

Abstract
Human arylamine N-acetyltransferase 1 (hNAT1) has become an attractive potential biomarker for estrogen-receptor-positive breast cancers. We describe here the mechanism of action of a selective non-covalent colorimetric biosensor for the recognition of hNAT1 and its murine homologue, mNat2, over their respective isoenzymes, leading to new opportunities in diagnosis. On interaction with the enzyme, the naphthoquinone probe undergoes an instantaneous and striking visible color change from red to blue. Spectroscopic, chemical, molecular modelling and biochemical studies reported here show that the color change is mediated by selective recognition between the conjugate base of the sulfonamide group within the probe and the conjugate acid of the arginine residue within the active site of both hNAT1 and mNat2. This represents a new mechanism for selective biomarker sensing and may be exploited as a general approach to the specific detection of biomarkers in disease.
Introduction
Diagnosis of breast cancer combines non-invasive examinations, such as mammography, ultrasound or magnetic resonance imaging and biopsy tests. At the present time, the analysis of biological samples allows the identification of tumor-specific biomarkers to stratify anti-target therapies [1]–[3]. A variety of chemical approaches have been developed to selectively detect and monitor biomolecules, and to generate novel molecular sensors for biological markers to facilitate diagnosis with improved accuracy [4]–[7].
Proteomic and microarray analyses have identified the overexpression of human arylamine N-acetyltransferase 1 (hNAT1) in estrogen-receptor-positive ductal and lobular breast cancers [8]–[10] and more recently in male breast cancers [11]; furthermore, this overexpression inversely correlates to tumor grade [12]. In addition to its catalytic role as an arylamine-metabolizing enzyme using acetyl coenzyme A (AcCoA) as cofactor [13] subsequent studies have assessed hNAT1 as a new biomarker to be developed as a novel diagnostic, prognostic and potential therapeutic target in breast cancers [10]–[11].
We have previously described a family of naphthoquinones as non-covalent competitive selective inhibitors of hNAT1 and its murine homologue mNat2 over other eukaryotic and prokaryotic isoforms [14]–[16]. Remarkably, an instantaneous distinctive color change from red to blue is observed upon binding of these naphthoquinone ligands, such as compound 1, to both hNAT1 and mNat2, which share more than 80% identity in amino acid sequence and are functionally homologous ( Figure 1 ) [16]–[17]. No such shift in the λmax of naphthoquinone 1 was observed in the presence of the other human and murine NAT enzymes despite the high number of identical residues (>70%) ( Figure 1 ), nor with NATs from prokaryotes [16]. Since hNAT1 is a candidate biomarker in breast cancer, it was reasoned that understanding the mechanism of recognition and color change between this family of naphthoquinone probes and hNAT1 could allow both the development of these probes for tumor subtype diagnosis and the application of this technology to other protein families.
Figure 1. Competitive inhibition of 1 towards mNat2 and active site differences in mammalian NATs.

(a) Left panel: Structure of compound 1; Right panel: Dixon plot shows competitive inhibition of mNat2 (9 ng) by 1 at different pABA concentrations (25 µM (circles), 50 µM (triangles), 100 µM (diamonds), and 250 µM (squares)). Initial rates of the mNat2 catalysed reaction were determined by monitoring the rate of hydrolysis of AcCoA (400 μM) (b) Summary table of active site differences of human and murine NATs and the effects of their interaction with 1. Blue and red columns indicate the color of 1 on interaction with the protein.
The observed color change was proposed to be caused by selective recognition of the conjugate base of the naphthoquinone mediated by an appropriate residue within the active site of hNAT1 or mNat2, since a similar bathochromic shift in the λmax of naphthoquinone 1 was also observed in an alkaline solution [16]. Kinetic studies with pure recombinant mNat2 have revealed a competitive mode of inhibition of 1 towards the arylamine substrate para-amino benzoate (Ki,app. = 420 nM), which is consistent with previous studies using another member of this naphthoquinone family ( Figure 1 and Figure S1 in File S1) [16].
We describe a set of spectroscopic, chemical, molecular modeling and biochemical studies to interrogate the key molecular interactions between hNAT1 or its murine homologue, mNat2, and naphthoquinone 1 which lead to the observed color change event.
Results and Discussion
Reduction of the naphthoquinone core of 1 has previously been examined and discounted as a possible mechanism for this color change [16]. Additionally, it was found that the visible spectrum of 1 when treated with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), a non-nucleophilic amidine base, is comparable to that of 1 in the presence of aqueous NaOH (Figure S2 in File S1). This strongly suggests that the observed color change follows an acid-base interaction rather than a potential nucleophilic addition mechanism to the electrophilic enone system within 1.
Based on the hypothesis that selective recognition of the conjugate base of 1 within the hNAT1 or mNat2 active site is responsible for the observed color change, we aimed to identify both the acidic proton within 1 and the suitably located basic residue within the enzyme active site.
An acid-base titration revealed the pKa of 1 to be ∼9.2 in 5% DMSO/95% H2O (Figure S3 in File S1), which is consistent with the deprotonation of the sulfonamide moiety of 1. Although the sulfonamide-NH could be identified in the 1H-NMR spectrum of 1 in 100% DMSO-d6 solution (δH = 9.05 ppm) [16], adding as little as 5% D2O to the DMSO-d6 solvent promoted rapid proton-deuteron exchange. Direct observation of the sulfonamide-NH resonance by 1H-NMR spectroscopy could not be carried out under aqueous alkaline conditions nor in the presence of a NAT isoform under the assay conditions required for protein integrity (5% DMSO-d6/95% D2O).
The corresponding 15N-labelled sulfonamide analogue of 1 was thus synthesized (compound 2) to monitor the sulfonamide-NH using 15N-NMR spectroscopy. The 15N-NMR spectrum of 2 under alkaline conditions (95% DMSO-d6/5% aqueous NaOD) was acquired and compared to the spectrum in 100% DMSO-d6 ( Figure 2a ). The 15N spectrum of 2 under neutral conditions displayed a sharp resonance at 104.7 ppm, whilst the spectrum under alkaline conditions showed a broader resonance at 166.3 ppm. The displacement of the 15N chemical shift is consistent with the electron density changing around the sulfonamide-15N in an alkaline environment. It was not however possible to acquire 15N-NMR spectra in the presence of hNAT1 or mNat2 under conditions in which the protein would be active (5% DMSO-d6/95% D2O) as 2 was insufficiently soluble.
Figure 2. Requirement for the acidic proton of sulfonamide-NH for the color change event.

(a) Upper: 15 N-NMR spectrum of 2 in 95% DMSO-d6/5% H2O. Image of 50 μL of 2 (2 mM) in DMSO with 10 μL of 20 mM Tris.HCl, pH 8. Lower: 15 N-NMR spectrum of 2 in 95% DMSO-d6/5% aq. NaOD (final NaOD concentration 75.8 mM). Image of 50 μL of 2 (2 mM) in DMSO with 10 μL of 4 mM NaOH, pH 13. (b) Comparison of the colorimetric and inhibitory properties of compounds 1, 3, 4 and 5 towards hNAT1 and mNat2. Colors of cells indicate the observed color of the relevant compound under the given conditions.
Therefore, an alternative approach was adopted, whereby a close analogue of 1 was synthesized incorporating an N-methyl-N-sulfonyl moiety to determine the effect of removing the possibility of sulfonamide deprotonation with minimal disruption to other structural and chemical properties of the molecule. Treatment of 1 with one equivalent of TMS-diazomethane produced a separable mixture of the N-methyl substituted species 3 and the O-methyl substituted species 4, which were isolated in 55% and 35% yields respectively (see File S1).
Compound 3 was found to be a good inhibitor of hNAT1 (IC50 = 5.8 µM), but a very poor inhibitor of mNat2 (IC50 >150 µM), whilst 4 inhibited both enzymes with reasonable potency (IC50 = 3.6 µM for hNAT1 and IC50 = 14.6 µM for mNat2) ( Figure 2b ). Visible spectra of the N-methylated species 3 show that no color change occurs either in aqueous NaOH solution or in the presence of hNAT1 or mNat2. Visible spectra of the O-methylated species 4 also do not show an instantaneous shift in λmax in the presence of mNat2 ( Figure 2b and Figures S4 and S5 in File S1). Neither 3 nor 4 was active against the other human NAT isoenzyme, hNAT2, as more than 90% of enzyme activity was retained at an inhibitor concentration of 30 µM.
Sulfonate ester 5 was also synthesized, since this compound was predicted to have similar steric and electronic properties to 1 but lacks an acidic sulfonamide-NH. As predicted, 5 does not undergo a color change in the presence of either aqueous NaOH or in the presence of either hNAT1 or mNat2 ( Figure 2b and Figure S6 in File S1). Compound 5 was observed to be a weak inhibitor of hNAT1 (IC50 = 93 µM) and a very poor inhibitor of mNat2 (IC50 >150 µM).
With structure-activity-relationship data in hand supporting the key role of sulfonamide deprotonation in the color change event, structural-based site-directed mutagenesis studies were next undertaken to establish which amino acid residue within the active site of hNAT1 or mNat2 is able to recognize the conjugate base of the sulfonamide moiety within 1.
X-Ray crystal structures of both human NAT enzymes are available [18]; hNAT1 (PDB:2PQT) was used to build a structural model for mNat2. 1 was computationally docked within the active site of hNAT1 ( Figure 3a ) and into the mNat2 structural model ( Figure 3b ). The results from both docking studies were compared and several distinctive interactions between 1 and hNAT1 or mNat2 were predicted.
Figure 3. Inhibitor binding pocket of hNAT1 and mNat2.
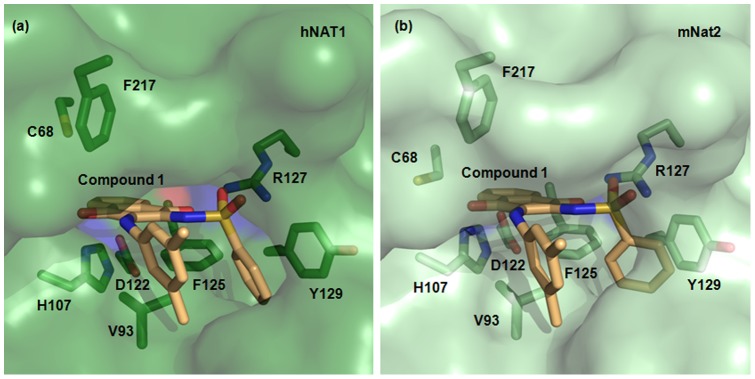
(a) The active site of hNAT1 crystal structure (PDB:2PQT) in surface representation with 1 docked in stick representation. The hNAT1 residues involved in inhibitor binding and selectivity are shown in stick representation and labeled with carbon in green, nitrogen in blue, oxygen in red, and sulfur in yellow. 1 is labeled with carbon atoms in light orange, nitrogen in blue, oxygen in red, and sulfur in yellow. (b) The active site of mNat2 structural model with docked compound 1 is shown using the same representation as in (a).
The close proximity of the sulfonamide functionality of the ligand and the Arg127 guanidine of both enzymes in the docking models suggested this residue was important for mutual recognition. In particular, both a sulfonamide oxygen atom and the carbonyl group at C1 of the naphthoquinone core of 1 appear to be able to form a hydrogen bond with the Arg127 guanidine group in the range of 2.5–3.0 Å. Meanwhile, the other sulfonamide oxygen appears close in space to the backbone amide carbonyl of Phe287 (2.5 Å).
Further notable potential interactions include the hydrophobic van der Waals interactions between the naphthoquinone core of 1 and the hydrophobic plane defined by the side chain arene of Phe125 and the isopropyl side chain of Val93 (3.5–4.0 Å); and additionally, the parallel-displaced π-π stacking between the C2 sulfonamide arene of 1 and the side chain arene of Tyr129 (4.2–4.8 Å).
The unique presence of the triad Phe125, Arg127 and Tyr129 in both hNAT1 and mNat2 compared to other eukaryotic and prokaryotic isoenzymes strongly supports the key role of these residues in selective ligand binding. This observation is consistent with previous findings on the crucial role played by the same residues on arylamine substrate preference [18]–[20]. Arg127 is not found in any of the other NAT enzymes investigated here (Figure S7 in File S1).
It was important to rule out the involvement of the catalytic Cys68 residue in mediating the color change event. Selective alkylation of the Cys68 thiolate within the mNat2 active site by incubating mNat2 with iodoacetamide inactivates the enzyme, but does not prevent the ligand 1 from changing color in the presence of the modified enzyme. This provides evidence to suggest that Cys68 is not the residue responsible for the color change (Figures S8 and S9 in File S1).
Arg127, which is likely to be protonated under the assay conditions (pH 8), is therefore anticipated to interact with the conjugate base of the sulfonamide moiety on binding of ligand 1, thereby driving the observed color change of 1 from red to blue, as the conjugate base of 1 is sequestered by the enzyme. From a chemical perspective, this accords with the pKaH of a free arginine guanidine (∼12.5) being higher than the pKa of 1 (∼9.2). The proposed mode of recognition between the protein and the ligand generates a strong bidentate ionic interaction between the two counterparts, an important feature contributing to selective recognition and stability of the protein/ligand complex.
Arg127 in mNat2 was therefore mutated by site-directed mutagenesis. Two different mutant mNat2 constructs were generated after single nucleotide mutation: the first encoding for a Gly at position 127 (mNat2_R127G), and the second for a Leu (mNat2_R127L, Figure S10 in File S1). By substituting Leu in place of Arg, a basic, charged group is removed with only a minor reduction in the predicted active site volume. Substitution for Gly significantly alters the overall size and shape of the active site. Moreover, Gly is the residue present at the same position in the other murine isoenzyme, mNat1. Wild type mNat2 (mNat2_WT) and the two mutants were successfully produced as recombinant proteins and purified with good yields (≥40 mg/L culture) consistent with previous studies on mNat2_WT (Figures S11 and S12 and Table S1 in File S1) [17].
Both site-directed mutants were found to be catalytically active: whilst the R127G mutant has a similar specific activity to the native protein with pABA as substrate (920 nmol/min/mg compared to 850 nmol/min/mg for mNat2_WT), the R127L mutant is less active (13 nmol/min/mg). Each of the mutants are also active against anisidine and showed no activity towards procainamide, like mNat2_WT, suggesting the proteins are appropriately folded to be catalytically active. AcCoA-hydrolysis assays were therefore carried out to investigate their kinetic properties towards naphthoquinone 1. The activity of each mNat2 mutant enzyme was tested with different concentrations of the inhibitor and IC50 curves were obtained and compared ( Figure 4 ). The IC50 value of 1 with mNat2_WT was 1.9 µM, whereas the IC50 values with mNat2_R127G and mNat2_R127L were higher, at 51.7 and 102.5 µM respectively.
Figure 4. The effects of mutating Arg127 of mNat2 in the interaction with 1.

(a) Visible spectra of compound 1 (15 μM) incubated under different conditions: 20 mM Tris.HCl at pH 8.0 (red line), with mNat2 variants (30 μM) in 20 mM Tris.HCl at pH 8.0 (mNat2_WT (light blue line), mNat2_R127G (orange line), mNat2_R127L (green line)) or of compound 1 in 80 mM NaOH at pH 13.75 (dark blue line). (b) Summary of interactions of 1 with hNAT1, mNat2 and the two engineered mNat2 mutants.
Full visible wavelength scans were obtained with 1 in the presence of the three mNat2 variants at pH 8.0. A bathochromic shift was observed with mNat2_WT, but no color change was observed with either mNat2_R127G or mNat2_R127L ( Figure 4 ). This supports the hypothesis that Arg127 is key in binding of the ligand to the enzyme and is required for the concomitant color change.
Conclusions
In summary, the combination of inhibitor structural studies, docking results and mutagenesis studies conducted indicate the color change observed is driven by selective recognition between the conjugate base of the sulfonamide-NH of the ligand and the Arg127 guanidininum of the enzyme. The high selectivity and striking colorimetric properties of the naphthoquinone probe 1 towards hNAT1 strongly support the further development of this family of naphthoquinones as selective inhibitors and colorimetric biosensors to target native overexpressed hNAT1 in breast tumors.
Materials and Methods
Chemicals and reagents
All chemicals were purchased from Sigma-Aldrich UK, TCI UK, Apollo Scientific UK, Alfa Aesar UK, Fluorochem UK or Fisher Scientific UK unless otherwise stated. Molecular biology reagents were obtained from Promega (Southampton, UK). Competent E. coli cells were purchased from Promega and Invitrogen (Carlsbad, USA). The pH of buffer solutions was adjusted at the appropriate temperature.
Chemical synthesis
The description of the methods for the chemical synthesis of compounds and their characterization data are detailed in the File S1.
Site-directed mutagenesis and transformation
The pET28b(+) plasmid vector containing the sequence of mNat2 [17] was isolated from 5 mL overnight bacterial cultures using a QIAprep spin miniprep kit (Qiagen). Site-directed mutagenesis was achieved using QuikChange II kit (Stratagene) to mutate one of the three nucleotides (CGT) encoding for Arg127 in mNat2. The reaction mixture (50 μL), which contained 5 μL 10× reaction buffer, 50 ng pET28b(+) plasmid as template, 125 ng of both R127G forward (5′-GCTGGGTTTGGAGGTTCCTACCAGATGTGGGAGCC-3′) and R127G reverse (5′-GGCTCCCACATCTGGTAGGAACCTCCAAACCCAGC-3′) primers, or 125 ng of both R127L forward (5′-GCTGGGTTTGGACTTTCCTACCAGATGTGGGAGCC-3′) and R127L reverse (5′-GGCTCCCACATCTGGTAGGAAAGTCCAAACCCAGC-3′) primers, and 1 μL dNTP mix, was subjected to thermo-cycling: one cycle of 2 min. at 95°C, 30 cycles of 1 min. at 95°C, 30 s at 60°C, and 1 min. at 72°C, an extra cycle at 72°C for 10 min., and a final cycle at 4°C for 5 min. The un-mutated parental DNA template present in the PCR product was digested by 10 U of Dpn I (New England Biolabs) for 1 h at 37°C. Mutant plasmids were verified by sequencing analysis (GeneService at Department of Biochemistry, University of Oxford, UK) to ensure the correct change before transformation by the heat shock method [21] into E. coli JM109 and E.coli Rosetta(DE3)pLysS strains, as previously described [17].
Protein production, purification and characterization
All recombinant mouse enzymes including the site-directed mutants of mNat2 were expressed with a hexa-histidine tag from E.coli Rosetta(DE3)pLysS strain transformed with the appropriate plasmid and the protein was then purified via Ni-NTA affinity chromatography (Qiagen) and thrombin cleavage of the His-tag, as previously described [17]. Details of the purification steps are shown in SDS page gels and in purification tables reported in the (Figures S11 and S12 and Table S1 in File S1). Pure recombinant hNAT1 was produced as previously described [22].
Enzymatic assays
NAT activity was determined as previously described [15],[23]; a full procedure is outlined in the (File S1).
Colorimetric properties of inhibitors
Visible spectra of each compound were recorded with a U-2001 spectrophotometer (Hitachi) using 1 mL plastic cells of 1 cm path-length (FisherBrand) or 50 μL UVettes® (Eppendorf). Concentrations of inhibitors and protein used are given in the appropriate figure legends. All spectra were blank-corrected.
Covalent modification of pure recombinant mNat2
To an aliquot of mNat2 (100 µL at 4 mg/mL in 20 mM Tris.HCl, pH 8) was added 5 µL 2-iodoacetamide (0.105 M solution in DMSO) to a final concentration of 5 mM 2-iodoacetamide. The aliquot was incubated at 4°C for 3 h, as previously described [18],[24]–[25]. Enzymatic activity was abolished and MS (MALDI) support covalent modification of Cys68 within mNat2, consistent with previous studies (Figures S8 and S9 in File S1, m/z 33988 (unmodified enzyme); 34046 (modified enzyme)) [18],[24]–[25].
Modelling structures
All images showing protein structures were generated using the software PyMOL (W. L. DeLano (2002) PyMOL, DeLano Scientific, San Carlos, CA). A structural model of mNat2 was generated based on the hNAT1 structure (PDB:2PQT) using the on-line software SwissModel, Automated mode (http://swissmodel.expasy.org/) [26]–[28], after removing the acetanilide molecule and restoring the thiol functionality of Cys68. The docking studies on 1 within hNAT1 and mNat2 active sites were conducted as follows. The ligand was first drawn as a 3D structure using the software ChemBio3D Ultra 12.0. The molecular editor Avogadro was used to predict the ground state conformation of the ligand. The analysis of the possible interactions between the protein and the ligand was performed using the licensed software GOLD [29]. A docking site was defined as a region of 10 Å within the active pocket of the enzyme and the ligand was then loaded into the software. The software gave different docked conformers and ranked the generated solutions using the GOLD Score Fitness function [29].
Supporting Information
Synthetic procedures, analytical data, NMR spectra and HPLC traces for all reported compounds, supplementary figures and tables (Figures S1–S12; Table S1) can all be found in File S1.
(DOCX)
Acknowledgments
The authors would like to thank Dr Barbara Odell (NMR Facility, Department of Chemistry, University of Oxford) for assistance in assigning the structure of 4, and Dr David Staunton (Biophysical Instrument Facility, Department of Biochemistry, University of Oxford) for performing the protein mass spectrometry experiments.
Funding Statement
This work was supported by: Cancer Research United Kingdom through a Cancer Medicinal Chemistry Studentship (NL), an Oxford Cancer Research Centre Prize DPhil Studentship (C38302/A12450, JEE) and a Small Molecule Cancer Drug Discovery Award (C17468/A9332; PTS, CEQ and ST). (http://www.cancerresearchuk.org/home/). The authors also thank Research Councils United Kingdom for a fellowship (AJR). (http://www.rcuk.ac.uk/). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Ransohoff DF (2003) Cancer. Developing molecular biomarkers for cancer. Science 299: 1679–1680. [DOI] [PubMed] [Google Scholar]
- 2. Dalton WS, Friend SH (2006) Cancer biomarkers–an invitation to the table. Science 312: 1165–1168. [DOI] [PubMed] [Google Scholar]
- 3. Kelloff GJ, Sigman CC (2012) Cancer biomarkers: selecting the right drug for the right patient. Nat Rev Drug Discov 11: 201–214. [DOI] [PubMed] [Google Scholar]
- 4. Wang BQ, Xia CH, Lv Y, Gao GG, Wu H (2011) Inorganic nanomaterial applications in cancer detection. Materials Technol 26: 236–242. [Google Scholar]
- 5. Nomura DK, Dix MM, Cravatt BF (2010) Activity-based protein profiling for biochemical pathway discovery in cancer. Nature Rev Cancer 10: 630–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Soh N (2008) Selective chemical labeling of proteins with small fluorescent molecules based on metal-chelation methodology. Sensors 8: 1004–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li N, Overkleeft HS, Florea BI (2012) Activity-based protein profiling: an enabling technology in chemical biology research. Curr Opin Chem Biol 16: 227–233. [DOI] [PubMed] [Google Scholar]
- 8. Adam PJ, Berry J, Loader JA, Tyson KL, Craggs G, et al. (2003) Arylamine N-acetyltransferase-1 is highly expressed in breast cancers and conveys enhanced growth and resistance to etoposide in vitro. Mol Cancer Res 1: 826–835. [PubMed] [Google Scholar]
- 9. Tozlu S, Girault I, Vacher S, Vendrell J, Andrieu C, et al. (2006) Identification of novel genes that co-cluster with estrogen receptor alpha in breast tumor biopsy specimens, using a large-scale real-time reverse transcription-PCR approach. Endocr Relat Cancer 13: 1109–1120. [DOI] [PubMed] [Google Scholar]
- 10. Wakefield L, Robinson J, Long H, Ibbitt JC, Cooke S, et al. (2008) Arylamine N-acetyltransferase 1 expression in breast cancer cell lines: a potential marker in estrogen receptor-positive tumors. Genes Chromosomes Cancer 47: 118–126. [DOI] [PubMed] [Google Scholar]
- 11. Johansson I, Nilsson C, Berglund P, Lauss M, Ringner M, et al. (2012) Gene expression profiling of primary male breast cancers reveals two unique subgroups and identifies N-acetyltransferase-1 (NAT1) as a novel prognostic biomarker. Breast Cancer Res 14: R31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bieche I, Girault I, Urbain E, Tozlu S, Lidereau R (2004) Relationship between intratumoral expression of genes coding for xenobiotic-metabolizing enzymes and benefit from adjuvant tamoxifen in estrogen receptor alpha-positive postmenopausal breast carcinoma. Breast Cancer Res 6: R252–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sim E, Fakis G, Laurieri N, Boukouvala S (2012) Arylamine N-acetyltransferases–from drug metabolism and pharmacogenetics to identification of novel targets for pharmacological intervention. Adv Pharmacol 63: 169–205. [DOI] [PubMed] [Google Scholar]
- 14. Westwood IM, Kawamura A, Russell AJ, Sandy J, Davies SG, et al. (2011) Novel small-molecule inhibitors of arylamine N-acetyltransferases: drug discovery by high-throughput screening. Comb Chem High Throughput Screen 14: 117–124. [DOI] [PubMed] [Google Scholar]
- 15. Russell AJ, Westwood IM, Crawford MH, Robinson J, Kawamura A, et al. (2009) Selective small molecule inhibitors of the potential breast cancer marker, human arylamine N-acetyltransferase 1, and its murine homologue, mouse arylamine N-acetyltransferase 2. Bioorg Med Chem 17: 905–918. [DOI] [PubMed] [Google Scholar]
- 16. Laurieri N, Crawford MH, Kawamura A, Westwood IM, Robinson J, et al. (2010) Small molecule colorimetric probes for specific detection of human arylamine N-acetyltransferase 1, a potential breast cancer biomarker. J Am Chem Soc 132: 3238–3239. [DOI] [PubMed] [Google Scholar]
- 17. Kawamura A, Westwood I, Wakefield L, Long H, Zhang N, et al. (2008) Mouse N-acetyltransferase type 2, the homologue of human N-acetyltransferase type 1. Biochem Pharmacol 75: 1550–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu H, Dombrovsky L, Tempel W, Martin F, Loppnau P, et al. (2007) Structural basis of substrate-binding specificity of human arylamine N-acetyltransferases. J Biol Chem 282: 30189–30197. [DOI] [PubMed] [Google Scholar]
- 19. Goodfellow GH, Dupret JM, Grant DM (2000) Identification of amino acids imparting acceptor substrate selectivity to human arylamine acetyltransferases NAT1 and NAT2. Biochem J 348 Pt 1: 159–166. [PMC free article] [PubMed] [Google Scholar]
- 20. Westwood IM, Kawamura A, Fullam E, Russell AJ, Davies SG, et al. (2006) Structure and mechanism of arylamine N-acetyltransferases. Curr Top Med Chem 6: 1641–54. [DOI] [PubMed] [Google Scholar]
- 21.Sambrook J, Fritsch EF, Maniatis T (1989) Molecular Cloning. A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
- 22. Dairou J, Atmane N, Dupret JM, Rodrigues-Lima F (2003) Reversible inhibition of the human xenobiotic-metabolizing enzyme arylamine N-acetyltransferase 1 by S-nitrosothiols. Biochem Biophys Res Commun 307: 1059–1065. [DOI] [PubMed] [Google Scholar]
- 23. Brooke EW, Davies SG, Mulvaney AW, Pompeo F, Sim E, et al. (2003) An approach to identifying novel substrates of bacterial arylamine N-acetyltransferases. Bioorg Med Chem 11: 1227–1234. [DOI] [PubMed] [Google Scholar]
- 24. Wang H, Vath GM, Gleason KJ, Hanna PE, Wagner CR (2004) Probing the mechanism of hamster arylamine N-acetyltransferase 2 acetylation by active site modification, site-directed mutagenesis, and pre-steady state and steady state kinetic studies. Biochemistry 43: 8234–8246. [DOI] [PubMed] [Google Scholar]
- 25. Wang H, Guo Z, Vath GM, Wagner CR, Hanna PE (2004) Chemical modification of hamster arylamine N-acetyltransferase 2 with isozyme-selective and nonselective N-arylbromoacetamido reagents. Protein J 23: 153–166. [DOI] [PubMed] [Google Scholar]
- 26. Arnold K, Bordoli L, Kopp J, Schwede T (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 2: 195–201. [DOI] [PubMed] [Google Scholar]
- 27. Kiefer F, Arnold K, Kunzli M, Bordoli L, Schwede T (2009) The SWISS-MODEL Repository and associated resources. Nucleic Acids Res 37: D387–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Peitsch MC (1995) Protein Modeling by E-Mail. Bio-Technology 13: 658–660. [Google Scholar]
- 29. Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD (2003) Improved protein-ligand docking using GOLD. Proteins-Structure Function and Genetics 52: 609–623. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Synthetic procedures, analytical data, NMR spectra and HPLC traces for all reported compounds, supplementary figures and tables (Figures S1–S12; Table S1) can all be found in File S1.
(DOCX)