


Abstract
We developed a novel simple cDNA normalization method [termed duplex-specific nuclease (DSN) normalization] that may be effectively used for samples enriched with full-length cDNA sequences. DSN normalization involves the denaturation–reassociation of cDNA, degradation of the double-stranded (ds) fraction formed by abundant transcripts and PCR amplification of the equalized single-stranded (ss) DNA fraction. The key element of this method is the degradation of the ds fraction formed during reassociation of cDNA using the kamchatka crab DSN, as described recently. This thermostable enzyme displays a strong preference for cleaving ds DNA and DNA in DNA–RNA hybrid duplexes compared with ss DNA and RNA, irrespective of sequence length. We developed normalization protocols for both first-strand cDNA [when poly(A)+ RNA is available] and amplified cDNA (when only total RNA can be obtained). Both protocols were evaluated in model experiments using human skeletal muscle cDNA. We also employed DSN normalization to normalize cDNA from nervous tissues of the marine mollusc Aplysia californica (a popular model organism in neuroscience) to illustrate further the efficiency of the normalization technique.
INTRODUCTION
cDNA libraries rich in clones containing complete coding sequences with 5′ and 3′ untranslated regions (UTRs) can be used to obtain entire sequence information for each transcript in a single cloning step, which is invaluable for high-throughput transcriptome analysis. A number of methods have been developed for cDNA library preparations enriched with full-length sequences (1–7). However, one well recognized obstacle of the efficient high-throughput analysis of these libraries is the differential abundance of various transcripts in any particular cell type. Usually, 10–20 abundant genes (several thousand mRNA copies per cell) account for at least 20% of the cellular mRNA mass, several hundred genes of medium abundance (several hundred mRNA copies per cell) comprise 40–60% of the mRNA mass, and several thousand rare genes (<10 mRNA copies per cell) may account for 20–40% of the mRNA mass (8). Hence, straightforward random sequencing of clones from standard cDNA libraries is inefficient for discovering rare transcripts, owing to the repeated occurrence of intermediately and highly abundant cDNA. Decreasing the prevalence of clones representing abundant transcripts before sequencing by normalization of cDNA libraries may significantly increase the efficiency of random sequencing and is essential for rare gene discovery.
The normalization process generally utilizes second-order reaction kinetics of reassociation of denatured DNA, so that relative transcript concentrations within the remaining single-stranded (ss) cDNA fraction are equalized to a considerable extent (9). The normalization methods described to date (8,10–18) are distinct from those used to isolate this ss fraction. The techniques proposed to achieve this goal include separation of the ss and double-stranded (ds) fractions using hydroxylapatite columns (10–12) or magnetic beads (8,16), digestion of the ds fraction by restriction endonucleases (14) and amplification of the ss fraction using suppression PCR (18–20). However, the majority of the earlier methods are unsuitable for normalization of full-length cDNA. Procedures based on the reassociation of amplified plasmid libraries (12,13,15,17) are not appropriate for long cDNA normalization, due to the loss of long inserts during cloning and amplification of the plasmid library. The suppression PCR-based method that is widely used in subtractive hybridization (18–21) is only applicable to short (restriction endonuclease-treated) cDNA fragments, since the suppression is not effective for long DNA molecules. The use of restriction endonucleases to digest the ds fraction (14) often results in loss of transcripts that tend to form secondary structures. Solid matrix-based methods (16,22) are generally not efficient enough, since the hybridization kinetics of nucleic acids immobilized on a solid phase are slower than those in solution (9).
The only method proposed for full-length cDNA normalization is the ‘normalization and subtraction of Cap-Trapper-selected cDNA’ (8). In this method, Cap-trapper-prepared first-strand cDNA is equalized during reassociation in the presence of biotinylated mRNA from the same source. Bio-RNA/cDNA hybrids are removed using magnetic beads. The Cap-Trapper method comprises multiple steps, involving the physical separation of the target cDNA fraction, and requires a large quantity of poly(A)+ RNA.
In this report, we describe a simple cDNA normalization method [termed duplex-specific nuclease (DSN) normalization] that utilizes the recently described (23) DSN from kamchatka crab (Paralithodes camtschaticus). Our technique efficiently normalizes cDNA samples enriched with full-length sequences, does not include laborious physical separation procedures and requires minimum hands-on time.
MATERIALS AND METHODS
DSN purification
The DSN enzyme was purified from kamchatka crab hepatopancreas, as described previously (23). The purified DSN is available commercially from Evrogen JSC.
Skeletal muscle cDNA preparation
First-strand cDNA was prepared from poly(A)+ skeletal muscle RNA (Clontech) using a SMART™ PCR cDNA Synthesis Kit (Clontech), according to the manufacturer’s protocol. SMART™ Oligo II and CDS primers (Clontech) were used for first-strand cDNA synthesis.
To prepare non-normalized skeletal muscle cDNA, first-strand cDNA was amplified with PCR primer provided in the SMART™ PCR cDNA Synthesis Kit (Clontech). The PCR mixture (50 µl) contained 1× Advantage 2 Polymerize mix (Clontech), 1× Advantage 2 PCR reaction buffer (Clontech), 200 µM dNTPs, 0.3 µM primer and 3 ng first-strand cDNA. Fourteen PCR cycles (95°C for 7 s, 65°C for 20 s, 72°C for 3 min) were performed. The resulting cDNA was used for preparing a plasmid non-normalized cDNA library and the normalization of amplified cDNA.
Preparation of amplified cDNA from central neurons of Aplysia californica
The central nervous systems (CNS) were dissected from eight molluscs, and giant metacerebral cells were acutely isolated and immediately subjected to total RNA isolation, followed by cDNA synthesis and amplification, as described by Matz (24). Briefly, first-strand cDNA was synthesized using the T-RSA (5′-CGCAGTCGGTACTTTTTTTTTTTTT-3′) primer. Following second-strand cDNA synthesis, the Lu4-St1RsaM adapter (5′-CGACGTGGACTATCCATGAACGCAACTCTCCGACCTCTCACCGAGTACG-3′) was ligated and cDNA was amplified by PCR with T-RSA and Lu4M (5′-CGTGGACTATCCATGAACGCA-3′) primers, using the Advantage 2 PCR Kit (Clontech). Twelve PCR cycles (95°C for 7 s, 65°C for 20 s, 72°C for 3 min) were performed.
First-strand cDNA normalization
Upon completion of first-strand cDNA synthesis, the reaction mixture was precipitated with ethanol and dissolved in milliQ water to a final cDNA concentration of ∼100 ng/µl. A 1.5 µl aliquot of this solution was mixed with 1 µl of 4× hybridization buffer (200 mM HEPES pH 7.5, 2 M NaCl, 0.8 mM EDTA) and 1.5 µl of milliQ water, overlaid with mineral oil, heated at 98°C for 3 min and allowed to hybridize at 70°C. After 4 h of incubation, 5 µl of 2× DSN buffer (100 mM Tris–HCl pH 8.0, 10 mM MgCl2, 2 mM dithiothreitol) preheated to 70°C was added to the reaction mixture. Next, 0.25 Kunitz units of DSN enzyme were added to the reaction and incubation was continued for 20 min. DSN was subsequently inactivated by the addition of 10 µl of 5 mM EDTA.
To amplify the normalized ssDNA fraction remaining after DSN treatment, 2 µl of reaction mixture was used for PCR in a 50 µl reaction volume containing 1× Advantage 2 Polymerize mix (Clontech), 1× Advantage 2 PCR reaction buffer (Clontech), 200 µM dNTPs and 0.3 µM CapM primer (5′-CAAGCAGTGGTATCAACGCAG-3′). Twenty PCR cycles (95°C for 7 s, 65°C for 20 s, 72°C for 3 min) were performed.
Normalization of SMART™-amplified cDNA
Amplified cDNA was purified using the QJ Aquich PCR Purification Kit (Qiagen), precipitated with ethanol and dissolved in milliQ water to a final cDNA concentration of 100 ng/µl. An aliquot (1 µl) of this solution containing ∼100 ng cDNA was mixed with 1 µl of 4× hybridization buffer and 2 µl of milliQ water, overlaid with mineral oil, denatured at 98°C for 3 min and allowed to renature at 70°C for 4 h. DSN treatment was performed as described in the section entitled ‘First-strand cDNA normalization’.
After DSN inactivation, milliQ water was added to the reaction mixture to a final volume of 40 µl. An aliquot (1 µl) of the resulting solution was used for PCR with the CapM primer. The PCR mixture (25 µl) contained 1× Advantage 2 Polymerize mix (Advantage 2 PCR Kit, Clontech), 1× reaction buffer (Advantage 2 PCR Kit, Clontech), 200 µM dNTPs and 0.3 µM ÑapM primer. Twenty PCR cycles (95°C for 7 s, 65°C for 20 s, 72°C for 3 min) were performed.
Normalization of amplified cDNA from A.californica
Amplified Aplysia cDNA was purified using a QJ Aquich PCR Purification Kit (Qiagen), precipitated with ethanol and dissolved in milliQ water to a final cDNA concentration of 100 ng/µl. An aliquot (1 µl) of this solution was utilized for normalization. Hybridization and DSN treatment were performed as described in the previous section, but at a modified hybridization time of ∼6 h. Following DSN inactivation, milliQ water was added to the reaction mixture to a final volume of 40 µl. An aliquot (1 µl) of the resulting solution was used for PCR with CapM-T-RSA (5′-AAGCAGTGGTATCAACGCAGCGCAGTCGGTACTTTTTTTTTTTTT-3′) and CapM-St1RsaM (5′-AAGCAGTGGTATCAACGCAGCCGACCTCTCACCGAGTACG-3′) primers to introduce the same sequences to the cDNA ends. The PCR mixture (50 µl) contained 1× Advantage 2 Polymerize mix (Clontech), 1× reaction buffer (Clontech), 200 µM dNTPs and 0.3 µM of each primer. Eighteen PCR cycles (95°C for 7 s, 65°C for 20 s, 72°C for 3 min) were performed.
The resulting product was subjected to ‘regulation of the average PCR product length’, as described by Shagin et al. (25). The amplified product was diluted five times with milliQ water. An aliquot (1 µl) of the solution was employed for re-amplification with the CapM primer. The PCR mixture (50 µl) contained 1× Advantage 2 Polymerize mix (Clontech), 1× reaction buffer (Clontech), 200 µM dNTPs and 0.3 µM CapM primer. Ten PCR cycles (95°C for 7 s, 65°C for 20 s, 72°C for 3 min) were performed.
Virtual northern blotting
Virtual northern blotting was performed as described by Franz et al. (26). Normalized and non-normalized skeletal muscle cDNA samples were resolved on agarose gels and transferred to Hybond-N membranes (Amersham). Gel electrophoresis and subsequent membrane transfer were performed according to standard protocols (27). Hybridization was performed with an optimized low-background protocol (28) using 32P-labeled gene-specific probes generated with Human Amplimer Sets (Clontech). The Prime-a-Gene Labeling System (Promega) was used for labeling.
Hybridization and PCR analysis of bacterial colonies
Normalized and non-normalized skeletal muscle cDNA samples were cloned into the pGEM-T-easy vector (Promega) according to the manufacturer’s instructions, and used for Escherichia coli transformation with the Bio-Rad Micropulser. For screening, colonies were grown on duplicate nylon filters and hybridized with 32P-labeled gene-specific probes (prepared as described above) according to standard protocols (27). Frequencies of the corresponding cDNA sequences in the libraries were calculated from the number of positive colonies observed among 7000–84 000 colonies from each library (depending on the gene).
For PCR analysis of the insert size distribution, 106 white colonies from each library were randomly picked and grown in 150 µl of Luria–Bertrani broth with ampicillin. One microliter of the bacterial cultures was used for PCR with standard M13 primers. PCR products were visualized on a 1.5% agarose gel, following ethidium bromide staining, alongside a 100 bp DNA ladder (Gibco-BRL). Sizes of the plasmid inserts were calculated, ranged and used for diagram drawing.
Aplysia californica EST sequencing and analysis
The products of non-normalized and normalized cDNA amplification were ligated into a pGEM-T vector (Promega) following the manufacturer’s protocol, and transformed into E.coli (JM109) cells. Ligation volumes and number of transformations were appropriately selected to ensure that ∼50 000 recombinant bacterial colonies were obtained for non-normalized (CNS_MCC) and normalized (CNS_MCC_N) libraries. Inserts in recombinant plasmids from randomly picked clones were sequenced. A total of 1150 raw sequences were collected from each library, resulting in 1121 non- vector expressed sequence tag (EST) sequences for the non-normalized library and 1130 sequences for the normalized one. Sequences obtained were submitted to DDBJ/EMBL/GenBank with accession numbers BF707524–BF708380, BF713631, BF713632, BI273615–BI273627 for sequences from the CNS_MCC library and CK327631–CK328797 for sequences from the CNS_MCC_N library, except 247 sequences from the non-normalized library and two sequences from the normalized library that corresponded to ribosomal RNAs. In the submitted set, direct and reverse sequences for 39 CNS_MCC_N clones were deposited separately.
Sequences were compared using SeqMan II software with the following construction parameters: match size, 50; maximum added gap length in contig, 20; maximum added gap length in sequence, 20; minimum match percentage, 90; maximum register shift difference, 10; last group considered, 2; gap penalty, 0.00; gap length penalty, 0.70; and consensus threshold, 75. The observed frequency distribution data were fitted to the Poisson distribution using software available online (at http://faculty.vassar.edu/lowry/poissonfit.html). Sequences were compared with the National Center for Biotechnology Information (NCBI; Bethesda, MD) non-redundant nucleotide database using the BLAST web service at NCBI (http://www.ncbi.nlm.nih.gov/BLAST).
RESULTS
Normalization strategy
The DSN normalization method involves the denaturation–reassociation of cDNA, degradation of the ds fraction formed by abundant transcripts and PCR amplification of the equalized ssDNA fraction. The key element of this method is the degradation of the ds fraction formed during reassociation of cDNA using the DSN enzyme. A number of specific features of DSN make it ideal for removing dsDNA from complex mixtures of nucleic acids. DSN displays a strong preference for cleaving dsDNA in both DNA–DNA and DNA–RNA hybrids compared with ss DNA and RNA, irrespective of the sequence length. Moreover, the enzyme remains stable over a wide range of temperatures and displays optimal activity at 55–65°C (23). Consequently, degradation of the ssDNA-containing fraction by this enzyme may occur at high temperatures, thereby avoiding loss of transcripts due to the formation of secondary structures and non-specific hybridization involving adapter sequences.
We developed DSN normalization for both first-strand cDNA [when poly(A)+ RNA is available] and amplified cDNA (when only total RNA can be obtained).
First-strand cDNA normalization
A scheme of the first-strand cDNA normalization procedure is depicted in Figure 1. The strategy includes preparation of first-strand cDNA and its normalization by reassociation in the presence of mRNA initially used for first-strand cDNA preparation. Such RNA is postulated as the ideal ‘driver’ for normalization, since it reflects the complexity of first-strand cDNA (8). Following reassociation, DNA in DNA–RNA hybrids is degraded by DSN. The remaining ss cDNA fraction is amplified by PCR.
Figure 1.

Scheme of DSN normalization of SMART™-prepared first-strand cDNA using the RNA ‘driver’. Black line, abundant transcript; grey line, intermediate transcript; dotted line, rare transcript.
Suitable cDNA for this procedure should contain known flanking sequences for subsequent PCR amplification. This first-strand cDNA may be prepared in several ways, including using the oligo-capping method (2), the Cap-Trapper method (4) and the template-switching effect approach (SMART™) (6,29,30). We selected the SMART™ method as a popular and simple way to prepare cDNA enriched with full-length sequences. This approach makes it possible to attach the same synthetic adapter sequence to both 5′ and 3′ ends of cDNA during synthesis.
Normalization of amplified cDNA
In a number of cases, the initial amount of biological material is limited, and only total RNA is available. For these situations, we modified our technology for amplified cDNA normalization: ds cDNA is denatured and subsequently allowed to rehybridize. Following reassociation, the ds DNA fraction (formed by mostly abundant transcripts) is degraded by DSN and the equalized ss fraction is amplified by PCR.
Hybridization
Similar to the majority of the previously described methods, DSN normalization is based on second-order solution hybridization kinetics. We used standard hybridization conditions well proven in the suppression of subtractive hybridization (20,21).
Additional enrichment of full-length sequences
PCR has a recognized tendency to amplify shorter fragments more efficiently than longer ones. This may result in the loss of rare long transcripts during PCR and a reduction in the average cDNA length. The use of a ‘long and accurate PCR system’ (31) provides only a partial solution to this problem. To effectively increase the proportion of long fragments in the cDNA sample, we incorporated a previously developed procedure for regulation of the average length of the complex PCR product (25) in our normalization protocol.
Normalization of skeletal muscle cDNA
The new method was evaluated by normalizing cDNA from the human skeletal muscle. SMART™-prepared first-strand cDNA and amplified cDNA were used for DSN normalization (see Materials and Methods for details). The normalized cDNA samples obtained were compared with non-normalized SMART™-prepared amplified skeletal muscle cDNA. Analysis of the normalized samples on an agarose gel revealed that the bands corresponding to abundant transcripts disappeared, while the average length of cDNA remained unchanged (Fig. 2).
Figure 2.
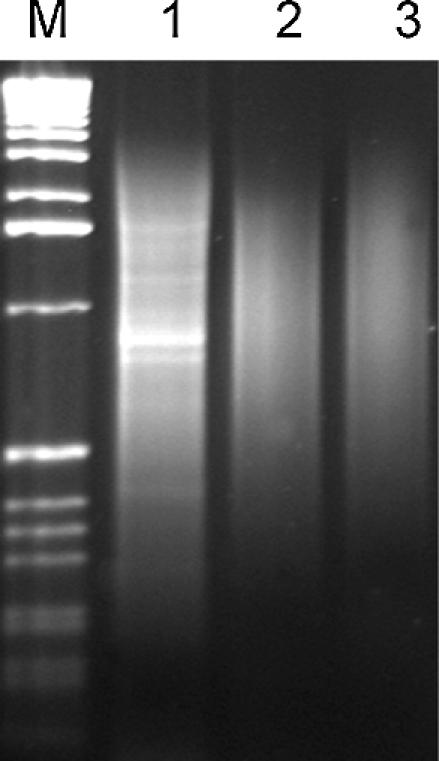
Agarose gel electrophoresis of non-normalized and normalized amplified SMART™-prepared cDNA from skeletal muscle. Lane 1, non-normalized cDNA; lane 2, normalized first-strand cDNA (FN); lane 3, normalized amplified cDNA (AN); lane M, 1 kb ladder (Gibco-BRL).
The efficiency of equalization and transcript integrity were systematically assessed by virtual northern blotting (Fig. 3) and colony hybridization (Table 1) of normalized and non-normalized cDNA samples with radioactive labeled fragments of several marker genes. A sharp decrease in the representation levels of abundant transcripts (GAPD, UBC, ACTB) was observed in normalized cDNA samples compared with non-normalized samples. The concentrations of the intermediate transcripts (RPL13A, YWHAZ, RPS9) did not change significantly in a sample obtained by normalization of amplified cDNA (AN), but decreased in a sample obtained by first-strand cDNA normalization (FN). Surprisingly, a 3- to 4-fold increase in the concentrations of rare transcripts (NFKB1, IGF2R, JUNC) was observed in the FN cDNA sample, whereas the representation levels of these transcripts were not affected in the AN sample. Moreover, virtual northern blot hybridization studies showed that the transcript lengths in normalized cDNA samples were similar to those in non-normalized cDNA.
Figure 3.

Virtual northern blot analysis of normalized and non-normalized skeletal muscle cDNA. Lane 1, normalized first-strand cDNA; lane 2, normalized amplified cDNA; lane 3, non-normalized amplified cDNA. DDBJ/EMBL/GenBank accession nos of marker genes employed are listed in table 1.
Table 1. Frequencies of cDNA sequences of the several genes in non-normalized, normalized first-strand (FN) and normalized amplified (AN) cDNA libraries.
| Gene | DDBJ/EMBL/GenBank accession no. | Positive colonies (%) | ||
|---|---|---|---|---|
| Non-normalized | FN | AN | ||
|
GAPD |
NM_002046 |
1.561 |
0.002 |
0.01 |
|
ACTB |
NM_001101 |
0.111 |
0.001 |
0.005 |
|
UBC |
NM_021009 |
0.95 |
0.003 |
0.007 |
|
YWHAZ |
NM_003406 |
0.044 |
0.009 |
0.03 |
|
RPS9 |
NM_001013 |
0.042 |
0.013 |
0.051 |
|
RPL13a |
NM_012423 |
0.046 |
0.019 |
0.079 |
|
NFKB1 |
NM_003998 |
0.004 |
0.011 |
0.005 |
|
IGF2R |
NM_000876 |
0.003 |
0.013 |
0.004 |
|
P53 |
NM_000546 |
<0.001 |
0.001 |
0.001 |
| JUNC | NM_002228 | <0.001 | 0.004 | 0.002 |
The preservation of the average insert size in the normalized AN and FN libraries in comparison with the non-normalized one was additionally confirmed by PCR of the 106 randomly picked clones from each library with standard plasmid primers. Insert sizes were calculated and used for diagram drawing (Fig. 4). We found that insert size distribution is not appreciably altered during normalization.
Figure 4.

Insert size distribution in the non-normalized (white bars) and normalized FN (gray bars) and AN (black bars) human skeletal muscle cDNA libraries.
DSN normalization of amplified cDNA from Aplysia’s neurons
Representative amplified cDNA was prepared from several giant metacerebral cells that were acutely isolated from the CNS of the sea slug A.californica using a suppression PCR-based method suitable for cDNA preparation from small amounts of starting material (24,32), and subjected to normalization following the protocol for amplified cDNA, with one modification. Specifically, since this cDNA was flanked by different adapter sequences, we performed the additional PCR step to introduce same adapter sequences into cDNA molecules. This allows facilitation of regulation of the average length of the PCR product (25). Non-normalized and normalized cDNAs were used for preparation of the non-normalized (CNS_MCC) and normalized (CNS_MCC_N) cDNA libraries. Next, 1150 randomly picked clones were sequenced from each library. Following elimination of vector-only sequences and unreliable data, 1130 clones (representing 1080 non-overlapping sequences) were collected from the normalized CNS_MCC_N library and 1121 clones (representing 745 non-overlapping sequences) were collected from the non-normalized CNS_MCC library. Among sequences collected from the normalized library, 1034 appeared only once, while 46 appeared more than once (43 appeared twice, two appeared three times and one appeared four times).
Analysis of the sequences obtained from the non- normalized CNS_MCC library revealed several cDNA types that were represented more frequently than the others. For example, 101 sequences corresponded to 16S mitochondrial rRNA, 108 to 18S rRNA and 31 to 28S rRNA (see Table 2 for the most represented sequences in the non-normalized library). To confirm normalization efficiency, we tested for cDNA sequences that were redundant in the non-normalized library. The occurrence of these sequences was significantly diminished after normalization (Table 2).
Table 2. Changes in the concentrations of the top represented sequences from amplified Aplysia cDNA after DSN normalization.
| Sequence name | Representation | |
|---|---|---|
| Non-normalized CNS_MCC cDNA library % | Normalized CNS_MCC_N cDNA library % | |
| 18S rRNA |
9.6 (108/1121) |
0.09 (1/1130) |
| 16S mitochondrial RNA |
9.0 (101/1121) |
0.09 (1/1130) |
| 28S rRNA |
2.8 (31/1121) |
<0.09 (0/1130) |
| Large (rrnL) subunit of mitochondrial ribosomal RNA homolog (BF708086) |
1.1 (12/1121) |
<0.09 (0/1130) |
| Beta tubulin (AY256661) |
1.0 (11/1121) |
<0.09 (0/1130) |
| Ferritin homolog (BF708039) |
0.8 (9/1121) |
<0.09 (0/1130) |
| Calmodulin (AY036120) |
0.8 (9/1121) |
<0.09 (0/1130) |
| Unknown 1 (BF707535) |
0.7 (8/1121) |
<0.09 (0/1130) |
| Unknown 2 (BF707854) |
0.7 (8/1121) |
<0.09 (0/1130) |
| Reductase-related protein (AF042739) |
0.6 (7/1121) |
<0.09 (0/1130) |
| 12S mitochondrial RNA |
0.45 (5/1121) |
<0.09 (0/1130) |
| Histone H3 homolog (BF708342) |
0.45 (5/1121) |
<0.09 (0/1130) |
| Cytochrome c oxidase subunit I (AF077759) | 0.45 (5/1121) | <0.09 (0/1130) |
Poisson statistics allows the approximate estimation of the total complexity (i.e. the total number of different sequences) of the library. Our data displayed the best fit to a Poisson distribution, with a mean µ of 0.09. Thus, a value of ∼13 000 unique clones was calculated for the normalized Aplysia library. This value is in agreement with the expected total number of different mRNA types in certain mollusc neurons.
DISCUSSION
The DSN normalization method works efficiently with both first-strand cDNA and ds amplified cDNA prepared using two different protocols. The sizes of individual cDNA sequences remain unchanged with this procedure. Consequently, the method may be used effectively for samples enriched with full-length cDNA sequences. Our technology appears to be considerably simpler than most normalization methods reported to date.
DSN normalization is a highly efficient procedure. In model experiments, the representation levels of abundant transcripts essentially decrease to those of rare transcripts. Theoretically, if ∼10 000–20 000 mRNA types are present in a sample, the levels of abundant genes should decrease by one or two orders of magnitude after equalization, the representation level of genes of medium abundance should decrease several-fold or remain the same, and the concentration of rare genes should increase ∼3-fold. As a result, the representation level of each gene in an ideally equalized library should be ∼0.01% (33). This prediction is generally consistent with data obtained from our experiments, particularly in the case of first-strand cDNA normalization. Moreover, in this case, the representation levels of several abundant transcripts are even lower than those of mRNA that was originally rare. We propose that this is due to the additional removal of abundant transcripts during DSN treatment. DSN cleaves DNA molecules in DNA–RNA hybrids, but does not affect RNA molecules. Therefore, as the DNA molecules within these hybrids are hydrolyzed, RNA molecules are released. These ‘recycled’ RNA molecules hybridize with corresponding abundant ssDNAs that remain within the solution, thereby causing further degradation.
Normalization of amplified cDNA is less efficient than that of first-strand cDNA, but also results in a considerable decrease in the abundant transcript concentrations. This procedure will undoubtedly aid rare transcript discovery when only limited amounts of total RNA are available for manipulation.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by Evrogen JSC, a grant from the Russian Foundation to Support Domestic Science (to S.L) and a grant from the physico-chemical biology program of RAS. This work was also supported in part by Packard, National Science Foundation and National Institutes of Health grants R01NS39103 and PO HG0028006 to (L.L.M.).
DDBJ/EMBL/GenBank accession nos+ BF707524–BF708380, BF713631, BF713632, BI273615–BI273627 and CK327631–CK328797
REFERENCES
- 1.Kato S., Sekine,S., Oh,S., Kim,N., Umezawa,Y., Abe,N., Yokoyama-Kobayashi,M. and Aoki,T. (1994) Construction of a human full-length cDNA bank. Gene, 150, 243–250. [DOI] [PubMed] [Google Scholar]
- 2.Maruyama K. and Sugano,S. (1994) Oligo-capping: a simple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides. Gene, 138, 171–174. [DOI] [PubMed] [Google Scholar]
- 3.Edery I., Chu,L., Sonenberg,N. and Pelletier,J. (1995) An efficient strategy to isolate full-length cDNAs based on an mRNA cap retention procedure (CAPture). Mol. Cell. Biol., 15, 3363–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carninci P., Kvam,C., Kitamura,A., Ohsumi,T., Okazaki,Y., Itoh,M., Kamiya,M., Shibata,K., Sasaki,N., Izawa,M. et al. (1996) High-efficiency full-length cDNA cloning by biotinylated CAP trapper. Genomics, 37, 327–336. [DOI] [PubMed] [Google Scholar]
- 5.Suzuki Y., Yoshitomo-Nakagawa,K., Maruyama,K., Suyama,A. and Sugano,S. (1997) Construction and characterization of a full length-enriched and a 5′-end-enriched cDNA library. Gene, 200, 149–156. [DOI] [PubMed] [Google Scholar]
- 6.Zhu Y.Y., Machleder,E.M., Chenchik,A., Li,R. and Siebert,P.D. (2001) Reverse transcriptase template switching, a SMART approach for full-length cDNA library construction. Biotechniques, 30, 892–897. [DOI] [PubMed] [Google Scholar]
- 7.Spiess A.N. and Ivell,R. (2002) A highly efficient method for long-chain cDNA synthesis using trehalose and betaine. Anal. Biochem., 301, 168–174. [DOI] [PubMed] [Google Scholar]
- 8.Carninci P., Shibata,Y., Hayatsu,N., Sugahara,Y., Shibata,K., Itoh,M., Konno,H., Okazaki,Y., Muramatsu,M. and Hayashizaki,Y. (2000) Normalization and subtraction of cap-trapper-selected cDNAs to prepare full-length cDNA libraries for rapid discovery of new genes. Genome Res., 10, 1617–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Young B.D. and Anderson,M. (1985) Quantitative analysis of solution hybridisation. In Hames,B.D. and Higgins,S.J. (eds) Nucleic Acid Hybridisation: A Practical Approach. IRL Press, Oxford, pp. 47–71. [Google Scholar]
- 10.Ko M. (1990) An ‘equalized cDNA library’ by the reassociation of short double-stranded cDNAs. Nucleic Acids Res., 18, 5705–5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patanjali S.R., Parimoo,S. and Weissman,S.M. (1991) Construction of a uniform-abundance (normalized) cDNA library. Proc. Natl Acad. Sci. USA, 88, 1943–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Soares M., Bonaldo,M., Jelene,P., Su,L., Lawton,L. and Efstratiadis,A. (1994) Construction and characterization of a normalized cDNA library. Proc. Natl Acad. Sci. USA, 91, 9228–9232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swaroop A., Xu,J., Agarwal,N. and Weissman,S.M. (1991) A simple and efficient cDNA library subtraction procedure: isolation of human retina-specific cDNA clones. Nucleic Acids Res., 19, 1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coche T. and Dewez,M. (1994) Reducing bias in cDNA sequence representation by molecular selection. Nucleic Acids Res., 22, 4545–4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li W.-B., Gruber,C.E., Lin,J.-J., Lim,R., D’Alessio,J.M. and Jessee,J.A. (1994) The isolation of differentially expressed genes in fibroblast growth factor stimulated BC3H1 cells by subtractive hybridization. Biotechniques, 16, 722–729. [PubMed] [Google Scholar]
- 16.Sasaki Y., Ayusawa,D. and Oishi,M. (1994) Construction of a normalized cDNA library by introduction of a semi-solid mRNA–cDNA hybridization system. Nucleic Acids Res., 22, 987–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bonaldo M.F., Lennon,G. and Soares,M.B. (1996) Normalization and subtraction: Two approaches to facilitate gene discovery. Genome Res., 6, 791–806. [DOI] [PubMed] [Google Scholar]
- 18.Luk'ianov K.A., Gurskaia,I.G., Matts,M.V., Khaspekov,G.L., D’iachenko,L.B., Chenchik,A.A., Il’evich-Stuchkov,S.G. and Luk’ianov,S.A. (1996) A method for obtaining the normalized cDNA libraries based on the effect of suppression of polymerase chain reaction. Bioorg. Khim. (Russ), 22, 686–690. [PubMed] [Google Scholar]
- 19.Luk'ianov S.A., Gurskaia,N.G., Luk’ianov,K.A., Tarabykin,V.S. and Sverdlov,E.D. (1994) Highly efficient subtractive hybridisation of cDNA. Bioorg. Khim. (Russ), 20, 701–704. [PubMed] [Google Scholar]
- 20.Diatchenko L., Lau,Y.-F.C., Campbell,A.P., Chenchik,A., Mogadam,F., Huang,B., Lukyanov,S., Lukyanov,K., Gurskaya,N., Sverdlov,E.D. et al. (1996) Suppression sabtracive hybridization, a method for generating differentially regulated or tissue-specific cDNA probes and libraries. Proc. Natl Acad. Sci. USA, 93, 6025–6030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gurskaya N.G., Diatchenko, L, Chenchik,A., Siebert,P.D., Khaspekov,G.L., Lukyanov,K.A., Vagner,L.L., Ermolaeva,O.D., Lukyanov,S.A. and Sverdlov,E.D. (1996) The equalizing cDNA subtraction based on selective suppression of polymerase chain reaction, cloning of the Jurkat cells’ transcripts induced by phytohemaglutinin and phorbol 12-myristate 13-acetate. Anal. Biochem., 240, 90–97. [DOI] [PubMed] [Google Scholar]
- 22.Tanaka T., Ogiwara,A., Uchiyama,I., Takagi,T., Yazaki,Y. and Nakamura,Y. (1996) Construction of a normalized directionally cloned cDNA library from adult heart and analysis of 3040 clones by partial sequencing. Genomics, 35, 231–235. [DOI] [PubMed] [Google Scholar]
- 23.Shagin D.A., Rebrikov,D.V., Kozhemyako,V.B., Altshuler,I.M., Shcheglov,A.S., Zhulidov,P.A., Bogdanova,E.A., Staroverov,D.B., Rasskazov,V.A. and Lukyanov,S. (2002) A novel method for SNP detection using a new duplex-specific nuclease from crab hepatopancreas. Genome Res., 12, 1935–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matz M.V. (2002) Amplification of representative cDNA samples from microscopic amounts of invertebrate tissue to search for new genes. Methods Mol. Biol., 183, 3–18. [DOI] [PubMed] [Google Scholar]
- 25.Shagin D.A., Lukyanov,K.A., Vagner,L.L. and Matz,M.V. (1999) Regulation of average length of complex PCR product. Nucleic Acids Res., 27, e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Franz O., Bruchhaus,I.I. and Roeder,T. (1999) Verification of differential gene transcription using virtual northern blotting. Nucleic Acids Res., 27, e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 28.Engler-Blum G., Meier,M., Frank,J. and Muller G. (1993) Reduction of background problems in nonradioactive Northern and Southern blot analysis enables higher sensitivity then 32P-based hybridizations. Anal. Biochem., 44, 235–244. [DOI] [PubMed] [Google Scholar]
- 29.Chenchik A., Zhu,Y.Y., Diatchenko,L., Li,R., Hill,J. and Siebert,P.D. (1998) In Siebert,P. and Larrick,J. (eds) Gene Cloning and Analysis by RT–PCR. Biotechniques Books, Natick, MA, pp. 305–319. [Google Scholar]
- 30.Matz M., Shagin,D., Bogdanova,E., Lukyanov,S., Diatchenko,L. and Chenchik,A. (1999) Amplification of cDNA ends based on template-switching effect and step-out PCR. Nucleic Acids Res., 27, 1558–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barnes W.M. (1994) PCR amplification of up to 35-kb DNA with high fidelity and high yield from lambda bacteriophage templates. Proc. Natl Acad. Sci. USA, 91, 2216–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lukyanov K., Diatchenko,L., Chenchik,A., Nanisetti,A., Siebert,P., Usman,N., Matz,M. and Lukyanov,S. (1997) Construction of cDNA libraries from small amounts of total RNA using the suppression PCR effect. Biochem. Biophys. Res. Commun., 230, 285–288. [DOI] [PubMed] [Google Scholar]
- 33.Moreno-Palanques R.F. and Fuldner,R.A. (1994) Construction of cDNA libraries. In Adams,M.D., Fields,C. and Venter,J.C. (eds) Automated DNA Sequencing and Analysis. Academic Press, London, UK, pp. 102–109. [Google Scholar]