

Abstract
Background and purpose
The phenotype of IBMPFD [inclusion body myopathy with Paget’s disease of the bone and frontotemporal dementia (FTD)] associated with valosin-containing protein(VCP) mutation is described in three families.
Methods
Probands were identified based on a pathological diagnosis of frontotemporal lobar degeneration with TDP-43-positive inclusions type IV. VCP sequencing was carried out. Clinical data on affected family members were reviewed.
Results
Ohio family: four subjects presented muscle weakness and wasting. (One subject had both neuropathic and myopathic findings and another subject showed only evidence of myopathy. The etiology of weakness could not be ascertained in the remaining two subjects.) Two individuals also showed Parkinsonism (with associated FTD in one of the two). The proband’s brain displayed FTLD-TDP type IV and Braak stage five Parkinson’s disease (PD). A VCP R191Q mutation was found. Pennsylvania family: 11 subjects developed IBMPFD. Parkinsonism was noted in two mutation carriers, whilst another subject presented with primary progressive aphasia (PPA). A novel VCP T262A mutation was found. Indiana family: three subjects developed IBMPFD. FTD was diagnosed in two individuals and suspected in the third one who also displayed muscle weakness. A VCP R159C mutation was found.
Conclusions
We identified three families with IBMPFD associated with VCP mutations. Clinical and pathological PD was documented for the first time in members of two families. A novel T262A mutation was found. One individual had PPA: an uncommon presentation of IBMPFD.
Keywords: frontotemporal dementia, genetic and inherited disorders, motor neuron disease, myopathies, neurodegenerative disorders (other than dementia), neuropathology, neuropsychology, Parkinson’s disease
Introduction
Inclusion body myopathy (IBM) with Paget’s disease of the bone (PDB) and frontotemporal dementia (FTD) (IBMPFD, OMIM 167320) is an autosomal dominant, multisystem degenerative disorder associated with mutations in the valosin-containing protein(VCP) gene [1]. VCP is a molecular chaperone, a member of the AAA (ATPase associated with diverse cellular activities) gene superfamily which is involved in multiple cellular processes including nuclear envelope reconstruction, Golgi and endoplasmic reticulum assembly, stress responses, programmed cell death, and protein degradation [2, 3].
The neuropathologic substrate of FTD in IBMPFD is frontotemporal lobar degeneration (FTLD) associated with TDP-43-immunoreactive inclusions type IV (FTLD-TDP type IV). Affected muscles display characteristic hallmarks of IBM: atrophic, angulated muscle fibers with frequent rimmed vacuoles, and TDP-43- and/or VCP-immunoreactive sarcoplasmic inclusions in the absence of inflammation. PDB can be suspected on the basis of characteristic radiologic findings or otherwise unexplained elevation in serum level of alkaline phosphatase and confirmed by the observation of typical findings of bone biopsy [4–7].
Phenotypic heterogeneity includes IBM in about 90% of patients, PDB in half the patients, and FTD in about one-third of cases [8]. Recently, VCP mutations have also been associated with sporadic amyotrophic lateral sclerosis (ALS), familial ALS, or familial FTD-ALS [9–12]. Finally, several affected individuals from IBMPFD families have been reported to manifest symptoms of Parkinsonism/Parkinson’s disease (PD) [1, 9, 13–17] although the pathological substrate of this association remains unclear.
We present clinicopathologic findings in three families with IBMPFD associated with VCP mutations. PD was observed in two families. A novel T262A mutation was found in a family in which one individual presented with primary progressive aphasia (PPA), an uncommon clinical presentation of IBMPFD.
Materials and methods
Subjects
Probands were identified amongst cases referred to the Indiana Alzheimer Disease Center (IADC) for neuropathologic examination of suspected FTLD and selected on the basis of a pathological diagnosis of FTLD-TDP type IV [6]. All cases with FTLD-TDP type IV pathology in the IADC neuropathologic series were found to be associated with a VCP gene mutation.
Pedigrees were reconstructed by means of interviews with family members and review of medical charts (Fig. 1). Informed consent for participation in the study was obtained from all subjects or next of kin according to the Internal Review Board approved protocol at the authors’ institution.
Figure 1.
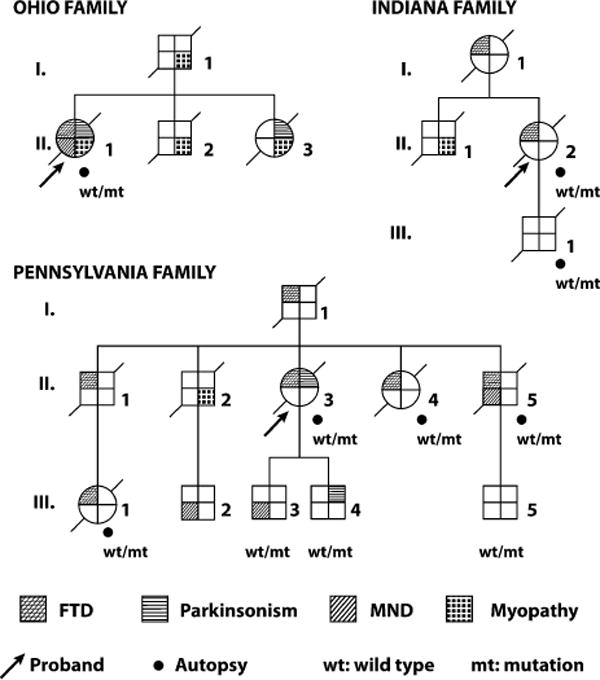
Pedigree of the Ohio, Pennsylvania, and Indiana families. The diagnosis of myopathy in subjects O.I.1, O.II.3, P.II.2, and I.II.1 indicated above is solely based on the clinical description of the examining clinicians, in patients with evidence of weakness and muscle wasting that did not meet established criteria for motor neuron disease. No neurophysiologic or pathological data are available for these subjects.
Clinical assessment
The proband from the Ohio (O) family was examined by JQ at the University of Cincinnati Department of Neurology. Proband and subject II.5 of the Pennsylvania (P) family were examined at the Alzheimer’s Disease Research Center (ADRC) of the University of Pittsburgh School of Medicine (STDeK). Subjects III.3 and III.4 were examined by MF at the Indiana Alzheimer Disease Center (IADC). Clinical information about additional family members who were examined elsewhere was obtained and reviewed.
A clinical diagnosis of upper motor neuron disease (MND) was given to patients who presented muscle weakness associated with spastic hypertone and/or hyperreflexia. A clinical diagnosis of lower MND was given to patients with evidence of muscle weakness, waste, and/or fasciculations associated with electrophysiological evidence of active denervation. ALS was diagnosed according to the Revised El Escorial diagnostic criteria [18].
A diagnosis of Parkinsonism was made in the presence of bradykinesia associated with at least one of the following: muscular rigidity, resting tremor, or postural instability not caused by primary visual, vestibular, cerebellar, or proprioceptive dysfunction [19, 20].
Neuropathologic studies
Brain and spinal cord of the proband from each family (as well as from subjects II.4, II.5, and III.1 from the Pennsylvania family) were available for neuropathologic examination, with the exception of spinal cord tissue from the Indiana family proband. Skeletal muscle tissue was available from the Ohio family proband’s autopsy. Sections were obtained from representative brain and spinal cord regions and processed for classic histology and immunohistochemistry (IHC) according to published protocols [21]. IHC was performed using antibodies specific for ubiquitin, TDP-43, phosphorylated tau, [beta]-amyloid, glial fibrillary acidic protein, a[beta] crystalline, phosphorylated neurofilament, VCP, and [alpha]-synuclein [22] (see Data S2).
Genetic studies
DNA was extracted from a frozen sample of cerebellum from subjects O.II.1, P.II.3, P.II.4, I.II.2, and I.III.1 using a standard protocol [23] and from blood from subjects P.II.5, P.III.1, P.III.3, P.III.4, and P.III.5. The entire VCP coding region and flanking intronic sequences were analyzed (see Data S2).
Results
Clinical and neuropathologic findings
Clinicopathologic findings are summarized in Table 1.
Table 1.
Summary of clinical and neuropathologic findings
| Subjects | Age at onset (years) | Age at death (years) | Presenting signs | Clinical signs along disease coursea | Fresh brain weight (g) |
|---|---|---|---|---|---|
| Ohio family (R191Q) | |||||
| I.1 | 53 | 68 | W | W | n.a. |
| II.1 | 63 | 70 | M, P | F, My, M, P | 930 |
| II.2 | 48 | 64 | My | My, B? | n.a. |
| II.3 | 51 | 63 | W | W, P | n.a. |
| Pennsylvania family (T262A) | |||||
| I.1 | n.a. | 55 | n.a. | F | n.a. |
| II.1 | 48 | 56 | F | F | n.a. |
| II.2 | 47 | 60 | W | W, F? | n.a. |
| II.3 | 55 | 65 | F | F, P | 980 |
| II.4 | 53 | 68 | F | F | 1000 |
| II.5 | 51 | 64 | F | F, M | 1150 |
| III.1 | 56 | 60 | F | F | 1130 |
| III.2 | n.a. | n.a. | M | M | n.a. |
| III.3 | 39 | n.a. | M | M | n.a. |
| III.4 | 44 | n.a. | P | P | n.a. |
| III.5 | 20s? | n.a. | F? | F? | n.a. |
| Indiana family (R159C) | |||||
| I.1 | 68 | 78 | F | F | n.a. |
| II.1 | 65 | 72 | W | W, F? | n.a. |
| II.2 | 73 | 78 | F | F | 1052 |
| III.1 | n.a. | 53 | n.a | n.a. | 1440 |
F, frontotemporal dementia; My, myopathy; M, motor neuron disease; W, weakness not otherwise specified; P, Parkinsonism; B, Paget’s disease of the bone; ?, unconfirmed finding; n.a., not applicable or not available.
Diagnoses are based on best clinical judgment, diagnostic criteria, and, when available, diagnostic evidence from neurophysiologic and/or pathological studies.
Ohio family
The proband (subject O.II.1)
At age 63, this woman started experiencing stiffness and cramps in her arms and legs. Neurological examination revealed hypomimia, dystonic posturing of the right arm, and the absence of deep tendon reflexes (DTRs). Electromyography (EMG) revealed mildly enlarged motor unit action potentials (MUAPs) at the majority of the muscles examined and fibrillation potentials in the solei. At age 64, she had developed rigidity, reduced rapid alternating movements, and decreased arm swing (R>L). Weakness and wasting of dorsal and plantar flexors of the feet were noted at age 65. Later, she developed difficulty performing house chores and job duties, managing her medications, and remembering family events. She became socially withdrawn, incontinent, and dependent for activities of daily living and fell repeatedly. Her language deteriorated, and she became unable to communicate verbally. She died at age 70.
At autopsy, severe symmetric frontotemporal atrophy, thinning of frontotemporal cortical ribbon and corpus callosum, and extensive enlargement of the lateral ventricles were noted. The basal ganglia, brainstem, and cerebellum were grossly unremarkable. Histologically, neuronal loss and gliosis were moderate in the frontal, cingulate, temporal, and parietal cortices, hippocampus, basal nuclei, substantia nigra, locus coeruleus, and dentate nucleus. There was extensive gliosis of the deep white matter of the frontal lobes. In the spinal cord, mild loss of motor neurons and occasional axonal spheroids were seen in the anterior horns (Fig. 2a). Numerous TDP-43-immunoreactive (IR) neuronal intranuclear inclusions (NII) were present in the frontal and temporal cortices, and more particularly abundant in cortical layer II. Neuronal cytoplasmic inclusions (NCI) and dystrophic neurites (DN) were also seen, although to a much lesser extent (Fig. 2b). Sparse NII were seen in the upper cortical layers of the parietal and occipital lobes. Occasional NII and NCI were observed in the dentate gyrus. NII were occasionally seen in the striatum and amygdala. No TDP-43-IR inclusions were seen in the subcortical white matter. Alpha-synuclein IHC revealed scattered Lewy bodies in the cingulate gyrus, as well as numerous Lewy bodies and Lewy neurites in the substantia innominata, substantia nigra, reticular formation, locus coeruleus, raphe nuclei, and medullary intermediate reticular zone (Fig. 2c–d). Lewy neurites were seen occasionally in the striatum. Tau-IHC revealed occasional tau-immunoreactive neurons and neuropil threads in the orbital cortex, caudate nucleus, amygdala, hippocampus, entorhinal cortex, locus coeruleus, and raphe nuclei. Aβ-protein IHC was negative in all sections examined. Histology and IHC of skeletal muscle tissue revealed atrophic and angulated muscle fibers with occasional rimmed vacuoles. VCP-IR deposits were seen in the sarcoplasm, but TDP-43-IR deposits were absent (Fig. 2e).
Figure 2.
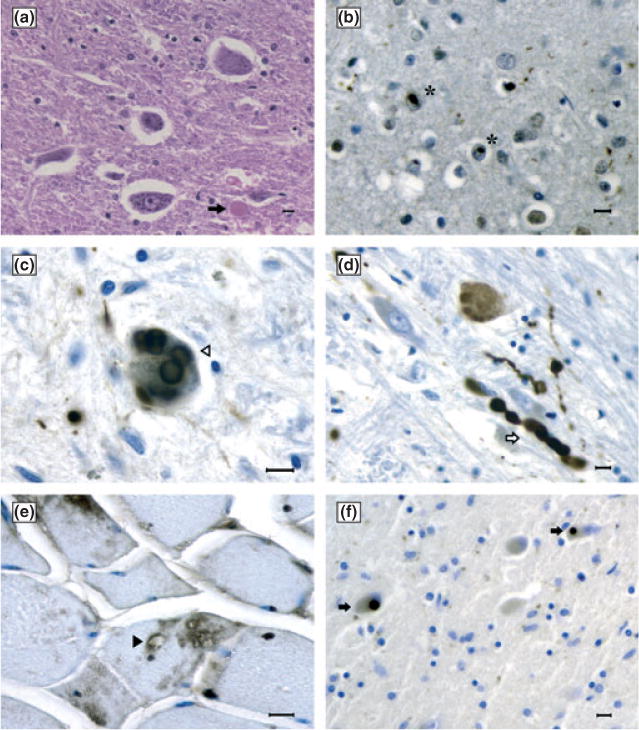
(a) Neuronal loss and a rare axonal spheroid (arrow) in the anterior horn of the cervical cord (hematoxylin and eosin). (b) TDP-43-immunoreactive (IR) neuronal intranuclear inclusion in the frontal cortex (stars). (c) Alpha-synuclein-IR Lewy bodies (open arrowhead) and Lewy neuritis (open arrow) (d) in the substantia nigra. (e) Sarcoplasmic vacuoles (solid arrowhead) and VCP-IR deposits in the muscle. (f) TDP-43-IR Lewy-body-like inclusions in the thalamus (solid arrows). (a)–(e) Ohio family’s proband. (f) Pennsylvania family’s proband. Bars: 10 μm.
Family history
Subject O.I.1 began experiencing progressive weakness in the four extremities at age 53. Neurological examination revealed marked atrophy of the biceps, forearms, and intrinsic muscles in the hands. DTRs were absent in the arms and diminished in the legs. He was diagnosed with myopathy and died at age 68. Subject O.II.2 was diagnosed with diabetes mellitus (DM) at age 44. At age 48, he started developing slowly progressive proximal muscle weakness in all four limbs. In the 2 years that followed, he became unable to climb stairs and also could not rise from the floor without help. He complained of low back pain and bilateral leg pain. Neurological examination revealed muscle wasting and weakness in the upper girdle, hip flexors, and quadriceps. DTRs were decreased in the upper extremities and absent in the lower extremities. Sensory examination revealed diminished pinprick, vibration, and position sensation distally in the lower extremities. CPK levels were normal. EMG revealed both enlarged and small polyphasic motor unit potentials in virtually all muscles as well as fibrillation potentials in half of the examined muscles, findings that were felt consistent with myopathy. Sensory nerve potentials were either reduced or absent. He was initially diagnosed with spinal muscular atrophy and diabetic polyneuropathy and later with myopathy. He died at age 64. Subject O.II.3 presented at age 51 with progressive muscle weakness in all extremities. DTRs were either reduced or absent throughout. She was diagnosed with myopathy. Later, she developed Parkinsonism and became unable to communicate, eat, or ambulate without assistance. She died at age 63.
Pennsylvania family
The proband (subject P.II.3)
At age 55, this woman presented with apathy, withdrawal, reduced empathy, forgetfulness, emotional lability, and reduced verbal output. She developed an increased appetite and gained considerable weight. She complained of altered olfaction and taste, poor sleep quality, and reduced libido. At age 58, her speech was remarkable for dysnomia, short sentences, and increasing monosyllabic (yes/no) answers. She manifested severe deficits of attention, concentration, short memory and episodic memory. She scored in the moderately demented range (115/144) on the Mattis Dementia Rating Scale although she retained awareness of her cognitive and behavioral changes. Physical examination revealed mildly generalized muscle hypertonia. At age 61, she developed disinhibition, obsessive hair combing, cogwheel rigidity in four limbs (more severe in the upper extremities, right>left), reduced arm swing, tremor of the upper limbs, and bradykinesia. DTRs were increased in the right upper extremity and symmetrically reduced in the lower extremities. Snout, glabellar, grasp, and palmomental reflexes were present. By age 62, she had become globally aphasic, dysphagic, unable to walk, and incontinent. She died at age 65, and an autopsy was performed.
There was severe atrophy of the frontal and temporal lobes and, to a lesser extent, parietal and occipital lobes. The cerebral ventricles were markedly enlarged. The cerebellum was unremarkable. Histologically, neuronal loss and gliosis were severe in the frontotemporal and entorhinal cortices, CA1 sector of the hippocampus, and subiculum. Moderate neuronal loss and gliosis were seen in the basal nuclei, thalami, dentate nucleus, substantia nigra, motor dorsal nucleus of the vagus nerve, hypoglossal and inferior olivary nuclei, and anterior horns of the spinal cord. The pons was not available for examination. Numerous TDP-43-IR NII and neurites were observed in the upper layers of the frontal, temporal, and entorhinal cortices. To a much lesser extent, NCI were seen in the cortex as well as in the thalamus, where they had a Lewy-body-like appearance (Fig. 2f). A few TDP-43-IR neurites were seen in the substantia nigra. Alpha-synuclein-IR neurites were seen in the substantia innominata and substantia nigra, together with a few tau-IR neurons and neurites. A few tau-IR neurons and neurofibrillary tangles were seen in the entorhinal cortex. Aβ-protein IHC was negative in all examined sections.
Family history
Subject P.I.1, who died of a myocardial infarction at age 55, had a history of personality changes and memory deficits. A paternal aunt had had multiple psychiatric hospitalizations and had died of a heart attack at age 72. The proband’s mother died demented of cardiorespiratory failure at age 76. The proband’s maternal grandmother was described as ‘bizarre’ and withdrawn from the family. She was severely demented and aphasic by the time she died at age 41. Amongst the proband’s siblings, three subjects developed FTD between the ages of 48 and 53; one of them met criteria for PPA. Another sibling was diagnosed with MND at age 48 and died demented at age 60. Amongst subjects in the third generation, two people were diagnosed with ALS, one with FTD at age 56 and another one with PD at age 44. Another subject was known to have shown behavioral abnormalities in his mid-20s. Neuropathologic examination of the brain of subjects P.II.4, P.II.5, and P. III.1 displayed findings consistent with a diagnosis of FTLD-TDP type IV. No evidence of MND or alpha-synuclein-IR inclusions was observed (for additional information, see Data S1).
Indiana family
The proband (subject I.II.2)
At age 73, this woman presented with a history of forgetfulness, dysexecutive symptoms, disinhibition, and marked preference for sweet food, leading to a 40-pound weight gain in a year. Clinical examination at age 75 revealed flat affect, memory loss, impaired abstract reasoning, as well as reduced speech initiation, attention and response inhibition. Her speech became monosyllabic. She developed agitation, delusions, and bowel and bladder incontinence. She died at age 78, and an autopsy was carried out (see Data S1).
There was moderate atrophy of the frontal, temporal, and parietal lobes, more pronounced at the level of the pre-frontal and superior temporal gyri. The hippocampus and parahippocampal gyrus were severely atrophied. The head of the caudate nucleus was flattened; the amygdala was moderately atrophic. There was marked atrophy of the centrum semiovale and corpus callosum. The cerebral ventricles were extensively enlarged. The cerebellum was unremarkable. The substantia nigra and locus coeruleus were moderately depigmented.
Histologically, neuronal loss and gliosis were moderate in the frontal, temporal, insular, cingulate, and entorhinal cortices, dentate gyrus, basal nuclei, thalamus, and dentate nucleus, as well as mild in the substantia nigra, locus coeruleus, motor dorsal nucleus of the vagus nerve, and hypoglossal and inferior olivary nuclei. Hippocampal sclerosis was noted. TDP-43-IR NII were numerous in the upper layers of the frontal, temporal, insular, and entorhinal cortices, together with a few NCI and DN. To a lesser extent, NII were also seen in the amygdala and dentate gyrus. Alpha-synuclein IHC was negative in all the examined sections. Tau-IR neurons were seen in the amygdala, CA2 sector of the hippocampus, entorhinal and transentorhinal cortex, and pons. A few neurofibrillary tangles were seen in the entorhinal cortex. A few Aβ-protein IR diffuse plaques but no neuritic plaques were seen in the frontal, entorhinal, and calcarine cortices, amygdala, and nucleus accumbens.
Family history
The proband’s mother (subject I.I.1) developed depression at age 68, followed by aggressiveness, paranoia, and forgetfulness. She had a slowly progressive disease course and died at age 78. The proband’s brother (subject I.II.1) was initially diagnosed with spinocerebellar degeneration at age 65, later revised to muscle dystrophy along the disease course. He was demented for the last 6 years of his life and died at age 72. A healthy son of the proband (I.III.1) died tragically at age 53 of a work-related traumatic head injury. His brain’s autopsy revealed mechanical trauma, ischaemia, and edema but no evidence of neurodegeneration.
Genetic results
A c.572G>A (p.R191Q) substitution was found in one allele of the Ohio family proband’s VCP gene. A c.784A>G (p.T262A) substitution was found in one allele of the Pennsylvania family proband’s VCP gene, as well as in subjects P.II.4, P.II.5, P.III.1, P.III.3, P.III.4, and P.III.5. This mutation is located in the heat-enhanced ATPase domain of the VCP protein. A c.475C>T (p.R159C) substitution was found in one allele of the Indiana family proband’s VCP gene, as well as in subjects I.III.1, I.III.2, and I.III.3.
Discussion
We report clinicopathologic findings in individuals from three families with IBMPFD associated with VCP mutations. In the Ohio family (R191Q), the proband presented with muscle weakness and wasting associated with neurogenic EMG findings and Parkinsonism, whilst dementia developed later. Muscle weakness was seen in all affected family members. Another subject developed Parkinsonism later in the disease course. The R191Q VCP mutation has been described in a family of German origin (family 13), in which all patients but one, who first developed PDB, presented with myopathy [1, 15]. Stojkovic et al. [24] reported an R191Q mutation carrier who developed PDB at age 40 and later manifested a myopathy. Johnson et al. [9] described an IBMPFD family (ITALS#1) with an R191Q mutation, in which a proband’s parent died at age 58 of dementia, Parkinsonism, Paget disease, and upper limb muscle weakness. No autopsy was carried out [9]. Finally, Shi et al. [25] reported an R191Q mutation in a subject with myopathy.
Frontotemporal dementia was the presenting sign in six subjects from the Pennsylvania family (T262A) (I.1, II.1, II.3, II.4, II.5, and III.1). Subject II.1’s clinical history was compatible with a diagnosis of PPA. Subject III.3 was diagnosed with ALS at age 39. Another individual (III.4) was diagnosed with PD at age 44. His mother (subject II.3) developed Parkinsonism in the late disease stage, and her autopsy revealed Lewy neurites in the substantia nigra. The T262A mutation, not previously reported, is located in the heat-enhanced ATPase domain of the protein. It has been suggested that mutations in this domain may be associated with a more severe clinical phenotype [1].
Two subjects from the Indiana family (R159C) presented with FTD, whilst another subject presented with muscle weakness. Another mutation carrier from this family (III.1) was asymptomatic when he died at age 53. Autopsy showed neither FTLD nor TDP-43-IR pathology. The R159C mutation has been described in an individual from an Italian family who presented with myopathy at age 50 and subsequently developed FTD at age 68 [26].
An interesting finding of this report is the observation of Parkinsonism in two siblings from the Ohio family (O.II.1 and O.II.3) and two subjects (P.II.3 and P.III.4) from the Pennsylvania family. Parkinsonism was the presenting sign of disease in subject O.II.1, who had neuropathologic evidence of Braak stage 5 PD, as well as in subject P.III.4 [27]. Alpha-synuclein-IR inclusions were found in the brain of subject P.II.3. Parkinsonism has been described in one subject (I.2) from Family 7, as well as in three subjects (II.6, III.6, and III.8) from Family 16 with IBMPFD associated with the R155H mutation [1, 15]. Neuropathologic examination of III.6 did not reveal Lewy bodies [15]. Forman et al. [28] described alpha-synuclein-IR Lewy bodies and neurites in neocortex, limbic regions, nucleus basalis, and brainstem of a subject from an IBMPFD family, who died at age 52 after an 11-year disease course of myopathy, without evidence of PDB, dementia, or Parkinsonism. Pirici et al. [16] and van der Zee et al. [17] reported a Belgian family (DR7) with IBMPFD associated with the R159H mutation. Three subjects in this family had Parkinsonism (II.3, II.4, and II.3). Another subject (III.4), who did not show signs of Parkinsonism, was found to have rare alpha-synuclein-negative Lewy-body-like inclusions in the thalamus and medullary reticular formation, similar to the ones described in this report in the Pennsylvania family proband [16, 17]. Chan et al. [14] reported a family with IBMPFD associated with the R159C mutation in which a mutation carrier presented PD at age 44 and later developed biopsy-proven IBM. Finally, Palmio et al. [29] reported a subject (III.7) from a large Finnish IBMPFD family who presented with PD associated with the P137L mutation.
To our knowledge, our study is the first to demonstrate the association between clinical and neuropathologic findings of PD in subjects with VCP mutation. The significance of this association is unclear. Whilst a common pathogenetic mechanism centered on a defective mechanism of protein degradation has been proposed, the molecular biology of this interaction remains unknown [30]. A recent VCP mutational screening in a large cohort of PD cases failed to identify proven pathogenic variants [31].
Johnson et al. [9] reported the association between familial ALS and VCP mutations and provided clinicopathologic evidence of ALS in an obligated carrier of the R155H mutation who developed hand weakness at age 53, died 39 months later, and had no neuropathologic findings of IBMPFD at autopsy. All of the other affected family members had clinical, electrophysiological, and/or pathological findings of slowly progressive myopathy with protracted clinical course of up to 30 years [9]. The authors also described a family (ITALS#1) in which at least four affected members and a distantly related individual from a different pedigree (Coriell#1) presented clinical signs of rapidly progressive upper and lower MND, consistent with a diagnosis of ALS. An R191Q VCP mutation was found in four of these subjects who underwent genetic testing. No CNS pathologic data were available for these subjects at the time of publication [9]. These findings differ in part from those seen in the Ohio family described in this report. The Ohio carriers of the R191Q mutation did not meet clinical criteria for ALS, and no TDP-43-IR inclusions were seen in the brainstem and spinal cord of the proband despite EMG findings suggestive of a neurogenic pattern of disease. However, neuronal loss and axonal spheroids were noted in her cervical cord at autopsy. Neurogenic, myopathic, mixed neurogenic/myopathic, and more complex or inconclusive EMG patterns have been described in association with VCP mutations [15, 32, 33]. These findings may represent different stages in the evolution of disease-related biologic changes in muscles and/or motor neurons. Consequently, we propose that EMG findings should cautiously be considered as a supportive sign in internationally validated criteria when making the clinical diagnosis of ALS in subjects with VCP mutation [34]. More recently, an R159H VCP mutation has been found in a subject with pathologically confirmed ALS and history of familial FTD [11], whilst an I151V VCP mutation has been found in a subject with sporadic ALS [12].
The absence of a systematic radiologic survey and alkaline phosphatase level screening prevented us from assessing the prevalence of PDB in the three families and reflects the limits of the retrospective nature of our study. It is possible that low back pain and bilateral leg pain in subject O.I.1 may have been an expression of PDB.
Finally, subject II.1 from the Pennsylvania family presented with PPA. Kovach et al. described a carrier of the R155H mutation (family 1, V-23) who presented with dysnomia, decreased comprehension, paraphasic errors, and fluent speech devoid of content words 6 years prior to the onset of myopathy [1, 35]. More recently, Kim et al. [36] reported an Asian family in which three members, carriers of an R155C mutation, presented with semantic dementia, which in one case (subject P3) was the presenting sign of disease.
In conclusion, we report clinicopathologic findings in three families with IBMPFD associated with VCP mutations. A novel T262A mutation was found in the Pennsylvania family, in which one individual had PPA, an uncommon presentation of IBMPFD. Clinicopathologic evidence of coexistent PD was documented for the first time in members of two families. Four subjects had a clinical diagnosis of MND: two of them met diagnostic criteria for ALS. The biologic base of the association between PD or MND and VCP mutations remains to be elucidated. If confirmed, it may prompt a reconsideration of the nomenclature used to designate this syndrome. In this sense, the term ‘VCP disease’ may be better suited to comprehend the multiple phenotypic expressions of this syndrome, beyond the ones originally described and summarized by the acronym ‘IBMPFD’.
Supplementary Material
Acknowledgments
The authors wish to thank Mrs Brenda Dupree and Mrs Rose Richardson for histology and immunohistochemistry as well as Mr Brad Glazier for editing of illustrations. Supported by PHS AG010133 and AG005133.
Footnotes
Disclosure of conflict of interest
The authors declare no other conflict of interests.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Data S1. Clinicopathologic information.
Data S2. Methods.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Watts GD, Wymer J, Kovach MJ, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36:377–381. doi: 10.1038/ng1332. [DOI] [PubMed] [Google Scholar]
- 2.Wang Q, Song C, Li CC. Molecular perspectives on p97-VCP: progress in understanding its structure and diverse biological functions. J Struct Biol. 2004;146:44–57. doi: 10.1016/j.jsb.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 3.Ju JS, Fuentealba RA, Miller SE, et al. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J Cell Biol. 2009;187:875–888. doi: 10.1083/jcb.200908115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mackenzie IR, Neumann M, Bigio EH, et al. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol. 2009;117:15–18. doi: 10.1007/s00401-008-0460-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neumann M, Mackenzie IR, Cairns NJ, et al. TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J Neuropathol Exp Neurol. 2007;66:152–157. doi: 10.1097/nen.0b013e31803020b9. [DOI] [PubMed] [Google Scholar]
- 6.Cairns NJ, Neumann M, Bigio EH, et al. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol. 2007;171:227–240. doi: 10.2353/ajpath.2007.070182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weihl CC. Valosin containing protein associated frontotemporal lobar degeneration: clinical presentation, pathologic features and pathogenesis. Curr Alzheimer Res. 2011;8:252–260. doi: 10.2174/156720511795563773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nalbandian A, Donkervoort S, Dec E, et al. The multiple faces of valosin-containing protein-associated diseases: inclusion body myopathy with paget’s disease of bone, frontotemporal dementia, and amyotrophic lateral sclerosis. J Mol Neurosci. 2011;45:522–531. doi: 10.1007/s12031-011-9627-y. [DOI] [PubMed] [Google Scholar]
- 9.Johnson JO, Mandrioli J, Benatar M, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68:857–864. doi: 10.1016/j.neuron.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kimonis VE, Fulchiero E, Vesa J, Watts G. VCP disease associated with myopathy, Paget disease of bone and frontotemporal dementia: review of a unique disorder. Biochim Biophys Acta. 2008;1782:744–748. doi: 10.1016/j.bbadis.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 11.Koppers M, van Blitterswijk MM, Vlam L, et al. VCP mutations in familial and sporadic amyotrophic lateral sclerosis. Neurobiol Aging. 2012;33:837.e7–837.e13. doi: 10.1016/j.neurobiolaging.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 12.DeJesus-Hernandez M, Desaro P, Johnston A, et al. Novel p.Ile151Val mutation in VCP in a patient of African American descent with sporadic ALS. Neurology. 2011;77:1102–1103. doi: 10.1212/WNL.0b013e31822e563c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spina S, Van Laar AD, Murrell JR, et al. Frontotemporal dementia associated with a valosin-containing protein mutation: report of three cases. FASEB J. 2008;22(Meeting Abstract Supplement):58.4.20. [Google Scholar]
- 14.Chan N, Le C, Shieh P, et al. Valosin-containing protein mutation and Parkinson’s disease. Parkinsonism Relat Disord. 2012;18:107–109. doi: 10.1016/j.parkreldis.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 15.Kimonis VE, Mehta SG, Fulchiero EC, et al. Clinical studies in familial VCP myopathy associated with Paget disease of bone and frontotemporal dementia. Am J Med Genet A. 2008;146A:745–757. doi: 10.1002/ajmg.a.31862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pirici D, Vandenberghe R, Rademakers R, et al. Characterization of ubiquitinated intraneuronal inclusions in a novel Belgian frontotemporal lobar degeneration family. J Neuropathol Exp Neurol. 2006;65:289–301. doi: 10.1097/01.jnen.0000205147.39210.c7. [DOI] [PubMed] [Google Scholar]
- 17.van der Zee J, Pirici D, Van Langenhove T, et al. Clinical heterogeneity in 3 unrelated families linked to VCP p.Arg159His. Neurology. 2009;73:626–632. doi: 10.1212/WNL.0b013e3181b389d9. [DOI] [PubMed] [Google Scholar]
- 18.Brooks BR, Miller RG, Swash M, et al. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293–299. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- 19.Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. 1992;55:181–184. doi: 10.1136/jnnp.55.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gelb DJ, Oliver E, Gilman S. Diagnostic criteria for Parkinson disease. Arch Neurol. 1999;56:33–39. doi: 10.1001/archneur.56.1.33. [DOI] [PubMed] [Google Scholar]
- 21.Takao M, Benson MD, Murrell JR, et al. Neuroserpin mutation S52R causes neuroserpin accumulation in neurons and is associated with progressive myoclonus epilepsy. J Neuropathol Exp Neurol. 2000;59:1070–1086. doi: 10.1093/jnen/59.12.1070. [DOI] [PubMed] [Google Scholar]
- 22.Piccardo P, Mirra S, Young K, Gearing M, Dlouhy SR, Ghetti B. A-synuclein accumulation in Gerstmann–Straussler–Scheinker disease (GSS) with prion protein gene (PRNP) mutation F198S. Neurobiol Aging. 1998;19:S172. (Abstract). [Google Scholar]
- 23.Murrell J, Farlow M, Ghetti B, Benson MD. A mutation in the amyloid precursor protein associated with hereditary Alzheimer’s disease. Science. 1991;254:97–99. doi: 10.1126/science.1925564. [DOI] [PubMed] [Google Scholar]
- 24.Stojkovic T, Hammouda el H, Richard P, et al. Clinical outcome in 19 French and Spanish patients with valosin-containing protein myopathy associated with Paget’s disease of bone and frontotemporal dementia. Neuromuscul Disord. 2009;19:316–323. doi: 10.1016/j.nmd.2009.02.012. [DOI] [PubMed] [Google Scholar]
- 25.Shi Z, Hayashi YK, Mitsuhashi S, et al. Characterization of the Asian myopathy patients with VCP mutations. Eur J Neurol. 2012;19:501–509. doi: 10.1111/j.1468-1331.2011.03575.x. [DOI] [PubMed] [Google Scholar]
- 26.Bersano A, Del Bo R, Lamperti C, et al. Inclusion body myopathy and frontotemporal dementia caused by a novel VCP mutation. Neurobiol Aging. 2009;30:752–758. doi: 10.1016/j.neurobiolaging.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 27.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 28.Forman MS, Mackenzie IR, Cairns NJ, et al. Novel ubiquitin neuropathology in frontotemporal dementia with valosin-containing protein gene mutations. J Neuropathol Exp Neurol. 2006;65:571–581. doi: 10.1097/00005072-200606000-00005. [DOI] [PubMed] [Google Scholar]
- 29.Palmio J, Sandell S, Suominen T, et al. Distinct distal myopathy phenotype caused by VCP gene mutation in a Finnish family. Neuromuscul Disord. 2011;21:551–555. doi: 10.1016/j.nmd.2011.05.008. [DOI] [PubMed] [Google Scholar]
- 30.Ju JS, Weihl CC. Inclusion body myopathy, Paget’s disease of the bone and fronto-temporal dementia: a disorder of autophagy. Hum Mol Genet. 2010;19:R38–R45. doi: 10.1093/hmg/ddq157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Majounie E, Traynor BJ, Chio A, et al. Mutational analysis of the VCP gene in Parkinson’s disease. Neurobiol Aging. 2012;33:209.e1–209.e2. doi: 10.1016/j.neurobiolaging.2011.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tucker WS, Jr, Hubbard WH, Stryker TD, et al. A new familial disorder of combined lower motor neuron degeneration and skeletal disorganization. Trans Assoc Am Physicians. 1982;95:126–134. [PubMed] [Google Scholar]
- 33.Kumar KR, Needham M, Mina K, et al. Two Australian families with inclusion-body myopathy, Paget’s disease of bone and frontotemporal dementia: novel clinical and genetic findings. Neuromuscul Disord. 2010;20:330–334. doi: 10.1016/j.nmd.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 34.Chaudhuri KR, Crump S, al-Sarraj S, Anderson V, Cavanagh J, Leigh PN. The validation of El Escorial criteria for the diagnosis of amyotrophic lateral sclerosis: a clinicopathological study. J Neurol Sci. 1995;129(Suppl):11–12. doi: 10.1016/0022-510x(95)00050-c. [DOI] [PubMed] [Google Scholar]
- 35.Kovach MJ, Waggoner B, Leal SM, et al. Clinical delineation and localization to chromosome 9p13.3–p12 of a unique dominant disorder in four families: hereditary inclusion body myopathy, Paget disease of bone, and frontotemporal dementia. Mol Genet Metab. 2001;74:458–475. doi: 10.1006/mgme.2001.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim EJ, Park YE, Kim DS, et al. Inclusion body myopathy with Paget disease of bone and frontotemporal dementia linked to VCP p.Arg155Cys in a Korean family. Arch Neurol. 2011;68:787–796. doi: 10.1001/archneurol.2010.376. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.