the genes to be recombined, PCR reassembly without primers, amplification of recombined gene products of the correct size from the primerless PCR pool, and ligation of the reassembled product into an acceptor vector. Although DNA shuffling has proven to be a highly effective method, numerous PCR and associated purification steps are required. Site-specific recombination has the advantage of utilizing portions of known sequence space that have already proven functional, but it is often an impractical method because it requires the engineering of restriction enzyme sites into the genes to be combined. Often finding unique sites is difficult and the process can become tedious when multiple chimeric products are desired. Other forms of site-specific recombination have been used to make libraries such as the n-CoDer antibody library in which CDRs were shuffled using specific primers and PCR reassembly (13), chain-shuffled scFv libraries in which the shuffling was performed by PCR utilizing homology in the linker region between the heavy and light chains (14), and chain-shuffled antibody libraries using Cre-catalyzed recombination of antibody heavy and light chains that are flanked by lox sites (8,9). In all of these libraries, the points at which gene exchange occur are fixed and previous knowledge of the gene sequences for the creation of PCR primer sets or engineering of specific sites is required.
A simple method for creating hybrid genes that does not require the extensive PCR steps of DNA shuffling, the pre-engineering of site-specific recombination methods, or ligation would be desirable. In this paper, we present a technique for creating large, chimeric antibody libraries using plasmid reconstruction by homologous recombination in the yeast Saccharomyces cerevisiae (15,16). This method allows for the coupling of diversity generation and protein production within the host organism, rather than having a separate in vitro diversity generation step. In vivo recombination in yeast has been used in the past to create hybrid genes (17–19) and has been successfully used to generate a library for the directed evolution of a heme peroxidase enzyme (19). However, this technique has not been exploited for the generation of antibody libraries. This absence can be explained by the fact that most in vitro antibody engineering is performed using phage, which relies upon the machinery of E.coli for gene propagation and protein production. Although intra- (Fig. 1A) and intermolecular recombination (Fig. 1B and C) have both been performed in E.coli to make hybrid genes, intermolecular recombination has generally been used for plasmid construction and not recombination (20). Intermolecular recombination is inherently more flexible than intramolecular recombination because only one (Fig. 1B and C) or none (Fig. 1D) of the genes to be recombined must be within a vector, while intramolecular recombination requires both genes to be within the same vector.
Figure 1.

Some common in vivo homologous recombination schemes. (A) Intramolecular recombination, where both genes to be recombined are located within the same plasmid. (B) Intermolecular recombination between a gene and a similar gene that is located within a plasmid and that has been cut at an internal restriction site. The acceptor gene within the plasmid determines the 5′ and 3′ ends of the gene hybrid. (C) Intermolecular recombination between a donor gene that contains a portion of homology on its 3′ end to the vector to which it is being inserted. The vector is cut at the 3′ end of the acceptor gene that is located within the plasmid. The acceptor gene determines the 5′ end of the hybrid gene generated. (D) Multiple gene hybrids are generated by genes that have homology at their 5′ and 3′ ends to the vector into which they are to be inserted. Homology within the genes allows for multiple recombination events and the order of the genes within the final hybrid is not fixed by the presence of a gene within the acceptor plasmid. This figure is adapted from Wang (20).
The most flexible scheme, presented in Figure 1D, allows for recombination anywhere in the genes to be shuffled and is most applicable to antibody engineering because none of the genes must be within the acceptor vector. Thus, pools of antibodies can be shuffled with no bias towards any individual acceptor antibody. To our knowledge, the crossover scheme depicted in Figure 1D has not been used to generate antibody libraries in E.coli. This fact can be explained by the proficiency of S.cerevisiae in homologous recombination in comparison with the inherent problems in the endogenous E.coli homologous recombination mechanism (21). For example, the E.coli endogenous homologous recombination mechanism is initiated by the cooperation between RecA and the enzyme RecBCD, which impedes the use of linear DNA because RecBCD is a vigorous exonuclease (21). In the cases where E.coli has been used to create hybrid genes (Fig. 1B and C), mutant strains must be used, which are not advantageous because they can exhibit genetic instabilities during library transformation and propagation. Although it is possible to exploit the yeast recombination method to make chimeric genes and then recover the plasmids containing the hybrid gene products for transfer to E.coli, this method is hampered by both the inefficient recovery of plasmids from yeast minipreps and the added difficulty of having to switch hosts. Not until the advent of yeast surface display did it become practical to exploit yeast in vivo homologous recombination methods for making chimeric antibody libraries.
This paper is the first report of in vivo homologous recombination for the creation of large, yeast-surface- displayed antibody libraries. Yeast surface display has already proven a powerful tool for affinity maturation by mutagenic PCR of antibodies (2,11) and T-cell receptors (3), and recently has been shown to be a valuable tool for the isolation of antibodies with novel specificities (22). Here, we demonstrate that homologous recombination shuffling in conjunction with yeast surface display greatly simplifies the creation of large, chimeric antibody libraries. First, we show that homologous recombination by electroporation reliably generates yeast-surface-displayed libraries of 107 clones and eliminates the need for a ligation step and an E.coli cloning step. Next, we use two scFvs that share 89.8% homology between them as a test case to demonstrate that yeast can shuffle genes in vivo by homologous recombination. The technique presented in this paper requires fewer PCR cycles than in vitro DNA shuffling, does not require a ligation step, and, unlike site-specific recombination methods, it allows for recombination anywhere that homology exists between the genes to be recombined. Moreover, the diversity generated is not limited to single crossovers; hybrid antibodies created by double and triple crossovers were easily obtained. This technique should prove to be an important addition to the aforementioned methods for the isolation of affinity-improved antibodies.
MATERIALS AND METHODS
DNA preparation for homologous recombination
The two scFvs used in the shuffling experiments were isolated from a previously constructed yeast-surface-displayed non-immune library (22). To test homologous recombination frequencies of a mutagenic PCR product, a third scFv from this library was amplified in a PCR reaction using a nucleotide analog mutagenesis procedure that has been described previously (12). All PCR products were inserted into the vector PCTCON (22), which had been restriction digested from NheI to BamHI (New England Biolabs) and gel purified using a gel purification kit (Qiagen) according to the manufacturer’s instructions. As described in the study by Raymond et al. (16), to obtain the best transformation efficiency, homologous recombination primers were designed so that the inserts would have an ∼50 bp overlap at each end with the cut acceptor vector. The primer used to make inserts with 5′ homology to the cut vector was 5′-CGACGATTGAAGGTAGATACCCATACGACGTTCCAGACTACGCTCTGCAG-3′, and the primer used to make inserts with 3′ homology to the cut vector was 5′-CAGATCTCGAGCTATTACAAGTCTTCTTCAGAAATAAGCTTTTGTTC-3′. To make either scFv 1 or scFv 2, which lacked 5′ homology to the cut vector, the primer 5′-GCTAGCCAGGTACAGCTGCAGC-3′ was used. To make either scFv 1 or scFv 2, which lacked 3′ homology to the cut vector, the primer 5′-AATTCCGGATAGGACGGTGAGCTTGG-3′ was used. All oligonucleotides were obtained from MWG-Biotech. PCR reactions were carried out on a Perkin Elmer DNA Thermal Cycler 480. A typical PCR reaction was carried out in a 100 µl volume using 10–100 ng of template, 1 µM primers, 0.2 mM of each dNTP, 6.25 U Taq (Invitrogen) and 2 mM MgCl2. Cycling conditions used were: one cycle at 94°C for 1 min followed by 35 cycles at 94°C for 1 min, 50°C for 1 min and 72°C for 2 min, followed by one cycle at 72°C for 10 min. PCR products were gel purified using a Qiagen kit. Insert fragments were concentrated with Pellet Paint (Novagen) to a concentration of 5 µg/µl according to the manufacturer’s instructions and cut backbone was likewise concentrated to a concentration of 1 µg/µl.
Preparation of electrocompotent yeast for homologous recombination
The method of yeast preparation closely follows that described by Meilhoc et al. (23). First, 50 ml of YPD was inoculated with the S.cerevisiae strain EBY100 (11) to an optical density (OD) of 0.1 from an overnight culture of EBY100 in YPD. Next, the cells were grown with shaking at 30°C to an OD of 1.3–1.5 (∼6 h of growth). Cells were harvested by centrifugation and resuspended in 50 ml of freshly prepared 10 mM Tris pH 8.0, 25 mM dithiothreitol (DTT) in YPD and shaken for 20 min at 30°C. The cells were washed once with 25 ml buffer E (10 mM Tris pH 7.5, 270 mM sucrose, 1 mM MgCl2) and again with 5 ml of E buffer. Finally, cells were suspended in buffer E to give 2 × 108 cells per 50 µl aliquot.
Homologous recombination protocol
The ratio of total insert fragment to cut acceptor vector was maintained at 10:1 for all transformations. For shuffling experiments where both scFvs were transformed together, half of the insert fragment pool consisted of one scFv and half of the insert fragment pool consisted of the other. One microgram (1 µl) of cut acceptor vector and 10 µg (2 µl) of insert were added to a 50 µl aliquot of electrocompotent yeast and incubated on ice for 5 min. Electroporation was carried out using a Bio-Rad Gene Pulser with a 0.2 cm cuvette (voltage 0.54 kV, capacitance 25 µF) giving a time constant of ∼18 ms. After pulsing, the cell aliquots were transferred to 1 ml of YPD media and incubated for 1 h at 30°C. Cells were then harvested at 3500 r.p.m. for 4 min and resuspended in SDCAA selective media (–Ura, –Trp). A small aliquot of cells was removed and plated on SDCAA plates to determine transformation efficiency.
DNA isolation and sequencing
Colonies from the SDCAA plates were grown in 5 ml of SDCAA overnight and the DNA was isolated using a Zymoprep kit (Zymo Research) according to the manufacturer’s protocol. Two microliters of Zymoprep DNA were used in an XL1-Blue (Stratagene) E.coli transformation according to the manufacturer’s instructions. Cells were plated on selective Luria–Bertani (LB) plates supplemented with 100 mg/l ampicillin. Colonies from these plates were grown overnight at 37°C in LB media plus 100 mg/l ampicillin and DNA was isolated using a Qiagen miniprep kit according to the manufacturer’s instructions. DNA was sequenced on a Applied Biosystems model 3730 DNA sequencer using version 3.0 Big Dye chemistry.
RESULTS
Creation of large yeast-surface-displayed libraries by homologous recombination
As has previously been reported, the yeast S.cerevisiae is highly efficient at reconstructing plasmids from a linearized plasmid and a PCR product that contains sufficient homology at the 3′ and 5′ ends (15,16). Figure 2 outlines the homologous recombination strategy. The first step involves the linearization of a yeast surface display plasmid, which contains an irrelevant gene flanked by a 5′ NheI site and a 3′ BamHI site, in a restriction digest. The second step involves the creation of a pool of PCR fragments that share 5′ and 3′ homology to the cut display vector. Because Raymond et al. have reported that overlaps of ≥50 base pairs on each end of the PCR product yield the highest number of recombinants (16), we made PCR fragments of mutagenic antibody library DNA that have ∼50 base pairs of homology to the display vector at their 5′ and 3′ ends. Co-transformation of these fragments with linearized plasmid allowed for the creation of large yeast-displayed libraries. Table 1 shows the results of two different transformations of mutagenic antibody library DNA using homologous recombination. Both experiments yielded a transformation efficiency of an order of magnitude 105/µg of insert DNA. Homologous recombination by electroporation simplifies library generation by completely eliminating the need for ligation of PCR products into a cut vector and the subsequent cloning into E.coli. Library sizes of 107 are routinely achieved without great effort.
Figure 2.

General outline of library construction using homologous recombination in yeast. First, PCR products are created with 5′ and 3′ homology to the vector into which they are to be inserted. Next, the PCR products are transformed together with a restriction-digested vector by electroporation, and yeast utilizes the homology between the cut vector and the PCR products to reconstruct whole plasmids.
Table 1. Transformation efficiencies of two different homologous recombination experiments.
| Trial | Insert to vector ratio | Micrograms of insert transformed | Number of transformed clones | Transformants per microgram of insert |
|---|---|---|---|---|
| 1 |
10:1 |
31 |
1.8 × 107 |
5.81 × 105 |
| 2 | 10:1 | 60 | 2.3 × 107 | 3.83 × 105 |
In vivo shuffling of antibody DNA by homologous recombination
It is our desire to extend the technique of homologous recombination in yeast to allow for the easy construction of chimeric scFv antibody libraries. To test if chimeras can be created by homologous recombination, a series of four different transformation experiments (Fig. 3) were performed using two different antibodies, termed scFv1 and scFv2, which shared 89.8% homology between them. These antibodies, which were isolated from a non-immune S.cerevisiae- displayed library (22), were chosen not only because they shared significant patches of homology, but also because they had numerous differences spread throughout the entire length of their sequences. These differences are critical for the determination of crossover points. In the experiments depicted in Figure 3A and B, the two genes to be recombined had homology with the linearized display vector at one end only. In principle, neither fragment alone could recreate a whole plasmid because each fragment was missing critical homology at one end, therefore only recombinants would be isolated after transformation. To determine if forcing recombination is necessary for the creation of hybrid genes, a third experiment was performed (Fig. 3C) using scFv1 and scFv2, where both genes contained 5′ and 3′ homology to the display vector. In a fourth experiment (Fig. 3D), scFv1 was transformed alone to verify that both 5′ and 3′ homology are necessary for plasmid reconstruction.
Figure 3.

In vivo homologous recombination experiments. (A) Homologous recombination when scFv 1 has 3′ homology to the display vector and scFv 2 has 5′ homology to the display vector. (B) Homologous recombination when scFv 1 has 5′ homology to the display vector and scFv 2 has 3′ homology to the display vector. (C) Homologous recombination when both scFv 1 and scFv 2 have 5′ and 3′ homology to the display vector. Solid lines indicate a crossover producing chimeric clones that have 5′ homology to scFv 1 and 3′ homology to scFv 2. Dashed lines indicate a crossover to produce chimeric clones with the opposite arrangement. A mixed path of one solid line crossover and one dashed line crossover without crossover between the two scFvs would produce a plasmid without a chimeric gene. (D) Homologous recombination when scFv 1 has 3′ homology to the display vector and scFv 2 is not present. This recombination occurs between the (gly4ser)3 linker that joins the scFv heavy and light chains and the (gly4ser)3 linker located immediately 5′ to the the ultimate location of the gene in the display plasmid.
Figure 4 shows the results of the in vivo homologous recombination shuffling experiments. For all of the results depicted in Figure 4, the crossover point in the chimeric antibody sequence was determined to be the last nucleotide of homology 5′ to a difference between the chimeric antibody sequence and whichever scFv sequence to which the chimeric antibody had homology up until that point. Because scFv1 and scFv2 share 60 base pairs of homology at the beginning of their sequences and 44 base pairs of homology at the end of their sequences, it is possible for crossovers in these regions to generate genes that have 100% sequence identity to one of the two original scFvs. It was our initial hypothesis that for the insertion into the display vector to be successful, the insert fragment needs both 5′ and 3′ homology to the cut plasmid. However, the results depicted in Figure 4D indicate that this hypothesis is false because 10 of the 12 clones sequenced were 100% homologous to scFv1, although scFv1 lacked the 5′ homology necessary for in vivo homologous recombination to occur. Apparently, the single-stranded end of the NheI cut plasmid can recombine with the blunt, double-stranded DNA of the scFv to be inserted. It is likely that the BamHI-restricted end of the vector can also recombine with blunt end DNA in a similar manner. This integration phenomena might be a form of illegitimate integration, defined as recombination involving little or no sequence homology, that has been previously reported to occur in S.cerevisiae (24). Thus, it is impossible to tell if a gene that has 100% homology to either scFv1 or scFv2 is a hybrid or if it was generated through illegitimate integration. In this paper, only genes that clearly contain portions of scFv1 and scFv2 will be considered chimeric.
Figure 4.

Results of homologous recombination shuffling experiments. scFv 1 is shown in blue, scFv 2 is shown in red, and the differences between them are indicated by black bars connecting the two. (A) Chimeric clones produced by homologous recombination of scFv 1 with 3′ homology to the cut vector and scFv 2 with 5′ homology to the cut vector. (B) Chimeric clones produced by homologous recombination of scFv 1 with 5′ homology to the cut vector and scFv 2 with 3′ homology to the cut vector. (C) Clones produced when both scFv 1 and scFv 2 have 5′ and 3′ homology to the cut vector. (D) Clones produced when only scFv 1 with 3′ homology to the cut vector is transformed.
Figure 4A (corresponding to the experiment depicted in Fig. 3A) shows that of the 48 clones sequenced, 12 were chimeric, and Figure 4B (corresponding to the experiment depicted in Fig. 3B) shows that of the 46 clones sequenced, 17 were chimeric. It is not surprising that all of the hybrids in Figure 4A shared both homology 5′ to the crossover point with scFv2 and homology 3′ to the crossover point with scFv1, because in this experiment scFv2 had 5′ homology to the cut plasmid and scFv1 had 3′ homology to the cut plasmid. As expected, the reverse is generally true for the experiment depicted in Figure 3B. We see that in all but two cases (clones 23 and 24) the chimeric clones shared homology 5′ to the crossover point with scFv 1 and homology 3′ to the crossover point with scFv 2 (Fig. 4B). Clones 23 and 24 may have been generated by a crossover event in the first 60 base pairs or may be the result of illegitimate integration. Figure 4A and B indicates that the crossover points are evenly distributed in the hybrids created. Only clones 6 and 18, generated by a crossover point immediately 5′ to CDR3 of the heavy chain, were found at a slightly higher frequency than other chimeras (three times and five times, respectively, compared with approximately one occurrence per chimera for the other hybrids).
In Figure 4C (corresponding to the experiment depicted in Fig. 3C), only five of 46 clones sequenced were chimeric. Thus, it appears that forcing the recombination as in the experiments depicted in Figure 3A and B yields a higher frequency of chimeras than when both genes can recombine with the cut vector at both their 5′ and 3′ ends. The presence of light-chain-only inserts (clones 33, 34 and 36) is not surprising when one considers that every scFv contains a 15 amino acid linker consisting of three repeats of four glycines and a serine [(gly4ser)3], which joins the variable heavy and variable light domains. This same linker is present immediately 5′ to the gene in the display plasmid and these linkers share 80% sequence homology. It is surprising that these clones were not found in the other two libraries, although there is every reason to believe that further sequencing would reveal the presence of light-chain-only insertions in both cases. It is important to note that clone 31 resulted from a crossover in a region of two base pairs of homology, indicating that on rare occasions crossovers can occur in regions of very limited homology. This result is in agreement with that of Mezard et al. (17) and demonstrates that the power of this technique is not limited by the need for long stretches of homology between the genes to be recombined.
Double (clone 24 and 32) and triple (clone 11 and 25) crossovers greatly increase the diversity potential of libraries generated by in vivo homologous recombination. To estimate this diversity, we applied the Poisson distribution to estimate the probability of obtaining a clone with x = 0, 1, 2… crossovers in a given sequence by using the following equation:
![]()
where λ is defined as the average number of crossovers per sequence in a given experiment. The value of λ was 0.29, 0.43 and 0.13 in the experiments depicted in Figure 3A–C, respectively. For each of these experiments the expected and actual number of clones obtained are listed in Table 2. As can clearly be seen, the number of clones experimentally obtained for x = 0 or x = 1 is well approximated by the Poisson distribution. The limited sampling size of between 45 and 50 clones per experiment explains the lack of agreement for x = 2 and x = 3. Table 3 gives the expected number of clones containing a given number of crossovers for a 1 × 107 library. Although in the second column this table gives an idea of the distribution of crossovers, it does not take into account the number of unique sequences, which is the true measure of library diversity. The following equation can be used to determine the number of unique sequences (u) obtained with a given number of crossovers (x) when y genes are crossed:
Table 2. Expected and experimentally determined number of clones with a given number of crossovers as predicted by a Poisson distribution.
| Number of crossovers per sequence (x) | scFv1 with 3′ homology + scFv2 with 5′ homology |
scFv1 with 5′ homology + scFv2 with 3′ homology |
scFv1 with 5′ and 3′ homology + scFv2 with 5′ and 3′ homology |
|||
|---|---|---|---|---|---|---|
| Actual number of sequences with x crossovers | Poisson calculated number of sequences with x crossovers | Actual number of sequences with x crossovers | Poisson calculated number of sequences with x crossovers | Actual number of sequences with x crossovers | Poisson calculated number of sequences with x crossovers | |
| 0 |
36 |
36 |
29 |
30 |
41 |
40 |
| 1 |
11 |
10 |
15 |
13 |
4 |
5 |
| 2 |
0 |
2 |
1 |
3 |
1 |
0 |
| 3 | 1 | 0 | 1 | 0 | 0 | 0 |
Table 3. Expected number of total clones and unique clones with a given number of crossovers as predicted by Poisson distribution for a 1 × 107 library.
| Number of crossovers per sequence (x) | Number of sequences with x crossovers in a 1 × 107 library with λ = 0.13 | Number of unique sequences assuming 20 distinct crossover points |
|||
|---|---|---|---|---|---|
| Recombine 2 genes | Recombine 3 genes | Recombine 4 genes | Recombine 100 genes | ||
| 0 |
8 777 137 |
2 |
3 |
4 |
100 |
| 1 |
1 144 844 |
40 |
120 |
240 |
198 000 |
| 2 |
74 664 |
380 |
2280 |
6840 |
186 219 000 |
| 3 |
3246 |
2280 |
27 360 |
123 120 |
1.1 × 1011 |
| Total | 1 × 107 | 2702 | 29 763 | 130 204 | 1.1 × 1011 |
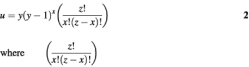
indicates the number of combinations with z crossover points. As shown in Table 3, for a cross of two genes with an arbitrarily chosen number of 20 distinct crossover points, this equation yields 40 unique one-crossover clones, 380 unique two-crossover clones and 2280 unique three-crossover clones. The data in Table 3 reveal the trend that the number of unique clones with multiple crossovers will become greater than the number of clones predicted to have multiple crossovers by the Poisson distribution as the number of genes crossed is increased. For crosses of small numbers of antibodies, the chimeras in the library created would be dominated by many copies of each unique single crossover clone, but the added diversity created by double and triple crossovers would be non-zero. For a cross of 100 genes, the potential theoretical diversity would be dominated by unique multiple crossover clones and would greatly exceed the diversity of multiple crossover clones predicted to be in the library by the Poisson distribution; however, the unique single crossover clones would still be oversampled in a 1 × 107 library. In addition, the number of double and triple crossover clones as predicted by the Poisson distribution, 77 910, is 39% of the maximum number of unique single crossover clones, 198 000, indicating that double and triple crossovers are a non-trivial component of the total diversity. Of course, clones with four or more crossovers may exist, but because none were isolated in the limiting sampling of this experiment they were not included in the analysis.
The possibility that forcing recombination reduced transformation efficiency below a level that was useful was considered. However, for all of the crosses performed here, the transformation efficiency was of an order of 104 to 105 per microgram of insert. Thus, forcing recombination does not appreciably affect transformation efficiency.
DISCUSSION
In this paper, we present a simple method for reliably producing large, yeast-surface-displayed chimeric antibody libraries. This method takes advantage of the homologous recombination pathway in yeast to reconstruct full plasmids from restriction-digested plasmids and PCR products that have 5′ and 3′ homology to that cut plasmid. Transformation of multiple PCR products that share homology can cause recombination events to yield chimeric gene products. Forcing recombination by making a PCR product of one gene with 5′ homology to a cut vector and making a PCR product of a second gene with 3′ homology to a cut vector yields greater numbers of chimeric gene products than when both genes to be recombined share both 5′ and 3′ homology to the vector into which they are to be inserted. The transformation efficiency for forced recombination was not appreciably lower than that of a single PCR product, with both 5′ and 3′ homology to the cut display plasmid.
Although forced recombination yields the greatest number of hybrid genes, this method may prove impractical because one must know the sequence of the 5′ or 3′ ends of the genes that are to be shuffled. In the case presented in this paper, the two genes to be recombined had known sequences that facilitated primer design. However, one may wish to shuffle one scFv gene against an entire library of scFvs or shuffle a batch of scFv genes that all bind a given antigen. Generic primer sets for all antibody heavy and light chains are known and it would be possible to force recombination by creating PCR products that lack 5′ or 3′ homology to the vector into which the genes are to be inserted. This method is rather tedious and in our experience has proven unnecessary. As our experiments demonstrate, not forcing recombination allows for the creation of hybrid clones, albeit at a lower rate than forced recombination. The number of chimeric clones obtained is only 2- to 3-fold less than in forced recombination and is therefore not low enough to cause concern that forcing recombination is necessary for this technique to be practical.
It is important to stress that the ability of as few as two nucleotides of homology to yield successful recombinants in conjunction with the presence of double and triple crossovers demonstrates that this technique has a vast potential for producing highly diverse libraries. Although it is true that most chimeras contain only one crossover, there will still be many unique clones in the library to ensure the necessary diversity for directed evolution experiments. It is likely that the unique diversity of libraries made by yeast in vivo homologous recombination will be sufficient for affinity improvements, even if the actual diversity of the libraries generated is orders of magnitude lower than the total number of transformants because the shuffling of antibodies is a rearrangement of diversity that has been maintained specifically because it is functional. The advantage of this technique is its simplicity and it is likely that many variations other than those presented here will emerge for the production of functional diversity in protein libraries.
Acknowledgments
ACKNOWLEDGEMENTS
The authors wish to thank the NIGMS/MIT Biotechnology Training Program (GM08334) for funding.
REFERENCES
- 1.Daugherty P.S., Chen,G., Iverson,B.L. and Georgiou,G. (2000) Quantitative analysis of the effect of the mutation frequency on the affinity maturation of single chain fv antibodies. Proc. Natl Acad. Sci. USA, 97, 2029–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boder E.T., Midelfort,K.S. and Wittrup,K.D. (2000) Directed evolution of antibody fragments with monovalent femtomolar antigen-binding affinity. Proc. Natl Acad. Sci. USA, 97, 10701–10705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holler P.D., Holman,P.O., Shusta,E.V., O’Herrin,S., Wittrup,K.D. and Kranz,D.M. (2000) In vitro evolution of a T cell receptor with high affinity for peptide/MHC. Proc. Natl Acad. Sci. USA, 97, 5387–5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stemmer W.P.C. (1994) Rapid evolution of a protein in-vitro by DNA shuffling. Nature, 370, 389–391. [DOI] [PubMed] [Google Scholar]
- 5.Stemmer W.P.C. (1994) DNA shuffling by random fragmentation and reassembly—in-vitro recombination for molecular evolution. Proc. Natl Acad. Sci. USA, 91, 10747–10751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rehberg E., Kelder,B., Hoal,E.G. and Pestka,S. (1982) Specific molecular activities of recombinant and hybrid leukocyte interferons. J. Biol. Chem., 257, 11497–11502. [PubMed] [Google Scholar]
- 7.Streuli M., Hall,A., Boll,W., Stewart,W.E.,II, Nagata,S. and Weissmann,C. (1981) Target cell specificity of two species of human interferon-alpha produced in Escherichia coli and of hybrid molecules derived from them. Proc. Natl Acad. Sci. USA, 78, 2848–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Waterhouse P., Griffiths,A.D., Johnson,K.S. and Winter,G. (1993) Combinatorial infection and in vivo recombination: a strategy for making large phage antibody repertoires. Nucleic Acids Res., 21, 2265–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sblattero D. and Bradbury,A. (2000) Exploiting recombination in single bacteria to make large phage antibody libraries. Nat. Biotechnol., 18, 75–80. [DOI] [PubMed] [Google Scholar]
- 10.Fromant M., Blanquet,S. and Plateau,P. (1995) Direct random mutagenesis of gene-sized DNA fragments using polymerase chain reaction. Anal. Biochem., 224, 347–353. [DOI] [PubMed] [Google Scholar]
- 11.Boder E.T. and Wittrup,K.D. (1997) Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol., 15, 553–557. [DOI] [PubMed] [Google Scholar]
- 12.Zaccolo M., Williams,D.M., Brown,D.M. and Gherardi,E. (1996) An approach to random mutagenesis of DNA using mixtures of triphosphate derivatives of nucleoside analogues. J. Mol. Biol., 255, 589–603. [DOI] [PubMed] [Google Scholar]
- 13.Soderlind E., Strandberg,L., Jirholt,P., Kobayashi,N., Alexeiva,V., Aberg,A.M., Nilsson,A., Jansson,B., Ohlin,M., Wingren,C. et al. (2000) Recombining germline-derived cdr sequences for creating diverse single-framework antibody libraries. Nat. Biotechnol., 18, 852–856. [DOI] [PubMed] [Google Scholar]
- 14.Marks J.D., Griffiths,A.D., Malmqvist,M., Clackson,T.P., Bye,J.M. and Winter,G. (1992) By-passing immunization: building high affinity human antibodies by chain shuffling. Biotechnology, 10, 779–783. [DOI] [PubMed] [Google Scholar]
- 15.Ma H., Kunes,S., Schatz,P.J. and Botstein,D. (1987) Plasmid construction by homologous recombination in yeast. Gene, 58, 201–216. [DOI] [PubMed] [Google Scholar]
- 16.Raymond C.K., Pownder,T.A. and Sexson,S.L. (1999) General method for plasmid construction using homologous recombination. Biotechniques, 26, 134–138, 140–131. [DOI] [PubMed] [Google Scholar]
- 17.Mezard C., Pompon,D. and Nicolas,A. (1992) Recombination between similar but not identical DNA sequences during yeast transformation occurs within short stretches of identity. Cell, 70, 659–670. [DOI] [PubMed] [Google Scholar]
- 18.Abecassis V., Pompon,D. and Truan,G. (2000) High efficiency family shuffling based on multi-step PCR and in vivo DNA recombination in yeast: statistical and functional analysis of a combinatorial library between human cytochrome p450 1a1 and 1a2. Nucleic Acids Res., 28, E88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cherry J.R., Lamsa,M.H., Schneider,P., Vind,J., Svendsen,A., Jones,A. and Pedersen,A.H. (1999) Directed evolution of a fungal peroxidase. Nat. Biotechnol., 17, 379–384. [DOI] [PubMed] [Google Scholar]
- 20.Wang P.L. (2000) Creating hybrid genes by homologous recombination. Dis. Markers, 16, 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muyrers J.P., Zhang,Y. and Stewart,A.F. (2001) Techniques: recombinogenic engineering—new options for cloning and manipulating DNA. Trends Biochem. Sci., 26, 325–331. [DOI] [PubMed] [Google Scholar]
- 22.Feldhaus M.J., Siegel,R.W., Opresko,L.K., Coleman,J.R., Feldhaus,J.M., Yeung,Y.A., Cochran,J.R., Heinzelman,P., Colby,D., Swers,J. et al. (2003) Flow-cytometric isolation of human antibodies from a nonimmune Saccharomyces cerevisiae surface display library. Nat. Biotechnol., 21, 163–170. [DOI] [PubMed] [Google Scholar]
- 23.Meilhoc E., Masson,J.M. and Teissie,J. (1990) High-efficiency transformation of intact yeast-cells by electric-field pulses. Biotechnology, 8, 223–227. [DOI] [PubMed] [Google Scholar]
- 24.Schiestl R.H. and Petes,T.D. (1991) Integration of DNA fragments by illegitimate recombination in Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA, 88, 7585–7589. [DOI] [PMC free article] [PubMed] [Google Scholar]