

Abstract
Spindle cell rhabdomyosarcoma (RMS) is a rare form of RMS with different clinical characteristics and behavior between children and adult patients. Its genetic hallmark remains unknown and it remains debatable if there is pathogenetic relationship between the spindle cell and the so-called sclerosing RMS. We studied two pediatric and one adult spindle cell RMS by next generation RNA sequencing and used FusionSeq for data analysis to detect novel fusions. An SRF-NCOA2 gene fusion was detected in a spindle cell RMS from the posterior neck in a 7 month-old child. The fusion matched the tumor karyotype and was further confirmed by fluorescence in situ hybridization (FISH) and by RT-PCR, which showed fusion of SRF exon 6 to NCOA2 exon 12. Additional 14 spindle cell (from 8 children and 6 adults) and 4 sclerosing (from 2 children and 2 adults) RMS were tested by FISH for the presence of abnormalities in NCOA2, SRF, as well as for PAX3 and NCOA1, identifying NCOA2 rearrangements in two additional spindle cell RMS from a 3 month-old and a 4 week-old child, both arising in the chest wall. In the latter tumor, TEAD1 was identified by rapid amplification of cDNA ends (RACE) to be the NCOA2 gene fusion partner. None of the adult tumors were positive for NCOA2 rearrangement. Despite similar histomorphology in adults and young children, these results suggest that spindle cell RMS is a heterogeneous disease genetically as well as clinically. Our findings also support a relationship between NCOA2-rearranged spindle cell RMS occurring in young childhood and the so-called congenital RMS, which often displays rearrangements at 8q13 locus (NCOA2).
Keywords: rhabdomyosarcoma, spindle cell, NCOA2, SRF, TEAD1, translocation, infantile
INTRODUCTION
Rhabdomyosarcoma (RMS) is the most common soft tissue sarcoma in childhood, representing 5–8% of all malignancies in children. On the basis of histological and molecular criteria, RMS is classified into two major subgroups, namely the more frequent embryonal rhabdomyosarcoma (ERMS, 60%) and the less common alveolar rhabdomyosarcoma (ARMS, 20%). A great majority of the ARMS are associated with recurrent chromosomal translocations (fusion-positive ARMS). Of these, 75% are t(2;13)(q35;q14) and 25% are t(1;13)(p36;q14), leading to the fusion of either PAX3 on 2q35 or the related transcription factor PAX7 on 1p36 to another transcription factor named FOXO1A on 13q14. The remaining 20% of ARMS are translocation-negative (fusion-negative ARMS) and form a more heterogeneous group, of which the unambiguous classification and discrimination from ERMS based on classical methods such as histology and immunohistochemistry remains challenging.
A rare form of RMS is the congenital form of ERMS, known to arise primarily in the genitourinary tract of developing fetuses and young children in the neonatal period. However the morphologic and genetic characteristics of the congenital and neonatal RMS are poorly understood, with conflicting data in the literature.
The spindle cell variant is an uncommon subtype of rhabdomyosarcoma, initially described in the paratesticular and head and neck regions of children and associated with a low malignant potential (Cavazzana et al., 1992; Leuschner et al., 1993). In adults, the preferred location of spindle cell RMS is the head and neck region, and in contrast with the pediatric counterpart they follow a more aggressive clinical course (Nascimento and Fletcher, 2005). A subset of spindle cell RMS may display areas of prominent hyaline sclerosis and pseudo-vascular growth pattern, suggesting morphologic overlap with the even less common sclerosing type RMS (Nascimento and Fletcher, 2005). As both spindle and sclerosing RMS have similar clinical presentations, it was suggested that they may represent a histologic spectrum of a single pathologic entity (Mentzel and Katenkamp, 2000; Mentzel, 2010). However, no genetic studies are available to address this hypothesis.
The goal of this study was to investigate a group of pediatric and adult spindle cell RMS by next generation RNA sequencing and to identify potential novel fusions, which can be then validated in larger cohorts of RMS.
MATERIAL AND METHODS
Patient selection
Archival material from adult and pediatric patients with diagnosis of spindle cell or sclerosing RMS was retrieved from Pathology files at Memorial Sloan-Kettering Cancer Center and Weill Medical College of Cornell University/New York-Presbyterian Hospital. Twenty-one cases were identified and the diagnosis was confirmed based on a constellation of morphologic appearance, immunohistochemical reactivity for desmin and myogenin, as well as the lack of known gene fusions. Next-generation RNA sequencing was performed on fresh frozen tissue of three cases (RMS1, RMS2 and RMS3). Formalin-fixed paraffin-embedded (FFPE) tissue was also available for the additional 18 cases for further evaluation and validation assays (Table 1). Furthermore, a control group of 4 embryonal rhabdomyosarcomas and 3 ectomesenchymomas [an infantile primitive sarcoma composed of both embryonal rhabdomyosarcoma and (ganglio)neuroblastoma components] were studied for comparison. The study was approved by the Institutional Review Board at each institution (IRB# 02-060 MSKCC and IRB# 1007011157 WCMC).
TABLE I.
Summary of Clinicopathologic Characteristics, FISH, and RT-CPR Results
| Clinicopathologic Characteristics |
FISH Results |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| RMS# | Age | Sex | Location | Histologic Type | NCOA2 | SRF | TEAD1 | NCOA1 | PAX3 | FOXO1 a |
| 1b | 13 y | F | Thigh | Spindle cell | − | − | − | |||
| 2b,c | 7 mo | M | Posterior neck | Spindle cell | + | + | ||||
| 3b | 21 y | M | Pelvis | Spindle cell | − | − | − | |||
| 4d | 4 wks | M | Chest wall | Spindle cell | + | − | + | |||
| 5 | 3 mo | F | Chest wall | Spindle cell | + | − | − | − | ||
| 6 | 2 y | F | Buttock | Spindle cell | − | − | − | |||
| 7 | 2 y | M | Abdominal/pelvic | Spindle cell | − | − | − | |||
| 8 | 2 y | M | Paratesticular | Spindle cell | − | − | − | |||
| 9 | 6 y | M | Abdominal/pelvic | Spindle cell | − | − | − | |||
| 10 | 10 y | F | Paraspinal | Spindle cell | − | − | − | |||
| 11 | 14 y | M | Flank | Spindle cell, post-RT | − | − | − | |||
| 12 | 30 y | M | Spermatic cord | Spindle cell | − | − | − | − | ||
| 13 | 31 y | M | Prostate | Spindle cell | − | − | − | |||
| 14 | 50 y | M | Diaphragm | Spindle cell | − | − | − | |||
| 15 | 60 y | M | Abdominal wall | Spindle cell | − | − | − | |||
| 16 | 71 y | M | Neck | Spindle cell | − | − | − | |||
| 17 | 76 y | M | Thigh | Spindle cell | − | − | − | |||
| 18 | 13 y | F | Chest wall | Sclerosing | − | − | − | − | ||
| 19 | 15 y | F | Infra-temporal fossa | Sclerosing | − | − | − | − | ||
| 20 | 34 y | F | Buccal/masticator space | Sclerosing | − | − | − | − | ||
| 21 | 39 y | M | Lower leg | Sclerosing | − | − | − | |||
Y, year; mo, months; wks, weeks; RT, radiation therapy.
Cases tested either for FOXO1 by FISH or by RT-PCR for PAX3/7-FOXO1 fusion transcripts.
Tested by RNA-Seq and data-analyzed by Fusion-Seq.
RNA-seq results were confirmed by the presence of a t(6;8)(p12;q11.2) translocation and by RT-PCR assay.
TEAD1-NC0A2 fusion was confirmed by RT-PCR as well by the presence of a t(8;11) translocation on conventional karyotyping.
Clinicopathologic features
Hematoxylin and eosin (H&E) stained slides from all cases were reviewed by two pathologists (JMM and CRA). The diagnosis of spindle cell RMS was defined as the presence of a predominantly spindle cell morphology, lacking overt nuclear pleomorphism and only focal evidence of rhabdomyoblastic differentiation. The sclerosing RMS were defined as relatively hypocellular lesions, showing an abundant hyalinized collagenous stroma, with embedded spindle or strap cells, arranged in a deceptive pseudovascular or tubular-like pattern. Immunohistochemical confirmation of desmin reactivity and at least focal myogenin staining was required for diagnosis. RT-PCR for PAX3/7-FOXO1 fusions (5 cases), or FISH for PAX3 and/or FOXO1 gene rearrangement (14 cases) was performed, and results were negative (Table 1).
RNA sequencing
Total RNA was prepared for RNA sequencing in accordance with the standard Illumina mRNA sample preparation protocol (Illumina). Briefly, mRNA was isolated with oligo(dT) magnetic beads from total RNA (10 μg) extracted from cases RMS1, RMS2, and RMS3. The mRNA was fragmented by incubation at 94°C for 2.5 min in fragmentation buffer (Illumina). To reduce the inclusion of artifact chimeric transcripts into the sequencing library, an additional gel size-selection step was introduced prior to the adapter ligation step(Quail et al., 2008). Size-ranges captured were 300-350 bp during the first size-selection step and then 400-450 bp for the second size-selection step after the ligation of the adapters. The adaptor-ligated library was then enriched by PCR for 15 cycles and purified. The library was sized and quantified using DNA1000 kit (Agilent) on an Agilent 2100 Bioanalyzer according to the manufacturer’s instructions. Paired-end RNA-sequencing at read lengths of 50 or 51 bp was performed with the Genome Analyzer IIx (Illumina). A total of about 268 million paired-end reads were generated, corresponding to about 27 billion bases (Supplementary Table 1).
Analysis of RNA sequencing results with FusionSeq
All reads were independently aligned with the CASAVA 1.8 software provided by Illumina against the human genome sequence (hg19) and a splice junction library, simultaneously. The splice junction library was generated by considering all possible junctions between exons of each transcript. We considered the University of California, Santa Cruz (UCSC) Known Genes annotation set (Hsu et al., 2006) to generate this library via RSEQtools, a computational method for processing RNA-seq data (Habegger et al., 2011). The mapped reads were converted into Mapped Read Format (Habegger et al., 2011) and analyzed with FusionSeq (Sboner et al., 2010) to identify potential fusion transcripts. FusionSeq is a computational method successfully applied to paired-end RNA-seq experiments for the identification of chimeric transcripts (Pflueger et al., 2011; Tanas et al., 2011; Pierron et al., 2012). Briefly, we first analyzed the paired-end reads by selecting those pairs that mapped to different genes [according to the UCSC Known Genes annotation set (Hsu et al., 2006)], thus indicating potential chimeric candidates. Spurious fusion transcript candidates were then eliminated through a cascade of filters, each talking different sources of noise in RNA-sequencing experiments: misalignment due to highly similar paralogous genes, ambiguous mapping resulting from repetitive regions, random pairing of transcript fragments generated during sample preparation, genes that resembled highly abundant ribosomal RNA, or PCR artifacts. Once we generated a confident list of fusion candidates, several statistics are computed to rank the candidates and prioritize the experimental validation. In these cases, we used the DASPER score (Difference between the observed and Analytically calculated expected SPER): a higher DASPER score indicates a greater likelihood that the fusion candidate is authentic and did not occur randomly. In addition, the candidate fusions were classified in terms of whether they represented two genes on different chromosomes (interchromosomal) or two genes on the same chromosome (intrachromosomal). See Sboner A, et al. (Sboner et al., 2010) for further details about FusionSeq.
Fluorescence in situ hybridization (FISH)
FISH on interphase nuclei from paraffin embedded 4-micron sections was performed applying custom probes using bacterial artificial chromosomes (BAC), covering and flanking PAX3, NCOA1, NCOA2, SRF, and TEAD1, (Supplementary Table 2). BAC clones were chosen according to USCS genome browser (http://genome.uscs.edu) and obtained from BACPAC sources of Children’s Hospital of Oakland Research Institute (CHORI) (Oakland, CA) (http://bacpac.chori.org). DNA from individual BACs was isolated according to the manufacturer’s instructions, labeled with different fluorochromes in a nick translation reaction, denatured, and hybridized to pretreated slides. Slides were then incubated, washed, and mounted with DAPI in an antifade solution. The genomic location of each BAC set was verified by hybridizing them to normal metaphase chromosomes. Two hundred successive nuclei were examined using a Zeiss fluorescence microscope (Zeiss Axioplan, Oberkochen, Germany), controlled by Isis 5 software (Metasystems). A positive score was interpreted when at least 20% of the nuclei showed a break-apart signal. Nuclei with incomplete set of signals were omitted from the score.
Rapid amplification of cDNA ends (RACE) and Reverse Transcriptase Polymerase Chain Reaction (RT-PCR)
Total RNA was extracted from frozen tumor tissue using the Trizol reagent according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA). The quality of RNA was assessed by RT-PCR for PGK housekeeping gene. One microgram of total RNA was reverse-transcribed to cDNA, followed by 5′-RACE, using the SMARTer™ RACE cDNA Amplification Kit (Clonetech, Mountain View, CA). Reverse transcribed mRNA was initiated by SMARTer II A Oligonucleotide with appropriate 5′-RACE CDS Primer A in a 10 μl reaction volume according to the manufacturer’s protocol. Primary PCR was performed by Advantage® 2 polymerase chain reaction (Clontech, Mountain View, CA) with the SMARTer™ RACE Universal Primer A Mix and reverse primer on NCOA2 exon 14 (5′- GGAACCCAGCAGCCAGCATC -3′). Nested PCR was performed with the SMARTer™ RACE Nested Universal Primer A and reverse primer on NCOA2 exon 13 (5′- TCAAGGGATGATAGGAAACCAAGG -3′). Amplified PCR products were being cloned by TOPO® TA Cloning® Kit for Sequencing with One Shot® TOP10 Chemically Competent E. coli (Invitrogen, Carlsbad, CA). The constructed plasmid DNA was sequenced using Sanger’s method. To confirm RACE results, RT-PCR was performed by SuperScript® First-Strand Synthesis System for RT PCR (Invitrogen, Carlsbad, CA). Primers used for RT-PCR validation included: SRF Exon 4 Fwd (5′-CAGCAGCACAGACCTCACGCAG-3′), TEAD1 exon 6 Fwd (5′-GCCACTGCCATTCATAACAAGC-3′), and NCOA2 exon 13 Rev (same as above). The PCR products were analyzed by electrophoresis. The amplified PCR products were sequenced using the Sanger’s method.
RESULTS
Patient’s Characteristics
Among the 17 spindle cell RMS included in the study, 10 occurred in children, while remaining 7 in adults (older than 18 years of age). Among the pediatric cases, three were infants presenting with truncal soft tissue masses: a 4-week male (chest wall), a 3-month old female (chest wall) and a 7 month-old male (posterior neck/thoracic paraspinal). The remaining 7 spindle cell RMS occurred in children between 2-14 years of age, arising preferentially in truncal (3 cases) or intra-abdominal/pelvic (3 cases) locations, with only one case occurring in extremities (thigh).
Most adult spindle cell RMS occurred in males, with a wide age distribution at diagnosis, range: 21-76 years old (mean 48 years). The most common anatomic location was intra-abdominal/pelvic (5 cases) with one case each occurring in the extremity (thigh) and head and neck. In contrast, three of the 4 sclerosing RMS were diagnosed in females, with ages at diagnosis: 13, 15 and 34 years (mean 20 years), while one case occurred in an 87 year-old man in the skull in the field of prior radiation. Three of the cases occurred in the head and neck area and one in the chest wall.
Histopathology
All 17 cases diagnosed as spindle cell RMS were composed predominantly of long, intersecting fascicles of spindle cells with scattered rhabdomyoblasts and easily identifiable mitoses. Necrosis was present in most cases but comprised a minor component of tumor volume. Eight cases demonstrated histological features resembling leiomyosarcoma, with elongated nuclei and fibrillary eosinophilic cytoplasm, including the case with SRF-NCOA2 fusion (RMS2) (Figure 1A, and 2A). Six cases featured distinctive herringbone-like areas reminiscent of ‘fibrosarcoma’, and the appearance of one case resembled malignant-peripheral nerve sheath tumor (MPNST) (Figure 2B to C). Four cases were predominantly hypocellular, composed of bland spindle cells and rare rhabdomyoblasts embedded in a densely hyalinized background. Focally they had a distinctive pseudovascular appearance (Figure 2 D) and were classified as sclerosing RMS. One case arising in the chest wall of a 4-week old child (RMS4, positive for NCOA2-TEAD1 fusion) showed bland, hypocellular areas reminiscent of infantile fibromatosis admixed with high grade, hypercellular components arranged in herringbone fascicles, with high mitotic activity and scattered pleomorphism (Figure 3A, B). An additional case arising in the chest wall of a 3-month old child (RMS5, positive for NCOA2 gene rearrangement) had spindle cell histomorphology at presentation, but after treatment, the tumor showed features of sclerosing RMS, and focally areas similar to solid variant of ARMS (Supplementary Figure 1A, B). All cases were positive for desmin and demonstrated multifocal positivity for myogenin (Figure 1 B, C). The reactivity for desmin was diffuse and strong in all except one case (RMS4), in which the staining pattern was quite patchy (Figure 3C). These morphologic features are consistent with previous descriptions of both spindle cell and sclerosing RMS in adults and children (Folpe et al., 2002; Chiles et al., 2004; Nascimento and Fletcher, 2005; Mentzel and Kuhnen, 2006).
Figure 1. Spindle cell rhabdomyosarcoma with SRF-NCOA2 fusion.

This tumor is from a 7-month old child who presented with a posterior neck mass (case RMS2). (A) It is composed of monotonous spindle cells arranged in long fascicles, resembling leiomyosarcoma. The tumor infiltrates the adjacent fibroadipose tissue (original magnification 100×). By immunohistochemistry, it is diffusely positive for desmin (B) and multifocal nuclear positivity for myogenin (C). (D) Schematic representation of SRF-NCOA2 fusion involving exons 12 and 6, respectively. The numbers indicate exons. (E) This result was confirmed by RT-PCR which identified an in frame fusion of the SRF exon 6 with exon 12 of NCOA2, by direct sequencing of the 705 bp amplified product (F). FISH assay demonstrated break-apart signals for both SRF (G) and NCOA2 (H) (Probes centromeric to SRF and NCOA2 are in red, probes telomeric to SRF and NCOA2 are in green). (I) Number of reads normalized per million mapped nucleotides demonstrated differential expression of NCOA2 5′ and 3′ to the breakpoint, which supports the orientation of fusion transcript.
Figure 2. Histomorphology of NCOA2-negative spindle cell and sclerosing rhabdomyosarcoma.
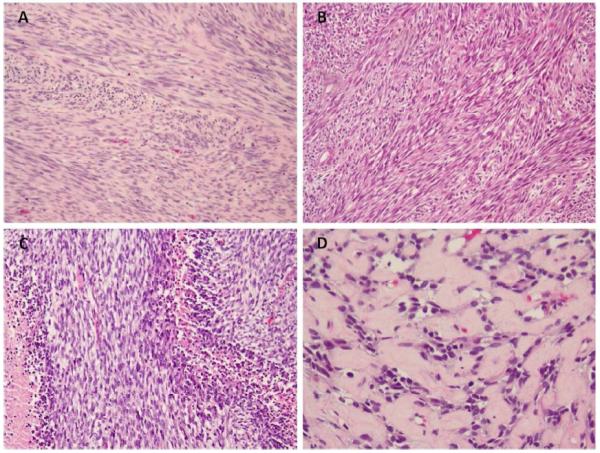
A variegated histological pattern is observed in spindle cell rhabdomyosarcoma including (A) smooth muscle-like appearance (case RMS3), (B) fibrosarcoma-like features (case RMS9), (C) occasionally mimicking MPNST (case RMS8) and (D) a hypocellular, densely collagenized background in a sclerosing RMS (RMS18). (A, original magnification 100×; B-C, original magnification 200×).
Figure 3. Spindle cell rhabdomyosarcoma with TEAD1-NCOA2 fusion.

Tumor from a 4 week-old baby boy who was born with a chest wall mass (case RMS4). (A) It is composed of short fascicles of spindle cells that have somewhat myofibroblastic appearance, infiltrating adjacent skeletal muscle. (B) A more cellular high grade component was also noted arranged in intersecting fascicles and associated with a high mitotic activity. Tumor cells demonstrate patchy positivity for Desmin and focal nuclear reactivity for myogenin (C) (A-C, original magnification 200×). (D) Partial karyotype showing a t(8;11) translocation. (E) Direct sequencing of the RT-PCR product confirmed the fusion of the TEAD1 exon 8 with NCOA2 exon 13. (F) FISH assay demonstrated TEAD1 and NCOA2 fusion-signal (highlighted with blue arrows; probes centromeric to NCOA2 in red, probes telomeric to TEAD1 in green).
Identification of SRF-NCOA2 as gene fusion candidate by FusionSeq and validation assays
The FusionSeq algorithm (Sboner et al., 2010) was applied to the aforementioned three frozen tumor samples that were RNA sequenced (RMS1, RMS2 and RMS3) (Table 1). Among the top candidate chimeric transcripts, we identified one containing exons 5 and 6 of SRF (c-fos serum response factor) located on 6p21, and exons 10, 11 and 12 of NCOA2 (nuclear receptor coactivator) located on 8q13 in rhabdomyosarcoma RMS2 (Figure 1D). Further review of clinical information on this patient revealed that at the time of diagnosis (2 years prior to elaboration of this manuscript), cytogenetic analysis performed at an outside hospital from the primary tumor biopsy showed a 46, XY, t(6;8)(p12, q11.2)[16] / 46, XY[4] in the twenty metaphases available (Mentrinoski et al., 2012). To validate the RNA sequencing and FusionSeq results, RT-PCR was performed using SRF exon 4 forward and the NCOA2 exon 13 reverse primes, which revealed a 705 bp amplified product on gel electrophoresis (Figure 1 F). Direct sequencing of the PCR product confirmed the fusion in frame of the SRF exon 6 with exon 12 of NCOA2 (Figure 1E). We then assessed for the presence of SRF and NCOA2 gene rearrangements by FISH. Break-apart signals were detected in both SRF and NCOA2 genes (Figure 1G, H). The number of reads normalized per million mapped nucleotides demonstrated differential expression of NCOA2 5′ and 3′ to the breakpoint (Figure 1I).
NCOA2 rearrangement is present in spindle cell rhabdomyosarcomas of infants
Among the remaining 14 spindle cell RMS and the 4 sclerosing RMS studied by FISH for the presence of gene abnormalities, NCOA2 break-apart signals were detected in two additional patients with spindle cell RMS, both located in the chest wall: a 4 week-old male infant (RMS4, Figure 3), and a 3 month-old girl (RMS5, Supplementary Figure 1). Thus, all three infantile spindle cell RMS studied showed the presence of NCOA2 gene rearrangements. However, the SRF rearrangement detected in RMS2 was not identified by FISH in the other two NCOA2-positive cases. Fresh frozen material available in both RMS4 and RMS5 was used to detect novel NCOA2-fusion partners by RACE. In RMS4, 5′RACE identified the fusion of fusion of TEAD1 exon 8 with NCOA2 exon 13, result which was then confirmed by RT-PCR (Figure 3E). This case was also successfully karyotyped and found to have a t(8;11) translocation (Figure 3D). These results were further validated by FISH using individual break-apart probes as well as fused-signal assay for TEAD1 and NCOA2 (Figure 3F). 5′RACE on RMS5 failed after few attempts.
None of the spindle cell RMS occurring in older children or adults or the sclerosing RMS showed NCOA2 gene abnormalities by FISH. All NCOA2-negative spindle and sclerosing RMS were further tested for gene rearrangements in PAX3 and NCOA1, as an alternative t(2;2)(p23;q35) translocation that has been previously described in rare cases of RMS (Sumegi et al., 2010), but no structural abnormalities were noted in any of our cases (Table 1). No abnormalities in NCOA2 or PAX3 were identified in any of the 4 ERMS and 3 ectomesenchymomas included in the control group.
DISCUSSION
The term spindle cell rhabdomyosarcoma was first used by the German-Italian Cooperative Sarcoma Study (Cavazzana et al., 1992) to distinguish it from the more common ERMS group, based on its distinctive clinicopathologic features and favorable outcome. In their study, comprising 21 cases (4% of 471 RMS studied), there was a male predominance, mean age at diagnosis of 6 years old, and the most common location being the paratesticular location (half of the cases), followed by the head and neck (a quarter of cases). The same demographic and clinicopathologic characteristics were then confirmed in a larger study of 800 RMS tumors from the Intergroup Rhabdomyosarcoma Study (IRS)(Leuschner et al., 1993). Spindle cell RMS was defined as a proliferation of bland spindle cells, with eosinophilic, fibrillary cytoplasm, resembling true smooth muscle differentiation. The cells were typically arranged in intersecting long fascicles, reminiscent of the ‘herringbone’ pattern of adult-type fibrosarcoma. In all tumors, evidence of rhabdomyoblasts with cross-striations were noted, as well as occasional areas of classic ERMS, composed of a mixture of spindle, stellate, and round cells embedded in a myxoid stroma. Immunohistochemically, the spindle cell RMS showed a uniform expression of skeletal muscle markers. At their last follow-up, 16 of the 21 patients were alive without disease, with one patient succumbing of disease 24 months after diagnosis (Cavazzana et al., 1992). Patients with spindle cell RMS of non-paratesticular sites had more extensive disease compared with patients having paratesticular lesions (Leuschner et al., 1993). In contrast with the favorable behavior in the pediatric age group, spindle cell RMS in adults was shown to follow an aggressive course and had a predilection for head and neck location (Rubin et al., 1998; Nascimento and Fletcher, 2005; Mentzel and Kuhnen, 2006).
Another level of complexity transpired by the possible relationship between the sclerosing RMS and spindle cell RMS. Sclerosing RMS in adults was first recognized in small series by Mentzel and Katenkamp (Mentzel and Katenkamp, 2000) and Folpe et al (Folpe et al., 2002). It was defined as a sclerosing, pseudovascular RMS in adults, showing an extensive hyalinized matrix, which may mimic osteosarcoma or angiosarcoma. Although the focal alveolar architecture and the primitive cytologic appearance suggested that it may be closely related to ARMS, the occasional presence of strap cells, the low level of myogenin expression and the absence of FOXO1-related fusions, pointed more toward an ERMS variant (Folpe et al., 2002). The anatomic location of these combined 7 cases showed an equal distribution for head and neck and limbs, with 3 cases each (Mentzel and Katenkamp, 2000; Folpe et al., 2002). Subsequently, sclerosing RMS was investigated in a large cohort of children and adolescents from the Intergroup Rhabdomyosarcoma Study (IRS)/Children Oncology Group (COG) (Chiles et al., 2004). The authors studied 13 sclerosing RMS patients, showing a 1:1 sex ratio and being equally distributed between head and neck and extremities. The predominant morphologic feature was the presence of undifferentiated small blue cells, lacking evidence of rhabdomyoblastic differentiation in all cases. The abundant hyaline matrix separated the tumor cells in either tubular pattern or single files, reminiscent of carcinomatous growth. Similar to the adult counterpart, sclerosing RMS in children showed a rather limited expression of myogenin, compared to ARMS. The clinical behavior of sclerosing RMS in pediatric age group was quite favorable, reminiscent of the outcome reported in the pediatric spindle cell RMS (Cavazzana et al., 1992). Subsequently, Mentzel et al suggested that due to overlapping morphologic features, spindle cell RMS and sclerosing RMS may represent a morphological spectrum of a distinct variant of RMS, separate from ERMS and ARMS, although unifying genetic abnormalities are still to be discovered (Mentzel, 2010).
From the genetic standpoint, the information available in both spindle and sclerosing RMS is limited and quite confusing based on mainly single case reports. One case of a pediatric head and neck spindle cell RMS showed an abnormal hypotriploid karyotype with numerous structural rearrangements (Gil-Benso et al., 1999). Another case of spindle cell RMS in the cheek of an 18 year-old girl, showed a 2q36-37 breakpoint in an unbalanced der(2)t(2;7)(q36-37;q3?), outside the 2q35 PAX3 locus (Debiec-Rychter et al., 2003). Similar 2q37 breakpoints have been reported in three additional cases of ERMS, including 2 cases of congenital RMS with t(2;11)(q37;q13) (Whang-Peng et al., 1986) and t(2;8)(q37;q13) (Hayashi et al., 1988) (see Table 2), respectively. The third case showed t(2;5)(q37.3;q31.3) in the peripheral lymphocytes of a patient with ERMS of the bladder (Moriyama et al., 1986).
Table 2.
Summary of Previously Reported ERMS with a t(2;8) Translocation or Involving the 8q12-13 Locus by Conventional Karyotyping
| Diagnosis | Age/Sex | Location | Morphologya | Karyotype | Reference |
|---|---|---|---|---|---|
| Congenital ERMS |
27 wks fetus/M | Cervical (cheek) |
Round and spindle cells (undifferentiated) |
t(2;8)(q35;q21.2)? | (Yoshino et al., 2005) |
| Congenital ERMS |
2 wk/M | Perineal gluteal/ rectal |
Round and spindle cells, rare strap cells (undifferentiated) |
t(2;8)(q35;q13) PAX3-NCOA2 |
(Meloni-Ehrig et al.,2009; Sumegi et al.,2010) |
| Congenital ERMS |
27 wks fetus (at birth)/F |
Upper eyelid | Spindle cells, rare Rblasts (no illustrations) |
t(2;8)(q35;q13) | (Hayashi et al., 1988) |
| ERMS | 8 mo/M | Pelvis/bladder | Round and spindle cells (undifferentiated) |
t(2;12;8)(q11.2;q22;q13) PAX3 rearrangement by FISH |
(Hosoi et al., 2009) |
| ERMS (botryoides) |
2 yrs/F | Abdominal | Round and spindle cells, myxoid stroma |
t(8;11)(q12-13;q21) | Calabrese et al., 1992) |
M, male; F, female; ERMS, embryonal rhabdomyosarcoma; Rblasts, rhabdomyoblasts, mo, months; wks, weeks; yrs, years.
Listed in parenthesis is the authors of this article interpretation of the morphology illustrated in the referenced publications.
From the IRS study, 3 of 13 sclerosing RMS had molecular/genetic analysis, showing the presence of a PAX3-FKHR fusion in one case, numerous marker chromosomes in the second, and extra copies of chromosomes 11 and 19 in a third (Chiles et al., 2004). Two other pediatric sclerosing RMS have been karyotyped showing numerous structural and numerical abnormalities, including double minute chromosomes, in one case (8 year-old girl, suprapubic/intra-abdominal), while a balanced t(5;20)(q31;p13) was identified in the other case (17 year-old girl, lower leg) (Zambrano et al., 2006). A closely located 20p12 break was described as part of a t(2;20)(q35;p12), possibly involving the PAX3 locus, in a 2 year-old girl with a buttock ERMS with an undifferentiated morphology (Ho et al., 2004). These results, although still limited in number and depth of analysis, remain inconclusive if spindle cell and sclerosing RMS represent a clinicopathologic continuum, or if they represent distinct pathologic entities or variants of ERMS.
Approximately 5-10% of patients with RMS are diagnosed during the first year of life, although RMS rarely occurs in congenital settings or during the neonatal period (Lobe et al., 1994; Rodriguez-Galindo et al., 2001). From the St. Jude Children’s Research Hospital experience, only four patients with neonatal RMS were treated between 1962-1999. All except one were ARMS, two of them presenting with multiple skin and subcutaneous metastatic nodules at the time of diagnosis and developed brain metastases early in their course (Rodriguez-Galindo et al., 2001). Three other similar cases of neonatal ARMS with metastases to the skin and brain have been reported in the literature (Grundy et al., 2001). Moreover, detailed molecular analysis using RT-PCR and FISH was unable to detect the presence of either t(2;13) or t(1;13) translocation in these three cases tested (Grundy et al., 2001), suggesting that the pathogenesis of this highly malignant and aggressive congenital tumor may be different than most ARMS in older children. In contrast, a similar pattern of molecular/genetic abnormalities is emerging in congenital/neonatal ERMS, with three cases reported so far showing a recurrent t(2;8) translocation (Hayashi et al., 1988; Yoshino et al., 2005; Meloni-Ehrig et al., 2009), and an additional case showing an identical translocation in an 8 month-old boy (Hosoi et al., 2009) (Table 2). In the case reported by Meloni et al. (Meloni-Ehrig et al., 2009), the t(2;8)(q35;q13) translocation was subsequently cloned as resulting in PAX3-NCOA2 fusion (Sumegi et al., 2010). Unfortunately, the histopathology of these congenital ERMS harboring a t(2;8)(q35;q13) is not well documented in these publications, with limited microscopic description provided and in some instances without accompanying illustrations (Hayashi et al., 1988). From the review of the provided microscopic images, the lesions show a rather primitive morphology with undifferentiated small rounded cells, with possible transition to oval/fusiform cells, separated by a thick fibrous stroma (Table 2)(Yoshino et al., 2005; Hosoi et al., 2009; Meloni-Ehrig et al., 2009). Thus, the exact subclassification of these cases into ARMS vs. ERMS vs. other categories remains far from straightforward. It is possible that the previously reported congenital fusion-negative ARMS (Grundy et al., 2001; Rodriguez-Galindo et al., 2001) represent similar lesions. To confound matters further, the additional case reported by Sumegi et al. with an identical t(2;8)(q35;q13), resulting in a PAX3-NCOA2 fusion, was classified as an ARMS, arising in the testis of a 2 year-old boy (Sumegi et al., 2010). In contrast, all 4 cases reported with t(2;2)(p23;q35), resulting in a PAX3-NCOA1, had an ARMS histology, although none of them presented before age of 5 years (Sumegi et al., 2010), (Wachtel et al., 2004).
In the current study we have identified recurrent NCOA2 gene rearrangements in congenital/infantile cases of spindle cell RMS involving SRF and TEAD1 genes. NCOA2 (nuclear receptor coactivator) is a member of the p160 steroid receptor coactivator gene family, which is composed of three members NCOA1 (SRC-1), NCOA2 (SRC-2), and SRC-3 (Xu and O’Malley, 2002). These coactivators do not encode for transcriptional factors, but interact with ligand-bound nuclear receptors to recruit histone acetyltransferases and methyltransferases to specific enhancer/promotor regions, which facilitates chromatin remodelling, assembly of general transcription factors, and transcription of target genes. In the COOH-terminal part, present in the fusion protein, there are two intrinsic transcriptional activation domains, TAD1 and TAD2. They are responsible for interaction with general transcriptional co-integrators such as p300 and CBP, as well as with histone methyltransferases CARM1 (coactivator-associated arginine methyltransferase 1) and PMRT1 (protein arginine methyltransferase 1) (Chen et al., 1999; Koh et al., 2001). Recurrent gene fusions involving NCOA2 have been demonstrated in other soft tissue tumors such as mesenchymal chondrosarcoma (HEY1-NCOA2) (Wang et al., 2012) and soft tissue angiofibroma (AHRR-NCOA2)(Jin et al., 2012), and in acute myeloid and biphenotypic leukemias (MYST3-NCOA2 and ETV6-NCOA2) (Troke et al., 2006; Strehl et al., 2008).
The interaction of NCOA2 with SRF on 6p21 and TEAD1 on 11p15.2 in the context of gene fusions present in spindle cell rhabdomyosarcoma is noteworthy. The serum response factor (SRF) is a member of the MADS (MCM1, Agamous, Deficiens, and serum response factor) box transcription factors and regulates skeletal, as well as cardiac and smooth muscle genes by binding a DNA sequence known as a CArG box (Owens et al., 2004). SRF is highly expressed in skeletal muscle and controls the expression of genes specifically expressed in skeletal muscle (dystrophin, muscle creatine kinase, myoD), including several genes encoding sarcomeric proteins (such as α skeletal actin, myosin light chain, tropomyosin) (Pipes et al., 2006). The combinatorial interactions between the MyoD family of basic helix–loop–helix transcription factors and the myocyte enhancer factor-2 (MEF2) family of MADS are involved in the early steps of skeletal muscle development (Molkentin et al., 1995). In mice lacking skeletal muscle SRF expression, the muscle fibers are formed but fail to undergo hypertrophic growth after birth and mice die during the perinatal period from severe skeletal muscle hypoplasia (Li et al., 2005). The myopathic phenotype of these mutant mice resembles that of mice expressing a dominant negative mutant of a myocardin (MYOCD) family member in skeletal muscle. These findings reveal an essential role for the partnership of SRF and MYOCD-related transcription factors in the control of skeletal muscle growth and maturation in vivo (Li et al., 2005).
TEAD1 (TEA domain family member 1) also designated as TEF-1 (Transcription Enhancer Factor 1) is constitutively expressed in cardiac and skeletal muscle. It acts as a key molecule of muscle development, and transactivates multiple target genes involved in cell proliferation and differentiation pathways (Qiu et al., 2011). Multiple transcription factors including SRF, MYOCD and TEAD1 have been shown to be involved in the control of muscle-specific gene transcription, although most of these transcription factors are not specific for a particular muscle cell type (Yoshida and Owens, 2005). Therefore, it has been proposed that expression of muscle-specific genes is controlled by unique combinations of transcription factors that are expressed ubiquitously or cell-selectively. In this regard, it is worth noting that TEAD1 family members interact with several transcription factors including SRF to control muscle-specific genes (Azakie et al., 2005).
In summary, our findings reveal that NCOA2 is consistently rearranged in a subset of congenital and infantile spindle cell RMS, being fused with key transcription factors involved in skeletal muscle differentiation, such as SRF and TEAD1. The results of this study also suggest that spindle cell RMS occurring in older children and adults, although sharing a close morphologic appearance, have different genetic abnormalities. Further studies are required to investigate if spindle cell and sclerosing RMS represent morphologic variation of the same clinicopathologic entity.
Supplementary Material
Acknowledgments
Grant support: PO1 CA047179-15A2 (CRA), P50 CA 140146-01 (CRA).
Footnotes
Conflict of interest: none
REFERENCES
- Azakie A, Lamont L, Fineman JR, He Y. Divergent transcriptional enhancer factor-1 regulates the cardiac troponin T promoter. Am J Physiol Cell Physiol. 2005;289:C1522–1534. doi: 10.1152/ajpcell.00126.2005. [DOI] [PubMed] [Google Scholar]
- Calabrese G, Guanciali Franchi P, Stuppia L, Rossi C, Bianchi C, Antonucci A, Palka G. Translocation (8;11)(q12-13;q21) in embryonal rhabdomyosarcoma. Cancer Genet Cytogenet. 1992;58:210–211. doi: 10.1016/0165-4608(92)90118-r. [DOI] [PubMed] [Google Scholar]
- Cavazzana AO, Schmidt D, Ninfo V, Harms D, Tollot M, Carli M, Treuner J, Betto R, Salviati G. Spindle cell rhabdomyosarcoma. A prognostically favorable variant of rhabdomyosarcoma. Am J Surg Pathol. 1992;16:229–235. doi: 10.1097/00000478-199203000-00002. [DOI] [PubMed] [Google Scholar]
- Chen D, Ma H, Hong H, Koh SS, Huang SM, Schurter BT, Aswad DW, Stallcup MR. Regulation of transcription by a protein methyltransferase. Science. 1999;284:2174–2177. doi: 10.1126/science.284.5423.2174. [DOI] [PubMed] [Google Scholar]
- Chiles MC, Parham DM, Qualman SJ, Teot LA, Bridge JA, Ullrich F, Barr FG, Meyer WH. Sclerosing rhabdomyosarcomas in children and adolescents: a clinicopathologic review of 13 cases from the Intergroup Rhabdomyosarcoma Study Group and Children’s Oncology Group. Pediatr Dev Pathol. 2004;7:583–594. doi: 10.1007/s10024-004-5058-x. [DOI] [PubMed] [Google Scholar]
- Debiec-Rychter M, Hagemeijer A, Sciot R. Spindle-cell rhabdomyosarcoma with 2q36 approximately q37 involvement. Cancer Genet Cytogenet. 2003;140:62–65. doi: 10.1016/s0165-4608(02)00647-7. [DOI] [PubMed] [Google Scholar]
- Folpe AL, McKenney JK, Bridge JA, Weiss SW. Sclerosing rhabdomyosarcoma in adults: report of four cases of a hyalinizing, matrix-rich variant of rhabdomyosarcoma that may be confused with osteosarcoma, chondrosarcoma, or angiosarcoma. Am J Surg Pathol. 2002;26:1175–1183. doi: 10.1097/00000478-200209000-00008. [DOI] [PubMed] [Google Scholar]
- Gil-Benso R, Carda-Batalla C, Navarro-Fos S, Pellin-Perez A, Llombart-Bosch A. Cytogenetic study of a spindle-cell rhabdomyosarcoma of the parotid gland. Cancer Genet Cytogenet. 1999;109:150–153. doi: 10.1016/s0165-4608(98)00163-0. [DOI] [PubMed] [Google Scholar]
- Grundy R, Anderson J, Gaze M, Gerrard M, Glaser A, Gordon A, Malone M, Pritchard-Jones K, Michalski A. Congenital alveolar rhabdomyosarcoma: clinical and molecular distinction from alveolar rhabdomyosarcoma in older children. Cancer. 2001;91:606–612. doi: 10.1002/1097-0142(20010201)91:3<606::aid-cncr1041>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Habegger L, Sboner A, Gianoulis TA, Rozowsky J, Agarwal A, Snyder M, Gerstein M. RSEQtools: a modular framework to analyze RNA-Seq data using compact, anonymized data summaries. Bioinformatics. 2011;27:281–283. doi: 10.1093/bioinformatics/btq643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Inaba T, Hanada R, Yamamoto K. Translocation 2;8 in a congenital rhabdomyosarcoma. Cancer Genet Cytogenet. 1988;30:343–345. doi: 10.1016/0165-4608(88)90208-7. [DOI] [PubMed] [Google Scholar]
- Ho RH, Johnson J, Dev VG, Whitlock JA. A novel t(2;20)(q35;p12) in embryonal rhabdomyosarcoma. Cancer Genet Cytogenet. 2004;151:73–77. doi: 10.1016/j.cancergencyto.2003.08.023. [DOI] [PubMed] [Google Scholar]
- Hosoi H, Kakazu N, Konishi E, Tsuchihashi Y, Hada S, Amaya E, Nakabayahi Y, Misawa-Furihata A, Tabata-Maruyama H, Iehara T, Sugimoto T, Yamane H, Yamasaki M, Shiwaku K, Yanagisawa A, Ono M, Tokiwa K, Iwai N, Hashiba M, Abe T, Sawada T. A novel PAX3 rearrangement in embryonal rhabdomyosarcoma. Cancer Genet Cytogenet. 2009;189:98–104. doi: 10.1016/j.cancergencyto.2008.10.016. [DOI] [PubMed] [Google Scholar]
- Hsu F, Kent WJ, Clawson H, Kuhn RM, Diekhans M, Haussler D. The UCSC Known Genes. Bioinformatics. 2006;22:1036–1046. doi: 10.1093/bioinformatics/btl048. [DOI] [PubMed] [Google Scholar]
- Jin Y, Moller E, Nord KH, Mandahl N, Von Steyern FV, Domanski HA, Marino-Enriquez A, Magnusson L, Nilsson J, Sciot R, Fletcher CD, Debiec-Rychter M, Mertens F. Fusion of the AHRR and NCOA2 genes through a recurrent translocation t(5;8)(p15;q13) in soft tissue angiofibroma results in upregulation of aryl hydrocarbon receptor target genes. Genes Chromosomes Cancer. 2012;51:510–520. doi: 10.1002/gcc.21939. [DOI] [PubMed] [Google Scholar]
- Koh SS, Chen D, Lee YH, Stallcup MR. Synergistic enhancement of nuclear receptor function by p160 coactivators and two coactivators with protein methyltransferase activities. J Biol Chem. 2001;276:1089–1098. doi: 10.1074/jbc.M004228200. [DOI] [PubMed] [Google Scholar]
- Leuschner I, Newton WA, Jr., Schmidt D, Sachs N, Asmar L, Hamoudi A, Harms D, Maurer HM. Spindle cell variants of embryonal rhabdomyosarcoma in the paratesticular region. A report of the Intergroup Rhabdomyosarcoma Study. Am J Surg Pathol. 1993;17:221–230. doi: 10.1097/00000478-199303000-00002. [DOI] [PubMed] [Google Scholar]
- Li S, Czubryt MP, McAnally J, Bassel-Duby R, Richardson JA, Wiebel FF, Nordheim A, Olson EN. Requirement for serum response factor for skeletal muscle growth and maturation revealed by tissue-specific gene deletion in mice. Proc Natl Acad Sci U S A. 2005;102:1082–1087. doi: 10.1073/pnas.0409103102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobe TE, Wiener ES, Hays DM, Lawrence WH, Andrassy RJ, Johnston J, Wharam M, Webber B, Ragab A. Neonatal rhabdomyosarcoma: the IRS experience. J Pediatr Surg. 1994;29:1167–1170. doi: 10.1016/0022-3468(94)90302-6. [DOI] [PubMed] [Google Scholar]
- Meloni-Ehrig A, Smith B, Zgoda J, Greenberg J, Perdahl-Wallace E, Zaman S, Mowrey P. Translocation (2;8)(q35;q13): a recurrent abnormality in congenital embryonal rhabdomyosarcoma. Cancer Genet Cytogenet. 2009;191:43–45. doi: 10.1016/j.cancergencyto.2009.01.010. [DOI] [PubMed] [Google Scholar]
- Mentrinoski M, Golden W, Bourne T, LeGallo R. Pediatr Dev Pathol. 2012 doi: 10.2350/12-04-1180-CR.1. Accepted. [DOI] [PubMed] [Google Scholar]
- Mentzel T. Spindle cell rhabdomyosarcoma in adults: a new entity in the spectrum of malignant mesenchymal tumors of soft tissues. Pathologe. 2010;31:91–96. doi: 10.1007/s00292-009-1249-6. [DOI] [PubMed] [Google Scholar]
- Mentzel T, Katenkamp D. Sclerosing, pseudovascular rhabdomyosarcoma in adults. Clinicopathological and immunohistochemical analysis of three cases. Virchows Arch. 2000;436:305–311. doi: 10.1007/s004280050451. [DOI] [PubMed] [Google Scholar]
- Mentzel T, Kuhnen C. Spindle cell rhabdomyosarcoma in adults: clinicopathological and immunohistochemical analysis of seven new cases. Virchows Arch. 2006;449:554–560. doi: 10.1007/s00428-006-0284-4. [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Black BL, Martin JF, Olson EN. Cooperative activation of muscle gene expression by MEF2 and myogenic bHLH proteins. Cell. 1995;83:1125–1136. doi: 10.1016/0092-8674(95)90139-6. [DOI] [PubMed] [Google Scholar]
- Moriyama M, Shuin T, Kubota Y, Satomi Y, Sugio Y, Kuroki Y. A case of rhabdomyosarcoma of the bladder with a (2;5) chromosomal translocation in peripheral lymphocytes. Cancer Genet Cytogenet. 1986;22:177–181. doi: 10.1016/0165-4608(86)90204-9. [DOI] [PubMed] [Google Scholar]
- Nascimento AF, Fletcher CD. Spindle cell rhabdomyosarcoma in adults. Am J Surg Pathol. 2005;29:1106–1113. [PubMed] [Google Scholar]
- Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- Pflueger D, Terry S, Sboner A, Habegger L, Esgueva R, Lin PC, Svensson MA, Kitabayashi N, Moss BJ, MacDonald TY, Cao X, Barrette T, Tewari AK, Chee MS, Chinnaiyan AM, Rickman DS, Demichelis F, Gerstein MB, Rubin MA. Discovery of non-ETS gene fusions in human prostate cancer using nextgeneration RNA sequencing. Genome Res. 2011;21:56–67. doi: 10.1101/gr.110684.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierron G, Tirode F, Lucchesi C, Reynaud S, Ballet S, Cohen-Gogo S, Perrin V, Coindre JM, Delattre O. A new subtype of bone sarcoma defined by BCORCCNB3 gene fusion. Nat Genet. 2012;44:461–466. doi: 10.1038/ng.1107. [DOI] [PubMed] [Google Scholar]
- Pipes GC, Creemers EE, Olson EN. The myocardin family of transcriptional coactivators: versatile regulators of cell growth, migration, and myogenesis. Genes Dev. 2006;20:1545–1556. doi: 10.1101/gad.1428006. [DOI] [PubMed] [Google Scholar]
- Qiu H, Wang F, Liu C, Xu X, Liu B. TEAD1-dependent expression of the FoxO3a gene in mouse skeletal muscle. BMC Mol Biol. 2011;12:1. doi: 10.1186/1471-2199-12-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail MA, Kozarewa I, Smith F, Scally A, Stephens PJ, Durbin R, Swerdlow H, Turner DJ. A large genome center’s improvements to the Illumina sequencing system. Nat Methods. 2008;5:1005–1010. doi: 10.1038/nmeth.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Galindo C, Hill DA, Onyekwere O, Pin N, Rao BN, Hoffer FA, Kun LE, Pappo AS, Santana VM. Neonatal alveolar rhabdomyosarcoma with skin and brain metastases. Cancer. 2001;92:1613–1620. doi: 10.1002/1097-0142(20010915)92:6<1613::aid-cncr1487>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Rubin BP, Hasserjian RP, Singer S, Janecka I, Fletcher JA, Fletcher CD. Spindle cell rhabdomyosarcoma (so-called) in adults: report of two cases with emphasis on differential diagnosis. Am J Surg Pathol. 1998;22:459–464. doi: 10.1097/00000478-199804000-00011. [DOI] [PubMed] [Google Scholar]
- Sboner A, Habegger L, Pflueger D, Terry S, Chen DZ, Rozowsky JS, Tewari AK, Kitabayashi N, Moss BJ, Chee MS, Demichelis F, Rubin MA, Gerstein MB. FusionSeq: a modular framework for finding gene fusions by analyzing paired-end RNA-sequencing data. Genome Biol. 2010;11:R104. doi: 10.1186/gb-2010-11-10-r104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strehl S, Nebral K, Konig M, Harbott J, Strobl H, Ratei R, Struski S, Bielorai B, Lessard M, Zimmermann M, Haas OA, Izraeli S. ETV6-NCOA2: a novel fusion gene in acute leukemia associated with coexpression of T-lymphoid and myeloid markers and frequent NOTCH1 mutations. Clin Cancer Res. 2008;14:977–983. doi: 10.1158/1078-0432.CCR-07-4022. [DOI] [PubMed] [Google Scholar]
- Sumegi J, Streblow R, Frayer RW, Dal Cin P, Rosenberg A, Meloni-Ehrig A, Bridge JA. Recurrent t(2;2) and t(2;8) translocations in rhabdomyosarcoma without the canonical PAX-FOXO1 fuse PAX3 to members of the nuclear receptor transcriptional coactivator family. Genes Chromosomes Cancer. 2010;49:224–236. doi: 10.1002/gcc.20731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanas MR, Sboner A, Oliveira AM, Erickson-Johnson MR, Hespelt J, Hanwright PJ, Flanagan J, Luo Y, Fenwick K, Natrajan R, Mitsopoulos C, Zvelebil M, Hoch BL, Weiss SW, Debiec-Rychter M, Sciot R, West RB, Lazar AJ, Ashworth A, Reis-Filho JS, Lord CJ, Gerstein MB, Rubin MA, Rubin BP. Identification of a disease-defining gene fusion in epithelioid hemangioendothelioma. Sci Transl Med. 2011;3:98ra82. doi: 10.1126/scitranslmed.3002409. [DOI] [PubMed] [Google Scholar]
- Troke PJ, Kindle KB, Collins HM, Heery DM. MOZ fusion proteins in acute myeloid leukaemia. Biochem Soc Symp. 2006:23–39. doi: 10.1042/bss0730023. [DOI] [PubMed] [Google Scholar]
- Wachtel M, Dettling M, Koscielniak E, Stegmaier S, Treuner J, Simon-Klingenstein K, Buhlmann P, Niggli FK, Schafer BW. Gene expression signatures identify rhabdomyosarcoma subtypes and detect a novel t(2;2)(q35;p23) translocation fusing PAX3 to NCOA1. Cancer Res. 2004;64:5539–5545. doi: 10.1158/0008-5472.CAN-04-0844. [DOI] [PubMed] [Google Scholar]
- Wang L, Motoi T, Khanin R, Olshen A, Mertens F, Bridge J, Dal Cin P, Antonescu CR, Singer S, Hameed M, Bovee JV, Hogendoorn PC, Socci N, Ladanyi M. Identification of a novel, recurrent HEY1-NCOA2 fusion in mesenchymal chondrosarcoma based on a genome-wide screen of exon-level expression data. Genes Chromosomes Cancer. 2012;51:127–139. doi: 10.1002/gcc.20937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whang-Peng J, Triche TJ, Knutsen T, Miser J, Kao-Shan S, Tsai S, Israel MA. Cytogenetic characterization of selected small round cell tumors of childhood. Cancer Genet Cytogenet. 1986;21:185–208. doi: 10.1016/0165-4608(86)90001-4. [DOI] [PubMed] [Google Scholar]
- Xu J, O’Malley BW. Molecular mechanisms and cellular biology of the steroid receptor coactivator (SRC) family in steroid receptor function. Rev Endocr Metab Disord. 2002;3:185–192. doi: 10.1023/a:1020016208071. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Owens GK. Molecular determinants of vascular smooth muscle cell diversity. Circ Res. 2005;96:280–291. doi: 10.1161/01.RES.0000155951.62152.2e. [DOI] [PubMed] [Google Scholar]
- Yoshino K, Takeuchi M, Nakayama M, Suehara N. Congenital cervical rhabdomyosarcoma arising in one fetus of a twin pregnancy. Fetal Diagn Ther. 2005;20:291–295. doi: 10.1159/000085088. [DOI] [PubMed] [Google Scholar]
- Zambrano E, Perez-Atayde AR, Ahrens W, Reyes-Mugica M. Pediatric sclerosing rhabdomyosarcoma. Int J Surg Pathol. 2006;14:193–199. doi: 10.1177/1066896906290558. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.