

Abstract
Plasmids containing a site-specific DNA interstrand cross-link (ICL) are invaluable tools for the investigation of ICL repair pathways at the biochemical and cellular level. We describe a procedure for preparation of plasmid DNA substrates containing a single ICL at a specific site. The procedure is versatile, leads to reliable yields of pure DNA substrate, and is suitable for the incorporation of virtually any type of DNA lesion into plasmids.
Keywords: Plasmid DNA substrate, Interstrand cross-link, Site-specific damage, BbsI restriction enzyme, Gel filtration, Ligation
1. Introduction
DNA interstrand cross-links (ICLs) are among the most cytotoxic DNA lesions, covalently linking together both strands of a DNA duplex. ICLs are formed by endogenous metabolites and by cross-linking agents used in antitumor therapy (1, 2). Despite their biological and clinical importance, the cellular responses to ICLs, particularly the mechanisms by which they are repaired, are not yet understood in detail. The investigation of ICL repair has been hampered by the heterogeneity of DNA lesions produced by treatment of cells with cross-linking agents, which yield mostly monoadducts and intrastrand cross-links (a covalent linkage between two bases on the same DNA strand) (3). The recent development of methods to generate oligonucleotides containing site-specific ICLs and their incorporation into plasmids has been extremely useful for the dissection of different ICL repair pathways using biochemical (4–6) and cell biological methods (7–10). However, the preparation of such substrates is technically difficult, generally resulting in low yields or nonhomogenous substrates that contain varying amounts of impurities. We report a procedure for preparing ICL-containing plasmids with high purity and yield that overcomes these limitations.
The challenge of incorporating duplex oligonucletides into plasmids in vitro lies in the low efficiency of the ligation reaction, risk of incorporating multiple copies of the oligonucleotide or linearized plasmid, and controlling the orientation of incorporation. Our method addresses these issues by inserting two BbsI sites in tandem within a plasmid (Fig. 1a). BbsI is a type IIS restriction enzyme that cleaves outside of a non-palindromic recognition sequence (Fig. 1b). After treatment with BbsI, the plasmid forms linear fragments (a vector and the excised fragment) with two different non-palindromic cohesive ends (Fig. 1a, c). Following linearization with BbsI, the plasmid vector is separated from the excised fragment by gel filtration, and the 4-nucleotide 5′ ends are ligated to the complimentary 5′ phosphorylated ends of an oligonucleotide insert containing a single ICL (Fig. 1d). We have found that the best yield of closed circular plasmid is obtained when very low concentrations of linearized plasmid vector (0.5–2 nM) and slight excess of the oligonucleotide insert (1.5–3×) are used. Under these conditions, the formation of multimeric species and the ligation of separate insert molecules at each end of the linear vector are reduced. Finally, the covalently closed circular plasmid is purified by ethidium bromide cesium chloride density gradient centrifugation. This design ensures the incorporation of a single molecule of the damage-containing oligonucleotide duplex in a controlled orientation into a closed circular plasmid. The non-palindromic sticky ends preclude intramolecular and intermolecular ligation amongst separate molecules of plasmid vector or insert, and are a key factor for the specificity of the ligation procedure.
Fig. 1.

Strategy for the construction of the site-specific, modified plasmid. (a) pSVRLuc plasmid containing 2 BbsI sites in tandem. (b) The recognition sequence (underlined) and cleavage site of the BbsI restriction enzyme. (c) The sequence of the tandem BbsI sites in the pSVRLuc plasmid. (d) The sequence of the cisplatin ICL-containing duplex oligonucleotide. The 5′ phosphorylated four nucleotide overhangs are shown in bold.
We have successfully used this method for the incorporation of different ICLs (cisplatin or nitrogen mustard-like), as well as intrastrand cross-links (cisplatin) into plasmids. The substrates obtained have been used, in their circular form, for in vitro investigations of replication-associated ICL repair (4, 5) and the repair of a transcription-blocking ICL in mammalian cells (using a reporter gene reactivation assay), or as a linear fragment for an in vitro transcription assay (Enoiu M, Jiricny J, Schärer OD, manuscript in revision). Herein, we describe the construction of the plasmid substrate used for in vitro replication assays in Xenopus laevis egg extracts (described in the accompanying Chapter 16 by Knipscheer et al.). Our procedure is versatile and can be used for any plasmid engineered to contain the tandem BbsI sites, as well as for the incorporation of duplex oligonucleotides containing virtually any type of DNA modification.
2. Materials
2.1. DNA Components
Plasmid DNA: pSVRLuc (5.6 kb) (Maxiprep) (see Note 1). This DNA and detailed sequence information is available upon request from the authors.
Oligonucleotides 5′ phosphorylated, for the preparation of the DNA duplex containing a cisplatin ICL (see Note 2).
Upper strand sequence: 5′pCCCTCTTCCGCTCTTCTTTC; bottom strand sequence: 5′pGCACGAAAGAAGAGCGGAAG (the cross-linked bases are indicated in bold) (Fig. 1d) (see Note 3). Dissolve the oligonucleotides to 2 mM in H2O.
2.2. Reagents and Buffers
Use ultrapure water (18 MΩ) for the preparation of solutions and in all reactions. All the reagents are of analytical grade.
Cisplatin: To obtain a 4 mM cisplatin solution, freshly dissolve 6 mg cisplatin in 5 mL H2O. Vortex until completely dissolved. Protect from light.
15.2 mM AgNO3:12.9 mg AgNO3 (MW: 169.9) in 5 mL H2O.
20 mM NaClO4, pH 5.2: 281 mg NaClO4·H2O (MW: 140.46) in 90 mL H2O. Adjust the pH to 5.2 with 1 % acetic acid and the volume to 100 mL. Dilute with an equal volume of H2O to obtain 10 mM NaClO4.
100 mM NaClO4: 1.4 g NaClO4·H2O in 100 mL H2O.
0.5 M NaCl: 2.92 g NaCl (MW: 58.44) in 100 mL H2O.
-
Mobile phases for anion exchange chromatography:
-
A1
10 mM Tris–HCl, pH 7.4: 1.21 g Tris in 800 mL H2O, adjust the pH to 7.4 with HCl and the volume to 1 L. Filter and degas.
-
B1
1 M NaCl, 10 mM Tris–HCl, pH 7.4: 1.21 g Tris and 58.4 g NaCl in 800 mL H2O, adjust the pH to 7.4 with HCl and the volume to 1 L. Filter and degas.
-
A2
10 mM NaOH: 0.4 g NaOH in 1 L H2O. Filter and degas.
-
B2
1 M NaCl, 10 mM NaOH: 0.4 g NaOH and 58.4 g NaCl in 1 L H2O. Filter and degas.
-
A1
BbsI (NEB R0539L) 5,000 U/mL (see Note 4).
Analytical restriction enzymes: EarI, BamHI.
T4 DNA Ligase (NEB M0202L) 400,000 U/mL.
10× Ligase buffer: 500 mM Tris–HCl, pH 7.5, 100 mM MgCl2, 10 mM DTT, 1 mM ATP, 25 μg/mL bovine serum albumin (BSA). Make 1 mL aliquots and store at −20 °C.
NEBuffer 2 (10×).
BSA 10 mg/mL (100×): Dilute 1/10 in H2O to obtain a 10× stock.
10 mM Tris–HCl, pH 8.0.
TE buffer: 10 mM Tris–HCl pH 8.0, 1 mM EDTA. Filter and degas for use as a gel filtration buffer.
DNA ladders: 1 kb DNA Ladder (NEB N3232) and 100 bp DNA Ladder (NEB 3231). Store at −20 °C.
1× Tris–acetate buffer (TAE): 40 mM Tris–acetate, 1 mM EDTA, pH 8.0. Prepare a 50× stock solution from 242 g Tris base, 57.1 mL glacial acetic acid, 100 mL 0.5 M EDTA, H2O to 1 L (pH 8.0).
1× Tris–borate buffer (TBE): 89 mM Tris–borate, 2 mM EDTA, pH 8.0. Prepare a 5× stock solution from 54 g Tris base, 27.5 g boric acid, 0.5 M EDTA, H2O to 1 L (pH 8.0).
Gel loading buffer (6×): 0.25 % Bromophenol blue, 0.25 % xylene cyanol FF, 30 % glycerol in water. Store at 4 °C.
Acrylamide/Bisacrylamide 29:1. Store at 4 °C.
Agarose, ultrapure.
10% Ammonium persulfate. To 1 g ammonium persulfate, add H2O to 10 mL. Store at 4 °C for up to 4 weeks.
N,N,N′,N′-Tetramethylethylenediamine (TEMED). Store at 4 °C.
Phenol:chloroform:isoamyl alcohol (25:24:1). Store at 4 °C.
1-Butanol.
Ethanol.
Chloroform.
Ethidium bromide 10 mg/mL (Sigma E 8751).
3 M sodium acetate pH 5.2: 40.8 g of sodium acetate·3H2O in 80 mL of H2O and set pH to 5.2 with 3 M acetic acid.
Cesium chloride.
2.3. Equipment and Consumables
Fast protein liquid chromatography (FPLC) system (e.g., ÄKTA FPLC from GE Healthcare).
Mono Q 5/50 GL column (GE Healthcare).
Gel filtration column HiLoad Superdex 75 prep grade 16/60. Store in 20 % ethanol at 4 °C.
UV spectrophotometer.
Ultracentrifuge (Sorvall Ultra Pro 80), Vertical rotor 65 V13.
PA Ultracrimp Tubes 6 mL (Sorvall 792730).
Ultracrimp sealing tool (Sorvall 79263).
High-speed centrifuge (Sorvall Evolution RC), Sorvall SS-34 fixed angle rotor.
Oak Ridge centrifuge tube (30 mL and 50 mL).
Bench-top centrifuge (Eppendorf 5804R), Swing-bucket rotor A-4-44.
Microcentrifuge (Eppendorf 5417R).
UV lamp (360 nm wavelength).
UV transilluminator.
Agarose gel electrophoresis system.
Bio-Rad Mini-PROTEAN 3 Electrophoresis system.
Water bath.
Thermomixer compact (Eppendorf).
Amicon Ultra-15 3K (centrifugal filter device).
Amicon Ultra-15 30K (centrifugal filter device).
Amicon Ultra-4 30K (centrifugal filter device).
0.47 μm Millipore filter.
21-gauge needle.
18-gauge needle attached to 1 mL syringe.
Qiagen Plasmid Maxi Kit. Kit includes QIAGEN-tip 500 columns as well as QBT and QC and QF buffers.
3. Methods
3.1. Preparation of the DNA Duplex Containing a Cisplatin ICL
This protocol is based on the procedure established by Hofr and Brabec (11).
-
Generation of monoaqua species from cisplatin:
Cisplatin can react with DNA only after a chloride ligand is replaced by H2O (see Note 5). The activation of cisplatin to monoaquamonochloro cisplatin is performed by precipitating chloride in a reaction with 0.95 molar equivalents of AgNO3 in 10 mM NaClO4, pH 5.2.
Mix in an Eppendorf tube 250 μL 4 mM cisplatin, 187.5 μL H2O, 500 μL 20 mM NaClO4, pH 5.2, and 62.5 μL 15.2 mM AgNO3. Incubate for 24 h at 37 °C in the dark. Filter through a 0.47 μm Millipore filter to remove the AgCl precipitate. Keep the solution on ice and use it immediately for the next step.
-
Platination of the single-stranded oligonucleotide to a cisplatin monoadduct: The single-stranded oligonucleotide (upper strand sequence, see Subheading 2) is reacted with monoaquamonochloro cisplatin at a cisplatin-to-DNA ratio of 3:1 in 10 mM NaClO4, pH 5.2.
Mix in an Eppendorf tube 45 μL 10 mM NaClO4 pH 5.2, 30 μL of 1 mM monoaquamonochloro cisplatin, and 5 μL of 2 mM oligonucleotide (10 nmol DNA) (see Note 6). Incubate for 12 min at 37 °C. Stop the reaction by adding 20 μL of 0.5 M NaCl (0.1 M final concentration). Keep on ice until next step.
-
Purification of the cisplatin monoadduct containing oligonucleotide: The cisplatin monoadduct is separated from the unreacted oligonucleotide and overplatinated by-products by anion exchange FPLC using a Mono Q column.
Connect the Mono Q 5/50 GL column to the ÄKTA purifier. Wash the column with H2O and then equilibrate with 0.1 M NaCl and 10 mM Tris–HCl, pH 7.4 (10 % buffer B1, 90 % buffer A1).
Dilute the DNA sample to 1 mL with 0.1 M NaCl and 10 mM Tris–HCl, pH 7.4 and inject it using a 1 mL syringe into the 2 mL sample loop. Start a three-step gradient: 10–37 % buffer B1 in 5 column volumes, 37–47 % buffer B1 in 40 column volumes, and 47–100 % buffer B1 in 1 column volume at a flow rate of 2 mL/min. Re-equilibrate to 10 % buffer B1. Collect 0.5 mL fractions during the second gradient step.
The cisplatin monoadduct elutes at about 0.4 M NaCl before the unreacted oligonucleotide (Fig. 2a). Pool the fractions corresponding to the monoadduct and measure the DNA concentration by UV (the contribution of cisplatin to the absorbance is ignored). The yield of the cisplatin monoadduct oligonucleotide is typically around 15–20 %.
Annealing of the cisplatin monoadduct containing oligonucleotide to the complementary strand: Add MgCl2 to the solution containing the cisplatin monoadduct oligonucleotide to a final concentration of 2 mM. Add 1.05 molar equivalents of the complementary oligonucleotide (bottom strand sequence, see Subheading 2). Mix and incubate for 24 h at 25 °C (protect from light).
Cross-linking reaction: The formation of the ICL is performed in 100 mM NaClO4. Desalt and exchange the buffer of the DNA sample by extensive washing with 100 mM NaClO4 in a centrifugal filter device Amicon Ultra-15 3K at 4 °C in a bench-top centrifuge (Eppendorf 5804R) at 2,500 × g at 4 °C (see Note 7). Repeat the washing step seven to eight times. Stop the last washing step when the volume of the sample is about 1 mL. Incubate for 48 h in the dark at 37 °C for the cross-linking reaction.
Purification of the cross-linked duplex: The cross-linked DNA is separated from the single-stranded oligonucleotides on the Mono Q column in an alkaline gradient. Equilibrate the Mono Q column with 0.1 M NaCl and 10 mM NaOH (10 % mobile phase B2, 90 % mobile phase A2). Dilute the DNA sample to 2 mL with 0.1 M NaCl and 10 mM NaOH. Inject the DNA sample into the 2 mL sample loop. Start a three-step gradient: 10–55 % B2 in 7 column volumes; 55–70 % B2 in 40 column volumes; and 55–100 % B2 in 1 column volume. Collect 0.5 mL fractions during the second step of the gradient. The cross-linked duplex elutes in a single peak after the single-stranded oligonucleotides (Fig. 2b).
Pool the fractions corresponding to the ICL. Concentrate using the centrifugal filter device Amicon Ultra-15 3K in a bench-top centrifuge at 2,500 × g at 4 °C and wash seven to eight times with 10 mM Tris–HCl, pH 7.4 containing 10 mM NaClO4. Concentrate to about 1 mL at the final washing step. Measure the DNA concentration using a UV spectrophotometer. Make aliquots of 60 pmol DNA and store at −80 °C. The typical yield of the ICL duplex is 4–5 % with respect to the single-stranded oligonucleotide used in the first platination reaction (see Note 8).
Fig. 2.
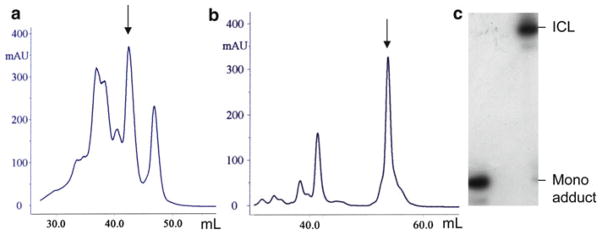
Purification of the cisplatin monoadduct and cisplatin ICL DNA. (a) Typical anion exchange chromatogram showing separation of the cisplatin monoadduct DNA (indicated by arrow) from the unreacted oligonucleotide (last peak) and the overplatinated by-products (early eluting peaks). (b) Typical anion exchange chromatogram showing separation of the cisplatin ICL (indicated by arrow ) from the single-stranded oligonucleotides. (c) Analysis of radiolabeled (32P) cisplatin ICL and cisplatin monoadduct DNA in denaturing polyacrylamide gel.
3.2. BbsI Digestion of the Plasmid DNA
Set up a BbsI digest by mixing in an Eppendorf tube 300 μg plasmid DNA (see Note 9), 30 μL BbsI (150 U), and 60 μL 10× NEB2, and add H2O to 600 μL total volume. Mix well (without vortexing) and incubate at 37 °C overnight in a Thermomixer.
Check BbsI digestion on an agarose gel. Prepare a 0.8 % mini agarose gel in 1×TAE; add ethidium bromide to a final concentration of 0.5 μg/mL and allow to polymerize completely. Mix 0.5 μL of the BbsI digestion mix (250 ng DNA) with 4.5 μL H2O and 1 μL 6× gel loading dye. Load digested sample on the agarose gel alongside a sample of the undigested plasmid (250 ng) and 1 kb DNA ladder. Run the gel at 5 V/cm until the bromophenol blue and xylene cyanol are separated by about 2 cm and take a photograph. The BbsI-digested plasmid should be linearized (Fig. 3a) (see Note 10).
-
Check by restriction analysis whether the BbsI digest was complete (see Note 11).
Place 2 μL of the BbsI digest mix (1 μg DNA) into each of the two Eppendorf tubes. Mix the first sample with 0.5 μL EarI (10 U), 1.8 μL 10× NEBuffer 2, and 15.7 μL H2O. Mix the second sample with 0.5 μL BamHI (10 U), 1.8 μL 10× NEBuffer 2, 2 μL 10× BSA, and 13.7 μL H2O. Incubate both tubes for 2 h at 37 °C.
Prepare a 12 % non-denaturing polyacrylamide gel using the BioRad Miniprotean 3 gel casting system. For a minigel with 1.5 mm spacers, prepare 10 mL of polyacrylamide gel: 3 mL 40 % Acrylamide (Acrylamide/Bisacrylamide 29:1), 2 mL 5×TBE, 4.93 mL H2O, 70 μL 10 % Ammonium persulfate, and 3.5 μL TEMED. Pour the gel and insert a comb with ten slots.
Add 4 μL 6× gel-loading buffer (without xylene cyanol) to the EarI and BamHI digests and load on the gel alongside the 100 bp DNA marker. Load 1× gel loading dye (with both marker dyes) in one slot to estimate the migration of the DNA fragments during the run (see Note 12). Run the gel in 1×TBE buffer at 1–8 V/cm until the bromophenol blue runs out of the gel.
-
Perform ethidium bromide post-staining by submerging the gel in 1×TBE containing 0.5 μg/mL ethidium bromide for 30–45 min shaking at room temperature. Wrap the gel in Saran Wrap and photograph with an ultraviolet transilluminator (see Note 13).
Only the expected fragments of 69 and 73 base pairs should be detected (Fig. 3b). If any residual fragments containing 24 extra base pairs are detected, add 1 μL BbsI (in 20 μL 1× NEB2) and continue the digest until completion (check again by EarI and BamHI restriction analysis). Do not continue to the next step until complete BbsI digestion is ensured (see Note 14).
Fig. 3.
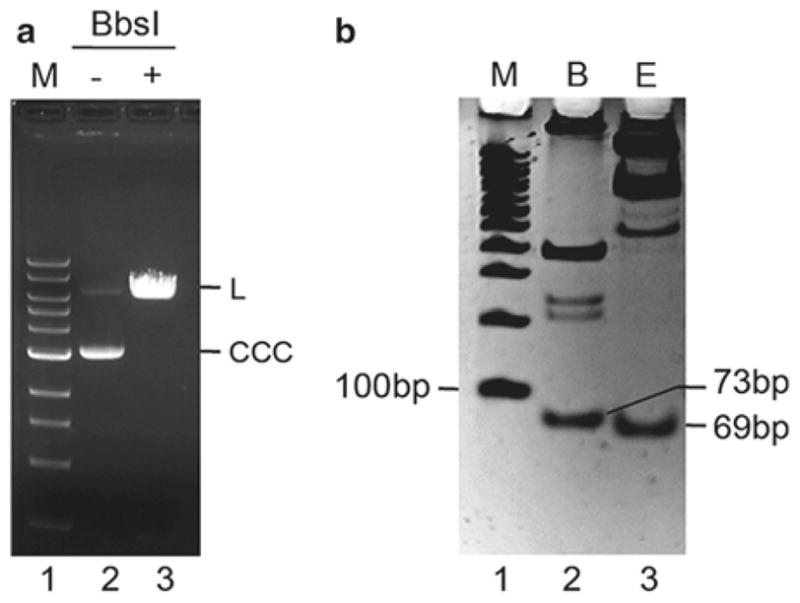
BbsI digest of the pSVRLuc plasmid. (a) Analyzing BbsI digestion by agarose gel. Lane 1: 1 kb DNA Ladder, lane 2: undigested plasmid, lane 3: BbsI-digested plasmid. CCC covalently closed circular form, L linear plasmid. (b) Restriction analysis of the BbsI-digested plasmid. The fragments released after BamHI (73 bp, lane 2) and EarI (69 bp, lane 3) digests were analyzed by 12 % non-denaturing PAGE (inverted mode picture). Lane 1: 100 bp DNA Ladder.
3.3. Purification of the Digested Plasmid
Deproteinize the BbsI-digested sample by extraction with phenol–chloroform–isoamyl alcohol. Add an equal volume of phenol–chloroform–isoamyl alcohol (25:24:1) to the BbsI reaction, vortex contents of the tube until an emulsion forms, and centrifuge at 12,000 × g for 15–20 s in a microfuge at room temperature. Transfer the aqueous phase (containing the DNA) to a fresh tube and repeat the extraction with phenol–chloroform–isoamyl alcohol. Extract once more with an equal volume of chloroform and save the aqueous phase.
Connect the HiLoad Superdex 75 prep grade 16/60 gel filtration column to the FPLC system at 4 °C. Wash the column with 1 column volume H2O at a flow rate of 1 mL/min (see Note 15), and then equilibrate with 2–3 column volumes of TE buffer (see Note 16).
Program the FPLC run as follows: flow rate 1 mL/min, UV detection: 260 nm, sample injection size of 1 mL, start of fractionation after 1 column volume, collecting 2 mL fractions, and stop the run after 3 column volumes.
Pre-warm the digested DNA sample at 37 °C, then load into a 1 mL syringe, and inject it into the sample loop (1 mL). Start the FPLC run.
The plasmid vector elutes first (Fig. 4). The excised DNA fragment is eluted later (usually after 2 column volumes) and is hardly seen in the chromatogram (Fig. 4) (see Note 17). The fractions containing the plasmid DNA are further analyzed using a UV spectrophotometer (260 nm) to accurately determine the DNA concentration (this allows for an estimation of the subsequent precipitation efficiency). Pool the relevant fractions and measure the volume.
Concentrate the DNA by ethanol precipitation. Transfer the diluted DNA solution to an Oak Ridge centrifuge tube (30 mL or 50 mL capacity, depending on the final volume obtained after addition of ethanol; see below). Add 1/10th volume of 3 M sodium acetate to adjust the salt concentration to 300 mM. Add 2 volumes of ice-cold ethanol, mix well, and store at −20 °C for 1 h or overnight.
Recover the DNA by centrifugation at 20,000 × g for 30 min at 4 °C. Carefully remove the supernatant without disturbing the pellet, and then wash the pellet with 10 mL 70 % ethanol. Centrifuge at 20,000 × g for 10 min at 4 °C. After removing the supernatant, air-dry the pellet until the last traces of fluid have evaporated (do not overdry the pellet).
Dissolve the DNA pellet in 0.5–1 mL 10 mM Tris–HCl pH 8.0 (warm up at 30 °C to help dissolve). Transfer the DNA to an Eppendorf tube.
Measure the DNA concentration using a UV spectrophotometer (260 nm). The yield of purified DNA is typically ~80 % of the BbsI-digested DNA loaded onto the column. The purified DNA can be stored at −20 °C.
Fig. 4.
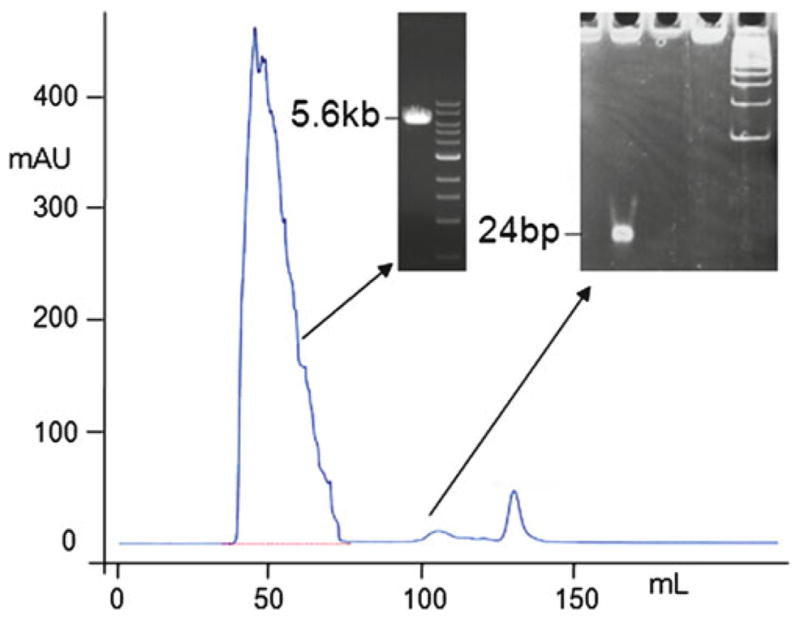
Purification of the BbsI-digested pSVRLuc plasmid. A typical gel filtration pro file shows the elution of the linearized plasmid after 1 column volume and of the excised fragment after 2 column volumes. The insets show the DNA corresponding to the peaks, analyzed by 1 % agarose gel (linearized plasmid) and by 8 % non-denaturing PAGE (24 bp oligonucleotide).
3.4. Ligation of the ICL-Containing Oligonucleotide to the Linearized Plasmid
Test the efficiency of the ligation in small-scale reactions. In general, we obtain good yields of the closed circular plasmid by using 0.5–1 nM plasmid vector and a 1.5× molar excess of the oligonucleotide DNA insert. However, since the efficiency of the ligation reaction might vary between different plasmid preparations, it is important to test the optimal amounts of DNA in small-scale reactions using various combinations of vector to insert ratios and concentrations (see Note 18). Set up ligation reactions containing 0.5, 1, and 2 nM plasmid vector, and 1.5 and 3× molar excess of the insert for each concentration of plasmid. Add 4 μL 10× Ligase buffer, 0.2 μL T4 DNA ligase, and H2O to 40 μL. Perform a control reaction in parallel without insert whenever a new preparation of the digested plasmid is used. Mix well and incubate overnight at 16 °C.
Load the same amount of DNA from each of the small-scale ligation reactions in a 0.8 % agarose gel and compare the yield of the closed circular DNA (Fig. 5a). In the control reaction without insert, only the linear form of the plasmid should be detected.
Scale up the ligation reaction using the DNA concentrations that give the best yield in the small-scale test reactions. The preparative ligation reaction can be set up with a volume of 20–50 mL depending on the desired yield of closed circular plasmid (see Note 19). For a typical ligation reaction (1 nM vector and 1.5× insert), mix in a 50 mL Falcon tube: 40 pmol pSVRLuc plasmid vector (148 μg), 60 pmol oligonucleotide insert, 4 mL 10× Ligase buffer, 10 μL T4 DNA ligase, and H2O to 40 mL. Mix by inverting the tube and incubate over-night at 16 °C in a water bath.
Take a reaction sample corresponding to 100 ng DNA and analyze on a 0.8 % agarose gel to check the efficiency of the preparative ligation reaction (Fig. 5b). Load an aliquot of the linearized plasmid vector without insert for comparison. The covalently closed circular plasmid should represent at least 20 % of the total DNA (typically ranging from 20 to 50 %).
-
Concentrate the DNA from the ligation reaction using one of the procedures below, depending on the volume of the ligation reaction.
For a ligation reaction volume up to 50 mL, concentrate the DNA solution to 0.7 mL using the centrifugal filter device Amicon Ultra-15 30K in a bench-top centrifuge at 4,000×g at 4 °C (see Note 20).
For a ligation reaction volume higher than 50 mL, concentrate the DNA using a QIAGEN-tip 500 column from a Qiagen Plasmid Maxi Prep kit. Pre-equilibrate the column with 10 mL of buffer QBT, apply the ligation reaction, and then wash the column with 30 mL of QC buffer. Elute with QF buffer (1.25 M NaCl, 50 mM Tris–HCl pH 8.5), collecting 1 mL fractions. Measure the DNA concentration of each fraction using a UV spectrophotometer, pool the concentrated fractions, and dialyze against TE.
Fig. 5.

Ligation of the ICL-containing oligonucleotide into the BbsI-digested plasmid vector. (a) Small-scale ligation reactions. The concentrations of the plasmid DNA and insert are indicated in [nM]. Lane 1: 1 kb DNA Ladder, lane 2: control ligation reaction without insert. CCC covalently closed circular form, L linear plasmid, N nicked plasmid, Mult: multimers. The same amount of total DNA was loaded for each ligation reaction. (b) Large-scale ligation reaction. A typical ligation reaction is shown in lane 2. Lane 1: 1 kb DNA Ladder, lane 3: control ligation reaction without insert. (c) Covalently closed circular plasmid containing the interstrand cross-link isolated from cesium chloride (Cs) gradient. A small amount of nicked plasmid may be formed during the purification procedure.
3.5. Purification of the Closed Circular Plasmid
-
The concentrated ligation solution (obtained in step 5(a) of the Subheading 3.4) is mixed with a cesium chloride ethidium bromide solution or alternatively, cesium chloride is dissolved directly into the DNA solution (obtained in step 5(b) of the Subheading 3.4).
Dissolve 12 g cesium chloride in 10.7 mL 10 mM Tris–HCl, pH 8.0, 1 mM EDTA buffer in a Falcon tube. Cover the tube with aluminum foil and add 80 μL 10 mg/mL Ethidium Bromide. Mix well. Load the 0.7 mL concentrated DNA sample into the 6 mL ultracentrifuge tube. Add the same volume of TE buffer to the balance tube. In the dark, fill the tubes with the CsCl/Ethidium bromide solution.
For every 1 mL of concentrated ligation solution, mix with 1 g of cesium chloride in a Falcon tube. In a separate Falcon tube, dissolve 15 g cesium chloride with 15 mL TE buffer. Load the CsCl-ligation solution into a 6 mL ultracentrifuge tube wrapped with aluminum foil. Add 30 μL 10 mg/mL ethidium bromide to the ultracentrifuge tube. Fill the remainder of the ultracentrifuge tube with the CsCl–TE solution. Fill a second ultracentrifuge tube with the CsCl–TE solution to serve as a balance.
Seal the tubes using the Ultracrimp sealing tool. Balance the tubes with the adaptors for Stepsaver 65V13 ultracentrifuge rotor (using different combinations of the adaptors) so that the difference in weight is less than 10 mg.
Centrifuge at 60,000 rpm (about 340,000 × g) for 16 h at 4 °C (see Note 21). Set the break off from 20,000 rpm (about 37,000×g) to 0.
Stop the ultracentrifuge run and wait until the rotor has come to a complete stop. This can take up to 1 h.
Take out the tube from the ultracentrifuge rotor carefully (without disturbing the gradient). In a darkroom, place the tube on a holder and visualize the bands using a UV lamp at 360 nm (try to minimize the exposure to UV light). Two bands of DNA should be visible: the upper band consists of linear and nicked DNA, and the lower band consists of closed circular DNA. Pierce the top of the tube with a 21-gauge needle, which serves as a vent (heating up the needle with a lighter can facilitate piercing). Pierce the tube with an 18-gauge needle attached to a 1 mL syringe just below the lower DNA band (make sure that the bevel of the needle is facing upwards toward the DNA band), and carefully extract the closed circular DNA. Avoid contamination with the upper DNA band.
Dilute the collected DNA to 2 mL with TE buffer in a 15 mL Falcon tube wrapped with aluminum foil.
Add an equal volume of water-saturated butanol to extract the ethidium bromide. Mix the two phases by inverting several times.
Centrifuge the mixture at 450 × g in a bench-top centrifuge for 2 min.
Remove the upper phase (butanol containing the ethidium bromide), and repeat the extraction (steps 7 and 8) four to six times to remove any remaining ethidium bromide.
-
Remove the CsCl by spin dialysis through an Amicon Ultra-4 30K centrifugal filter device. Concentrate to about 100 μL by spinning at 4,000 × g, 4 °C in the bench-top centrifuge.
Add 3.9 mL TE buffer and spin to concentrate as above. Repeat this buffer exchange step four more times. Perform a last buffer exchange with 10 mM Tris–HCl pH 7.5 and stop the spin when the volume of the DNA sample is about 200 μL.
Transfer the DNA sample to an Eppendorf tube. Measure the concentration using a UV spectrophotometer and analyze the DNA by agarose gel electrophoresis (Fig. 5b) (see Note 22). The final yield of the closed circular plasmid varies between 20 and 50 % of the linear vector DNA included in the ligation reaction. In general, we obtain 30–50 μg of closed circular plasmid from a 40 mL ligation reaction.
Dilute the DNA to 10 ng/μL with 10 mM Tris–HCl, pH 7.5 containing 10 mM NaClO4 (10 mM NaClO4 is added for cisplatin ICL-containing plasmids, but not for other types of ICLs). Make single-use aliquots and store at −80 °C.
Acknowledgments
This work was supported by the New York State Office of Science and Technology and Academic Research NYSTAR (C040069), the Association for International Cancer Research (AICR, Nr. 00-225), the Swiss Cancer League (OCS-01413-080-2003), the National Institutes of Health (GM080454 and CA092584 to O.D.S. and GM62267, HL098316, and GM80676 to J.C.W.), and the American Cancer Society (postdoctoral fellowship PF-10-146-01-DMC to D.T.L.).
Footnotes
The plasmid purity is important for the efficiency of the BbsI digest. We use the Qiagen Maxiprep kit and pay attention to completely dry out residual ethanol, since traces of ethanol inhibit BbsI.
We have also incorporated duplexes containing nitrogen mustard-like ICLs (12, 13) into the same plasmid using identical procedures with very similar yields and efficiency.
Use 5′ phosphorylated, highly pure oligonucleotides. We use chemically phosphorylated and HPLC- or PAGE-purified oligonucleotides. Oligonucleotides phosphorylated enzymatically (T4 Polynucleotide kinase) can also be used, but since enzymatic phosphorylation is less efficient, the yield of closed circular plasmid after ligation may be significantly decreased.
For BbsI, storage at −80 °C is recommended for periods longer than 30 days. We make aliquots (30 μL aliquot for digestion of 300 μg DNA) and store them at −80 °C.
We have found that some batches of cisplatin are extremely reactive and do not need activation prior to the cisplatination reaction. We recommend testing each new batch of the reagent in small-scale platination reactions (using 2 nmol DNA) in order to find out whether the activation of cisplatin is needed or not.
The platination reaction can be scaled up to 20 nmol single-stranded oligonucleotide (keep the final DNA concentration to 125 μM). For optimal resolution during the anion exchange chromatography step, it is important not to load more than 20 nmol DNA on the Mono Q column. For the preparation of higher amounts of cisplatin monoadduct DNA, make several reactions and inject them separately on the Mono Q column.
The desalting and buffer exchange step can be also done by dialysis against 0.1 M NaClO4, using a Spectra/Por Biotech RC MWCO 3.5K membrane, 4 h at 4 °C.
The presence of the cisplatin ICL within the DNA duplex can be further confirmed by radioactive labeling (32P) in a phosphate exchange reaction and analysis of the size by denaturing PAGE (Fig. 2c).
The BbsI digest reaction can be scaled up to 500 μg DNA using 0.5 U BbsI/μg DNA.
This step may be skipped since it does not discern whether BbsI cleavage occurred at one site or at both sites. The next step (restriction analysis of the digested DNA) is absolutely needed and sufficient to make sure that BbsI digest was complete.
The choice of the analytical restriction enzyme depends on the particular plasmid. Use two restriction enzymes that cut close to the BbsI sites and generate fragments of 50–300 bp on the BbsI-linearized plasmid. This size ensures that incompletely digested BbsI DNA fragments (+24 bp) can easily be detected.
For the pSVRLuc, EarI digest generates a 69 bp fragment (83 bp if BbsI digest is incomplete), and BamHI generates a 73 bp fragment (97 bp if BbsI digest is incomplete). BamHI and EarI are not unique sites in pSVRLuc, but the fragments generated by cleavage at the other sites are larger and do not interfere with the detection of the fragments of interest.
In a 12 % nondenaturing polyacrylamide gel, bromophenol blue runs at ~20 bp and xylene cyanol runs at ~70 bp DNA fragments.
Choose a UV exposure long enough to make sure that besides the expected fragments, no fragments containing an additional 24 base pairs, generated from an incomplete BbsI digest, can be detected (even very faint bands). Visualizing the picture in inverted mode often confers a better sensitivity.
In general, BbsI digestion goes to completion, and the presence of two BbsI sites might contribute to the activity of the enzyme, as known for some type IIS enzymes. However, it is very important to make sure that the digest goes to completion, since partially digested DNA can re-ligate, leading to contamination of the ICL-containing plasmid with uncross-linked parental DNA.
The gel filtration column has to be free of any contaminants in order to avoid the degradation of single-stranded overhangs. Prior to use, perform a rigorous cleaning of the column by washing at a flow rate of 0.8 mL/min with 4 column volumes of 1 M NaOH, 4 column volumes of H2O, then 0.5 column volumes of 30 % isopropanol, followed by 2 column volumes of H2O.
The equilibration of the gel filtration column with the running buffer can be done overnight at a flow rate of 0.2 mL/min.
If the excised fragment is not detected in the FPLC profile, fractions eluting after 2 column volumes can be pulled, ethanol precipitated, and analyzed by 10–20 % native PAGE with ethidium bromide post-staining (Fig. 4). The detection of the short DNA fragment will ensure that it has been efficiently removed by gel filtration and is particularly important when performing the procedure for the first time.
It is important to accurately determine the concentration of both the digested plasmid vector and the oligonucleotide insert by UV absorbance, since the efficiency of the ligation reaction is critically dependent on DNA concentration.
The ligation reaction can be scaled up to a volume higher than 50 mL if necessary (for the preparation of larger amounts of substrate, or for combinations of plasmid vectors and inserts that give lower yields of closed circular form than usual). However, it is important to stay well below the maximum binding capacity of the QIAGEN-tip 500 column (500 μg).
In order to decrease the time needed for the concentration of the DNA sample after the preparative ligation reaction, deproteinization by phenol–chloroform–isoamyl alcohol extraction (2×), followed by chloroform extraction (1×), and finally butanol extraction can be performed before loading into the Amicon Ultra-15 30K filter device. We have found that removal of the Ligase and BSA from the ligation reaction significantly increases the flow rate.
The ultracentrifugation can also be done for more than 16 h.
The quality of the ICL-containing plasmid preparation can be further analyzed using several methods. A fragment containing the ICL can be cut out from the plasmid, radiolabeled using a phosphate exchange reaction, and analyzed in denaturing polyacrylamide gel. Alternatively, use the restriction digest analysis at the unique SapI site that is inactivated by the cisplatin ICL.
References
- 1.Noll DM, Mason TM, Miller PS. Formation and repair of interstrand cross-links in DNA. Chem Rev. 2006;106:277–301. doi: 10.1021/cr040478b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schärer OD. DNA interstrand cross-links: natural and drug-induced DNA adducts that induce unique cellular responses. Chembiochem. 2005;6:27–32. doi: 10.1002/cbic.200400287. [DOI] [PubMed] [Google Scholar]
- 3.Guainazzi A, Schärer OD. Using synthetic DNA interstrand crosslinks to elucidate repair pathways and identify new therapeutic targets for cancer chemotherapy. Cell Mol Life Sci. 2010;67:3683–3697. doi: 10.1007/s00018-010-0492-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knipscheer P, Räschle M, Smogorzewska A, Enoiu M, Ho TV, Schärer OD, Elledge SJ, Walter JC. The fanconi anemia pathway promotes replication-dependent DNA inter-strand cross-link repair. Science. 2009;326:1698–1701. doi: 10.1126/science.1182372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Räschle M, Knipsheer P, Enoiu M, Angelov T, Sun J, Griffith JD, Ellenberger TE, Schärer OD, Walter JC. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell. 2008;134:969–980. doi: 10.1016/j.cell.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang N, Lu X, Zhang X, Peterson CA, Legerski RJ. hMutSbeta is required for the recognition and uncoupling of psoralen interstrand cross-links in vitro. Mol Cell Biol. 2002;22:2388–2397. doi: 10.1128/MCB.22.7.2388-2397.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen X, Do H, Li Y, Chung WH, Tomasz M, de Winter JP, Xia B, Elledge SJ, Wang W, Li L. Recruitment of fanconi anemia and breast cancer proteins to DNA damage sites is differentially governed by replication. Mol Cell. 2009;35:716–723. doi: 10.1016/j.molcel.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shen X, Jun S, O’Neal LE, Sonoda E, Bemark M, Sale JE, Li L. REV3 and REV1 play major roles in recombination-independent repair of DNA interstrand cross-links mediated by monoubiquitinated proliferating cell nuclear antigen (PCNA) J Biol Chem. 2006;281:13869–13872. doi: 10.1074/jbc.C600071200. [DOI] [PubMed] [Google Scholar]
- 9.Wang X, Peterson CA, Zheng H, Nairn RS, Legerski RJ, Li L. Involvement of nucleotide excision repair in a recombination-independent and error-prone pathway of DNA interstrand cross-link repair. Mol Cell Biol. 2001;21:713–720. doi: 10.1128/MCB.21.3.713-720.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng H, Wang X, Warren AJ, Legerski RJ, Nairn RS, Hamilton JW, Li L. Nucleotide excision repair- and polymerase eta-mediated error-prone removal of mitomycin C interstrand cross-links. Mol Cell Biol. 2003;23:754–761. doi: 10.1128/MCB.23.2.754-761.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hofr C, Brabec V. Thermal and thermodynamic properties of duplex DNA containing site-specific interstrand cross-link of antitumor cisplatin or its clinically ineffective trans isomer. J Biol Chem. 2001;276:9655–9661. doi: 10.1074/jbc.M010205200. [DOI] [PubMed] [Google Scholar]
- 12.Angelov T, Guainazzi A, Schärer OD. Generation of DNA interstrand cross-links by post-synthetic reductive amination. Org Lett. 2009;11:661–664. doi: 10.1021/ol802719a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guainazzi A, Campbell AJ, Angelov T, Simmerling C, Schärer OD. Synthesis and molecular modeling of a nitrogen mustard DNA interstrand crosslink. Chemistry. 2010;16:12100–12103. doi: 10.1002/chem.201002041. [DOI] [PMC free article] [PubMed] [Google Scholar]