

Abstract
Nitrosothiols are increasingly regarded as important participants in a range of physiological processes, yet little is known about their biological generation. Nitrosothiols can be formed from the corresponding thiols by nitric oxide in a reaction that requires the presence of oxygen and is mediated by reactive intermediates (NO2 or N2O3) formed in the course of NO autoxidation. Because the autoxidation of NO is second order in NO, it is extremely slow at submicromolar NO concentrations, casting doubt on its physiological relevance. In this paper we present evidence that at submicromolar NO concentrations the aerobic nitrosation of glutathione does not involve NO autoxidation but a reaction that is first order in NO. We show that this reaction produces nitrosoglutathione efficiently in a reaction that is strongly stimulated by physiological concentrations of Mg2+. These observations suggest that direct aerobic nitrosation may represent a physiologically relevant pathway of nitrosothiol formation.
Abbreviations: GSNO, S-nitrosoglutathione; DTPA, diethylenetriaminepentaacetic acid; DEA/NO, diethylamine NONOate (diethylammonium (Z)-1-(N,N-diethylamino)diazen-1-ium-1,2-diolate); PROLI/NO, proline NONOate (1-(hydroxyl-N,N,O-azoxy)-l-proline, disodium salt); DNIC, dinitrosyl–iron complex; SOD, superoxide dismutase; oxy-Hb, oxyhemoglobin; met-Hb, methemoglobin; TEA, triethanolamine; DAN, 2,3-diaminonaphthalene; DTT, dithiothreitol; NAC, N-acetylcysteine; β-ME, β-mercaptoethanol
Keywords: S-nitrosation, Nitrosoglutathione, Nitric oxide, Free radicals
Graphical abstract
Highlights
-
•
The mechanism of aerobic nonenzymatic S-nitrosation is revisited.
-
•
Nitrosothiol formation is not autoxidation-mediated at physiological NO levels.
-
•
Direct S-nitrosation is stimulated by divalent cations such as Mg2+.
-
•
Direct nonenzymatic S-nitrosation is fast enough to be physiologically relevant.
Nitric oxide (NO) exhibits a vast range of functions in signal transduction and the immune response in mammalian tissues [1]. Many functions in signal transduction are mediated by the NO-sensitive (soluble) isoform of guanylate cyclase [2]. However, under some conditions NO is converted to compounds with distinct properties that may alter its (patho)physiological impact [3].
Nitrosothiols are endogenously occurring formal adducts of protein or low-molecular-weight thiols with the one-electron oxidized form of nitric oxide, NO+ [4]. Evidence is mounting that nitrosothiols may perform distinct functions in biology [5,6]. Because nitrosothiols release NO under certain conditions and are generally more stable than NO, they may function as a storage and transport pool of NO. Nitrosothiols also exhibit biological actions completely different from those of NO, because nitrosation of specific cysteine residues may alter protein function. Hence, S-nitrosation is increasingly regarded as a posttranslational modification akin to phosphorylation [7–9].
Despite these potential (patho)physiological ramifications there is no consensus about the way in which nitrosothiols are generated cellularly [8,10–13]. In the laboratory nitrosothiols are synthesized at low pH from the corresponding thiols and nitrous acid. Biologically, similar reactions may occur under special conditions, such as in the stomach or in activated macrophages. At physiological pH nonenzymatic formation of nitrosothiols may be catalyzed by transition metals such as copper ions [14]. In view of the extremely low in vivo free copper concentration and the reversible nature of the reaction—copper ions also catalyze the decomposition of nitrosothiols [4,8,15]—the physiological relevance of this reaction is questionable. Nitrosothiols can also be formed by decomposition of low-molecular-weight dinitrosyl–iron complexes (DNICs) with thiolate ligands [16,17]. As DNICs are formed in vivo, these compounds constitute serious candidates as biological nitrosating agents. However, because the mechanism of biological DNIC formation has not been elucidated and because DNICs may also catalyze nitrosothiol breakdown, the exact role of DNICs in biological nitrosothiol formation is unclear. In addition to these nonenzymatic processes, several enzymes have been implicated in nitrosothiol formation, such as the copper protein ceruloplasmin [18] or hemoproteins [8,19]. Nitrosothiols will also be formed when NO and O2− are cogenerated at similar rates [11,20], which may be physiologically relevant under some conditions, for instance, when partly uncoupled nitric oxide synthase generates NO and O2− simultaneously [21].
The best studied pathway of nitrosothiol formation is the aerobic reaction of NO with glutathione (GSH). Anaerobically, NO does not form S-nitrosoglutathione (GSNO), but in the presence of oxygen GSNO is generated in a reaction that is first order in O2 and second order in NO [22–24]. The reaction starts with the rate-limiting formation of NO2 from NO and O2 (Eq. (1)). According to one hypothesis [23,25], NO2 then reacts with another molecule of NO to form the strong nitrosating agent N2O3, which reacts with GSH to GSNO and nitrite (Eqs. (2) and (3)). Alternatively, it has been proposed [11,24,26] that NO2 reacts directly with GSH to produce nitrite and a glutathiyl radical (GS•) that instantaneously combines with NO to GSNO (Eqs. (4) and (5)):
| 2NO+O2→2NO2 | (1) |
| NO2+NO↔N2O3 | (2) |
| N2O3+GSH→GSNO+H++NO2− | (3) |
| NO2+GSH→GS•+H++NO2− | (4) |
| NO+GS•→GSNO | (5) |
Because of the low (submicromolar) physiological concentrations of NO, this pathway is expected to be too slow to make an impact. In 1997 it was reported that, for low NO concentrations (≤1 µM), aerobic nitrosothiol formation was not due to NO autoxidation but involved a direct reaction between NO and the thiol [27]. However, later studies could not confirm that mechanism [17,28]. In the present study we demonstrate that the aerobic nitrosation of glutathione by submicromolar NO is first order in NO and proceeds more efficiently than previously thought, making it a serious candidate as a participant in nitrosothiol formation in vivo after all.
Materials and methods
All reagents were obtained from Merck (Vienna, Austria) or Sigma (Vienna, Austria), except for diethylamine NONOate (DEA/NO), proline NONOate (PROLI/NO), and GSNO, which were purchased from Enzo Life Sciences (Lausen, Switzerland). Stock solutions were made in ultrapure water (Barnstead, resistance >18 MΩ cm−1), except for DEA/NO and PROLI/NO, which were dissolved in 10 mM NaOH; GSH, which was dissolved in 1 M NaOH; and GSNO, which was dissolved in 10 mM HCl.
Determination of NO and nitrosothiols with the NO-sensitive electrode
Nitric oxide was measured with a Clark-type electrode from Iso-NO (WPI, Berlin, Germany) according to a published method [29]. Unless indicated otherwise, experiments were performed in open stirred vessels. Reactions were started by the introduction of DEA/NO (or PROLI/NO, 1 µM) in a total volume of 0.5 ml of 50 mM triethanolamine (TEA) buffer (pH 7.4) and GSH (routinely 1 or 2 mM), 5 mM MgCl2, 1000 U/ml superoxide dismutase (SOD), and 0.1 mM diethylenetriaminepentaacetic acid (DTPA). After complete decay of the NO signal, CuSO4 (4 mM) was added to measure nitrosothiol formation [30]. When indicated, MgCl2 was omitted or replaced by CaCl2, MnCl2, ZnCl2, or NaCl. Some experiments were performed in 50 mM potassium phosphate (KPi) or Tris–HCl instead of TEA and some experiments were performed in the presence of 1 mM NAD+ or NADH. Dependence of the NO curves on the concentration of DEA/NO was investigated between 10 nM and 1 µM. The effect of the GSH concentration was studied between 1 µM and 5 mM. At the highest GSH concentration, we increased the CuSO4 concentration to 10 mM, because copper ion-induced NO release from GSNO is slow in the presence of excess GSH [15].
Some experiments were performed in closed vessels with reaction volumes of 0.5 or 1.8 ml. Completely filled closed vessels were also used to determine the effect of O2. For these experiments solutions were bubbled with argon before use. Reaction mixtures were then incubated in septum-sealed, completely filled vessels and bubbled with argon for 15 min. Subsequently, the gas supply line was removed and experiments were started by addition of the NO donor. Because the covering of the sample under these conditions is not completely airtight, slow readmission of air into the sample occurs.
Calibration of the electrode was performed daily with NaNO2/KI [29]. Pre- and post-Cu2+ peaks are presented as micromolar NO based on those calibrations. To quantify the concentration of GSNO detected by CuSO4 addition, calibration curves were determined with authentic GSNO in concentrations between 0.1 and 2.0 µM. GSNO stock solutions were prepared in 10 mM HCl and used immediately. The concentration of the stock solutions was checked spectrophotometrically at 340 nm after 10-fold dilution in 50 mM KPi (pH 7.4). Calibration curves were linear over the full concentration range (R=0.999).
Quantification of NO released by DEA/NO
To determine how much NO is released by DEA/NO under the present experimental conditions, we measured the conversion of oxyhemoglobin (oxy-Hb) to methemoglobin (met-Hb) spectrophotometrically from the absorbance difference between 420 and 401 nm according to a published procedure [31], but with 50 mM TEA (pH 7.4) instead of KPi as the buffer.
Determination of NO, GSNO, and nitrite with the NO analyzer
A nitric oxide analyzer NOA 280 (Sievert Instruments, USA) was used to determine the amounts of NO, GSNO, and nitrite by chemiluminescence detection according to a published method [32]. Briefly, samples (500 µl) were injected in a purging vessel filled with KI/I2 (45 mM/10 mM) in glacial acetic acid. Under these conditions both nitrite and GSNO are reduced to NO. In parallel, a second set of samples was incubated with 10% of a solution of sulfanilamide (5% in 1 N HCl) for 1 min to scavenge nitrite and measure only the remaining GSNO. Calibration curves with authentic GSNO (0.1–2 µM) and nitrite (0.1–1 µM) were measured daily in sample buffer.
Alternatively, GSNO and GSNO+nitrite were measured by addition of 4 mM CuSO4 and 4 mM CuSO4+KI/I2 (45 mM/10 mM), respectively, whereas NO was determined in the absence of these substances.
Fluorimetric determination of nitrite in the presence of 2,3-diaminonaphthalene
Nitrite was determined using 12.6 µM 2,3-diaminonaphthalene (DAN) to form the fluorescent product 1-(H)-naphthotriazole [33]. Briefly, samples were incubated for 20 min with DAN under acidic conditions. After stabilization with 1 N sodium hydroxide, formation of 1-(H)-naphthotriazole was measured using an LS50B luminescence spectrometer (PerkinElmer, UK) with excitation and emission at 365 and 425 nm, respectively. Alternatively, a commercial assay (Nitrite/Nitrate Assay Kit, Cat. No. 06239; Sigma–Aldrich) based on the same principle was used. Calibration curves were measured daily with authentic nitrite (0.1–1 µM) in sample buffer.
Determination of nitrite, nitrate, and GSNO by HPLC
In some experiments we increased the sensitivity by separating the reaction products by HPLC (Merck–Hitachi D-6000; Vienna, Austria). For nitrite/nitrate determination the same commercial nitrite/nitrate-kit described above was used according to the manufacturer’s instructions. HPLC analysis was then performed as described before [34]. The mobile phase (53% Na2HPO4 15 mM, pH 7.5; 47% methanol) was pumped through a Lichrospher column (RP-18; 5 µm) at a flow rate of 1 ml/min. Samples (40 µl) were injected and measured by fluorescence (380 nm excitation, 405 nm emission). Calibration curves were measured for nitrite and nitrate (0.1–10 µM) in sample buffer.
For GSNO determination the mobile phase (20 mM K2HPO4, 50 µM neocuproine, 50 µM DTPA, pH 7.5) was pumped through a Lichrospher column (RP-18; 5 µm) at a flow rate of 1 ml/min. Samples (100 µl) were injected and measured by UV–Vis absorption at 338 nm [21]. Calibration curves with authentic GSNO (0.1–2 µM) were measured in sample buffer.
Results
Efficient GSNO formation from micromolar DEA/NO in the presence of GSH
Injection of 1 µM DEA/NO into a reaction mixture containing 1000 U/ml SOD, 5 mM MgCl2, and 100 µM DTPA in 50 mM TEA (pH 7.4) gave rise to a strong signal at the NO-sensitive electrode peaking at approximately 0.74 µM (Fig. 1A). When the experiment was repeated in the presence of 2 mM GSH we obtained a much smaller signal. Because we suspected that the decrease in peak height might be due to nitrosothiol formation, we added CuSO4 at the end of the reaction to promote NO release (Fig. 1A). No signal was detected upon addition of CuSO4 in the absence of GSH, but a strong signal evolved in its presence, suggesting that the apparent attenuation of NO release from DEA/NO in the presence of GSH is due to GSNO formation.
Fig. 1.

Effect of GSH on NO release from DEA/NO before and after addition of CuSO4. (A) The NO release curves from DEA/NO, added at t=0, in the absence and presence of GSH, and the effect of CuSO4 added after 12 min (indicated by the arrow). (B) Summary of several such experiments (n=5). (C) A similar experiment except that SOD was omitted. Experimental conditions: 1 µM DEA/NO, 2 mM GSH as indicated, 4 mM CuSO4, 1000 U/ml SOD (except for (C)), 0.1 mM DTPA, 5 mM MgCl2, and 50 mM TEA (pH 7.4) in 0.5 ml at 37 °C.
When the same experiment was performed in the absence of SOD, similar results were obtained, except that the peak height after CuSO4 addition was considerably smaller (Fig. 1C). In the absence of SOD interpretation of the results is confounded by the potential formation of superoxide, peroxynitrite, and its metabolites (see below). Consequently, we chose to include SOD in the majority of our studies.
Fig. 1 also illustrates that the post-Cu signal decays more rapidly than the pre-Cu signal. This phenomenon, which can also been seen in previous studies [30], is probably due to reductive nitrosylation of NO to NO2− with H2O as the nucleophile, a reaction known to be catalyzed by Cu2+ [35]. It was not investigated further in this study.
Determination of the NO-to-DEA/NO stoichiometry
DEA/NO has been observed to release between 1 and 2 equivalents of NO [36]. To determine how much NO is released by DEA/NO under the present experimental conditions, we measured the conversion of oxy-Hb to met-Hb spectrophotometrically. We found that 1 µM DEA/NO released 1.46±0.02 µM NO.
To directly correlate these observations with the amount of GSNO formed from DEA/NO under the same conditions (1 mM GSH), we determined GSNO in parallel with the NO electrode and found 1.01±0.04 µM, which corresponds to a GSNO-to-NO stoichiometry of 0.69±0.03.
Comparison with other thiols
To investigate if the phenomena described above are specific for GSH, we performed similar experiments in the presence of several other thiols at 1 mM (Table 1). GSH, N-acetylcysteine (NAC), β-mercaptoethanol (β-ME), cysteine (Cys), and dithiothreitol (DTT) all lowered the height of the initial NO peak, whereas GSSG had no effect. NO formation after CuSO4 addition was about 30% smaller with NAC or β-ME than with GSH, whereas no or hardly any NO was observed with GSSG, DTT, and Cys.
Table 1.
Effects of various thiols on the pre- and post-Cu2+ NO peak.
| Thiol | NO peak height (µM) |
|
|---|---|---|
| Before Cu2+ | After Cu2+ | |
| − | 0.69±0.02 | n.d. |
| GSH | 0.42±0.04 | 0.69±0.04 |
| GSSG | 0.74±0.01 | n.d. |
| Cys | 0.45±0.03 | n.d. |
| DTT | 0.51±0.05 | 0.065±0.001 |
| NAC | 0.43±0.08 | 0.49±0.09 |
| β-ME | 0.38±0.02 | 0.51±0.01 |
NO peak heights from 1 µM DEA/NO before and after addition of 4 mM CuSO4 were measured in the absence (−) or presence of various thiols at 1 mM: glutathione (GSH), oxidized glutathione (GSSG), cysteine (Cys), dithiothreitol (DTT), N-acetylcysteine (NAC), or β-mercaptoethanol (β-ME). Experimental conditions: 1 µM DEA/NO, 1 mM thiol as indicated, 4 mM CuSO4, 1000 U/ml SOD, 0.1 mM DTPA, 5 mM MgCl2, and 50 mM TEA (pH 7.4) in 0.5 ml at 37 °C. n.d., not detectable.
NO autoxidation is a minor process under the conditions of this study
The decay rate of the NO peak in Fig. 1 was considerably faster than previously reported and too fast to be explained by autoxidation [36,37]. Unlike previous studies, this study was performed in open, stirred vessels with DEA/NO concentrations of 1 µM or less in a reaction volume of 0.5 ml. We hypothesized that under those conditions NO disappears mainly by diffusion out of the reaction vessel. In that case NO disappearance should be first order in NO, in contrast to the second-order dependence of NO autoxidation. Although the observed decay curves indeed showed no sign of second-order behavior, it might be argued that the second-order decay is masked by the simultaneously continuing first-order NO release by DEA/NO. However, we could fit the observed traces if we assumed that NO disappeared by escape to the atmosphere, but not if we assumed NO disappeared by autoxidation (see Supplementary Figs. S9A and S9B). The nature of NO disappearance will affect the way in which the NO peak height varies with the DEA/NO concentration. If NO disappears in a first-order reaction, as is the case when NO escape predominates, the peak height will increase linearly with the DEA/NO concentration. If, on the other hand, NO is consumed in a second-order process such as autoxidation, the relation between peak height and DEA/NO will deviate from linearity, particularly at higher concentrations (see also the simulations in Supplementary Fig. S2). Moreover, in the case of a second-order process the peak will be reached earlier at higher DEA/NO concentrations, whereas this time is constant for a first-order process (see Supplementary Fig. S2). Therefore, to distinguish between both possibilities, we varied the DEA/NO concentration and observed an excellent linear correlation between the height of the NO peak and the DEA/NO concentration (Figs. 2A and B), which strongly suggests that the decay phase is caused by a first-order process. Moreover, the time at which the maximal NO concentration was reached hardly changed between 50 and 1000 nM DEA/NO (Figs. 2A and 2C).
Fig. 2.

Correlation between NO peak heights and DEA/NO concentrations. (A) NO time traces observed with the NO electrode with 30, 200, 400, and 1000 nM DEA/NO in the absence of GSH. (B) Peak heights observed with a range of DEA/NO concentrations between 30 nM and 1 µM (n=3). Peak heights are determined by the relative rates of NO release and NO consumption/escape. See main text and the supplementary material for details. Data were fitted (dashed lines) to the equation [NO]peak=C2·[DEA/NO]C1, in which C1 and C2 are variables. The observed value for C1 is very close to 1 (0.96±0.02), indicating a linear relationship between peak height and DEA/NO concentration. Please note that the apparent absolute correlation between observed peak heights and DEA/NO concentrations (1 µM NO for 1 µM DEA/NO) is accidental: DEA/NO can release up to 2 equivalents of NO, and observed peak heights are necessarily lower than the released NO concentrations. (C) Dependence of the time at which the NO concentration is maximal as a function of the DEA/NO concentration. See main text and the supplementary material for details. The observations suggest a first-order disappearance of NO. Experimental conditions: 1 µM DEA/NO, 4 mM CuSO4, 1000 U/ml SOD, 0.1 mM DTPA, 5 mM MgCl2, and 50 mM TEA (pH 7.4) in 0.5 ml at 37 °C.
To corroborate that our choice of experimental conditions (0.5 ml reaction volume, ≤1 µM DEA/NO, stirred open vessels) caused a shift from NO autoxidation to NO escape, and to investigate if this shift affects GSH nitrosation, we performed a number of control experiments. Simply closing off the vessel while keeping the sample volume at 0.5 ml hardly affected the DEA/NO-derived NO signal (Fig. 3A). This is explained by the fact that under these conditions the vessel contains a void volume of ~1.3 ml, which, with a solubility for NO of ~2 mM, implies that less than 2% of NO will remain in solution at equilibrium. However, when the volume of the sample was increased to completely fill the vessel, the NO peak was markedly higher, and decay of the signal was considerably slower (Fig. 3A), suggesting a shift in NO decay from relatively fast escape to the gas phase toward autoxidation. However, GSH still caused a pronounced decrease in the NO peak height and an increase in the apparent decay rate in closed vessels and gave rise to sizeable NO signals after CuSO4 addition (Fig. 3B).
Fig. 3.

Effect of closing the reaction vessel on NO decay kinetics. (A) Closing the vessel increases NO peak height and decreases the decay rate. The continuous line shows the NO release curve in an open vessel (reaction volume 0.5 ml). Experimental conditions were as in Fig. 1 in the absence of GSH. The dotted line shows the NO release curve in a closed vessel under otherwise identical conditions. The dashed line shows the NO release curve in a closed vessel with a reaction volume of 1.8 ml, leaving no void volume. (B) Closing the vessel does not impede the nitrosation of GSH. The blue curves show the NO release curves in open vessels (0.5 ml reaction volume) in the absence and presence of GSH. The red curves show the corresponding curves in closed vessels (1.8 ml reaction volume). In the presence of GSH, CuSO4 was added at the indicated times to measure GSNO formation. Experimental conditions: 1 µM DEA/NO, 2 mM GSH as indicated ((B) only), 4 mM CuSO4, 1000 U/ml SOD, 0.1 mM DTPA, 5 mM MgCl2, and 50 mM TEA (pH 7.4) in 0.5 ml at 37 °C.
Because NO release from DEA/NO is rather slow (t1/2~16 and 2.2 min at 22 and 37 °C, respectively [36,38]), NO formation continues during the decay phase, and decay kinetics are difficult to interpret. PROLI/NO releases NO much faster (t1/2~1.8 s at 37 °C [39]), allowing observation of NO decay without interference from NO release. When we repeated some of the experiments with PROLI/NO, closing the vessel caused a shift from fast first-order toward slower second-order decay (results not shown), which confirms that under open-vessel conditions NO escape outcompetes autoxidation.
NO autoxidation is not involved in GSNO formation under the conditions of this study
In the case of autoxidation-mediated GSNO formation, rate-limiting NO autoxidation (Eq. (1)) precedes the reaction with GSH (Eqs. (2)–(5)). Consequently, in line with reported observations [24], GSH should not affect the NO formation/consumption curve (see also the simulations in Supplementary Fig. S1). However, as illustrated by Figs. 1 and 3, we consistently observed that GSNO formation, measured as CuSO4-mediated NO release, is associated with a strong decrease in the NO peak originating from DEA/NO. The present results are therefore inconsistent with the involvement of NO autoxidation in GSH nitrosation. Rather, the data suggest a direct reaction between NO and GSH.
The correlation between GSNO yield and DEA/NO concentration should allow conclusions about the kinetic order of the nitrosation process. If nitrosation were first order in NO, competition between nitrosation and the alternative reaction—NO escape, which is also first order in NO—would not be affected by the concentration of the NO donor, the relative GSNO yield should remain constant, and the absolute GSNO yield should increase linearly with the DEA/NO concentration. By contrast, if nitrosation involved autoxidation, nitrosation would become more competitive at higher NO concentrations, the relative yield of GSNO should increase with the DEA/NO concentration, and the corresponding peak height should exhibit a stronger than linear dependence on the DEA/NO concentration (see also the simulations in Supplementary Fig. S3). We measured the height of the CuSO4-mediated NO peak in the presence of GSH as a function of the DEA/NO concentration and found an approximately linear relationship (Fig. 4), which suggests that GSH nitrosation is first order in NO and does not involve NO autoxidation.
Fig. 4.

Correlation between post Cu2+-peak heights and DEA/NO concentrations. (A) NO time traces observed with the NO electrode with 50, 200, 400, and 1000 nM DEA/NO in the presence of GSH. Individual traces were shifted horizontally and vertically for clarity. (B) Peak heights observed with a range of DEA/NO concentrations between 50 nM and 1 µM (n=3). Peak heights correspond to the amount of GSNO formed at the time of CuSO4 addition. See main text and the supplementary material for details. Data were fitted (dashed lines) to the equation [NO]peak=C2·[DEA/NO]C1, in which C1 and C2 are variables. The fit is linear with C1 close to 1 (1.14±0.02), suggesting a linear relationship between peak height and DEA/NO concentration. The apparent absolute correlation between observed peak heights and DEA/NO concentrations (1 µM NO for 1 µM DEA/NO) is accidental: it does not reflect a one-to-one correlation between NO and GSNO, because DEA/NO can produce more than 1 equivalent of NO. Experimental conditions: 1 µM DEA/NO, 2 mM GSH, 4 mM CuSO4, 1000 U/ml SOD, 0.1 mM DTPA, 5 mM MgCl2, and 50 mM TEA (pH 7.4) in 0.5 ml at 37 °C.
Evaluation of the influence of gas-phase and electrode reactions
In addition to a direct reaction of GSH with NO and autoxidation-mediated GSNO formation, there are a couple of other possibilities that need to be considered. There is a remote possibility that NO diffuses out of solution, reacts with O2 in the gas phase to NO2/N2O3, and diffuses back into solution to nitrosate GSH. However, the observation, discussed above, that GSH still lowers the NO peak and is still nitrosylated in a completely filled vessel (Fig. 3B), argues against that scenario. As a further test we performed the experiment in a completely filled open vessel, while blowing argon over the surface. Under these conditions formation of nitrosating species in the gas phase and rediffusion into the solution are prevented, yet GSNO formation was only moderately lower (0.51±0.06 vs 0.87±0.02 µM, n=4, not shown). The lower GSNO formation and slower decay of the NO signal (not shown) are probably caused by diffusion of the coreactant O2 out of the reaction vessel. Indeed, when we repeated the experiment with air instead of argon, no significant drop in GSNO yield was observed (0.91±0.17 vs 1.08±0.16 µM, n=2, not shown).
Reactions at the electrode constitute an additional potentially confounding factor, particularly in view of the small reaction volumes in this study. Oxidation of NO at the electrode might produce NO+ that could conceivably nitrosate GSH directly. However, from the sensitivity of the electrode (1 pA/nM, [36]) it can be estimated that in a 0.5-ml reaction volume the electrode reaction consumes only 0.12% NO per minute, which is negligible on the time scale of the experiments. This calculation also rules out the electrode reaction as a significant sink for NO.
Evidence for a direct reaction of GSH with NO but not with the NO donor
We also considered the possibility that GSH reacts directly with DEA/NO, because that would also result in a decrease in the DEA/NO-induced NO peak. Such a reaction should not be affected by the NO scavenger oxy-Hb, because it would not require the intermediacy of NO. We therefore performed an experiment with 1 µM DEA/NO and 1 mM GSH in the presence of 1 µM oxy-Hb. Under these conditions hardly any NO formation was observed after DEA/NO addition, and CuSO4 did not induce any NO release, whereas subsequent addition of 2 µM GSNO caused a pronounced signal (Fig. 5A). These observations rule out that GSNO is formed in a direct reaction between the thiol and the DEA/NO.
Fig. 5.

GSH does not react directly with the NONOates. (A) Effect of oxy-Hb on the GSNO yield from 1 µM DEA/NO and 1 mM GSH. In the presence of oxy-Hb (1 µM; 10 s) the NO peak observed after addition of DEA/NO (1 µM; 90 s) was very small, and no GSNO formation was evident after CuSO4 addition (4 mM; 506 s), whereas subsequent GSNO administration (2 µM; 570 s) yielded a pronounced peak. Experimental conditions: 1 mM GSH, 1000 U/ml SOD, 0.1 mM DTPA, 5 mM MgCl2, and 50 mM TEA (pH 7.4) in 0.5 ml at 37 °C; at the indicated times 1 µM oxy-Hb, 1 µM DEA/NO, 4 mM CuSO4, and 2 µM GSNO were added. (B) Effect of the time of GSH administration on the yield of GSNO from PROLI/NO and GSH. In the presence of 1 mM GSH the NO peak derived from 1 µM PROLI/NO was considerably smaller than in the absence of GSH, and a strong NO signal, originating from GSNO, was observed after CuSO4 addition (compare the dotted and dashed traces in the absence and presence of GSH, respectively). If GSH was added 50 s after PROLI/NO, at a time when all PROLI/NO should be decomposed, CuSO4 addition still caused a sizeable NO signal (continuous trace). Experimental conditions: 1 µM PROLI/NO, 1000 U/ml SOD, 0.1 mM DTPA, 4 mM CuSO4, 5 mM MgCl2, and 50 mM TEA (pH 7.4) in 0.5 ml at 37 °C; GSH (1 mM) was absent (dotted trace), present (dashed trace), or added at the indicated time (continuous trace).
In another experiment we monitored NO profiles when 1 mM GSH was administered before or after the addition of 1 µM PROLI/NO. Because PROLI/NO decays very fast, addition of GSH after PROLI/NO should no longer produce GSNO if a direct reaction with the donor were involved. As shown in Fig. 5B, GSNO formation was still substantial if GSH was added 50 s after PROLI/NO, which rules out a direct reaction between the NONOate and the thiol. The experiment also illustrates how GSH caused a massive acceleration of NO consumption, providing further evidence for a direct reaction between GSH and NO as the main route to GSNO formation.
Effects of NAD+, O2, SOD, and buffer on GSH nitrosation by NO
Experiments under quasi-anaerobic conditions (see Materials and methods) yielded much higher NO peaks and considerably diminished copper-catalyzed GSNO decomposition (not shown), indicating that the formation of GSNO from DEA/NO in the presence of GSH is O2 dependent.
NAD+ has been reported to serve as an electron acceptor during direct nitrosation [27]. However, experiments in the presence of NAD+ or NADH indicated that these compounds had little effect on NO peak height or GSNO formation (Supplementary Fig. S10). Although these results do not invalidate the observation by Gow et al. that NAD+ can substitute for O2 under anaerobic conditions [27], they do demonstrate that NAD+ is a poor substitute, because 1 mM NAD+ was unable to compete with ~0.2 mM O2, suggesting a rate constant at least 2 orders of magnitude smaller. Lack of reactivity of NAD+ with NO/GSH has also been reported by Hogg and collaborators [28].
By scavenging O2−, SOD prevents extra consumption of NO by O2− and by the products of homolysis of peroxynitrite (•OH and NO2•), which was proposed to be the cause of additional NO consumption in previous studies [28]. Consequently, the presence of SOD under our standard conditions greatly simplifies the interpretation of the results. In the absence of GSH, omission of SOD did not affect the height of the NO peak (0.70±0.04 and 0.68±0.01 with and without 1000 U/ml SOD, respectively, see also Fig. 1). Surprisingly, omission of SOD did not significantly affect the height of the NO peak in the presence of 1 mM GSH (0.42±0.04 and 0.37±0.03 with and without 1000 U/ml SOD, respectively), which was unexpected, as O2− is probably formed during direct aerobic nitrosation (see Discussion). In line with expectation, however, omission of SOD markedly decreased Cu2+-mediated NO release (from 0.69±0.04 to 0.28±0.03 µM, respectively, Fig. 6).
Fig. 6.

Effect of SOD on pre- and post-Cu2+ DEA/NO-derived NO peaks in the presence of GSH. NO peak heights were determined from traces as in Fig. 1. Experimental conditions: 1 µM DEA/NO, 1 mM GSH, 4 mM CuSO4, 1000 U/ml SOD as indicated, 0.1 mM DTPA, 5 mM MgCl2, and 50 mM TEA (pH 7.4) in 0.5 ml at 37 °C; n≥6.
To investigate the influence of the buffer we repeated the experiments in KPi (50 mM, pH 7.4). This did not affect the height of the NO peak after DEA/NO addition in the presence of 1 mM GSH (0.43±0.05 and 0.45±0.06 µM NO with TEA and KPi buffer, respectively), but substantially lowered Cu2+-induced NO release (from 0.69±0.04 to 0.37±0.03). With Tris buffer (50 mM, pH 7.4) we obtained results similar to those from TEA (not shown).
Effect of Mg2+on nitrosothiol formation
Additionally, we looked into the potential effect of Mg2+ on formation of NO and GSNO. In the presence of GSH, omission of Mg2+ yielded a considerably higher (2.3±0.4-fold) NO peak (i.e., before CuSO4 addition) and a much lower (0.27±0.08-fold) GSNO-derived peak (i.e., after CuSO4 addition), indicating that Mg2+ stimulates GSNO formation (Fig. 7). Additional experiments demonstrated that Ca2+ stimulated GSNO formation to the same extent (Fig. 7), whereas equimolar Na+ had no effect (not shown). GSNO formation was also stimulated by Mn2+, although in this case the interpretation is complicated by the fact that Mn2+ slowed down NO release from DEA/NO in the absence of GSH (not shown). An even stronger inhibition of DEA/NO decomposition precluded determination of the effect of Zn2+ (not shown). Omission of Mg2+ had similar effects with NAC or β-ME instead of GSH as the thiol (Supplementary Fig. S12).
Fig. 7.

Effects of Mg2+ and Ca2+ on DEA/NO decomposition and GSNO formation. Shown are the average peak heights (n=5) observed immediately after DEA/NO addition in the absence and presence of GSH, as well as the peak height after CuSO4 addition in the presence of GSH with and without 5 mM MgCl2 or CaCl2. Experimental conditions: 1 µM DEA/NO, 2 mM GSH as indicated, 4 mM CuSO4, 1000 U/ml SOD, 0.1 mM DTPA, and 50 mM TEA (pH 7.4) in 0.5 ml at 37 °C.
Effect of the GSH concentration on GSNO formation
The effects of the GSH concentration on the height of the DEA/NO-derived NO peaks before and after CuSO4 addition are shown in Fig. 8A. Both the decrease in the NO peak height before and the GSNO-mediated increase after Cu2+ addition were biphasic. A steep initial phase, up to about 50 µM GSH, resulted in an ~0.2 µM decrease in the DEA/NO-induced NO peak and the formation of ~0.2 µM GSNO from 1 µM DEA/NO. At higher concentrations of GSH, the NO peak and the GSNO peak continued to fall and rise, respectively, resulting in a further ~0.2–0.3 µM decrease in the NO peak and an ~0.6 µM increase in the GSNO peak at 2 mM GSH. Extrapolation of the observations suggests complete disappearance of the NO peak and complete conversion of NO to GSNO at saturating GSH concentrations. When MgCl2 was omitted from the reaction mixture, similar observations were made at low GSH concentrations, but the effects at high GSH concentrations were absent (Fig. 8A).
Fig. 8.

Effect of the GSH concentration on the nitrosation yield. Shown are the peak heights before (NO, circles) and after CuSO4 addition (GSNO-derived, squares) for 1 µM (A) DEA/NO or (B) PROLI/NO in the presence (black symbols) and absence (white symbols) of 5 mM MgCl2 for GSH concentrations between 0 and 2 mM. Experimental conditions: 1 µM (A) DEA/NO or (B) PROLI/NO, GSH as indicated, 4 mM CuSO4, 1000 U/ml SOD, 0.1 mM DTPA, 5 mM MgCl2 as indicated, and 50 mM TEA (pH 7.4) in 0.5 ml at 37 °C. Curves were fitted to one or two hyperbolic functions, as appropriate. See the supplementary material for details.
With 1 µM PROLI/NO instead of DEA/NO, the effect of GSH was monophasic, with the NO peak (before CuSO4 addition) decreasing from ~1.2–1.3 to 0.6–0.7 µM, and the GSNO peak (after CuSO4 addition) increasing from 0 to ~0.6 µM at 2 mM GSH (Fig. 8B). Extrapolation again suggests complete disappearance of the initial NO peak and complete conversion to GSNO at saturating GSH concentrations with an EC50 of ~2 mM GSH. These observations are in line with expectations for a direct reaction between NO and GSH, but not for autoxidation-mediated nitrosation, providing further support for a direct reaction. The biphasic character of the GSH concentration dependence is surprising. Principally, a decrease in nitrosating efficiency at higher GSH concentrations might be explained by a shift away from GSH nitrosation toward oxidation, but such an effect would not be expected to be as abrupt as it seems to be here, and it is unclear why the phenomenon is apparent with DEA/NO only in the presence of Mg2+. Further studies will be required to resolve these issues.
Effect of the GSH concentration on the rate of NO disappearance
To determine the effect of the concentration of GSH on the rate of NO disappearance, we added various concentrations of GSH to the DEA/NO reaction mixture when the NO concentration had decayed to approximately 0.6 µM. As is evident from Fig. 9A, the rate of NO decay increased with the GSH concentration, confirming that GSH consumes NO in a direct reaction. We determined the difference between the NO decay rates before and after GSH addition (to account for GSH-independent NO disappearance) and divided the resulting GSH-induced decay rates by the NO concentration at the time of GSH addition (to correct for variations in that parameter, with actual values varying between 0.41 and 0.68 µM NO). The apparent (first-order) rate constants thus obtained are plotted against the concentration of GSH in Fig. 9B, which illustrates how NO decay is accelerated when the GSH concentration increases. The dependence can be fitted linearly (dotted line), although the data are too noisy to rule out alternative interpretations. By dividing all observed rates by the NO and GSH concentrations and averaging the results, we can estimate an apparent second-order rate constant of 34±6 M−1 s−1.
Fig. 9.

Effect of the GSH concentration on the NO decay rate. (A) The effect of the addition of various concentrations of GSH on the NO release curve. NO released from 1 µM DEA/NO in the absence of GSH was measured as for Fig. 1. At the time indicated by the arrows GSH (0.2, 0.5, or 2.0 mM as indicated) was added to the reaction mixture. (B) The apparent pseudo-first-order rate constant as a function of the GSH concentration. Pseudo-first-order rate constants were obtained by dividing the GSH-induced NO decay rate, i.e., the difference between the rates before and after GSH addition, by the NO concentration at the time of GSH addition (which varied between the individual experiments). The dotted line is the best linear fit. An apparent second-order rate constant of 34±6 M−1 s−1 was calculated as the average of the quotients of the apparent pseudo-first-order rate constant and the GSH concentration for all experiments. Experimental conditions: 1 µM DEA/NO, GSH as indicated, 4 mM CuSO4, 1000 U/ml SOD, 0.1 mM DTPA, 5 mM MgCl2, and 50 mM TEA (pH 7.4) in 0.5 ml at 37 °C.
Effects of GSH on NO consumption with 0.1 mM DEA/NO
According to our hypothesis, one of the reasons that autoxidation plays a smaller part in NO disappearance in this study is the submicromolar concentration range of DEA/NO applied here. To test this assumption, we repeated the experiment with 0.1 mM DEA/NO. Under these conditions, in line with previous observations [24], GSH no longer affected the kinetics of NO formation and decay, although it still caused Cu2+-induced NO release (Fig. 10). This demonstrates that autoxidation takes over at higher NO concentrations as the main nitrosating pathway (see also Supplementary Fig. S3). Significantly, this phenomenon is accompanied by a decrease in efficiency of nitrosation, judging from the Cu2+-induced peak values of 0.9 and 27 µM at 1 and 100 µM DEA/NO, respectively, which correspond to nitrosation levels of 60 and 18%. Moreover, omission of Mg2+ had no effect at all at 100 µM DEA/NO, indicating that the effect of Mg2+ is specific for the direct reaction observed at submicromolar NO concentrations.
Fig. 10.

Effect of GSH on NO peak heights obtained with a high concentration of DEA/NO. Shown are pre- and post-Cu2+ peak heights for 100 µM DEA/NO in the absence and presence of 1 mM GSH and 5 mM MgCl2. Experimental conditions: 100 µM DEA/NO, 1 mM GSH as indicated, 4 mM CuSO4 as indicated, 1000 U/ml SOD, 0.1 mM DTPA, 5 mM MgCl2 as indicated, and 50 mM TEA (pH 7.4) in 0.5 ml at 37 °C.
Determination of NO, nitrite, and GSNO with the NO analyzer
As an alternative to the electrochemical studies described thus far, we determined NO, nitrite, and GSNO formation with the NO analyzer (Table 2). In the absence of GSH, incubation of 1 µM DEA/NO (in the presence of Mg2+ and SOD) for 12 min yielded 80±22 nM NO (measured by direct injection of the sample in the analyzer) and 607±28 nM nitrite (measured after treatment of the sample for 1 min with KI/I2). In the presence of 1 mM GSH we obtained 24±5 nM NO and 666±23 nM GSNO (measured after treatment of the sample with 4 mM CuSO4). The total sum of metabolites (NO, nitrite, and GSNO) amounted to 1216±37 nM (measured by the combined treatment of the sample with KI/I2 and CuSO4), from which we calculate a yield for nitrite of 525±34 nM. Strikingly, the presence of GSH had only a moderate effect on the nitrite yield, whereas the total yield of metabolites greatly increased.
Table 2.
Summary of yields of nitrite and GSNO from 1 µM DEA/NO in the absence and presence of 1 mM GSH observed with various assay methods.
| GSH | Mg2+ omitted | SOD omitted | KPi | Vessel closed | NO (µM) | NO2− (µM) | GSNO (µM) |
|---|---|---|---|---|---|---|---|
| − | I: 0.07±0.02 | ||||||
| − | II: 0.08±0.02 | II: 0.61±0.03 | |||||
| − | III: 0.60±0.05 | ||||||
| − | IV: 0.56±0.08 | ||||||
| − | V: 0.66±0.05 | ||||||
| − | VI: 0.43±0.02 | ||||||
| − | ⁎ | III: 0.37±0.06 | |||||
| − | ⁎ | V: 0.66±0.10 | |||||
| − | ⁎ | III: 0.48±0.05 | |||||
| − | ⁎ | III: 1.40±0.01 | |||||
| − | ⁎ | VI: 1.36±0.07 | |||||
| + | I: 0.02±0.01 | I: 0.94±0.05 | |||||
| + | II: 0.02±0.01 | II: 0.53±0.03 | II: 0.67±0.02 | ||||
| + | III: 0.53±0.05 | III: 0.69±0.04 | |||||
| + | IV: 0.71±0.10 | ||||||
| + | V: 0.74±0.09 | ||||||
| + | VI: 0.38±0.05 | VI: 1.20±0.20 | |||||
| + | ⁎ | I: 0.32±0.05 | |||||
| + | ⁎ | III: 0.36±0.01 | III: 0.25±0.01 | ||||
| + | ⁎ | V: 0.36 | |||||
| + | ⁎ | VI: 0.49±0.06 | |||||
| + | ⁎ | I: 0.42±0.06 | |||||
| + | ⁎ | VI: 0.56±0.07 | VI: 0.58±0.07 | ||||
| + | ⁎ | I: 0.51±0.01 | |||||
| + | ⁎ | III: 0.46±0.08 | III: 0.42±0.01 | ||||
| + | ⁎ | VI: 0.36±0.11 |
The following methods were used: NO electrode (method I), NO analyzer with KI/I2 and CuSO4 (method II), NO analyzer with sulfanilamide and KI/I2 (method III), fluorimeter with DAN (method IV), fluorimeter with commercial nitrite/nitrate kit (method V), and HPLC (method VI). See Materials and methods for further details. Experimental conditions: 1 µM DEA/NO, 1 mM GSH as indicated, 1000 U/ml SOD as indicated, 0.1 mM DTPA, and 5 mM MgCl2 as indicated in 50 mM triethanolamine–HCl (or 50 mM KPi) buffer (pH 7.4) in 0.5 ml at 37 °C; yields were determined after 12 min incubation in open or closed vessels as indicated.
We also determined GSNO and nitrite by the method introduced by Feelisch and co-workers [32]. With this method we obtained 531±52 nM nitrite and 694±37 nM GSNO, in good agreement with the results described above. When we applied this method to samples that were incubated in the absence of Mg2+, we obtained 372±6 nM nitrite (and no GSNO), in the absence of GSH, and 358±10 nM nitrite and 248±4 nM GSNO in its presence. No additional signals were observed after treatment with Hg2+, indicating that no other metabolites (nitrosamines, nitrosylated hemes) were formed. These results confirm the stimulatory effect of Mg2+ on GSNO formation observed with the NO electrode. Replacing TEA buffer with 50 mM KPi (pH 7.4) yielded 482±48 nM nitrite (and no GSNO) in the absence of GSH and 458±76 nM nitrite with 422±11 nM GSNO in its presence. These results corroborate the stronger nitrosation in TEA compared to phosphate observed with the electrode.
Determination of nitrite by fluorimetry
As a further method to quantify nitrite formation we applied a standard fluorimetric assay [33], which yielded 560±79 (n=4) and 713±97 nM (n=3) in the absence and presence of 1 mM GSH, respectively. A ready-to-use commercial kit yielded 665±49 and 737±93 nM in the absence and presence of 1 mM GSH, respectively (n=3). In the absence of Mg2+ the corresponding values were 656±102 and 360±19 nM, respectively. We also attempted to determine nitrate with the commercial kit, but the method proved not sensitive enough under the present conditions.
HPLC analysis of GSNO formation
In addition to the determinations of GSNO by the NO-sensitive electrode and the NO analyzer, we also measured GSNO by HPLC. Under our standard conditions (1 µM DEA/NO, 1 mM GSH, 5 mM Mg2+, 1000 U/ml SOD) we obtained 1198±202 nM GSNO. As with the other detection methods, omission of Mg2+ caused a large decrease in the nitrosothiol yield to 488±61 nM GSNO. Exchanging TEA buffer with phosphate diminished GSNO formation to 363±108 nM, whereas omission of SOD (in TEA buffer) reduced the GSNO yield to 579±74 nM (Table 2).
HPLC analysis of nitrite/nitrate formation
Finally, we determined nitrite/nitrate by HPLC with fluorimetric detection, after precolumn derivatization with DAN. Under standard conditions we obtained 425±24 and 376±50 nM nitrite in the absence and presence of 1 mM GSH, respectively. When the vessel was kept closed the yield of nitrite in the absence of GSH increased to 1361±70 nM, confirming the loss of NO to the atmosphere during incubation in a stirred open vessel. In the presence of GSH, omission of SOD caused a modest increase in the nitrite yield to 564±74 nM (Table 2).
Summary of product yields observed with various methods
Table 2 summarizes the yields of the main reaction products GSNO and nitrite as measured with various methods. The yield of the third potential reaction product nitrate could not be determined with sufficient accuracy because of a high background signal. Under standard conditions nitrite yields varied between 0.4 and 0.6 µM in the absence and 0.4 and 0.7 µM in the presence of GSH. In view of reported problems with batch-wise assays of nitrite and nitrate [40], we suspect the lower values (0.4 µM) are the more reliable. In the absence of GSH, nitrite was the only product observed, but we always found far less nitrite than the 1.5 µM NO that is released by 1 µM DEA/NO, confirming the large contribution that NO escape from solution makes to NO disappearance under the conditions of this study. In line with that assessment, the yield increased to 1.4 µM when the vessel was closed.
In the presence of GSH a similar yield of nitrite was accompanied by a large yield of GSNO with values between 0.7 and 1.2 µM, depending on the method used. Because of reported problems with iodine-based assays [41], we believe the higher yields observed with the NO electrode and by HPLC (0.9 and 1.2 µM, respectively) are the more reliable ones. The total product yield increased in the presence of GSH (to values between 1.2 and 1.6 µM depending on the method of detection used), which can be ascribed to the ability of GSH to capture NO before it escapes to the atmosphere. Significantly, autoxidation-mediated nitrosothiol formation would not have an effect on total product yield. The yield of GSNO measured with the NO electrode and by HPLC amounts to 64–82% of the total amount of NO produced, whereas for autoxidation-mediated nitrosation the theoretical maximal GSNO yield is only 50%, and the GSNO yield could never exceed the nitrite yield (see Eqs. (1)–(5)).
Discussion
At submicromolar NO concentrations nitrosothiols are formed from a direct reaction between NO and the thiol
According to the consensus view, aerobic nitrosothiol formation in the presence of NO involves a reaction between the thiol and NO2 and/or N2O3, which are formed as short-lived intermediates in the course of NO autoxidation (Eqs. (1)–(5)). However, the present data clearly demonstrate that at submicromolar concentrations the main route toward nitrosation involves a direct reaction of NO with the thiol:
-
(i)
The maximal NO concentration observed after introduction of DEA/NO decreases in the presence of the thiol. Similarly, the rate of decay of the NO is increased in the presence of GSH. The rate-limiting step in the autoxidation of NO is the reaction sequence represented by Eq. (1). Consequently, the reactions involving GSH (Eqs. (3) and (4)) cannot influence the kinetics of NO formation and decay (see also Supplementary Fig. S1), and the observation of such effects in this study rules out autoxidation-mediated nitrosation. Keszler et al. [28] ascribed a similar effect tentatively to radical reactions, mainly between NO and O2−, formed subsequent to thiyl radical formation in Eq. (4). However, the present studies were performed in the presence of SOD, ruling out such a scenario. Moreover, because of the second-order dependence of autoxidation on the NO concentration, one would predict the lowering of the NO peak by GSH to become more pronounced when the NO concentration increases, whereas the effect actually disappears at higher [NO].
-
(ii)
We observed a linear relationship between the GSNO-derived NO peak (after CuSO4 addition) and the DEA/NO concentration. Autoxidation-mediated nitrosation is expected to become more efficient at higher NO concentrations (see also Supplementary Fig. S3).
-
(iii)
The inhibition of nitrosation by oxy-Hb demonstrates that the reaction must involve free NO, ruling out a reaction of GSH with the NO donor.
-
(iv)We obtained yields of GSNO amounting to 64–82% of the NO formed. Because autoxidation-mediated nitrosation produces GSNO and nitrite in equal amounts (see the overall reaction in Eq. (6)), the yield of this process cannot exceed 50%:
2NO+1/2O2+GSH→GSNO+H++NO2− (6)
Mechanistic implications
This study implies a direct reaction between NO and GSH. A direct reaction between NO and thiols was previously proposed by Gow et al. [27]. According to their hypothesis, binding of NO to GSH results in a radical intermediate GSN•OH, which will be oxidized to GSNO in the presence of suitable oxidizing agents (Eqs. (7) and (8), in which A represents an electron acceptor). Under aerobic conditions, O2 will be reduced to O2− (Eq. (9)):
| NO+GSH↔GSN•OH | (7) |
| GSN•OH+A→GSNO+H++A− | (8) |
| GSN•OH+O2→GSNO+H++O2− | (9) |
Evidence was presented that anaerobically NAD+ can substitute for O2. Importantly, it was suggested that this mechanism accounts for GSNO formation at low (≤1 µM) concentrations of NO, with the conventional autoxidation-based mechanism predominating at higher concentrations (≥50 µM):
Gow et al. [27] reported the direct reaction to be second order in NO, yet at the same time noted that at higher NO concentrations (>50 µM) the familiar autoxidation-mediated process predominated. They suggested that NO autoxidation becomes more prominent at high NO concentrations because of the second-order dependence of autoxidation on the concentration of NO [27]. However, with both pathways (autoxidation and direct nitrosation) apparently exhibiting second-order dependence on [NO], the shift from direct to autoxidation-mediated nitrosation cannot be explained in this way. Mechanistically, the second-order character of the novel reaction was attributed to a rapid reaction between O2−, formed in the aerobic reaction (Eq. (9)), and a second NO molecule to produce peroxynitrite. However, as was also recently noted by Keszler et al. [28], such a scenario (a fast reaction following a slow initial step) would change the net stoichiometry (from 1 NO/GSNO to 2 NO/GSNO) but not the order of the reaction. The present data, however, vindicate the earlier study and support a mechanism along the lines proposed by Gow et al. The simplest way out of the conundrum is to assume that the reaction is actually first order in NO, as is observed by us and not ruled out by the results in Gow et al. [27].
In further confirmation of the reaction scheme represented by Eqs. (7) to (9), we also found the reaction to be dependent on O2. We should point out, however, that the data cannot discriminate the order in which NO and O2 react, so that a reaction between NO and a preformed GSOOH/GSOO− complex is also conceivable (Eqs. (10) and (11)). A third attractive possibility is that the reaction starts with the formation of a complex between NO and O2 (Eqs. (12) and (13)). The potential physiological relevance of the ONOO• complex, despite its low binding constant, has been argued [42]:
| O2+GSH↔GSOOH | (10) |
| GSOOH+NO→GSNO+H++O2− | (11) |
| O2+NO↔ONOO• | (12) |
| ONOO•+GSH→GSNO+H++O2− | (13) |
Reaction sequence (7)+(9) yields superoxide as a coproduct, which under our standard conditions is dismutated by SOD (Eq. (14)):
| 2O2−+2H++SOD→H2O2+O2+SOD | (14) |
In the absence of SOD rapid formation of peroxynitrite from O2− and NO would be expected to lower the NO peak, accelerate NO decay, and decrease the GSNO yield. We did indeed observe the last two effects, but the initial NO peak was hardly affected. Further studies will be required to resolve this issue.
GSH nitrosation vs GSH oxidation
The reaction between NO, O2, and GSH is known to yield GSSG in addition to GSNO by dimerization of GS• radicals (Eq. (15)) and, more importantly, by oxidation according to Eqs. (16) and (17). Determination of GSSG under the present conditions, with only micromolar levels of NO and millimolar concentrations of GSH, is impracticable. However, the overall reactions corresponding to GSSG formation (Eqs. (18) and (19)) differ from the overall reactions for autoxidation and for autoxidation-mediated nitrosation (Eqs. (20) and (21)) by consuming only two instead of four molecules of NO, while sharing the same rate-limiting step (NO2 formation). Consequently, the oxidative pathways should result in slower NO consumption and higher pre-Cu NO peaks. Obviously, Reactions (15)–(21) play no significant role under the present conditions:
| GS•+GS•→GSSG | (15) |
| GS•+GSH↔GSSG•−+H+ | (16) |
| GSSG•−+O2→GSSG+O2− | (17) |
| 2NO+O2+2GSH→GSSG+2H++2NO2− | (18) |
| 2NO+3O2+4GSH→2GSSG+4H++2NO2−+2O2− | (19) |
| 4NO+O2+2H2O→4H++4NO2− | (20) |
| 4NO+O2+2GSH→2GSNO+2H++2NO2− | (21) |
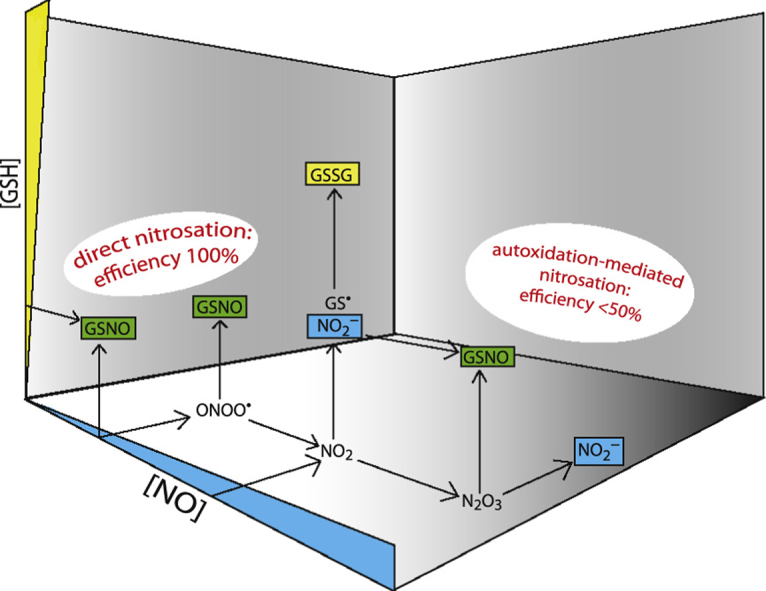
In autoxidation-mediated GSH oxidation, product distribution between GSNO and GSSG formation will be determined by competition for GS•, formed in Reaction (4), between NO (Reaction (5)), on one hand, and GS• and GSH (Reactions (15) and (16)) on the other. Consequently, lower NO levels are expected to result in dramatically diminished nitrosation. Indeed, with NO concentrations in the (sub)micromolar range, rather than the higher levels investigated in prior studies, autoxidation-mediated nitrosation is predicted to be almost negligible (~2%, see Supplementary Fig. S6). Instead, increased efficiency of nitrosation was observed in this study. This makes sense when one considers that distribution between direct nitrosation and autoxidation-mediated processes is determined by competition for NO (or possibly ONOO•) between GSH (nitrosative pathway) and NO (autoxidative pathway). Fig. 11 illustrates how nitrosation (direct and autoxidation mediated) and oxidation depend on the concentrations of NO and GSH.
Fig. 11.

Illustration of the way in which GSH nitrosation and oxidation are expected to depend on the concentrations of NO and GSH. At low (micromolar) [NO] and high (millimolar) [GSH] direct nitrosation will outcompete autoxidation, and a yield of 100% GSNO may be expected. Without direct nitrosation GSNO yields are expected to approach 0% under such conditions. When the NO concentration is raised, autoxidation will become more prominent, resulting in a mixture of oxidation and nitrosation with a limiting value of 50% for the GSNO yield at high [NO]. Please note that superoxide is assumed to be scavenged effectively; in the absence of SOD the maximal GSNO yield is expected to be 50% at both high and low [NO] (see also Supplementary Fig. S7).
Assessment of possible alternative effects of SOD
By eliminating O2− from the reaction mixture (Eq. (14)), SOD greatly simplifies the system and consequently limits the number of possible interpretations of the observations. However, SOD has been reported to catalyze other reactions that might affect the results. One potentially confounding factor is the ability of SOD to catalyze the decomposition of GSNO in the presence of GSH [43]. To investigate the relevance of this reaction under the present experimental conditions, we incubated GSNO (1 µM) in the presence of GSH (2 mM) with and without of SOD (10 µM) and measured the NO concentration continuously with the NO-sensitive electrode (results not shown). No NO formation was evident in the presence or absence of SOD, and similar NO peak heights were obtained when CuSO4 was added after 10 min (0.64±0.01 and 0.62±0.03 µM in the presence and absence of SOD, respectively), indicating that under our standard conditions (micromolar DEA/NO and SOD, millimolar GSH) SOD-catalyzed nitrosothiol decomposition is negligible. By contrast, when we repeated the experiment with 0.1 instead of 2 mM GSH, SOD did lower post-Cu2+ peak heights (from 0.43±0.01 to 0.20±0.03 µM, not shown). These observations agree with previous reports showing that GSNO decomposition catalyzed by SOD [43] or free Cu2+ ions [15] is stimulated by GSH at substoichiometric concentrations, but is gradually blocked when the GSH concentration is raised above the Cu2+ concentration. In summary, SOD-catalyzed GSNO decomposition is insignificant under the standard conditions of this study, but may have lowered nitrosothiol yields under conditions of submillimolar GSH (as in the experiments of Fig. 8). However, even then inclusion of SOD always caused a net increase in the nitrosation yield (results not shown), indicating that under those conditions SOD still acted mainly by scavenging superoxide.
SOD has also been reported to oxidize nitroxyl (HNO) to NO [44,45]. One should therefore account for the remote possibility that nitrosation takes place by Reactions (22) and (5). Reaction (22) is exceedingly unfavorable, but the reaction might be pulled toward the right if Reaction (5) is fast enough. In that case increased nitrosation in the presence of SOD might be ascribed to Reaction (23), because that would increase the maximal yield from 50 to 100%. However, this scenario can be ruled out on the same grounds that autoxidation-mediated nitrosation becomes negligible at low NO-to-GSH ratios, as GSH will outcompete NO for GS• (Reactions (5) and (16)):
| NO+GSH↔HNO+GS• | (22) |
| NO+GS•→GSNO | (5) |
| HNO+SOD→NO+H++e−+SOD | (23) |
Alternative thiols, alternative oxidants, and the effect of divalent cations
Direct nitrosation was not specific for GSH, but GSH was the most effective of the thiols tested. The very small Cu2+-mediated NO peaks observed with DTT and Cys are explained by the instability of the nitrosothiols that are formed with these compounds [46,47].
Although in this study O2 served as an obligatory coreactant, it is conceivable that other electron acceptors might fulfill this role. The original suggestion by Gow et al. [27] that NAD+ serves as an alternative electron acceptor was not confirmed here, perhaps because NAD+ is an obligate two-electron acceptor; physiological one-electron acceptors may provide better alternatives. An interesting candidate was recently suggested by Broniowska et al. [48], who reported stimulation of nitrosothiol formation in the presence of ferricytochrome c.
The striking stimulation of nitrosothiol formation by divalent cations has to the best of our knowledge not been reported before. The effect is not specific for Mg2+ or GSH, because similar results were obtained with Ca2+ or NAC. It is, however, peculiar to the direct nitrosation reaction, as it disappeared when the reaction was studied at higher DEA/NO concentrations, at which autoxidation-mediated nitrosation predominates. The underlying mechanism is currently unclear. Conceivably, divalent cations catalyze nitrosothiol formation in their capacity as Lewis acids by forming complexes not unlike the DNICs that have been proposed to play an important role in nitrosothiol formation [16]. The low solubility of Mg2+ in phosphate buffer is probably the cause of the smaller GSNO yields in KPi. The high intracellular concentration of Mg2+ suggests that stimulation of nitrosothiol formation by Mg2+ may be physiologically relevant.
Physiological implications
The central observation of this study is that GSH can be directly nitrosated by DEA/NO-derived NO with high efficiency. With an initial concentration of 1 µM DEA/NO, approximately 1 µM NO could be set free by CuSO4. With a NO–DEA/NO stoichiometry of ~1.5 [49], this corresponds to a level of nitrosation of ~2/3. This implies that nitrosation with 1 mM GSH was twice as fast as escape from solution, which we estimated to be ~0.005 s−1. If we accept the GSNO yield from the determinations by HPLC (1.2 µM), the rate of nitrosation might be 4× faster than the escape rate, in fair agreement with the fitting parameters of the observed NO time curves at 2 mM GSH (Supplementary Fig. S9). The rate of direct nitrosation in the presence of 2 mM GSH can thus be estimated at ~0.01–0.03 s−1.
Although the mechanism of endogenous cellular nitrosothiol formation is still a matter of dispute, there seems to be some consensus that NO/O2-mediated nitrosation is too slow to be physiologically relevant [3,5,6,8,10–12,18,20,24,48]. However, most previous studies were carried out at unphysiologically high NO concentrations. A recent analysis has estimated physiological NO concentrations to be 5 nM or less [50]. From the present results it can be estimated that in the presence of 1 mM GSH a steady-state concentration of 1 nM NO will give rise to a rate of GSNO formation of ~10 pM/s or 36 nM/h. For comparison, autoxidation-mediated nitrosation is predicted to produce ~1.4×10−3 pM/s GSNO under the same conditions (with ~0.2 mM O2 and a rate constant for autoxidation of 7×106 M−2 s−1 [36]). Even taking into account the described acceleration in hydrophobic compartments (300-fold [51]), autoxidation-mediated nitrosation would be a minor process (~0.4 pM/s).
In view of present uncertainties regarding nitrosothiol concentrations in vivo it is difficult to assess the physiological relevance of the predicted rate for the direct reaction reported here, but it would probably fit reported values at the lower end of the spectrum [12,41]. Indeed, the formation of physiologically relevant levels of nitrosothiols (specifically of S-nitrosoalbumin) by a reaction involving O2 and low fluxes of NO in plasma and even in whole blood has been reported [52], which strongly suggests that the reactions observed in this study may be operative in vivo. Conversely, the present observations offer a mechanistic explanation for the paradoxical results reported in that study. Summarized, our results put direct nitrosation by NO back in contention as a serious candidate for biological nitrosothiol formation.
Acknowledgments
This work was supported by the Austrian Science Fund: P23135 (to A.C.F.G.) and P20669, P21693, and P24005 (to B.M.).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.freeradbiomed.2013.04.034.
Appendix A. Supporting information
Supporting material
References
- 1.Gao Y. The multiple actions of NO. Pflugers Arch. Eur. J. Physiol. 2010;459:829–839. doi: 10.1007/s00424-009-0773-9. [DOI] [PubMed] [Google Scholar]
- 2.Friebe A., Koesling D. The function of NO-sensitive guanylyl cyclase: what we can learn from genetic mouse models. Nitric Oxide. 2009;21:149–156. doi: 10.1016/j.niox.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 3.Thomas D.D., Ridnour L.A., Isenberg J.S., Flores-Santana W., Switzer C.H., Donzelli S., Hussain P., Vecoli C., Paolocci N., Ambs S., Colton C.A., Harris C.C., Roberts D.D., Wink D.A. The chemical biology of nitric oxide: implications in cellular signaling. Free Radic. Biol. Med. 2008;45:18–31. doi: 10.1016/j.freeradbiomed.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Butler A.R., Rhodes P. Chemistry, analysis, and biological roles of S-nitrosothiols. Anal. Biochem. 1997;249:1–9. doi: 10.1006/abio.1997.2129. [DOI] [PubMed] [Google Scholar]
- 5.Gaston B. Nitric oxide and thiol groups. Biochim. Biophys. Acta. 1999;1411:323–333. doi: 10.1016/s0005-2728(99)00023-7. [DOI] [PubMed] [Google Scholar]
- 6.Hogg N. The biochemistry and physiology of S-nitrosothiols. Annu. Rev. Pharmacol. Toxicol. 2002;42:585–600. doi: 10.1146/annurev.pharmtox.42.092501.104328. [DOI] [PubMed] [Google Scholar]
- 7.Mannick J.B., Schonhoff C.M. Nitrosylation: the next phosphorylation? Arch. Biochem. Biophys. 2002;408:1–6. doi: 10.1016/s0003-9861(02)00490-3. [DOI] [PubMed] [Google Scholar]
- 8.Miersch S., Mutus B. Protein S-nitrosation: biochemistry and characterization of protein thiol–NO interactions as cellular signals. Clin. Biochem. 2005;38:777–791. doi: 10.1016/j.clinbiochem.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 9.Derakhshan B., Hao G., Gross S.S. Balancing reactivity against selectivity: the evolution of protein S-nitrosylation as an effector of cell signaling by nitric oxide. Cardiovasc. Res. 2007;75:210–219. doi: 10.1016/j.cardiores.2007.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akaike T. Mechanisms of biological S-nitrosation and its measurement. Free Radic. Res. 2000;33:461–469. doi: 10.1080/10715760000301001. [DOI] [PubMed] [Google Scholar]
- 11.Schrammel A., Gorren A.C.F., Schmidt K., Pfeiffer S., Mayer B. S-nitrosation of glutathione by nitric oxide, peroxynitrite, and •NO/O2•−. Free Radic. Biol. Med. 2003;34:1078–1088. doi: 10.1016/s0891-5849(03)00038-8. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y., Hogg N. S-nitrosothiols: cellular formation and transport. Free Radic. Biol. Med. 2005;38:831–838. doi: 10.1016/j.freeradbiomed.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 13.Lancaster J.R., Jr. Protein cysteine thiol nitrosation: maker or marker of reactive nitrogen species-induced nonerythroid cellular signaling? Nitric Oxide. 2008;19:68–72. doi: 10.1016/j.niox.2008.04.028. [DOI] [PubMed] [Google Scholar]
- 14.Stubauer G., Giuffrè A., Sarti P. Mechanism of S-nitrosothiol formation and degradation mediated by copper ions. J. Biol. Chem. 1999;274:28128–28133. doi: 10.1074/jbc.274.40.28128. [DOI] [PubMed] [Google Scholar]
- 15.Gorren A.C.F., Schrammel A., Schmidt K., Mayer B. Decomposition of S-nitrosoglutathione in the presence of copper ions and glutathione. Arch. Biochem. Biophys. 1996;330:219–228. doi: 10.1006/abbi.1996.0247. [DOI] [PubMed] [Google Scholar]
- 16.Vanin A.F. Dinitrosyl iron complexes with thiolate ligands: physico-chemistry, biochemistry and physiology. Nitric Oxide. 2009;21:1–13. doi: 10.1016/j.niox.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 17.Bosworth C.A., Toledo J.C., Jr, Zmijewski J.W., Li Q., Lancaster J.R., Jr. Dinitrosyliron complexes and the mechanism(s) of cellular protein nitrosothiol formation from nitric oxide. Proc. Natl. Acad. Sci. USA. 2009;106:4671–4676. doi: 10.1073/pnas.0710416106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inoue K., Akaike T., Miyamoto Y., Okamoto T., Sawa T., Otagiri M., Suzuki S., Yoshimura T., Maeda H. Nitrosothiol formation catalyzed by ceruloplasmin: implication for cytoprotective mechanism in vivo. J. Biol. Chem. 1999;274:27069–27075. doi: 10.1074/jbc.274.38.27069. [DOI] [PubMed] [Google Scholar]
- 19.Jia L., Bonaventura C., Bonaventura J., Stamler J.S. S-nitrosohaemoglobin: a dynamic activity of blood involved in vascular control. Nature. 1996;380:221–226. doi: 10.1038/380221a0. [DOI] [PubMed] [Google Scholar]
- 20.Espey M.G., Thomas D.D., Miranda K.M., Wink D.A. Focusing of nitric oxide mediated nitrosation and oxidative nitrosylation as a consequence of reaction with superoxide. Proc. Natl. Acad. Sci. USA. 2002;99:11127–11132. doi: 10.1073/pnas.152157599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mayer B., Pfeiffer S., Schrammel A., Koesling D., Schmidt K., Brunner F. A new pathway of nitric oxide/cyclic GMP signaling involving S-nitrosoglutathione. J. Biol. Chem. 1998;273:3264–3270. doi: 10.1074/jbc.273.6.3264. [DOI] [PubMed] [Google Scholar]
- 22.Wink D.A., Nims R.W., Darbyshire W.F., Christodoulou D., Hanbauer I., Cox G.W., Laval F., Laval J., Cook J.A., Krishna M.C., DeGraff W.G., Mitchell J.B. Reaction kinetics for nitrosation of cysteine and glutathione in aerobic nitric oxide solutions at neutral pH: insights into the fate and physiological effects of intermediates generated in the NO/O2 reaction. Chem. Res. Toxicol. 1994;7:519–525. doi: 10.1021/tx00040a007. [DOI] [PubMed] [Google Scholar]
- 23.Kharitonov V.G., Sundquist A.R., Sharma V.S. Kinetics of nitrosation of thiols by nitric oxide in the presence of oxygen. J. Biol. Chem. 1995;270:28158–28164. doi: 10.1074/jbc.270.47.28158. [DOI] [PubMed] [Google Scholar]
- 24.Goldstein S., Czapski G. Mechanism of the nitrosation of thiols and amines by oxygenated •NO solutions: the nature of the nitrosating intermediates. J. Am. Chem. Soc. 1996;118:3419–3425. [Google Scholar]
- 25.Keshive M., Singh S., Wishnok J.S., Tannenbaum S.R., Deen W.M. Kinetics of S-nitrosation of thiols in nitric oxide solutions. Chem. Res. Toxicol. 1996;9:988–993. doi: 10.1021/tx960036y. [DOI] [PubMed] [Google Scholar]
- 26.Jourd’heuil D., Jourd’heuil F.L., Feelisch M. Oxidation and nitrosation of thiols at low molecular exposure to nitric oxide: evidence for a free radical mechanism. J. Biol. Chem. 2003;278:15720–15726. doi: 10.1074/jbc.M300203200. [DOI] [PubMed] [Google Scholar]
- 27.Gow A.J., Buerk D.G., Ischiropoulos H. A novel reaction mechanism of the formation of S-nitrosothiol in vivo. J. Biol. Chem. 1997;272:2841–2845. doi: 10.1074/jbc.272.5.2841. [DOI] [PubMed] [Google Scholar]
- 28.Keszler A., Zhang Y., Hogg N. Reaction between nitric oxide, glutathione, and oxygen in the presence and absence of protein: how are S-nitrosothiols formed? Free Radic. Biol. Med. 2010;48:55–64. doi: 10.1016/j.freeradbiomed.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mayer B., Klatt P., Werner E.R., Schmidt K. Kinetics and mechanism of tetrahydrobiopterin-induced oxidation of nitric oxide. J. Biol. Chem. 1995;270:655–659. doi: 10.1074/jbc.270.2.655. [DOI] [PubMed] [Google Scholar]
- 30.Pfeiffer S., Schrammel A., Schmidt K., Mayer B. Electrochemical determination of S-nitrosothiols with a Clark-type nitric oxide electrode. Anal. Biochem. 1998;258:68–73. doi: 10.1006/abio.1998.2562. [DOI] [PubMed] [Google Scholar]
- 31.Wenzl M.V., Beretta M., Griesberger M., Russwurm M., Koesling D., Schmidt K., Mayer B., Gorren A.C.F. Site-directed mutagenesis of aldehyde dehydrogenase-2 suggests three distinct pathways of nitroglycerin biotransformation. Mol. Pharmacol. 2011;80:258–286. doi: 10.1124/mol.111.071704. [DOI] [PubMed] [Google Scholar]
- 32.Feelisch M., Rassaf T., Mnaimneh S., Singh N., Bryan N.S., Jourd’heuil D., Kelm M. Concomitant S-, N-, and heme-nitros(yl)ation in biological tissues and fluids: implications for the fate of NO in vivo. FASEB J. 2002;16:1775–1785. doi: 10.1096/fj.02-0363com. [DOI] [PubMed] [Google Scholar]
- 33.Misko T.P., Schilling R.J., Salvemini D., Moore W.M., Currie M.G. A fluorometric assay for the measurement of nitrite in biological samples. Anal. Biochem. 1993;214:11–16. doi: 10.1006/abio.1993.1449. [DOI] [PubMed] [Google Scholar]
- 34.Wölkart G., Pang X., Stessel H., Kirchengast M., Brunner F. Chronic endothelin-A receptor antagonism is as protective as angiotensin converting enzyme inhibition against cardiac dysfunction in diabetic rats. Br. J. Pharmacol. 2007;151:1187–1197. doi: 10.1038/sj.bjp.0707325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ford P.C., Fernandez B.O., Lim M.D. Mechanisms of reductive nitrosylation in iron and copper models relevant to biological systems. Chem. Rev. 2005;105:2439–2455. doi: 10.1021/cr0307289. [DOI] [PubMed] [Google Scholar]
- 36.Schmidt K., Desch W., Klatt P., Kukovetz W.R., Mayer B. Release of nitric oxide from donors with known half-life: a mathematical model for calculating nitric oxide concentrations in aerobic solutions. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1997;355:457–462. doi: 10.1007/pl00004969. [DOI] [PubMed] [Google Scholar]
- 37.Ford P.C., Wink D.A., Stanbury D.M. Autoxidation kinetics of aqueous nitric oxide. FEBS Lett. 1993;326:1–3. doi: 10.1016/0014-5793(93)81748-o. [DOI] [PubMed] [Google Scholar]
- 38.Hrabie J.A., Klose J.R., Wink D.A., Keefer D.A. New nitric oxide-releasing zwitterions derived from polyamines. J. Org. Chem. 1993;58:1472–1476. [Google Scholar]
- 39.Saavedra J.E., Southan G.J., Davies K.M., Lundell A., Markou C., Hanson S.R., Adrie C., Hurford W.E., Zapol W.M., Keefer L.K. Localizing antithrombotic and vasodilatory activity with a novel, ultrafast nitric oxide donor. J. Med. Chem. 1996;39:4361–4365. doi: 10.1021/jm960616s. [DOI] [PubMed] [Google Scholar]
- 40.Tsikas D. Methods of quantitative analysis of the nitric oxide metabolites nitrite and nitrate in human biological fluids. Free Radic. Res. 2005;39:797–815. doi: 10.1080/10715760500053651. [DOI] [PubMed] [Google Scholar]
- 41.Gow A., Doctor A., Mannick J., Gaston B. S-nitrosothiol measurements in biological systems. J. Chromatogr. B. 2007;851:140–151. doi: 10.1016/j.jchromb.2007.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Czapski G., Goldstein S. The role of the reactions of •NO with superoxide and oxygen in biological systems: a kinetic approach. Free Radic. Biol. Med. 1995;19:785–794. doi: 10.1016/0891-5849(95)00081-8. [DOI] [PubMed] [Google Scholar]
- 43.Jourd’heuil D., Laroux F.S., Miles A.M., Wink D.A., Grisham M.B. Effect of superoxide dismutase on the stability of S-nitrosothiols. Arch. Biochem. Biophys. 1999;361:323–330. doi: 10.1006/abbi.1998.1010. [DOI] [PubMed] [Google Scholar]
- 44.Murphy, M.E.; Sies, H. Reversible conversion of nitroxyl anion to nitric oxide by superoxide dismutase. Proc. Natl. Acad. Sci. USA 88: 10860-10864. [DOI] [PMC free article] [PubMed]
- 45.Fukuto, J.M.; Cisneros, C.J.; Kinkade, R.L. A comparison of the chemistry associated with the biological signaling and actions of nitroxyl (HNO) and nitric oxide (NO). J. Inorg. Biochem. 118: 201-208. [DOI] [PubMed]
- 46.Tullett J.M., Rees D.D., Shuker D.E.G., Gescher A. Lack of correlation between the observed stability and pharmacological properties of S-nitroso derivatives of glutathione and cysteine-related peptides. Biochem. Pharmacol. 2001;62:1239–1247. doi: 10.1016/s0006-2952(01)00750-x. [DOI] [PubMed] [Google Scholar]
- 47.Liebeskind S., Korth H.-G., de Groot H., Kirsch M. Dependence of product formation from decomposition of nitroso-dithiols on the degree of nitrosation: evidence that dinitroso-dithiothreitol acts solely as an nitric oxide releasing compound. Org. Biomol. Chem. 2008:2560–2573. doi: 10.1039/b801583j. [DOI] [PubMed] [Google Scholar]
- 48.Broniowska K.A., Keszler A., Basu S., Kim-Shapiro D.B., Hogg N. Cytochrome c-mediated formation of S-nitrosothiol in cells. Biochem. J. 2012;442:191–197. doi: 10.1042/BJ20111294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maragos C.M., Morley D., Wink D.A., Dunams T.M., Saavedra J.E., Hoffman A., Bove A.A., Isaac L., Hrabie J.A., Keefer L.K. Complexes of •NO with nucleophiles as agents for the controlled biological release of nitric oxide: vasorelaxant effects. J. Med. Chem. 1991;34:3242–3247. doi: 10.1021/jm00115a013. [DOI] [PubMed] [Google Scholar]
- 50.Hall C.N., Garthwaite J. What is the real physiological NO concentration in vivo? Nitric Oxide. 2009;21:92–103. doi: 10.1016/j.niox.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu X., Miller M.J.S., Joshi M.S., Thomas D.D., Lancaster J.R., Jr. Accelerated reaction of nitric oxide with O2 within the hydrophobic interior of biological membranes. Proc. Natl. Acad. Sci. USA. 1998;95:2175–2179. doi: 10.1073/pnas.95.5.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marley R., Patel R.P., Orie N., Ceaser E., Darley-Usmar V., Moore K. Formation of nanomolar concentrations of S-nitrosoalbumin in human plasma by nitric oxide. Free Radic. Biol. Med. 2001;31:688–696. doi: 10.1016/s0891-5849(01)00627-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting material