

Abstract
Cardiovascular effects of chronic AZT treatment on SD male rats (185 g) fed either a normal Mg diet (0.1% MgO) or a high Mg diet (0.6% MgO) were examined. AZT treatment (1 mg/ml drinking water) for 3 weeks led to a 5.5-fold (0.88 ± 0.11 nmol/min/106 cells, P < 0.05) elevation in neutrophil basal activity of O2− production versus controls (0.16 ± 0.03 nmol/min, assayed ex vivo as SOD-inhibitable cytochrome c reduction). Concomitantly, plasma 8-isoprostane and PGE2 levels rose 2.1-fold and 3-fold (both P < 0.05), respectively, compared to control; however, RBC GSH decreased 28% (P < 0.02) with GSSG content increased 3-fold, indicative of systemic oxidative stress. High Mg diet substantially attenuated the AZT-induced neutrophil activation by 70% (0.26 ± 0.05 nmol/min, P < 0.05); reduced plasma 8-isoprostane and PGE2 to levels comparable to normal; and RBC GSH was restored back to 92% of control. AZT alone caused moderate, but significant vascular inflammatory lesions in the heart (assessed by H&E staining). Immunohistochemical staining revealed significantly higher (about 4-fold) infiltration of CD11b positive cells (WBC surface marker) in the atria and ventricles of AZT-treated rats. However, these inflammatory pathological markers were minimal in samples of rats treated with AZT plus high Mg diet. Moreover, AZT alone significantly (P < 0.02) decreased rat weight gain by 21% at 3 weeks; Mg-supplementation completely prevented (P < 0.05) the weight gain loss due to AZT intake. It is concluded that high dietary Mg may provide beneficial effects against AZT toxicity due to its systemic antioxidative/anti-inflammatory properties.
Keywords: AZT toxicity, Neutrophil activation, Oxidative stress, Cardiac inflammation, Mg-supplementation
Introduction
Azidothymidine (AZT, Zidovudine) was the first approved nucleoside reverse transcriptase inhibitor (NRTIs) drug showing clinical efficacy against the human immunodeficiency virus (HIV) [1, 2]; it inhibits in vitro replication of some retroviruses including HIV, and is a thymidine analog in which the 3′-OH group is replaced by an azido (−N3) group [3]. Unfortunately, its use has also been associated with considerable toxicity, causing macrocytic anemia, neutropenia, and myopathy [1, 2, 4–6]. Chronic AZT toxicity is generally attributed to damaged mitochondrial mtDNA synthesis resulting in mitochondrial myopathy with ultra-structural changes in heart and skeletal muscle in both animals and humans [4–7]. However, Szabados et al. [8] reported that short-term treatment of rats with AZT increased reactive oxygen species (ROS) and peroxynitrite production in the rat heart before any significant changes were observed in mtDNA content. These findings suggested that ROS-mediated processes could be important contributors to the cardiomyopathy resulting from AZT treatment. Since AZT therapy may generate or amplify free radical toxicity, this argues in favor of employing antioxidant supplementation [9–11] during AZT treatment. De la Asuncion et al. [9] showed that mice treated with AZT alone experienced both a moderate loss of skeletal muscle total glutathione (GSH + GSSG) and a 5-fold increase in the GSSG/GSH ratio; this index of glutathione oxidation was substantially attenuated by supra-nutritional doses of the antioxidant vitamins C and E. High-dietary supplementation of Mg has been shown to provide beneficial effects against oxidative vascular pathogenesis in diabetes and cyclosporine-induced nephrotoxicity in rats [12, 13]. Others [14] also showed that Mg-supplements attenuated high cholesterol-induced atherogenesis in a rabbit model. Therefore, we sought to determine if a higher supplement of Mg in the diet of rats provided beneficial effects against AZT-mediated cardiovascular and systemic toxicity in vivo.
Materials and Methods
Animal Model, AZT Treatment and Diets
All animal experiments were guided by the principles for the care and use of laboratory animals as recommended by the US Department of Health and Human Services and approved by The George Washington University Animal Care and Use Committee. Male SD rats (185 g) were fed either a normal Mg (0.1% w/w MgO) or a high Mg (0.6% MgO, Teklad Lab., Madison, WI) diet. Use of MgO avoids potential confounding effects presented by the counter-ions found in other inorganic and organic Mg salts. AZT was administered orally in drinking water (1 mg/ml) for up to 3 weeks [15]. Plasma Mg levels were determined by atomic absorption spectroscopy (AA).
Systemic Oxidative Indices and Neutrophil Activity
At sacrifice, whole blood was obtained by cardiac puncture. Plasma 8-isoprostane and PGE2 (as PGE-metabolites) were assayed by EIA kits obtained from Cayman Chemical (Ann Arbor, MI) [16]. RBC reduced (GSH) and oxidized (GSSG) glutathione levels were determined enzymatically by the DTNB-GSSG reductase method [17, 18]. Neutrophils were isolated by a step-gradient centrifugation method [16, 19]. Superoxide anion production from neutrophils (0.5–0.75 × 106/ml) with or without phorbol myristate acetate (PMA, 100 ng/ml) stimulation was assayed in a phosphate buffer (pH 7.6) containing 5 mM glucose, 1 mM CaCl2, 1 mM MgCl2, 75 μM cytochrome c ± 50 μg SOD. The superoxide released was estimated as SOD-inhibitable reduction of cytochrome c using the extinction coefficient: E550 = 2.1 × 104 M−1 cm−1.
Histochemical Analysis
Cardiac tissue was rapidly excised, rinsed, quickly embedded in OCT compound, and frozen at −70°C until used. CD11b positive cells in the frozen tissue sections (Cryo-sections, 5–10 μm) were visualized by indirect, immunohistochemical staining using mouse anti-rat CD11b antibody (1:500, Chemical International, Temecula, CA) and Vectastain Elite ABC kit immunoperoxidase system (Vector Laboratories Inc., Burlingame, CA) [20]. In the negative controls, the primary antibody was omitted and they were included in all assays. Samples were examined under an Olympus BX60 microscope and multiple digital images were taken with a digital camera (Evolution Colour MP; Media Cybernetics, Silver Spring, MD). CD11b positive cells were counted in the micrographs and quantification was normalized based on standardized area obtained with Adobe Photoshop. Hematoxylin–Eosin (H&E) staining was also performed for overall morphology assessment.
Statistics
Data were reported as means ± SEM of five animals per group. Statistical difference was evaluated by Student's t-test between groups, and significance set at P < 0.05.
Results
Neutrophil Activation by AZT and Effect of Mg-Supplementation
It was suggested that AZT treatment may produce systemic production of ROS [8, 10]. Since neutrophils (PMNs) can be a major cellular source for ROS generation, we examined the relative short-term (3 weeks) effect of AZT treatment in rats on the basal free radical activity of PMNs. As represented by Fig. 1, without stimulation, PMNs isolated from untreated rats receiving normal dietary Mg displayed only a low level of basal superoxide anion generating activity (0.198 ± 0.071 nmol/min/106 cells). However, the activity of the PMNs was markedly and significantly elevated 4.92-fold by AZT treatment in the normal Mg rats (0.975 ± 0.11 nmol/min/106 cells, P < 0.01). Importantly, dietary supplementation with high MgO (6-fold higher than normal dietary level) effectively attenuated this AZT-induced elevation in basal activity by 70% (0.422 ± 0.08 nmol/min/106 cells); high Mg alone had no significant effect on basal neutrophil activity. As expected, in the presence of PMA, PMNs from the untreated Mg-normal rats were stimulated and generated a 5.5-fold higher superoxide production rate (Fig. 1). However, PMN samples from AZT-treated rats did not respond further to PMA stimulation. Nevertheless, the responses of PMNs from the high Mg + AZT or the high Mg alone groups to PMA challenge were comparable to that of the untreated Mg normal group.
Fig. 1.

AZT promotes basal neutrophil O2− generation, which is blocked by high dietary Mg-supplementation. Rats receiving AZT (1 mg/ml) in drinking water for 3 weeks were fed either a normal Mg or a 6× higher Mg diet. Neutrophils were isolated from the whole blood by a step gradient centrifugation method. The ability of the neutrophils to generate superoxide anions ±100 nM PMA was determined by SOD inhibitable cytochrome reduction. Values are means ± SEM from five rats; * P < 0.05 versus Ctl and Hi Mg + AZT group; # P < 0.01 versus Ctl and <0.05 versus Hi Mg + AZT rats
AZT-Mediated Changes in RBC Glutathione Status
Previously, we showed that AZT at a dose of 0.7 mg/ml (in drinking water) for 21 days produced only a modest (but non-significant) decrease in total glutathione (GSH + GSSG) in mice receiving normal dietary Mg [15]. In the current study, we examined the effect of AZT administration (1 mg/ml for 21 days) on RBC glutathione status (both GSH and GSSG levels). As indicated by Fig. 1a. AZT alone induced a 28% significant (P < 0.01) decrease in GSH in the RBCs from normal Mg rats, and promoted a 2-fold (P < 0.05) elevation of the GSSG content (118 + 21 vs. 58 ± 14 nmol/gm Hgb for the control). As a percentage of the total glutathione, GSSG normally constitutes less than 1% (0.8 ± 0.2%) in control rats; with the administration of AZT, the GSSG rose significantly (P < 0.01) to 2.2 ± 0.4% of the total RBC total glutathione. When supplemented with high Mg, the AZT-induced GSH decrease was significantly attenuated; the GSH level was restored to 96% of normal controls (Fig. 2). In addition, the RBC GSSG contents of the high Mg + AZT rats, either expressed as absolute quantity or as % of total GSH + GSSG, remained within the normal ranges.
Fig. 2.
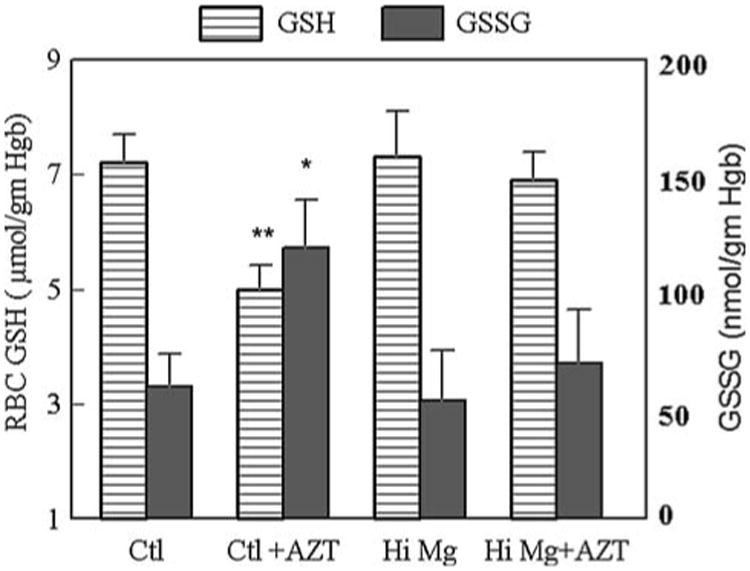
Changes in the glutathione status in RBCs obtained from normal Mg or high Mg rats ± AZT. GSH and GSSG were determined by the enzymatic “recycling” method. Other conditions are described in Fig. 1. * P < 0.05 versus Ctl and Hi Mg + AZT group; # P < 0.01 versus Ctl and <0.05 versus Hi Mg + AZT rats
Effects of AZT and High Mg on Systemic Lipid Peroxidation Indices
As a quantitative index of systemic oxidative stress, levels of F2-like isoprostanes, which derive from non-enzymatic peroxidation of polyunsaturated fatty acids [21], were determined by an 8-Isoprostane Enzyme Immunoassay. The results (Fig. 3a) indicate that AZT treatment of normal Mg rats induced a 2.2-fold higher level of 8-isoprostane (106 ± 13 vs. 49 ± 4 pg/ml for the controls), and this was significantly attenuated in the high Mg + AZT group. High Mg diet alone had no effect on the basal level of plasma 8-isoprostane. Previously, we found that AZT promoted COX-derived vasoactive prostanoid products such as TXA2 or PGE2 [15]. In current study, we also found that AZT treatment of normal Mg rats caused a 3.6-fold higher (P < 0.01) plasma PGE2-derived metabolites (PGEM). Again, when fed a high Mg-diet, the enhanced PGEM level induced by AZT was attenuated 70% (P < 0.05).
Fig. 3.

AZT enhances circulating 8-isoprostane (a) and PGE2 (b) in normal Mg rats and the attenuating effects of high Mg diet. Both plasma 8-isoprostane and PGE2-metabolites were determined by EIA Immunoassay kits. Other conditions are as described in Fig. 1; * P < 0.05 and # P < 0.01 versus Ctl and <0.05 versus Hi Mg + AZT group
Effect of AZT on Cardiac Morphology/Pathology
Histochemical analysis of cardiac tissue revealed that AZT treatment caused moderate, but noticeable vascular inflammatory lesions in the atria (by H&E staining) and ventricles (Fig. 4b) of AZT + normal Mg control rats. Immunohistochemical staining revealed further infiltration of monocytes/WBCs (4B, based on cell surface marker CD11b positivity) in these hearts. However, the appearance of this inflammatory marker was substantially reduced in hearts from the AZT + high Mg group (Fig. 4c). Using quantitative morphometric analysis, it was estimated that the relative number of CD11b positive cells (brown staining cells per sq. micron) significantly increased 4- to 5-fold in both atria and ventricles from the normal Mg + AZT group compared to the controls. These increases in CD11b positive cells in both atria (1.70 ± 0.21 vs. 0.43 ± 0.03 positive cells/sq. micron × 10,000) and ventricles (1.55 ± 0.15 cells vs. 0.0.34 ± .032 positive cells/sq. micron × 10,000) were almost completely attenuated by high Mg-supplementation in the AZT treated groups (reduced to 0.52 ± 0.07 [atria] and 0.34 ± 0.04 cells [ventricles]/same area). High Mg alone (without AZT) had no effects on the levels of positive CD11b cells in either atria or ventricles (Fig. 5).
Fig. 4.
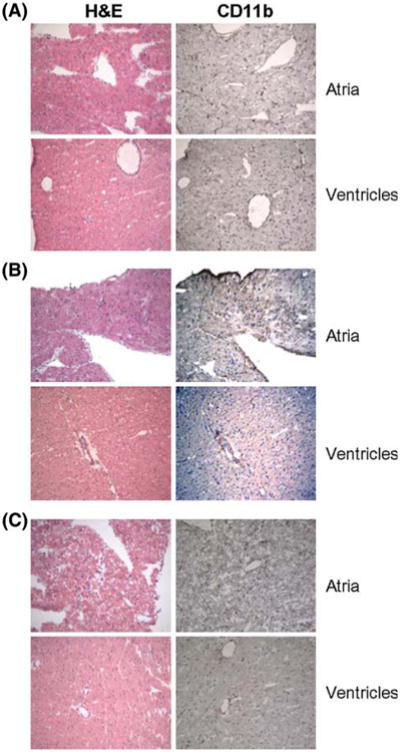
Micrographs showing H&E (left) and immunohistochemical staining (brown-dark) for CD11b of atrial and ventricular sections from a control, b control + AZT; c high Mg + AZT treated rat hearts. All tissues were obtained after 3 weeks of AZT treatment. Other conditions are as described in Fig. 1 and in “Materials and Methods” section. Magnification, 20× (color online)
Fig. 5.
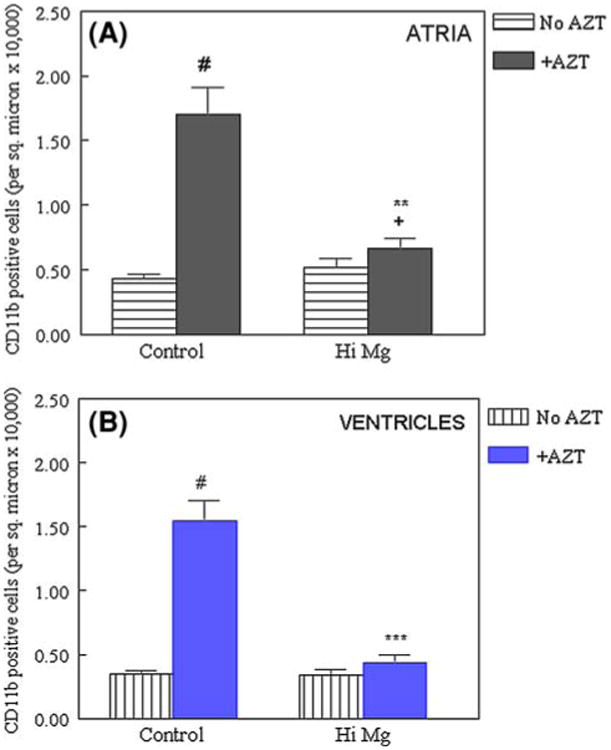
Numerical density of CD11b positive cells in the a atria and b ventricles from AZT treated rats with control or high Mg diets. Data are derived from 7 to 8 different fields including all five animals per group; # P < 0.01 versus Ctl, + P < 0.05 versus Ctl, ** P < 0.01, *** P < 0.001 versus #
Effect of AZT and Mg Supplementation on Weight Gain and Circulating Mg Levels
We used changes in animal weight gain rate as a gross indication of drug toxicity. AZT alone (in normal Mg group) significantly (P < 0.05) decreased the rate of animal weight gain by 21% (68 ± 6 g vs. 86 ± 5 g/3 weeks) (Table 1); however, Mg-supplementation completely prevented (90 ± 5 g, P < 0.05 vs. Ctl + AZT) this lower weight gain induced by AZT intake. These differences in weight gain occurred even though the amount of AZT consumed by the normal Mg and high Mg groups were comparable (Table 1). It was worth noting that Mg supplementation alone or with AZT treatment resulted in significantly higher (33%) circulating Mg levels (Table 1).
Table 1. Effect of high Mg-diet on plasma Mg and weight gain in AZT-treated rats.
| Rat groups | Initial body wt. (g) | Wt. gained (g/3 weeks) | AZT consumption (mg/kg/day) | Plasma Mg (mM) |
|---|---|---|---|---|
| Normal Ctl | 276 ± 18 | 86 ± 5 | – | 0.796 ± 0.05 |
| + AZT | 265 ± 8 | 68 ± 6* | 95 ± 7.7 | 0.794 ± 0.04 |
| Mg-Suppl. | 274 ± 12 | 87 ± 7 | – | 1.06 ± 0.08* |
| AZT ± Mg-Sup | 279 ± 13 | 90 ± 5+ | 98 ± 7.5 | 1.05 ± 0.06* |
Rats were fed either normal Mg diet (0.1% MgO) or Mg-Suppl diet (0.6% MgO) for 3 weeks ± AZT administration (1 mg/ml in drinking water) Data = means ± SE of five rats
P < 0.05 versus Ctl
P < 0.025 versus AZT
Discussion
In this study, we found that AZT alone caused enhanced basal free radical generation from circulating neutrophils in the normal Mg-rats; this elevation was suppressed by high dietary Mg. This elevated ROS generation from neutrophils may contribute substantially to systemic oxidative stress caused by AZT administration. Previous studies reported that AZT administration in rodents resulted in elevated ROS generation in various tissues, such as the heart, skeletal muscle, liver, and endothelium [7–10]. Since the mitochondrion is the main subcellular target of AZT toxicity [9, 22, 23], it is generally believed that this organelle contributed to the bulk of the elevated ROS generation; both superoxide anions and hydrogen peroxide could derive from AZT-induced partial inhibition of the respiratory electron transport chain in the mitochondria [22, 23]. However, in isolated neutrophils, it is presumed that the NADPH-oxidase system is one of the main sources of superoxide anion generation [24]. It remains to be determined if both the NADPH-oxidase and mitochondrial systems of the neutrophils contribute to the elevated superoxide due to AZT treatment. Interestingly, we found that while the neutrophils displayed elevated basal super-oxide activity, their response to PMA stimulation appeared to be blunted. Since PMA activates neutrophil through protein kinase C (PKC) stimulation [25], we speculate that this PKC activation step, which is Ca-dependent, might be impaired. Further study is required to confirm if this signaling cascade might be altered by AZT treatment.
In association with elevated ROS generation from circulating PMNs, and perhaps from other tissues, it was not surprising to observe changes in other indices of oxidative stress: elevated plasma 8-isoprostane levels, decreased RBC GSH, and increased GSSG. In normal Mg rats, increased PGE2 metabolite was induced by AZT treatment. In our previous studies, dietary Mg-deficiency in rodents resulted in systemic oxidative stress accompanied by elevated COX-derived vasoactive prostanoids such as PGI2, TBX, and PGE2 [16, 18–20]. The PGE2 increase may be attributed to a generalized oxidative activation of the endothelium when exposed to AZT. The toxic effects of AZT on the heart were evident from the appearance of micro-lesion formation (H&E staining), which may be due to the well-known AZT-induced mitochondrial myopathy [8, 9]. However, part of the injury may be a consequence of increased infiltration of inflammatory WBCs (macro-phages/monocytes/neutrophils). Once a CD11b-dependent adhesion interaction between neutrophils and myocytes has been established [26], infiltrating WBCs may become activated and induce direct oxidative injury to cardiac myocytes.
Although AZT cardiotoxicity in HIV patients has been well documented [1, 6, 7], the potential beneficial effects of Mg-supplementation has never been explored. In a historical review of patients with coronary artery disease, it was shown that Mg-supplements improved myocardial metabolism, inhibited tissue calcium accumulation and lessened myocardial cell death [27]. In our own study [28], we found that acute Mg supplementation provided significant protection against post-ischemic cardiac dysfunction and oxidative injury. Others have observed that dietary Mg-supplementation significantly lowered blood pressures in hypertensive patients [29] and in spontaneously hypertensive rats [30]. Since it has been reported that AZT (1 mg/ml) could increase systolic blood pressure in the rat [31], the cardioprotective effects observed here might be contributed, in part, by the anti-hypertensive effects of Mg-supplementation.
In this study, we have demonstrated that dietary supplementation with high Mg suppressed the endogenous activation of circulating neutrophils and the associated indices of oxidative stress. Recent evidence strongly suggests that Ca influx is required for both priming and activation of NADPH oxidase activity in neutrophils [24]. In this study, high dietary Mg resulted in significantly higher (33%) circulating total Mg levels. Since Mg is thought to be a natural “calcium antagonist” [32], the anti-calcium effect of high circulating Mg may contribute to suppression of the Ca-mediated priming and activation of NADPH oxidase in neutrophils from AZT-treated rats. At the cardiac tissue level, the beneficial effects of Mg were further manifested by the attenuation of the WBC infiltration and cardiac inflammatory pathology. A study by Asai et al. [33] indicated that Mg-supplementation attenuated macrophage infiltration in the kidney of animals exposed to cyclosporine A, by inhibiting adhesion molecule elevation. In the current study, it may be presumed that the suppressed WBC infiltration in the heart of the high Mg + AZT animals was also a consequence of the higher circulating Mg. Since Ca is required for COX activation and subsequent PGE2 production [34], one may assume that attenuation of this process was also mediated by the anti-Ca effect of high Mg. Interestingly, the protective effect reported here is similar to that reported in an antioxidant supplementation study (vitamin E + C) of an AZT myopathic mouse model [8]. In that study, AZT treatment decreased skeletal muscle GSH and the GSH/GSSG ratio; antioxidant treatment significantly blunted changes in both of these indices [8]. Previously, we showed that the cardiac pathologic effects induced by AZT were potentiated by dietary Mg-deficiency [15]. Using an isolated endothelial cell model [35], we showed that cellular injury mediated by AZT or select AZT-metabolites was iron-dependent, and that subnormal extracellular Mg (<0.5 mM) augmented and higher Mg >1 mM) attenuated this injury. We suggest that high Mg2+ may be able to competitively displace “catalytic iron” from membrane phospholipid binding sites, and thus able to prevent site-specific hydroxyl radical formation [35]. The oxidative toxicity of AZT may also be related to its influence on tissue distribution of prooxidant metals. Pollack and Weaver [36] reported that AZT treatment (oral, 1–2 mg/ml) of normal mice for 1–4 weeks led to 50% elevations in non-heme liver iron content and a 20% increase in peritoneal macrophage iron. They suggested that AZT interfered with heme synthesis, which may cause upregulation of transferrin receptor synthesis leading to increased cellular iron uptake. Our pilot experiment (N = 3) indicated that AZT treatment (1 mg/ml drinking water) caused modest increases in hepatic (∼10%) and cardiac (∼29%) tissue iron content after 3 weeks of exposure. In a different study, we showed that longer treatment (6 weeks) at a lower dose of AZT (0.5 mg/ml) significantly elevated (1.72-fold, P < 0.05) rat liver iron content [37].
In conclusion, we believe that the protective effects of Mg may be partially related to its anti-peroxidative properties. This observation may have potential clinical relevance to HIV patients, who frequently exhibit significant hypomagnesemia [38] as well as disturbances in iron metabolism and tissue distribution [39]. The presence of either or both conditions in HIV patients receiving AZT treatment may pose a further risk of oxidative stress due to iron-driven ROS production. While the mechanisms remain to be further explored, this study clearly demonstrates the protective effects of high dietary Mg against AZT-induced oxidative stress, cardiac pathology, and systemic inflammation in rats. Thus, our findings suggest that Mg-supplementation may warrant consideration as an effective adjunct therapy to combat NRTI toxicity in HIV patients.
Acknowledgments
The authors wish to thank Kenny Landgraf for his excellent technical assistance. This study was supported by NIH R21-AT003993 & NIH RO1-HL-65178.
References
- 1.Schroder JM, Bertram M, Schnabel R, Pfaff U. Nuclear and mitochondrial changes of muscle fibers in AIDS after treatment with high doses of zidovudine. Acta Neuropathologica. 1992;85:39–47. doi: 10.1007/BF00304632. [DOI] [PubMed] [Google Scholar]
- 2.Lewis W, Copeland WC, Day BJ. DNA depletion, oxidative stress and mutation: Mechanisms of dysfunction from NRTIs. Laboratory Investigation. 1998;81:777–790. doi: 10.1038/labinvest.3780288. [DOI] [PubMed] [Google Scholar]
- 3.Komarov AM, Hall JM, Weglicki WB. Azidothymidine promotes free radical generation by activated macrophages and hydrogen peroxide-iron-mediated oxidation in a cell-free system. Biochimica et Biophysica Acta. 2004;1688:257–264. doi: 10.1016/j.bbadis.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 4.Lewis W, Gozalex B, Chomyn A, Papoian T. Zidovudine induces molecular biochemical and ultrastructural changes in rat skeletal muscle mitochondria. The Journal of Clinical Investigation. 1992;89:1354–1360. doi: 10.1172/JCI115722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lamperth L, Dalakas MC, Dagani F, Anderson J, Ferrari R. Abnormal skeletal and cardiac muscle mitochondria induced by zidovudine. Laboratory Investigation. 1991;65:742–751. [PubMed] [Google Scholar]
- 6.Arnaudo E, Dalakas MC, Shanske S, Moraes CT, Dimauro S, Schon EA. Depletion of muscle mitochondrial DNA in AIDS with zidovudine-induced myopathy. Lancet. 1991;337:508–510. doi: 10.1016/0140-6736(91)91294-5. [DOI] [PubMed] [Google Scholar]
- 7.Dalakas M, Illa Y, Peseshkpour GH, Laukatis JP, Cohen B, Griffin JL. Mitochondrial myopathy caused by long-term zidovudine therapy. The New England Journal of Medicine. 1990;322:1098–1105. doi: 10.1056/NEJM199004193221602. [DOI] [PubMed] [Google Scholar]
- 8.Szabados E, Fischer G, Toth K, Csete B, Nemeti B, Trombitas K, et al. Role of reactive oxygen species and poly-ADP-ribose polymerase in the development of AZT-induced Cardiomyopathy in rats. Free Radical Biology and Medicine. 1999;26:309–317. doi: 10.1016/S0891-5849(98)00199-3. [DOI] [PubMed] [Google Scholar]
- 9.De la Asuncion JG, del Olmo ML, Sastre J, Millan A, Pellin A, Pallardo FV, et al. AZT treatment induces molecular and ultrastructural oxidative damage to muscle mitochondria. Prevention by antioxidant vitamins. The Journal of Clinical Investigation. 1998;102:4–9. doi: 10.1172/JCI1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sutliff RL, Dikalov S, Weiss D, Parker J, Raidel S, Racine AK, et al. Nucleoside reverse transcriptase inhibitors impair endothelium-dependent relaxation by increasing superoxide. The American Journal of Physiology. 2002;283(6):H2363–H2370. doi: 10.1152/ajpheart.00151.2002. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, Watson RR. Is vitamin E supplementation a useful agent in AIDS therapy? Progress in Food & Nutrition Science. 1993;17:351–375. [PubMed] [Google Scholar]
- 12.Soltani N, Keshavarz M, Minaii B. Effects of oral Mg on plasma glucose and pathological changes in the aortic and pancreas of diabetic rats. Clinical and Experimental Pharmacology and Physiology. 2005;32:604–610. doi: 10.1111/j.0305-1870.2005.04238.x. [DOI] [PubMed] [Google Scholar]
- 13.Asai T, Nakatani T, Yamanka S. Mg supplementation prevents experimental chronic cyclosporine nephrotoxicity. Transplantation. 2002;74:784–791. doi: 10.1097/00007890-200209270-00009. [DOI] [PubMed] [Google Scholar]
- 14.Altura BT, Brust M, Bloom S, Barbour RL, Stempak JG, Altura BM. Magnesium dietary intake modulates blood lipid levels and atherogenesis. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:1840–1844. doi: 10.1073/pnas.87.5.1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mak IT, Goldfarb MG, Weglicki WB, Haudenschild CC. Cardiac pathologic effects of AZT in Mg-deficent mice. Cardiovascular Toxicology. 2004;4:169–178. doi: 10.1385/CT:4:2:169. [DOI] [PubMed] [Google Scholar]
- 16.Mak IT, Kramer JH, Chmielinska JJ, Khalid MH, Landgraf KM, Weglicki WB. Inhibition of neutral endopeptidase potentiates neutrophil activation during Mg-deficiency in the rat. Inflammation Research. 2008;57:300–305. doi: 10.1007/s00011-007-7186-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mak IT, Stafford RE, Weglicki WB. Loss of red cell glutathione during Mg deficiency: Prevention by vitamin E, D-propranolol, and chloroquine. The American Journal of Physiology. 1994;267:C1366–C1370. doi: 10.1152/ajpcell.1994.267.5.C1366. [DOI] [PubMed] [Google Scholar]
- 18.Mak IT, Komarov AM, Wagner TL, Stafford RE, Dickens BF, Weglicki WB. Enhanced NO production during Mg deficiency and its role in mediating red blood cell glutathione loss. The American Journal of Physiology. 1996;271:C385–C390. doi: 10.1152/ajpcell.1996.271.1.C385. [DOI] [PubMed] [Google Scholar]
- 19.Mak IT, Kramer JH, Weglicki WB. Suppression of neutrophil and endothelial activation by substance P receptor blockade in the Mg-deficient rat. Magnesium Research. 2003;16:91–97. [PubMed] [Google Scholar]
- 20.Chmielinska JJ, Tejero-Taldo MI, Mak IT, Weglicki WB. Intestinal and cardiac inflammatory response shows enhanced endotoxin receptor (CD14) expression in magnesium deficiency. Molecular and Cellular Biochemistry. 2005;278:53–57. doi: 10.1007/s11010-005-2733-9. [DOI] [PubMed] [Google Scholar]
- 21.Morrow JD, Frei B, Longmire AW, Gaziano JM, Lynch SM, Shyr Y, et al. Increase in circulating products of lipid peroxidation (F2-isoprostanes) in smokers. Smoking as a cause of oxidative damage. The New England Journal of Medicine. 1995;332:1198–1203. doi: 10.1056/NEJM199505043321804. [DOI] [PubMed] [Google Scholar]
- 22.Lund KC, Wallace KB. Adenosine 3′5′-cAMP-dependent phosphoregulation of mitochondrial complex I is inhibited by nucleoside reverse transcriptase inhibitors. Toxicology and Applied Pharmacology. 2008;226:94–106. doi: 10.1016/j.taap.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lund KC, Peterson LL, Wallace KB. Absence of a universal mechanism of mitochondrial toxicity by nucleoside analogs. Antimicrobial Agents and Chemotherapy. 2007;51:2531–2539. doi: 10.1128/AAC.00039-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brechard S, Tschirhart EJ. Regulation of superoxide production in neutrophils: Role of calcium influx. Journal of Leukocyte Biology. 2008;84:1223–1237. doi: 10.1189/jlb.0807553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kurihara H, Murayama Y, Warbington ML, Champagne CME, Van Dyke TE. Calcium-dependent protein kinase C activity of neutrophils in localized juvenile periodonitis. Infection and Immunity. 1993;61:3137–3142. doi: 10.1128/iai.61.8.3137-3142.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Entman ML, Youker K, Shoji T, Kukielka G, Shappell SB, Taylor AA, et al. Neutrophil induced oxidative injury of cardiac myocytes: A compartmented system requiring CD11b/CD18-ICAM-1 adherence. The Journal of Clinical Investigation. 1992;90:1335–1345. doi: 10.1172/JCI115999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shechter M. Does magnesium have a role in the treatment of patients with coronary artery disease. American Journal of Cardiovascular Drugs. 2003;3:231–239. doi: 10.2165/00129784-200303040-00001. [DOI] [PubMed] [Google Scholar]
- 28.Murthi SB, Wise RM, Weglicki WB, Komarov AM, Kramer JH. Mg-gluconate provides superior protection against postischemic dysfunction and oxidative injury compared to Mg-sulfate. Molecular and Cellular Biochemistry. 2003;245:141–148. doi: 10.1023/A:1022840704157. [DOI] [PubMed] [Google Scholar]
- 29.Kawano Y, Matsuoka H, Takishita S, Omae T. Effects of magnesium supplementation in hypertensive patients. Hypertension. 1998;32:260–265. doi: 10.1161/01.hyp.32.2.260. [DOI] [PubMed] [Google Scholar]
- 30.Touyz RM, Milne FJ. Magnesium supplementation attenuates, but not prevents, development of hypertension in spontaneously hypertensive rats. American Journal of Hypertension. 1999;12:757–765. doi: 10.1016/S0895-7061(99)00064-3. [DOI] [PubMed] [Google Scholar]
- 31.Papparella I, Ceolotto G, Berto L, Cavalli M, et al. Vitamin C prevents zidovudine-induced NADPH oxidase activation and hypertension in the rat. Cardiovascular Research. 2007;73:432–438. doi: 10.1016/j.cardiores.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 32.Altura BT, Altura BM. Role of magnesium ions in contractility of blood vessels and skeletal muscles. Magnesium Bulletin. 1981;3:102–106. [Google Scholar]
- 33.Asai T, Nakatani T, Yamanaka S, Tamada S, Kishimoto T, Tashiro K, et al. Magnesium supplementation prevents experimental chronic cyclosporine a nephrotoxicity via renninangiotensin-system independent mechanism. Tansplantation. 2002;74:754–755. doi: 10.1097/00007890-200209270-00002. [DOI] [PubMed] [Google Scholar]
- 34.Choudlary S, Kumar A, Kale RK, Raisz LG, Pilbeam CC. Extracellular calcium induces COX-2 in osteoblasts via a PKA pathway. Biochemical and Biophysical Research Communications. 2004;322:395–402. doi: 10.1016/j.bbrc.2004.07.129. [DOI] [PubMed] [Google Scholar]
- 35.Mak IT, Nedelec LF, Weglicki WB. Pro-oxidant properties and cytotoxicity of AZT-monophosphate and AZT. Cardiovascular Toxicology. 2004;4:109–115. doi: 10.1385/CT:4:2:109. [DOI] [PubMed] [Google Scholar]
- 36.Pollack S, Weaver J. AZT-induced siderosis. American Journal of Hematology. 1993;43:230–233. doi: 10.1002/ajh.2830430314. [DOI] [PubMed] [Google Scholar]
- 37.Kramer JH, Dadgar S, Hall J, Mak IT, Weglicki WB. Rat tissue iron content is differentially altered by AZT and Mg-deficiency. Journal of Molecular and Cellular Cardiology. 2004;36:631. Abs. [Google Scholar]
- 38.Bogden JD, Baker H, Frank O, Perez G, Kemp F, Bruening K, et al. Micronutrient status and human immunodeficiency virus (HIV) infection. Annals of the New York Academy of Sciences. 1990;587:189–195. doi: 10.1111/j.1749-6632.1990.tb00146.x. [DOI] [PubMed] [Google Scholar]
- 39.Savarino A, Pescarmona GP, Boelaert JR. Iron metabolism and HIV infection: Reciprocal interactions with potentially harmful consequences? Cell Biochemistry and Function. 1999;17:279–287. doi: 10.1002/(SICI)1099-0844(199912)17:4<279∷AID-CBF833>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]