

Abstract
Campylobacter species.are phenotypically diverse in many aspects including host habitats and pathogenicities, which demands comprehensive characterization of the entire Campylobacter genus to study their underlying genetic diversification. Up to now, 34 Campylobacter strains have been sequenced and published in public databases, providing good opportunity to systemically analyze their genomic diversities. In this study, we first conducted genomic characterization, which includes genome-wide alignments, pan-genome analysis, and phylogenetic identification, to depict the genetic diversity of Campylobacter genus. Afterward, we improved the tetranucleotide usage pattern-based naïve Bayesian classifier to identify the abnormal composition fragments (ACFs, fragments with significantly different tetranucleotide frequency profiles from its genomic tetranucleotide frequency profiles) including horizontal gene transfers (HGTs) to explore the mechanisms for the genetic diversity of this organism. Finally, we analyzed the HGTs transferred via bacteriophage transductions. To our knowledge, this study is the first to use single nucleotide polymorphism information to construct liable microevolution phylogeny of 21 Campylobacter jejuni strains. Combined with the phylogeny of all the collected Campylobacter species based on genome-wide core gene information, comprehensive phylogenetic inference of all 34 Campylobacter organisms was determined. It was found that C. jejuni harbors a high fraction of ACFs possibly through intraspecies recombination, whereas other Campylobacter members possess numerous ACFs possibly via intragenus recombination. Furthermore, some Campylobacter strains have undergone significant ancient viral integration during their evolution process. The improved method is a powerful tool for bacterial genomic analysis. Moreover, the findings would provide useful information for future research on Campylobacter genus.
Introduction
Campylobacter is a genus of Gram-negative bacteria that are spiral-shaped microaerophilic and motile [1]. Campylobacter spp.have unipolar or bipolar flagella, and generally colonize different mucosal surfaces. The genus Campylobacter was first proposed by Sebald et al. in 1963 [2]. Since then, the number of Campylobacter species discovered has significantly increased. The genus comprised 17 species with validly published names, including six recognized subspecies, in 2004 [3], [4]. At present, 23 species are recorded in the National Center for Biotechnology Information (NCBI) Taxonomy Division. Members of the genus Campylobacter colonize diverse host habitats, from livestocks to humans [5]–[11], indicating that their genomes diversify to adapt to various host environments. Campylobacter spp. are significantly diverse with regard to their pathogenicity. Some species definitely cause human disease. C.jejuni is one of the most important food-borne pathogen in the world, and its infection is a leading cause of acute bacterial diarrhea in humans in many developed countries [12], [13]. Campylobacter coli, Campylobacter upsaliensis, and Campylobacter lari are also associated with human gastroenteritis [6], [7], [14]. Some species are causative agents of pericarditis/myocarditis [15], [16] and Guillain–Barre Syndrome in humans [17]–[19]. Other Campylobacter species are related to severe animal diseases; for instance, Campylobacter fetus can cause abortion in animals [20]–[22] and bovine genital campylobacteriosis [23], [24]. However, not all Campylobacter spp. are pathogenic, suggesting that their phenotypic diversities in terms of pathogenicity may result from genomic diversities within the Campylobacter genus. Thus, comprehensive characterization of their genetic diversities that contribute to their phenotypic diversities should be conducted, and the underlying mechanisms should be determined.
Since the publication of the Campylobacter genome for the C. jejuni strain 11168, 34 Campylobacter genomes have been sequenced. Thus, systemic analysis and comparison of the entire Campylobacter genus should be conducted to illustrate its genomic diversity and provide insights into its mechanisms for genomic diversity. Although several reports have analyzed a single or few genomes [25]–[33], to our knowledge, intensive and combined investigation on the genotypic diversity of all Campylobacter genomes has not been conducted yet. In the present study, 34 genomes of the Campylobacter genus were downloaded and systemically analyzed to describe their genetic diversities. After depiction, the possible mechanisms of their genetic diversity were investigated. The genome of C. jejuni contain high fraction of intraspecies-originated abnormal composition fragments (ACFs), whereas that of the other Campylobacter members have relatively high fraction of intragenus-originated ACFs. Moreover, ancient bacteriophage integration in some organisms contributed to their genomic evolution.
Materials and Methods
Collection of Campylobacter Genomic Data
Thirty-four Campylobacter genomes were downloaded from the NCBI website. Among these genomes, 14 were complete, while the rest were drafts (Table S1). Several control genomes including Campylobacterales bacterium GD 1 (draft, GI: 254456697); Methanothermobacter thermautotrophicus str. Delta H (complete, GI: 15678031), which is an archaeal genome; and Escherichia coli str. K12 substr. DH108 (complete, GI: 169887498) were also collected from NCBI.
Genome-wide Alignments
NUCleotide MUMmer (NUCmer) was used to assess the genome-wide alignments according to reference [34]. To ensure that all contigs were aligned, all maximal matches were used as alignment anchors (-maxmatch). The delta encoded alignment files produced by NUCmer were filtered using the delta-filter utility, leaving only the alignments that form the longest consistent set (-q –r –l 200). A summary of all the alignments produced by NUCmer was generated using the show-coords utility. Afterward, we developed in-house Perl scripts to calculate the pairwise-aligned genome percentages for each reference.
Gene de novo Prediction
Gene de novo prediction was conducted for some draft genomes such as C. jejuni CG8486, which have no protein coding gene information in NCBI. Considering the consistency of the gene prediction among all collected genomes, protein-coding genes for all listed genomes were de novo-predicted or re-predicted using Glimmer 3.02 with default parameters [35]. To facilitate subsequent analysis, tRNA-coding genes were re-predicted by tRNAscan-SE (version 1.23) [36], while rRNA-coding genes were re-predicted by RNAmmer (version 1.2) [37]. Only small subunit ribosomal RNA (SSU rRNA) sequences with RNAmmer score above 1700 were used for further analysis.
SSU rRNA Tree for the Campylobacter Genus
Among the several SSU rRNA sequences in specific Campylobacter genomes having an RNAmmer score above 1700, only the first SSU rRNA sequence was selected for the phylogenetic tree analysis. The size of all selected sequences was approximately 1500 bp, except for the representative sequence from Campylobacter curvus 525.92, which has an intervening sequence having a length of approximately 200 bp. After trimming the intervening sequence, all pooled sequences were subjected to multiple alignments using the software pyNAST [38]. The subprogram phyml of TreeBeST (http://treesoft.sourceforge.net/treebest.shtml) was used to construct a phylogenetic tree with default parameters. Non-parametric bootstrap analysis with a thousand re-sampling was conducted to obtain the bootstrap values for all branches.
Protein Family Construction and Pan-genome Analysis
All protein coding gene sequences were translated into protein sequences according to Codon Table 11 from NCBI. The resultant protein sequences were clustered into gene families using BLASTclust. Protein or gene sequences were automatically and systematically clustered according to the “fifty-fifty” rule based on pairwise matches. For example, two sequences were clustered in the same family if the best local alignment between these clusters covers at least 50% of the length of both sequences and contains at least 50% identities. The in-house Perl scripts were then used to analyze the gene family distribution among the strains of Campylobacter genus. The protein sequences of the repeat genes of an organism and the core genes of Campylobacter genus plus the total representative protein sequences were extracted to conduct alignments against the Cluster of Orthologous Groups (COG) database [39] using the local alignment tool BLASTP. Only the alignments with e-value ≥1e-5 and coverage ≥50% of both query and subject sequences were used for COG annotation. Functional enrichment difference between repeat genes and total genes, as well as between core genes and total genes were evaluated by chi-square test.
All protein sequences of the entire core genes were extracted from the results. When one organism had at least two genes in one gene family, only orthologous protein sequences with the best hit for homologous genes in other organisms were chosen to construct the genome-wide phylogenetic tree. After manual adjustment to search for conserved regions, all orthologous protein sequences for each core gene family, which refers to the family with at least one gene from each of the Campylobacter strain, were pooled for independent multiple alignments using the MUSCLE software [40]. Conserved sequences shared by all organisms were concatenated to construct a neighbor-joining phylogenetic tree using the TreeBeST software with default parameters. Bootstrap values of all branches were obtained after 1000 times of resampling. Gene families shared by four close species, namely, C.jejuni, C. coli, C. lari, and C. upsaliensis, were obtained and represented in Venn diagram.
Microevolution Analysis of C. jejuni
To identify further the phylogenetic relationships between different strains of C. jejuni, single nucleotide polymorphisms(SNPs) were used for phylogenetic analysis. Repeat genome fragments of C. jejuni NCTC11168 were previously identified using a combinatory approach based on RepeatMasker and self-Blast [41]. Each of the other strains was then aligned against the reference C. jejuni NCTC 11168 using NUCmer [34]. The delta-encoded alignment files generated by NUCmer were filtered using the delta-filter utility, and raw SNPs were detected using the show-snps utility. The program BLAT [42] was used to map raw SNP calls against their genomes for verification, while SNPs localized in repetitive genomic fragments were filtered out. The filtered SNPs were concatenated and aligned by ClustalW 2.0 [43]. Phylogenetic relationships based on the aligned sequences were inferred using neighbor-joining methodologies implemented in TreeBeST (default parameters) with 1000 bootstrap pseudoreplicates.
Genome Composition Analysis
Genomic composition, namely, tetranucleotide usage patterns (TUP), was analyzed for all the 34 collected Campylobacter genomes and the outgroup C. bacterium GD 1 genome. The overlapping oligonucleotides (length, 4) of the genomes were quantified as observed tetranucleotide frequencies by shifting the window at a step length of one nucleotide. The corresponding expected values were calculated according to a previous method [44]. The differences between the observed and expected values were transformed into z-values for each tetranucleotide [45]. The similarity between genomes was assessed by calculating the Pearson correlation coefficient for the 256 tetranucleotide-derived z-values. Hierarchical cluster analysis was conducted using Ward’s method.
Identification of ACFs
To exclude the similarity between genomes of the Campylobacter genus, a relatively distant genome from an archaea, M. thermautotrophicus str. Delta H, was used as control. All genomes were first split into contigs at positions with ambiguous nucleotides (“N”). The resultant contigs were further split into fragments (S) with length of 500 bp and 250 bp overlap. ACFs were then assessed by calculating the fold as previously described [46]: Fold = P(S:Gi)/P(S:G), where P is the probability calculated by the naïve Bayesian classifier (in this study, motif length is 4); G i is training genome; G is the testing genome, from which sequence S is derived; and S is a fragment with length of 500 bp for investigating abnormal composition.
A cutoff for identification of the ACFs was determined through comparison with M. thermautotrophicus. Any fragment above the cutoff was considered as an ACF (7, logarithmically transformed). To detect ancient viral or phage element integration into the Campylobacter genomes, at least two consecutive ACFs were concatenated into genomic regions and subjected to comparison with virus or phage homologous sequences. In addition, the gene types of the genomic regions were also analyzed.
Identification of Viral and Phage Homologous Protein-coding Genes
All Campylobacter proteins were aligned against the NR-virus database using BLASTP. The NR-virus database was in-house constructed by extracting all virus sequences from NCBI. All hits with e-value ≤1e-5 and cover at least 50% of the length of viral sequences were used, and their corresponding query sequences were considered as virus homologous sequences.
The virus database was not exclusively developed for bacterial analysis and may lack some useful information. Phage DNA in Campylobacter is better defined. We found that numerous sequences that were previously annotated as phage-related sequences according to published genomes were not included in the set of our virus homologous sequences. To identify the largest possible bacteriophage sequences in Campylobacter strains, phage homologous sequence identification was conducted. All phage-related Campylobacter proteins were obtained from NCBI and pooled together as phage database. A total of 285 phage-related sequences from the NCBI database were included in the phage database. All the predicted or re-predicted protein-coding sequences were then aligned against the phage database, following the same procedure as that for the detection of virus homologous proteins.
Results and Discussion
Genome-wide Alignments of Campylobacter Genomes
To explore the diversity of Campylobacter genomes, we conducted genome-wide alignments for all collected Campylobacter genomes to provide an overview of the percentage of the conserved genomic regions between pairwise genomes. As shown in Figure 1 , the Campylobacter genus exhibited genomic diversity at genus level despite the absence of conserved genomic fragments between genomes, such as Campylobacter rectus RM3267 and C. coli JV20 (for pairwise identity values see Table S2). Aside from the obvious genus differences, we also detected slight genomic differences between Campylobacters at the species level. For instance, the genomic similarity between C. fetus subsp. fetus 82–40 and C. fetus venerealis Azul-94 is 0.7 (calculated using the total length of C. fetus subsp. venerealis Azul-94 as the denominator) or 0.81 (calculated using the total length of C. fetus 82–40 as the denominator), similar to that of a previous study [32]. The diversity among the Campylobacter strains occurs not only at the genome level but also at the protein level, which was shown by a previous report using alignments of protein coding sequences (CDS) [47]. Therefore, some mutation events such as horizontal gene transfer (HGT) occurred after speciation from their ancestor.
Figure 1. Genome-wide alignment heatmap of the genus Campylobacter.

This figure represents the pairwise genome-wide alignment similarity of the 34 genomes in this study. The similarity ratios were calculated using the total length of the genome fragments preserved between any two organisms divided by the length of the total genomes of the organism in row. The ratio is used as a basis for color intensity. Table S1 displays the absolute ratios in detail.
Phylogenetic Analysis Based on SSU rRNA Sequences
All draft genomes such as that of C. jejuni subsp. jejuni strains CG8486, CG8421, and DFVF1099 have SSU rRNA sequences that meet the SSU rRNA prediction criteria (Table S1). The length of all predicted SSU rRNA sequences are approximately 1500 bp. However, the sequence from C. curvus 525.92 has a length of 1701 bp because of the intervening sequences with length of approximately 200 bp, similar to that in a documented report [48]. Therefore, the intervening sequence of the C. curvus 525.92 representative SSU rRNA was trimmed before being pooled together with other representative SSU rRNA sequences to construct the phylogenetic tree.
The phylogeny based on SSU rRNA (rRNA tree) shows that C. coli is hardly distinguishable from C. jejuni, confirming that they are phenotypically and genotypically similar ( Figure 2 ). This result is consistent with that by a previous study, in which the phylogeny was inferred from rpoB or SSU rRNA sequences [47]. However, C. jejuni is clearly different from C. coli in the groEL-based tree [49]. Furthermore, the Campylobacter genus can split into two main lineages, i.e., the jejuni lineage, which includes C. jejuni, C. coli, C. lari, and C. upsaliensis; and the non-jejuni lineage, which includes C. fetus, Campylobacter concisus, Campylobacter gracilis, Campylobacter hominis, C. curvus, C. rectus, and Campylobacter showae. Despite the discrepancies among different phylogenetic trees (rRNA tree, core gene tree in this study, and previously documented trees [26], [47], [49]), C. jejuni, C. coli, C. lari, and C. upsaliensis are always in a common cluster using different sequences or methods, suggesting that these four species are genetically close and the global topology of the jejuni lineage is always essentially identical, whereas that of the non-jejuni lineage is more or less diverse. In addition, the jejuni lineage is not limited to these four species; some other species, such as Campylobacter insulaenigrae, Campylobacter helveticus [47], and Campylobacter hyoilei [49], may also be included in this lineage.
Figure 2. Maximum likelihood phylogenetic tree of Campylobacters based on small subunit ribosomal RNA (SSU rRNA) sequences.

The tree is rooted with SSU rRNA sequence of Campylobacterales bacterium GD 1, a member of the common order of Campylobacterales considered as a relative close outgroup. Absolute bootstrap values of 1000 simulations are shown beside the major branches to indicate the stability of the branching. Only bootstrap values greater than 500 are shown. The scale bar represents the 0.06 nucleic acid substitutions per site.
Pan-genome Analysis
Protein-coding genes were predicted using GLIMMER 3.02 [35]. Collectively, 64686 genes were predicted for all the 34 collected genomes. Among these genes, C. fetus subsp. venerealis Azul-94 harbors the most number of protein-coding genes with 2447, whereas C. lari RM2100 harbors the fewest protein-coding genes with 1545 (Table S3). A total of 13167 gene families were obtained after clustering using BLASTclust according to the “fifty-fifty” rule mentioned earlier. Among the 13167 gene families, 348 (2.64%) are core gene families at the genus level, while the other 12819 (97.36%) are dispensable or unique gene families. Core gene families comprise 12181 protein coding genes, accounting for 18.83% of the total genes. By contrast, the dispensable or unique gene families comprise 52505 genes, accounting for approximately 81.17% of the total genes.
The protein families of each Campylobacter species are shown in Figure 3 . Genes from the same organism with more than one gene in a gene family were defined as repeat genes. All Campylobacter species have fewer repeat protein-coding genes than E. coli str. K12 substr. DH108; hence, Campylobacter genomes are more compact than that of E. coli. In addition, the number of gene families in Campylobacter species is smaller than that in E. coli; some species even comprise approximately half of E. coli, indicating that E. coli is more complex than the Campylobacter genus. Strains of the Campylobacter genus have 27 to 86 genes families with repeat genes (Table S4), whereas E. coli harbors 242 gene families with repeat genes. The average copy number of repeat genes in the repeat gene families range from 2.16 to 2.80, indicating that the majority of the copies of the repeat genes in the gene families of Campylobacter genus are two. Similarly, E. coli has 2.36 average copies of repeat genes. The repeat genes may have indispensable and key roles in Campylobacter strains. For example, flagellum-related genes are repeated in C. jejuni ICDCCJ07001 because flagellum is very important for its mobility, adherence, and pathogenesis. Repeat genes are also utilized for intra-genomic homologous rearrangements [50]. We also determined whether COG is enriched in repeat genes and found that no functional enrichment occurred compared with total genes (p = 0.059). Nevertheless, core genes showed significantly functional enrichment (p = 2.2e-16), indicating enrichment for translation, ribosomal structure, and biogenesis function (COG_J) ( Figure 4 ).
Figure 3. Gene families of genus Campylobacter.

Escherichia coli (E. coli) str. K12 substr. DH108 (GenBank accession CP000948) was used as a control. The values above each bar (in percent) were calculated as the average number of gene families divided by the average number of total genes.
Figure 4. Spider charts of COG enrichment.
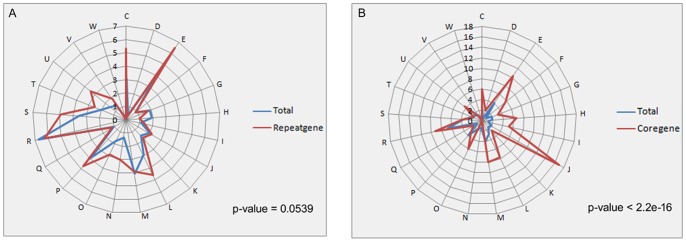
Chi-square tests were applied to test their differences. The blue line shows the COG enrichment of the total genes, whereas the red lines indicate that of repeat genes (A) and core genes (B). A. COG enrichment of the repeat genes compared with that of the total genes. Repeat genes have no significant differences compared with the total genes (p = 0.0539). B. COG enrichment of core genes compared with that of total genes. COG enrichment of core genes and that of total genes (p<2.2e-16) was significantly different between COG enrichment.
Given that evolutionary constraints are multidimensional [51], analysis of a single gene is insufficient to fully understand the phylogenetic relationship of Campylobacters. Single gene-based phylogenetic analysis may not reflect their natural relationship because HGT commonly exists in prokaryotes [52], [53]. Thus, we used a genome-wide phylogenetic analysis to definitively determine their relationship. Genome-scale core protein sequence-based method was applied to construct phylogenetic trees for all the 34 collected genomes. Only the orthologous protein sequence with the best hit for homologous genes was used to construct the trees if an organism had multiple copies in one protein family. The conserved regions of all the core proteins were concatenated to construct a phylogenetic tree. A total of 349 core proteins were composed of 84457 amino acid residues (including gaps in some organisms), which accounted for 11.88% (C. rectus RM3267) to 20.22% (C. fetus subsp. venerealis Azul-94) of their total proteins. The neighbor-joining phylogenetic tree based on the conserved region of the core protein sequences (core gene tree) showed similar two lineage clades with the phylogenetic analysis based on SSU rRNA sequences (rRNA tree). However, the intra-lineage relationship of the core gene tree was somewhat slightly inconsistent with that of the rRNA tree ( Figures 2 and 5 ). Thus, further study is still needed to identify the natural relationship of intra-lineage organisms. Based on the core gene tree, the two strains of C. coli were separated from C. jejuni organisms. Therefore, the phylogenetic relationship reflected by the core gene tree was more reliable than that reflected by the rRNA tree. The core gene tree can be used as species tree for all of the collected Campylobacter species in this study.
Figure 5. Neighbor-joining phylogenetic analysis of Campylobacters based on their core genes.

Only the conserved regions of the orthologous core protein sequences were used to construct this genome-wide tree. Bootstrap values (in percent) of 1000 simulations are indicated at all branches. Bar represents 0.06 amino acid substitutions per site.
We speculated that a large fraction of gene families may be shared by C. jejuni, C. coli, C. lari, and C. upsaliensis because they were constantly embraced in the jejuni lineage. As expected, 1074 gene families were shared by these four species, accounting for 26.9%, 54.13%, 71.74%, and 52.67% of their total gene families, respectively ( Figure 6 ), indicating that these four species possessed close evolutionary relationship. For C. jejuni, the relatively low fraction of gene families entirely shared by these four species was partially due to the large amount of pan-genome resulting from too many strains (up to 21 strains). The fractions of the unique gene families of C. jejuni, C. coli, C. lari, and C. upsaliensis were 55.28%, 15.63%, 14.96%, and 29.72% respectively, indicating that C. jejuni had a large amount of auxiliary genes. For each species, the majority of the gene families were shared by the other three species ( Figure 6 ), confirming the close relationship of these four species. We did not perform a cross-relationship analysis of gene families for the members in non-jejuni lineage because of its very diverse lineage.
Figure 6. Four-way Venn diagram for gene families of four Campylobacters.

C. jejuni, C. coli, C. lari, and C.upsaliensis from jejuni lineage (based on core gene tree) are shown. The absolute numbers of the core and the dispensable and unique gene families of the four species are all shown.
Microevolution Determination for C. jejuni
Although the interspecies phylogenetic relationship can be obtained through core gene-based methodology, the precise intraspecies relationship of C. jejuni needs further analysis. Both the core gene tree and the rRNA tree had low resolution for the microevolution (intraspecies phylogenetic relationship) of C. jejuni because of the high similarity between strains of C. jejuni at the genus level. Therefore, SNP information was applied to identify their microevolution relationships. To our knowledge, this study is the first to show the microevolution of C. jejuni. A total of 37734 filtered SNPs were obtained, accounting for approximately 0.02% of each genome. Phylogenetic analysis was conducted based on the concatenated SNP sequences. Figure 7 shows the microevolution relationship of C. jejuni. Almost all of the branches had 100% bootstrap support values, indicating that the SNP sequence-based phylogenetic tree was robust and may reflect the natural relationship of C. jejuni organisms. As expected, the resolution of the SNP-based tree within the C. jejuni was much higher than those of both the core gene tree and rRNA tree. The combination with the core gene tree may determine a comprehensive relationship of all the collected Campylobacter genomes. Performing other analyses, such as later gene transfers, is useful when using these two phylogenies as combined control.
Figure 7. Neighbor-joining phylogeny of C. jejuni based on SNP data.

The tree is rooted to the outgroup Campylobacterales bacterium GD 1. The node support (in percent) after 1000 bootstrap replications is indicated. The scale bar represents 0.06 nucleic acid substitutions per site.
Identification of ACFs
Campylobacters are genetically diverse according to the aforementioned comprehensive genomic analysis and characterization of all the collected genomes within the genus Campylobacter. However, a question on how Campylobacters evolved to such diversity is raised. To uncover some clues to this question, we focused on the ACF analysis. The genome-scale TUP of all 34 Campylobacter genomes was initially computed, and their relationship was determined ( Figure 8 ) to identify the genomic fragments with abnormal composition. An alternative figure was also provided to clearly show their relationship based on the TUP-derived z-values (Figure S1, TUP-derived tree) because the genome composition showed somewhat evolutionary implications [54]. The topologies of the TUP-derived tree were significantly different from those of the core gene tree and the rRNA tree (Figures S1, 2, and 5). Considering that the core gene tree is an inference tree, the most possible explanation for their incongruities was that all Campylobacter spp.were classified into two lineages, similar to that of the core gene tree or rRNA tree. However, C. fetus and C. hominis, which were included in the non-jejuni lineage in the core gene tree or rRNA tree, were transformed from non-jejuni lineage to jejuni lineage. The reason for this phenomenon is not clearly elucidated and is awaiting further investigation. One possible reason is that TUP evolved more rapidly than SSU rRNA and other core genes in these two species [54]. This hypothesis was partially confirmed based on our results ( Figure 9 ), in which the two C. fetus genomes possessed significantly different percentages of ACFs, inferring a rapid TUP evolution. In addition, some minor intralineage discrepancies were observed between TUP-derived tree and core gene tree or rRNA tree (Figures S1, 2, and 5) because of the different evolutionary rates and later gene transfers.
Figure 8. Tetranucleotide usage pattern (TUP) heatmap of the genus Campylobacter.

The rows and columns stand for strains. Both rows and samples are clustered using ward linkage hierarchical clustering to highlight strain groups with similar genomic signature. The tetranucleotide-derived z-values were predicted using maximum order Markov chain model. Pearson correlations for z-scores were used for clustering and as a basis for color intensity. TUPs of Campylobacterales bacterium GD 1 was used as a control.
Figure 9. Abnormal composition fragment percentages of Campylobacter strains under different folds.

M. thermautotrophicus str. Delta H (NC_000916.1) was used as control. Fold values (see Methods) were logarithmically transformed. The red dashed line represents the cutoff set in this study for the identified abnormal composition fragments.
Clues for Genomic Diversification
After identifying the global genomic composition features of Campylobacter genomes, we subsequently used the modified Naïve Bayesian classifier [46] to search for abnormal genomic fragments. We improved this method using two main aspects: (1) We set a more rigorous cutoff to guarantee that all predicted ACFs under this cutoff were absolutely abnormal enough for HGT prediction; (2) The ACFs were not directly called as HGTs because not all ACFs were HGTs and only certain ACFs that satisfy other additional requirements can be called HGTs. The prediction is very reliable and useful when using the improved Naïve Bayesian classifier.
In a previous study [46], the cutoff for the identification of ACF, called HGTs, was set to 0 (logarithmically transformed). By contrast, we found that almost 100% of the entire genomes for some strains, such as C. jejuni subsp. jejuni 84–25, had an abnormal composition if the cutoff was set to 0 ( Figure 9 ), which seemed impossible. The genome of an archaea M. thermautotrophicus str. Delta H [55] was also included in this study to obtain a more reasonable cutoff ( Figure 9 ). The line representing M. thermautotrophicus str. Delta H was constantly horizontal and intersected the lines of the Campylobacters genus at 7 (logarithmically transformed), which can be set for the identification threshold of the ACFs. Previous studies reported that 18% and 24% of the genes may be horizontally transferred in E. coli [56] and Thermotoga maritime [57], respectively. Under the cutoff of 7, a collection of fragments (500 bp) was identified as ACFs, accounting for 1.15% to 13.21% of their genomes ( Figure 10 ), comparable to previous studies [57], [58]. These results suggested that our selective threshold was reasonable and rigorous, and the resultant ACFs were reliable. In addition, the most possible origins of these fragments were also analyzed. As shown in Figure 10 , some species, for example C. rectus, which only had one strain in this study, had no intraspecies origin. C. jejuni harbored high fraction of intraspecies-derived ACFs, whereas other Campylobacter strains had high fraction of intragenus-derived ACFs, which may be imported from other species at the same genus other than the same species. Therefore, high intraspecies recombination or HGT events may mainly occur in C. jejuni, whereas high intragenus recombination or HGT events may mainly occur in other Campylobacter strains. To some extent, this phenomenon can explain why Campylobacters were genetically and phenotypically diverse.
Figure 10. Source of abnormal composition fragments.

The values (in percentage) above the bars were calculated as the total abnormal composition fragments divided by the total fragments of the genomes. C.u. = Campylobacter upsaliensis; C.j. = Campylobacter jejuni; C.g. = Campylobacter gracilis; C.j. ICDCCJ = Campylobacter jejuni ICDCCJ07001; C.j. doylei = Campylobacter jejuni doylei 269.97. Strain names of all the other species with only one strain were neglected for simplicity. “intraspecies” stands for abnormal composition fragments coming from other strains of the same species, intragenus means abnormal composition fragments derived from other species of the same genus and intergenus represents abnormal composition fragments originated from other genus.
De novo re-prediction genes including rRNA, tRNA, and protein-coding genes were formerly carried out. Consecutive fragments, which possessed at least two fragments with fold larger than 7 (logarithmically transformed), were concatenated into genomic regions (GRs). The gene types of GRs were subsequently analyzed (summarized in Table S4). Most of the rRNA- and tRNA-coding genes considered as housekeeping genes had abnormal compositions, indicating that the abnormal composition genes were horizontally transferred or acted as housekeeping genes through vertical inheritance during the evolutionary process. Therefore, we preferred the ACFs than the horizontally transferred genes in this study [46]. The results also showed that some protein-coding genes were also housekeeping genes rather than horizontally transferred genes (Table S3). A total of 3783 gene families had abnormal composition genes. In this study, the gene families residing equal to or larger than 90% of Campylobacter genomes were defined as housekeeping gene families. The genes from the housekeeping gene families were defined as Campylobacter housekeeping genes. A total of 3738 protein-coding gene families were considered to have abnormal fragments according to our modified method. Among these families, 669 were housekeeping gene families, accounting for 17.68% (669/3783) of the total gene families with abnormal composition genes. Other 82.32% (3114/3783) of gene families may be horizontally transferred genes (Table S5). Table S4 shows the percentage and absolute number of the housekeeping protein-coding genes and the horizontally transferred protein-coding genes. Housekeeping genes were not horizontally transferred genes, but possibly transferred to the common ancestor of all Campylobacters. Therefore, some housekeeping genes may be transferred a long time ago, at least before Campylobacter species differentiation.
Campylobacter genomes were reported to harbor phage-like genomic fragments [30] or undergo recombination under bacteriophage predation [59]. Therefore, virus or phage may have an important role in Campylobacter genome diversification. Viral and phage homoplasies of Campylobacters were thus identified. We initially thought that almost all virus-related and phage-related protein-coding genes should be included in the abnormal composition gene list because these homoplasies were foreign. Unexpectedly, for almost all the genomes, the vast majority of the virus-related and phage-related protein-coding genes were not included in the abnormal composition gene list; some genomes had no virus- or phage-related genes with abnormal composition (Table S6). Intergenomic differences were generally higher than intragenomic differences [60], and nearly all the native GRs within a certain genome had similar but organism-specific genomic composition. Consequently, GRs recently introduced from other organisms showed unusual sequence characteristics and can be distinguished from the recipient genome. However, imported GRs will be ameliorated to reflect the genomic characteristics of the recipient genome other than the donor genome after a long period of mutation because the introgressed genes were subjected to the same mutational processes affecting all genes in the recipient genome [61]. Viral or phage genes with similar genomic composition to their genomes were suggested to have transferred into the host genomes of genus Campylobacter a long time ago, indicating that an ancient viral integration occurred in the Campylobacter genomes. However, other genomes still recently possess abilities to acquire viral or phage genomic fragments (Table S6). For example, in C. concisus 13826, the majority of the phage-related genes may have been recently transferred into its genome because 76.47% of the phage-related genes were included in the abnormal composition gene list. This result implied that some genomes still have viral or phage integration abilities. This phenomenon explains, to some extent, why Campylobacter genomes are diverse. Therefore, we concluded that some Campylobacter genomes had undergone certain ancient vital integration during their evolution.
Supporting Information
Clustering result based on tetranucleotide usage patterns of the genus Campylobacter . Bar represents the clustering height. Campylobacterales bacterium GD 1 (termed Campylobacterales in picture) was used as the outgroup. Pearson correlations for tetranucleotide-derived z-values were used for clustering the genomes using ward linkage hierarchical algorithm.
(TIF)
Campylobacter genomes examined. Thirty-four Campylobacter genomes collected for analysis were listed with their genomic information including GenBank GI number, mark for genome in this study, contig number, total length, and SSU rRNA number.
(XLSX)
Genome-wide alignment matrix of genus Campylobacter . Genome-wide alignment similarity ratio in the table cells was computed as the length of genome fragments preserved between any two organisms against the length of total genomes of the organism in the row.
(XLSX)
Gene classification with abnormal composition. Genes with abnormal composition were classified into groups of different gene types including rRNA, tRNA, and protein-coding genes (coding sequences, CDS). The CDS was further divided into housekeeping CDS and HGT CDS. The gene number and the percentage to total gene number were listed for these genes.
(XLSX)
Gene families containing duplicated genes in genus Campylobacter . For each Campylobacter organism, the number of gene families containing duplicated genes and the total number of duplicated genes, together with average gene copies, (average gene number of all its gene families containing repeat genes) are shown.
(XLSX)
Gene family information of abnormal composition genes. The numbers in table cells represent the number of homologous genes in the corresponding genome. The Gene ID of the first column had the longest sequence in the gene family.
(XLSX)
Quantitative composition of Campylobacter virus-related and phage-related genes containing abnormal composition fragments. The percentage is equal to the number of virus- or phage-related genes with abnormal composition divided by the number of the total virus- or phage-related genes.
(XLSX)
Funding Statement
This work was supported by The Natural Science Foundation of Hunan Province, China (10JJ2012). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Gilbreath JJ, Cody WL, Merrell DS, Hendrixson DR (2011) Change is good: variations in common biological mechanisms in the epsilonproteobacterial genera Campylobacter and Helicobacter. Microbiol Mol Biol Rev 75: 84–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sebald M, Veron M (1963) [Base DNA Content and Classification of Vibrios]. Ann Inst Pasteur (Paris) 105: 897–910. [PubMed] [Google Scholar]
- 3.On SL (2001) Taxonomy of Campylobacter, Arcobacter, Helicobacter and related bacteria: current status, future prospects and immediate concerns. Symp Ser Soc Appl Microbiol: 1S-15S. [DOI] [PubMed]
- 4. Gorkiewicz G, Feierl G, Schober C, Dieber F, Kofer J, et al. (2003) Species-specific identification of campylobacters by partial 16S rRNA gene sequencing. J Clin Microbiol 41: 2537–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Moore JE, Madden RH (1998) Occurrence of thermophilic Campylobacter spp. in porcine liver in Northern Ireland. J Food Prot 61: 409–413. [DOI] [PubMed] [Google Scholar]
- 6. Kramer JM, Frost JA, Bolton FJ, Wareing DR (2000) Campylobacter contamination of raw meat and poultry at retail sale: identification of multiple types and comparison with isolates from human infection. J Food Prot 63: 1654–1659. [DOI] [PubMed] [Google Scholar]
- 7. Quinones B, Guilhabert MR, Miller WG, Mandrell RE, Lastovica AJ, et al. (2008) Comparative genomic analysis of clinical strains of Campylobacter jejuni from South Africa. PLoS One 3: e2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bourke B, Chan VL, Sherman P (1998) Campylobacter upsaliensis: waiting in the wings. Clin Microbiol Rev 11: 440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Atanassova V, Ring C (1999) Prevalence of Campylobacter spp. in poultry and poultry meat in Germany. Int J Food Microbiol 51: 187–190. [DOI] [PubMed] [Google Scholar]
- 10. Tresierra-Ayala A, Bendayan ME, Bernuy A, Pereyra G, Fernandez H (1994) Chicken as potential contamination source of Campylobacter lari in Iquitos, Peru. Rev Inst Med Trop Sao Paulo 36: 497–499. [DOI] [PubMed] [Google Scholar]
- 11. Miller WG, Englen MD, Kathariou S, Wesley IV, Wang G, et al. (2006) Identification of host-associated alleles by multilocus sequence typing of Campylobacter coli strains from food animals. Microbiology 152: 245–255. [DOI] [PubMed] [Google Scholar]
- 12. Gibson JR, Owen RJ (1998) Campylobacter infections : species identification and typing. Methods Mol Med 15: 407–418. [DOI] [PubMed] [Google Scholar]
- 13. Miljkovic-Selimovic B, Lavrnic D, Moric O, Ng LK, Price L, et al. (2010) Enteritis caused by Campylobacter jejuni followed by acute motor axonal neuropathy: a case report. J Med Case Rep 4: 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bullman S, O’Leary J, Corcoran D, Sleator RD, Lucey B (2012) Molecular-based detection of non-culturable and emerging campylobacteria in patients presenting with gastroenteritis. Epidemiol Infect 140: 684–688. [DOI] [PubMed] [Google Scholar]
- 15. Abbass K, Emig M, Bernstein JM (2011) Sepsis and pericarditis caused by Campylobacter fetus: case report and literature review. J Ark Med Soc 108: 88–89. [PubMed] [Google Scholar]
- 16. Uzoigwe C (2005) Campylobacter infections of the pericardium and myocardium. Clin Microbiol Infect 11: 253–255. [DOI] [PubMed] [Google Scholar]
- 17. Kuwabara S (2004) Guillain-Barre syndrome: epidemiology, pathophysiology and management. Drugs 64: 597–610. [DOI] [PubMed] [Google Scholar]
- 18.Heikema AP, Jacobs BC, Horst-Kreft D, Huizinga R, Kuijf ML, et al.. (2012) Siglec-7 specifically recognizes Campylobacter jejuni strains associated with oculomotor weakness in Guillain-Barre syndrome and Miller Fisher syndrome. Clin Microbiol Infect. [DOI] [PubMed]
- 19. Islam Z, Gilbert M, Mohammad QD, Klaij K, Li J, et al. (2012) Guillain-Barre syndrome-related Campylobacter jejuni in Bangladesh: ganglioside mimicry and cross-reactive antibodies. PLoS One 7: e43976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sahin O, Plummer PJ, Jordan DM, Sulaj K, Pereira S, et al. (2008) Emergence of a tetracycline-resistant Campylobacter jejuni clone associated with outbreaks of ovine abortion in the United States. J Clin Microbiol 46: 1663–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bulgin MS, Ward AC, Sriranganathan N, Saras P (1984) Abortion in the dog due to Campylobacter species. Am J Vet Res 45: 555–556. [PubMed] [Google Scholar]
- 22. Welsh RD (1984) Campylobacter jejuni abortion in a heifer. J Am Vet Med Assoc 185: 549–551. [PubMed] [Google Scholar]
- 23. Mshelia GD, Amin JD, Egwu GO, Woldehiwet Z, Murray RD (2012) The prevalence of bovine venereal campylobacteriosis in cattle herds in the Lake Chad basin of Nigeria. Trop Anim Health Prod 44: 1487–1489. [DOI] [PubMed] [Google Scholar]
- 24. Vasquez LA, Ball L, Bennett BW, Rupp GP, Ellis R, et al. (1983) Bovine genital campylobacteriosis (vibriosis): vaccination of experimentally infected bulls. Am J Vet Res 44: 1553–1557. [PubMed] [Google Scholar]
- 25. Zhang M, He L, Li Q, Sun H, Gu Y, et al. (2010) Genomic characterization of the Guillain-Barre syndrome-associated Campylobacter jejuni ICDCCJ07001 Isolate. PLoS One 5: e15060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Friis C, Wassenaar TM, Javed MA, Snipen L, Lagesen K, et al. (2010) Genomic characterization of Campylobacter jejuni strain M1. PLoS One 5: e12253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pearson BM, Gaskin DJ, Segers RP, Wells JM, Nuijten PJ, et al. (2007) The complete genome sequence of Campylobacter jejuni strain 81116 (NCTC11828). J Bacteriol 189: 8402–8403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Russell RG, Blaser MJ, Sarmiento JI, Fox J (1989) Experimental Campylobacter jejuni infection in Macaca nemestrina. Infect Immun 57: 1438–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Parkhill J, Wren BW, Mungall K, Ketley JM, Churcher C, et al. (2000) The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature 403: 665–668. [DOI] [PubMed] [Google Scholar]
- 30. Fouts DE, Mongodin EF, Mandrell RE, Miller WG, Rasko DA, et al. (2005) Major structural differences and novel potential virulence mechanisms from the genomes of multiple campylobacter species. PLoS Biol 3: e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hepworth PJ, Ashelford KE, Hinds J, Gould KA, Witney AA, et al. (2011) Genomic variations define divergence of water/wildlife-associated Campylobacter jejuni niche specialists from common clonal complexes. Environ Microbiol 13: 1549–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Moolhuijzen PM, Lew-Tabor AE, Wlodek BM, Aguero FG, Comerci DJ, et al. (2009) Genomic analysis of Campylobacter fetus subspecies: identification of candidate virulence determinants and diagnostic assay targets. BMC Microbiol 9: 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lefebure T, Bitar PD, Suzuki H, Stanhope MJ (2010) Evolutionary dynamics of complete Campylobacter pan-genomes and the bacterial species concept. Genome Biol Evol 2: 646–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Delcher AL, Phillippy A, Carlton J, Salzberg SL (2002) Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res 30: 2478–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Delcher AL, Bratke KA, Powers EC, Salzberg SL (2007) Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23: 673–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schattner P, Brooks AN, Lowe TM (2005) The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res 33: W686–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, et al. (2007) RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35: 3100–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, et al. (2010) PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26: 266–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, et al. (2003) The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kent WJ (2002) BLAT–the BLAST-like alignment tool. Genome Res 12: 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, et al. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948. [DOI] [PubMed] [Google Scholar]
- 44. Schbath S, Prum B, de Turckheim E (1995) Exceptional motifs in different Markov chain models for a statistical analysis of DNA sequences. J Comput Biol 2: 417–437. [DOI] [PubMed] [Google Scholar]
- 45. Teeling H, Meyerdierks A, Bauer M, Amann R, Glockner FO (2004) Application of tetranucleotide frequencies for the assignment of genomic fragments. Environ Microbiol 6: 938–947. [DOI] [PubMed] [Google Scholar]
- 46. Sandberg R, Winberg G, Branden CI, Kaske A, Ernberg I, et al. (2001) Capturing whole-genome characteristics in short sequences using a naive Bayesian classifier. Genome Res 11: 1404–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Korczak BM, Stieber R, Emler S, Burnens AP, Frey J, et al. (2006) Genetic relatedness within the genus Campylobacter inferred from rpoB sequences. Int J Syst Evol Microbiol 56: 937–945. [DOI] [PubMed] [Google Scholar]
- 48. Etoh Y, Yamamoto A, Goto N (1998) Intervening sequences in 16S rRNA genes of Campylobacter sp.: diversity of nucleotide sequences and uniformity of location. Microbiol Immunol 42: 241–243. [DOI] [PubMed] [Google Scholar]
- 49. Karenlampi RI, Tolvanen TP, Hanninen ML (2004) Phylogenetic analysis and PCR-restriction fragment length polymorphism identification of Campylobacter species based on partial groEL gene sequences. J Clin Microbiol 42: 5731–5738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wassenaar TM, Fry BN, van der Zeijst BA (1995) Variation of the flagellin gene locus of Campylobacter jejuni by recombination and horizontal gene transfer. Microbiology 141 (Pt 1): 95–101. [DOI] [PubMed] [Google Scholar]
- 51. Koonin EV, Aravind L, Kondrashov AS (2000) The impact of comparative genomics on our understanding of evolution. Cell 101: 573–576. [DOI] [PubMed] [Google Scholar]
- 52. Ko KS, Lee HK, Park MY, Lee KH, Yun YJ, et al. (2002) Application of RNA polymerase beta-subunit gene (rpoB) sequences for the molecular differentiation of Legionella species. J Clin Microbiol 40: 2653–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Acinas SG, Marcelino LA, Klepac-Ceraj V, Polz MF (2004) Divergence and redundancy of 16S rRNA sequences in genomes with multiple rrn operons. J Bacteriol 186: 2629–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pride DT, Meinersmann RJ, Wassenaar TM, Blaser MJ (2003) Evolutionary implications of microbial genome tetranucleotide frequency biases. Genome Res 13: 145–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Smith DR, Doucette-Stamm LA, Deloughery C, Lee H, Dubois J, et al. (1997) Complete genome sequence of Methanobacterium thermoautotrophicum deltaH: functional analysis and comparative genomics. J Bacteriol 179: 7135–7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lawrence JG, Ochman H (1998) Molecular archaeology of the Escherichia coli genome. Proc Natl Acad Sci U S A 95: 9413–9417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Swift J, Butts CA, Cheung-Lau J, Yerubandi V, Dmochowski IJ (2009) Efficient self-assembly of Archaeoglobus fulgidus ferritin around metallic cores. Langmuir 25: 5219–5225. [DOI] [PubMed] [Google Scholar]
- 58. Todorovic S, Rodrigues JV, Pinto AF, Thomsen C, Hildebrandt P, et al. (2009) Resonance Raman study of the superoxide reductase from Archaeoglobus fulgidus, E12 mutants and a ‘natural variant’. Phys Chem Chem Phys 11: 1809–1815. [DOI] [PubMed] [Google Scholar]
- 59. Scott AE, Timms AR, Connerton PL, Loc Carrillo C, Adzfa Radzum K, et al. (2007) Genome dynamics of Campylobacter jejuni in response to bacteriophage predation. PLoS Pathog 3: e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Deschavanne PJ, Giron A, Vilain J, Fagot G, Fertil B (1999) Genomic signature: characterization and classification of species assessed by chaos game representation of sequences. Mol Biol Evol 16: 1391–1399. [DOI] [PubMed] [Google Scholar]
- 61. Lawrence JG, Ochman H (1997) Amelioration of bacterial genomes: rates of change and exchange. J Mol Evol 44: 383–397. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Clustering result based on tetranucleotide usage patterns of the genus Campylobacter . Bar represents the clustering height. Campylobacterales bacterium GD 1 (termed Campylobacterales in picture) was used as the outgroup. Pearson correlations for tetranucleotide-derived z-values were used for clustering the genomes using ward linkage hierarchical algorithm.
(TIF)
Campylobacter genomes examined. Thirty-four Campylobacter genomes collected for analysis were listed with their genomic information including GenBank GI number, mark for genome in this study, contig number, total length, and SSU rRNA number.
(XLSX)
Genome-wide alignment matrix of genus Campylobacter . Genome-wide alignment similarity ratio in the table cells was computed as the length of genome fragments preserved between any two organisms against the length of total genomes of the organism in the row.
(XLSX)
Gene classification with abnormal composition. Genes with abnormal composition were classified into groups of different gene types including rRNA, tRNA, and protein-coding genes (coding sequences, CDS). The CDS was further divided into housekeeping CDS and HGT CDS. The gene number and the percentage to total gene number were listed for these genes.
(XLSX)
Gene families containing duplicated genes in genus Campylobacter . For each Campylobacter organism, the number of gene families containing duplicated genes and the total number of duplicated genes, together with average gene copies, (average gene number of all its gene families containing repeat genes) are shown.
(XLSX)
Gene family information of abnormal composition genes. The numbers in table cells represent the number of homologous genes in the corresponding genome. The Gene ID of the first column had the longest sequence in the gene family.
(XLSX)
Quantitative composition of Campylobacter virus-related and phage-related genes containing abnormal composition fragments. The percentage is equal to the number of virus- or phage-related genes with abnormal composition divided by the number of the total virus- or phage-related genes.
(XLSX)