

Abstract
The Notch pathway plays a pivotal role in regulating cell fate decisions in many stem cell systems. However, the full repertoire of Notch target genes in vivo and the mechanisms through which this pathway activity is integrated with other signaling pathways are largely unknown. Here, we report a transgenic mouse in which the activation of the Notch pathway massively expands the neural stem cell (NSC) pool in a cell context-dependent manner. Using this in vivo system, we identify direct targets of RBPJ/N1ICD in cortical NSCs at a genome-wide level through combined ChIP-Seq and transcriptome analyses. Through a highly conservative analysis of these data sets, we identified 98 genes that are directly regulated by N1ICD/RPBJ in vivo. These include many transcription factors that are known to be critical for NSC self-renewal (Sox2, Pax6, Tlx, and Id4) and the transcriptional effectors of the Wnt, SHH and Hippo pathways, TCF4, Gli2, Gli3, Yap1, and Tead2. Since little is known about the function of the Hippo-Yap pathway in NSCs, we analyzed Yap1 expression and function in NSCs. We show that Yap1 expression is restricted to the stem cell compartment in the developing forebrain and that its expression is sufficient to rescue Notch pathway inhibition in NSC self-renewal assays. Together, the results of this study reveal a previously underappreciated complexity and breadth of Notch1 targets in vivo and show direct interaction between Notch and Hippo-Yap pathways in NSCs.
Keywords: Neural stem cells, Notch pathway, Hippo-Yap pathway, transcriptional targets, ChIP-seq
Introduction
In the developing nervous system, neural stem cells (NSCs) in the neuroepithelium give rise to neurons and glia in a spatio-temporally controlled manner. During early stages of brain development, the naïve neuroepithelium expands laterally through symmetric stem cell divisions. As the brain matures, the symmetric divisions are gradually replaced by asymmetric divisions that generate a stem cell and a lineage-restricted progenitor cell or a postmitotic neuron, resulting in thickening of the cortex radially 1. Hence, regulation of the symmetric to asymmetric division transition is a pivotal step in NSC maturation. This transition is regulated by both cell-intrinsic and -extrinsic pathways, including a large number of signaling pathways originating within the brain and its surrounding tissues.
Classical studies in Drosophila development have shown that Notch signaling plays a critical role in specifying the appropriate numbers and types of cells arising from an equivalent pool of precursor cells 2. Notch function is mediated through cell-cell communication among adjacent cells (juxtacrine signaling). When a ligand (DELTA and JAGGED family members) binds to a NOTCH receptor (NOTCH 1-4 in mammals), NOTCH is cleaved through highly regulated step-wise processes and the intracellular domain of NOTCH (NICD) is released from the membrane, and then enters the nucleus and forms a transcriptional complex with RBPJ. RBPJ is a DNA-binding protein that is engaged in a transcriptional repressor complex in the absence of NICD. The current model is that when NICD binds to RBPJ, it displaces the repressive co-factors bound to RBPJ and recruits a transcriptional activator complex, which initiates transcription of its downstream target genes 3.
In mice, loss-of-function mutations in Notch pathway genes result in reduced numbers of neural stem/progenitor cells, precocious neurogenesis, and induction of apoptosis 4–8. In contrast, previous in vivo gain-of-function studies report mixed and inconsistent findings, depending on the methods used to manipulate the N1ICD levels and the numbers and types of cells affected by the manipulation 6, 9–11. Nevertheless, Notch signaling is elevated in many human cancers and implicated in regulating self-renewal of stem cell-like cells in tumors 12–14.
While the Notch pathway has been implicated in diverse biological processes, from angiogenesis to dendrite morphogenesis to tumorigenesis to stem cell maintenance, mechanistic details of its varied functions in different cellular contexts and biological processes remain to be resolved. Because NICD functions as a transcriptional co-factor to RBPJ, arguably the most informative approach to gaining insights into the mechanisms of Notch function is to identify its direct downstream target genes in different contexts. Currently, Hes and Hey gene family members are the best-characterized and most widely accepted direct targets of the NICD/RBPJ complex 15. More recent studies have suggested that GFAP, BLBP, SOX2 and p53 are also downstream targets of Notch in the brain 16–18. To date, however, these studies have been performed either on a gene-by-gene basis or in manipulated cells in culture 19–23. Hence, the full repertoire of genes that are regulated by direct binding of NICD/RBPJ in vivo is unknown.
Here we report a genome-wide analysis of direct RBPJ/N1ICD targets in NSCs in vivo, combining chromatin immunoprecipitation coupled to high-throughput sequencing (ChIP-seq) with transcriptome analyses. We use a mouse strain in which elevation of N1ICD level induces massive expansion of the NSC pool but not progenitor cells. Using very stringent criteria, we report 98 genes that are directly regulated by N1ICD/RPBJ in NSCs. These include many of the known transcription factors that control NSC proliferation and self-renewal, suggesting that Notch1 is a “master regulator” of NSCs. Surprisingly, Notch1 also directly activates expression of transcriptional effectors of the Wnt, SHH, and Hippo pathways. Since little is known about the function of Hippo-Yap pathway in NSC regulation, we examined YAP1 expression and function in NSCs. We show that ectopic Yap1 expression is sufficient to compensate for Notch signaling inhibition in NSCs. In summary, this study reveals an unexpected complexity and breadth of the transcriptional network downstream of N1ICD/RBPJ in vivo. It also suggests that activities of multiple signaling pathways converge to regulate an overlapping set of transcription factors, effectively integrating multiple signaling inputs into a single, discrete cell fate decision.
Materials and Methods
Please refer to Supplementary Methods for details.
Mouse strains
Gt(ROSA)26Sortm1(Notch1)Dam/J(stock# 8159), FVB-Tg(GFAP-cre)25Mes/J (stock # 4600), 129P2(Cg)-Foxg1<tm1(cre)Skm>/J (stock# 6084), B6.129S4-Gt(ROSA)26Sortm1Sor/J (stock# 3474), B6;129P-Tg(Neurog1-cre/ESR1)1Good/J (stock# 8529), and Tg(dlx6a-cre)1Mekk/J (Stock# 8199). All mouse work was performed according to the protocols approved by The Jackson Laboratory ACUC. All phenotype analyses were performed comparing at least three pairs of littermate control and transgenic embryos.
Neural stem cell culture
Single cells from freshly dissociated cortex or basal ganglia were cultured at 1 cell/μl density in NSC medium (DMEM/F12 + B27+ bFGF(10ng/ml) + EGF (20ng/ml)), unless noted otherwise. Neurospheres are counted 6–7 days later.
Histology and immunofluorescence analysis
BrdU-birthdating was performed by injecting pregnant dames at E11.5 and harvesting embryos for analysis at E13.5. Standard immunofluorescence protocols were used with antibodies listed on the figures.
RT-PCR
Realtime RT-PCR was performed using standard protocols using BioRad iQ5. For PCR conditions and primer sequences, see Supplementary Methods section. For statistical analysis, t-tests were used.
ChIP-Seq
Control and GFAP-Cre;N1ICD cortices (three each from littermates) were pooled together before the immunoprecipitation step with an antibody against RBPJ (Santa Cruz # SC-28713). High-throughput sequencing was performed using Illumina Genome Analyzer II. Peaks were identified as regions of the genome where significantly more reads align from the ChIP compared to input DNA samples (Bernoulli p-value < 0.05 for at least 20 consecutive bp).
Microarrays
RNA samples were isolated from 5 wildtype and 5 GFAP-Cre:N1ICD E15.5 cortices and were hybridized to Affymetrix Gene ST 1.0 arrays. Differentially expressed genes were identified by an analysis of variance (ANOVA) model 24 and by fold change (q-value < 0.05, and fold change > 1.5).
RNA-Seq
Pools of RNA from the wildtype and GFAP-Cre:N1ICD samples that were used for Affymetrix microarray analysis were sequenced using an Illumina Genome Analyzer II. Expression levels were quantified as RPKM values 25, and differentially expressed genes were identified by modeling read distributions between samples as a Bernoulli random variable (FDR adjusted p-value < 0.05).
Acute deletion of RBPJ in NSCs
NSCs from E15.5 RBPJfl/fl embryos were isolated and cultured in NSC medium. Small neurosphere cells at passage 0 or 1 were transduced with adenovirus expressing EGFP (control) or Cre-IRES-EGFP. RNA was harvested 48 hours after the addition of virus.
Results
Notch1 promotes symmetric stem cell divisions to expand the NSC pool in vivo
It has been shown that endogenous Notch activity level is high in NSCs but low in neural progenitor cells 26. To test whether high-level Notch pathway activity is sufficient to confer and/or maintain stemness in the developing nervous system, we conditionally expressed an activated form of the NOTCH1 receptor (N1ICD) in maturing neuroepithelium at three different stages. To do so, we used the Gt(ROSA)26Sortm1(Notch1)Dam/J (herein called N1ICD) transgenic mouse, which contains a single-copy insertion of the N1ICD transgene in the ROSA26 locus 27.
First, to determine the functional consequence of sustaining high levels of Notch activity in NSCs, we utilized Foxg1-Cre 28 mice to drive N1ICD expression in early NSCs. Foxg1-Cre is activated in the murine cortex at E9, when the neuroepithelium is entirely composed of NSCs that expand rapidly through symmetric stem cell divisions. Elevated and sustained Notch signaling in Foxg1-cre;N1ICD brains resulted in a dramatic expansion of the neuroepithelium (Fig. 1A), similar to what has been observed by activating Wnt signaling in the forebrain 29. Expression analyses of markers for NSCs (SOX2, PAX6), progenitors (TBR2), and neurons (TBR1, TuJ1, NeuN) indicated expansion of the NSC pool and reduction of neural progenitors and neurons at all ages examined (Supplementary Fig. 1 and not shown). Interestingly, the proportion of BrdU+ (S-phase) cells was reduced in the Foxg1-cre;N1ICD transgenic cortex, compared to the control cortex (Supplementary Fig. 1), suggesting that elevated Notch signaling does not necessarily enhance proliferation. These data suggest that maintaining high-level Notch1 activity in NSCs is sufficient to drive symmetric stem cell divisions and that switch to asymmetric divisions requires downregulation of Notch activity.
Figure 1. Notch activation expands the NSC pool in a dose-dependent manner.

(A) H&E staining of Foxg1-cre;N1ICD embryos at E13.5 shows greatly expanded neuroepithelium containing NSCs compared to control littermates. Arrows point to cortex. (B) Coronal sections of littermate control and GFAP-Cre; N1ICD embryos at E15.5 with 1 copy or 2 copies of the transgene, stained with H&E. Quantification of the lateral width and radial thickness of littermate cortices shows statistically significant changes in the transgenic cortex (p-values <0.004). Dashed lines: apical surface of the cortical epithelium, solid bars: radial thickness of cortex. (C) Western blot analyses with antibodies against NOTCH1, BLBP, SOX2 and PAX6 show increased expression in GFAP-Cre;N1ICD cortices at E15.5. N1ICD en: endogenous N1ICD, N1CID tg: transgenic N1ICD. (D) Marker analysis for progenitors (TBR2), neurons (TBR1) and NSCs (PAX6) at E15.5. (E) Quantification of NSCs (SOX2+, PAX6+) or progenitors (TBR2+) and neurons (TBR1+, NeuN+) in equivalent areas of control and transgenic cortices (n=3). (F) Neurosphere assays show increased number of NSCs isolated from E15.5 cortices of GFAP-Cre;N1ICD compared to control littermates, cultured with bFGF, EGF, or bFGF+EGF. (G) Relative expression levels of FGFR3 in control and GFAP-Cre; N1ICD cortices at E15.5, measured by realtime RT-PCR. VZ: ventricular zone, SVZ: subventricular zone. Error bar: standard deviation of the mean. Also see Supplementary Figure 1.
Notch1 expands the NSC pool in a dose-dependent manner in vivo
Previously Yang et al., reported that transgenic expression of N1ICD in the developing nervous system results in increased apoptosis and premature gliogenesis 9; however, we did not observe any increased apoptosis or gliogenesis in Foxg1-cre; N1ICD brains (not shown). To test whether these discrepancies between the two studies are due to differences in expression levels or the cell types in which N1ICD is expressed, we activated the N1ICD transgene using GFAP-Cre mice 30. In these mice, Cre recombinase is activated in the cortical NSCs and progenitors in the VZ from E12.5 onward. H&E analyses of GFAP-Cre;N1ICD double transgenic embryos with one copy of the N1ICD transgene showed an obvious expansion of the neuroepithelium compared to control littermates from E13.5 onward (Fig. 1B). We did not observe any precocious gliogenesis or apoptosis in cells over-expressing N1ICD (not shown). To test whether this is due to different levels of N1ICD expression, we generated embryos in which two copies of the transgene are expressed. GFAP-Cre;N1ICD embryos with two copies of the transgene consistently showed significantly greater lateral expansion of the neuroepithelium than those with a single copy (Fig. 1B, 0.0004<p<0.05). At the same time, GFAP-Cre;N1ICD transgenic cortices were significantly thinner than those of control littermates (Fig. 1B), suggesting a decreased generation of progenitors and neurons. Western blot analysis of NSC markers confirmed increased levels of SOX2, PAX6, and BLBP proteins in transgenic cortices (Fig. 1C). Immunofluorescence analyses with markers showed a significantly increased number of NSCs (SOX2, PAX6) and decreased numbers of progenitor cells (TBR2) and neurons (TBR1, MAP2, β-III-tubulin, NeuN) in transgenic cortices at E15.5 (Fig. 1D,E and not shown). Hence, elevation of Notch activity in NSCs and progenitors did not expand both proliferating cell types; rather, it specifically increased the NSC number while reducing the progenitor number. Again, we did not observe increased apoptosis or premature astrocytogenesis in GFAP-Cre;N1ICD brains with high doses of the transgene (not shown).
To functionally test whether GFAP-Cre;N1ICD cortices contain increased numbers of NSCs, freshly dissociated cells from E15.5 control and transgenic cortices were plated at a clonal density (1 cell/μl). We plated them in normal NSC medium containing both bFGF (basic Fibroblast Growth Factor) and EGF (Epidermal Growth Factor) or in medium containing either bFGF or EGF alone. As shown in Figure 1F, significantly higher numbers of neurospheres formed in the transgenic culture grown in normal NSC medium, and even more dramatic differences were observed in cultures grown in bFGF alone. Interestingly, while the number of sphere-forming cells was consistently higher, the size of each neurosphere was smaller in the GFAP-Cre;N1ICD cultures (not shown). This is consistent with our in vivo observation that high-level Notch activity prevents formation of highly proliferative neural progenitor cells (Fig. 1, Supplementary Fig. 1). Increased self-renewal of bFGF-responsive NSCs in the transgenic cultures is consistent with increased expression of FGFR3 (Fibroblast Growth Factor Receptor 3) in the transgenic cortices at E15.5 (Fig. 1G). No significant differences were observed between the control and transgenic neurospheres cultured in EGF alone (Fig. 2E). Together, these observations suggest that high-levels of Notch activity cooperate with the FGF but not EGF pathway to expand the NSC pool.
Figure 2. N1ICD expression in neural progenitors is not sufficient to confer stem cell phenotype.

(A) dlx6a-cre;ROSA-EYFP reporter analysis shows that CRE is active in proliferating progenitors (PCNA+) in the SVZ. (B) Proliferation (BrdU/PH3) and neurogenesis (TuJ1 and BrdU birthdating: inject E11.5-analysis at E13.5) are unaltered in dlx6a-cre;N1ICD. (C) Self-renewal assay of neurospheres isolated from basal ganglia of control and dlx6a-cre;N1ICD embryos confirm no significant change in NSC numbers. (n=3) (D) N1ICD expression and target gene (Hes1 and Hes5) activation in the basal ganglia of dlx6a-cre;N1ICD. (E) No significant changes in proliferation (PH3+ cells), progenitors (TBR2+), or stem cells (PAX6+) are observed in Ngn1-creER;N1ICD cortices treated with Tamoxifen at E11.5 and harvested at E13.5. PH3: phosphorylated Histone3, BG: basal ganglia, arrows point to positive cells in VZ and SVZ.
Notch1 activation is insufficient to confer NSC phenotype to neural progenitors
To formally test whether the increased number of NSCs in GFAP-Cre;N1ICD brains resulted from selective expansion of the NSC pool and not from conversion of neural progenitors to NSCs, we activated N1ICD expression only in committed neural progenitors and differentiating neurons in the VZ and SVZ of developing brain. As shown in Fig. 2A, dlx6a-cre31 is active in proliferating progenitors (PCNA+, MASH1+) and maturing neurons in the basal ganglia SVZ from E11.5 onwards. In dlx6a-Cre;N1ICD embryos, there was no significant change in proliferation as determined by BrdU and phosphorylated histone3 (PH3) staining (Fig. 2B). BrdU birthdating and a neuronal marker (TuJ1) analyses indicate that neurogenesis is also grossly normal (Fig. 2B). Isolation of NSCs from E13.5 basal ganglia showed that the number of NSCs also had not changed (Fig. 2C), despite high levels of the transgene expression and elevation of Hes1 and Hes5 expression (Fig. 2D). We made similar observations using Ngn1-CreER mice to activate N1ICD expression in cortical progenitors (Fig. 2E). These observations are in stark contrast to the same transgene expression only in NSCs in Foxg1-Cre;N1ICD embryos (Fig. 1). The fact that ectopic N1ICD expression is not sufficient to confer the stem-cell phenotype to committed progenitors or enhance progenitor cell proliferation indicates that the lateral expansion of the neuroepithelium in GFAP-Cre;N1ICD brain is due to selective expansion of NSCs. Together, our observations suggest that Notch function is highly cell context-specific in vivo and is insufficient to confer stemness to non-stem cells.
Identification of Notch1 targets in vivo at the genome level
To begin to understand the molecular mechanisms through which Notch1 maintains “stemness” in NSCs, we sought to identify direct downstream targets of the N1ICD/RBPJ transcriptional complex by taking advantage of the in vivo system in which N1ICD expression expands the NSC pool (Fig. 3A). First, E15.5 cortices from littermate control and GFAP-Cre;N1ICD embryos were micro-dissected and their transcriptomes were analyzed by RNA-seq (Supplementary Table 1) and Affymetrix Gene Chip (Supplementary Table 2) methods. As anticipated, RNA-seq revealed a greater number of differentially expressed genes due to greater signal resolution (Fig. 3B). The overlap between the two methods of transcriptome analysis was 280 genes (Fig. 3B, Supplementary Table 3). Both methods indicated that known canonical Notch target genes, Hes1, Hes5, Hey1, and Hey2, were higher expressed in GFAP-Cre;N1ICD than control cortices, validating our analysis. In addition, GFAP-Cre;N1ICD cortices expressed higher levels of known NSC regulators, including Pax6, Sox2, Nr2e1 (Tlx), and Id4, and lower levels of neurogenesis regulators, Neurod1 and Neurod4 (Supplementary Table 3). Also, the list of 280 genes included key components of the Notch, Wnt, SHH, TGFβ, FGF, and Hippo pathways, including Dll3, Fgf14, FGFR3, Fzd2, Fzd8, Fzd9, Gli2, Gli3, Notch1, SFRP2, SFRP3, Smo, TCF4 (Tcf7l2), Tead2, TAZ(Wwtr1), and Yap1 (Supplementary Table 3).
Figure 3. ChIP-seq and transcriptome analyses to identify N1ICD/RBPJ targets in vivo.

(A) A schema of experimental design: littermate wildtype and GFAP-Cre;N1ICD cortices were isolated and analyzed by RNA-seq and Affymetrix St 1.0 microarrays and ChIP-seq. (B) Numbers of differentially expressed genes measured by RNA-seq and Affymetrix microarray methods. (C) Numbers of genes within 15kb of significant peaks in wildtype and GFAP-Cre;N1ICD cortices. When the window is expanded to 50kb, a total of 8343 genes are associated with significant peaks in wildtype cortices. (D) Distribution of genomic regions bound by RBPJ. (E) Representative histogram presentations of aligned ChIP-seq reads in wildtype and transgenic cortices. Significant peaks are marked with bars below each histogram. Consensus RBPJ binding sites from earlier studies are marked with dots. (F) Independent ChIP validation of selected target sequences by realtime PCR. Untr6 and Untr17 are negative control regions. Also see Supplementary Figure 2.
To identify which of these genes are directly regulated by the NICD/RBPJ complex, E13.5 cortices from wildtype embryos were processed with a standard ChIP protocol to pull down chromosomal regions that are bound by the N1ICD/RBPJ transcriptional complex. Since RBPJ protein directly contacts DNA, we used an antibody against RBPJ that was successfully used in another ChIP study 32. A total of 5719 genes (9056 peaks) were identified to be within 15kb of significant peaks (Fig. 3C, p<0.05; see Methods for details). In GFAP-Cre;N1ICD cortices, 8640 genes (19, 261 peaks) were associated with peaks above the cutoff threshold, and of these, 4838 genes (6693 peaks) overlapped with the wildtype cortex (Figure 3C, Supplementary Tables 4, 5). Interestingly, over 85% of the RBPJ binding sites were located within 15kb of a coding sequence, and they are scattered approximately evenly in up/downstream, intronic, and exonic sequences (Fig. 3D), similar to other genome level ChIP studies of transcription factors 33, 34. The general pattern of binding sites was nearly identical between wildtype and GFAP-Cre;N1ICD transgenic cortices (Fig. 3E), indicating high reproducibility of these binding patterns. The known Notch1 target genes Hes1, Hes5, Hey1, Hey2, and Nrarp were all bound by N1ICD/RBPJ (Fig. 3E, Supplementary Fig. 2), validating our approach. In addition, we observed significant binding to many known NSC-regulating genes, including Pax6, Sox2, Nr2e1(Tlx), Msi, Bmi, Trp53, Ctnnb1(β-catenin), and Id4, as well as progenitor genes, Tbr2 and Neurogenin1 (Fig. 3E, Supplementary Fig. 2, Supplementary Tables 4,5). We also observed direct binding to many components of the Wnt, SHH, and FGF pathways (Supplementary Fig. 2, Supplementary Tables 4,5). We confirmed specificity and reproducibility of the ChIP pull down by randomly selecting 17 peaks associated with NSC maturation and testing them in an independent ChIP preparation. All 17 regions were validated by quantitative realtime PCR analysis (Fig. 3F).
To identify direct targets of N1ICD/RBPJ with the highest level of confidence, we focused on the genes that show direct binding and differential expression by N1ICD expression. By intersecting the list of genes that showed differential expression levels in both transcriptome analyses and significant DNA binding by RBPJ in wildtype cortices, we identified 98 genes that are most likely direct, endogenous targets of the N1ICD/RBPJ complex (Fig. 4A). Of these 98 genes, 77 were up-regulated and 21 were down-regulated in GFAP-Cre;N1ICD transgenic cortices compared to control littermates (Table 1). GO term analysis of these genes indicated that they include regulators of stem cell maintenance, axon guidance, synaptic transmission, cytoskeleton, and transcription (Supplementary Table 6 and 7), consistent with known functions of the Notch pathway in the nervous system 4. Gene set analysis of these 98 genes revealed that angiogenesis and the Notch pathway are among the most significantly enriched pathways (Fig. 4B). This is consistent with a large number of studies that indicate a critical role of the Notch pathway genes in angiogenesis 35, 36. Interestingly, the Wnt pathway is also significantly enriched (Fig. 4B). N1ICD/RBPJ complex directly regulates transcription of many Wnt pathway components (Fig. 4C, Table 1, Supplementary Fig. 2). In addition, components of the SHH and FGF signaling pathways were identified as direct targets of the N1ICD/RBPJ complex and validated by realtime RT-PCR analysis (Fig. 4C, Table 1). Remarkably, these targets include critical transcriptional effectors of the pathways, including Tcf7l2(Tcf4), Gli2 and Gli3.
Figure 4. Direct targets of N1ICD/RBPJ include transcription factors of the WNT, SHH, and Hippo pathway.

(A) Venn diagram summarizing the overlap among ChIP-seq and transcriptome analyses to identify 98 genes with highest confidence of being direct targets of N1ICD/RBPJ complex. (B) Gene set analysis 73 of most enriched pathways represented by the 98 target genes. (C) RT-PCR validation of selected genes in wildtype and N1ICD;GFAP-Cre cortices at E15.5 (student t-test, 0.0001<p< 0.04). (D) Endogenous NOTCH1 target gene expression in control vs. RBPJ−/− NSCs, measured by realtime RT-PCR. (n=7, p< 0.05 for all except Tbr2, p<0.17) (E) Realtime RT-PCR analysis of C17-2 cells transiently transfected with N1ICD (CaN1) or EGFP control expression vector (p<0.04, n=3). Also see Supplementary Figure 3.
Table 1.
Direct targets of N1ICD/RBPJ in NSCs in vivo
| Up-regulated | n | genes |
|---|---|---|
| transcription | 24 | Btg2, Gli2, Gli3, Hes1, Hes5, Hey1, Id4, Jun, Nfatc4, Notch1, Nr2e1 (Tlx), Pax6, Prdm16, Sall1, Sox2, Sox21,Sox9, Tcf3 (E2A/E47), Tcf7l2 (Tcf4), Tcfap2c, Tead2, Yap1, Zfhx4, Zhx2 |
| Notch pathway | 5 | Hes1, Hes5, Hey1,Notch1, Nrarp |
| Wnt pathway | 8 | Fzd2, Fzd8, Fzd9, Nrarp, Ppap2b, Sfrp1, Slc9a3r1 (NHERF1), Tcf7l2 (Tcf4) |
| SHH pathway | 4 | Cdon, Gli2, Gli3, Smo |
| Hippo pathway | 2 | Tead2, Yap1 |
| other signaling | 15 | AI464131, Angptl2, Cntfr, Dusp16, E130112L23Rik, Epb4.1l5, Fgfr3, Grik2, Igfbp5, Jun, Ltbp3,Prdm16, Rcn3, Rlbp1, Spata13 |
| calcium binding/signaling | 6 | Casq1,Grik2, Ltbp3, Rcn3, Rhbdl3, Ttyh1 |
| kinase/phosphatase | 5 | Alk, Axl, Camk2d, D8Ertd82e, Ppap2b |
| cell adhesion | 8 | Cdon, Celsr1, Epb4.1l5, Jub, Lamb2, Megf10, Tns3, Ttyh1 |
| extracellular matrix/membran | 7 | Bcan, Igdcc4, Igfbp5, Kcnj10, Lamb2, Mfap2, Ptgfrn |
| apoptosis | 3 | Aldoc, Bcl2l11 (Bim), Ddit4 |
| metabolism | 5 | Dbi, Aldoc, Ddit4, Prdm16, Slc27a1 |
| stress response | 3 | Hhipl1, HSPB6, Nfatc4 |
| other | 7 | 1190002N15Rik,2010011I20Rik, Cbs, Chst3, Myo10, Pgpep1, Pgpep1, Ston2 |
|
| ||
| down-regulated | n | genes |
|
| ||
| transcription | 4 | Bcl11b, Fezf2, Nr4a3, Sox5 |
| Notch pathway | 1 | Dll3 |
| other signaling | 6 | Abr, Adamts3, Agap2, Cck, Cnih2, Dab1 |
| cell adhesion | 1 | Dab1 |
| extracellular matrix/membran | 3 | Adamts3, Islr2, Tnr |
| calcium binding/signaling | 1 | Necab3 |
| apoptosis | 2 | Bcl11b, Cck |
| stress response | 1 | Necab3 |
| neural differentiation | 5 | Accn1, Islr2, Slc17a7, Slit1, Sox5 |
| other | 4 | Opcml, Sh3gl2, St3gal1, Unc13a |
Interestingly, a previously reported consensus sequence for RBPJ from in vitro studies is not enriched in the peaks we identified, although they are present in nearby sites (denoted by circles in Fig. 3,5, Supplementary Fig. 2). Analysis of de novo enriched sequence motifs within the significant peaks identified a GC-rich motif (Supplementary Fig. 3) within 311 of the 347 WT peaks near up-regulated genes, and in 42 of 61 peaks near down-regulated genes. To test whether there are distinct co-factor binding sites near up- and down-regulated genes, we analyzed sequences within 15kb of the transcription start sites of the up- and down- regulated genes. We found significant enrichment near the down-regulated genes for HIF1, E2F, and GAL4 (permutation p-value < 0.001) binding sites; and near the up-regulated genes we found enrichment for NMYC (p<0.001) and others (not shown).
Figure 5. Yap1 is a significant downstream target of N1ICD in NSCs.

(A) RT-PCR validation of increased Yap1 and Tead2 expression levels in GFAP-Cre;N1ICD cortices. (B) A histogram representation of RBPJ binding in Yap1 and Tead2 genomic regions in wildtype and GFAP-Cre;N1ICD cortices in vivo. (C) Yap1 and Tead2 RNA levels in control (Adeno-EGFP) and RBPJ-deleted (Adeno-Cre-EGFP) neurospheres (n=7, Yap1 p<0.0007, Tead2 p<0.08). (D) Realtime RT-PCR analysis of control (EGFP) and N1ICD (CaN1) transfected C17-2 cells at <24hr after transfection (n=3). (E) Immunofluorescence analyses of YAP1 in wildtype forebrain at E13.5 at low magnification. (F) Double immunofluorescence analysis with antibodies against YAP1 (green) and TBR2 (red) show radial glial fiber and nuclear YAP1 expression in some of the VZ cells. Most TBR2+ cells only contain cytoplasmic YAP1. Arrows point to nuclear and cytoplasmic YAP1+ cells; arrowheads point to only cytoplasmic YAP1+ cells. (G,H) Nuclear YAP1 expression in wildtype NSCs cultured in low (G) and high (H) densities (YAP1: green, SOX2: red). Arrows point to strong nuclear YAP1 and arrowheads point to weaker nuclear YAP1 expressing cells. (I) YAP1 protein is present in the nucleus and cytoplasm in E15.5 wildtype cortex. (J) DAPT (10μM) treatment of control (pBABE-Puro) and Yap1 over-expressing (pBABE-Yap1-puro) NSCs show that the reduced NSC self-renewal by Notch inhibition can be rescued by Yap1 over-expression (p=0.05). (K) Realtime RT-PCR analysis of control (pBABE-puro) and Yap1-expressing cells (pBABE-Yap1-puro). All gene expression levels shown are normalized to the control sample levels. Scale bar= 50μm.
To validate that the canonical Notch pathway modulates the endogenous expression of newly identified target genes, we measured the expression level changes of selected target genes after acutely inactivating the Notch pathway in NSCs. To do this, we isolated NSCs from E15.5 cortices of RBPJf/f, a conditional deletion allele of RBPJ embryos 37, and used Adeno-Cre-EGFP or Adeno-EGFP (control) virus transduction to acutely delete RBPJ expression. As shown in Fig. 4D, previously identified Notch targets, Hes1 and Hes5, as well as newly identified Notch targets, Sox2, Pax6, and Id4, showed significantly reduced expression levels in Cre-transduced NSCs, confirming that these are endogenous targets of the canonical Notch pathway in NSCs.
To complement the loss-of-function analysis, we transiently transfected an N1ICD expression vector in murine NSC cell line C17-2 38. RNA was harvested 24 hours after transfection (within 12 hrs of N1ICD expression onset), and analyzed by realtime RT-PCR. We observed significant up-regulation of Hes1, Sox2, and Gli2 in N1ICD-transfected cells compared to control GFP-transfected cells (Fig. 4E, p<0.04), indicating that N1ICD is sufficient to induce expression of these genes. These transient loss- and gain-of-function analyses provide corroborating evidence that the genes we have identified are directly regulated by N1ICD/RBPJ at the transcriptional level in NSCs and that the increased transcript level we observed in GFAP-Cre;N1ICD cortices is not simply due to an increased number of NSCs in transgenic embryos.
Yap1 is a significant downstream target of N1ICD in NSCs
An unanticipated finding from our genome-level analysis is the large number of NOTCH1 targets that are integral components of other signaling pathways (Table 1). In particular, we were intrigued by the interaction between the Notch and Hippo pathways, since little is known about the function of the Hippo pathway in NSCs. Hippo pathway is a cascading kinase pathway and the activation of the Hippo pathway results in phosphorylation and cytoplasmic retention of the YAP1 protein39. When the pathway is inactive, YAP1 translocates to the nucleus and form a transcriptional activation complex with TEAD proteins and YAP1/TEAD complex activates downstream target genes that promote proliferation and survival 40, 41. In chick neural tube, Yap1 expression induces neural progenitor proliferation 42, suggesting that Hippo-Yap1 pathway is involved in neural cell proliferation and survival in vertebrates. Since YAP1 is a rate limiting transcription factor in the Hippo pathway 43–45, we analyzed its expression and function in NSCs in the developing brain.
First, we validated that activation of Notch results in elevated Yap1 and Tead2 RNA levels in the GFAP-Cre;N1ICD cortices by realtime RT-PCR analysis (Fig. 5A, p< 0.01). In addition, RBPJ binds to regulatory regions of Yap1 and Tead2 in both wildtype and GFAP-Cre;N1ICD cortices (Fig. 5B), indicating that they are endogenous targets of N1ICD. To test whether endogenous Yap1 and Tead2 expression depends on the canonical Notch pathway, we acutely deleted RBPJ expression from NSCs in culture and measured the consequent Yap1 and Tead2 expression levels (Fig. 5C). Similar to other Notch target genes (Fig. 4D), Yap1 expression level was significantly reduced in RBPJ−/− NSCs (p<0.0007), while the reduction in Tead2 level did not quite reach statistical significance (p<0.0881). To test whether acute N1ICD expression is sufficient to induce Yap1 and Tead2 expression, we transiently transfected C17-2 cells with N1ICD and measured Yap1 and Tead2 levels within 12 hours after the transgene expression (<24hr after transfection). As shown in Fig. 5D, N1ICD is sufficient to induceYap1 expression in these cells although it appears other factors are needed to activate Tead2 in these cells. Together, these results suggest that Yap1 is a direct target of N1ICD.
To test whether YAP1 protein is expressed and is functional in mammalian NSCs, we analyzed YAP1 expression pattern by immunofluorescence analysis on wildtype mouse brains at E13.5 and E15.5. As shown in Fig. 5E, YAP1 expression is high in the stem cell compartment (VZ) of wildtype cortex and basal ganglia and significantly lower in the progenitor and differentiation zones (SVZ and mantle). Double labeling with a marker for neural progenitors (TBR2) suggests that most progenitor cells in the SVZ have very low to undetectable levels of nuclear YAP1, in contrast to many cells in the VZ that have both nuclear and cytoplasmic YAP1 (Fig. 5F). Consistent with its expression in NSCs, YAP1 immuno-reactivity is observed in radial glial fibers (Fig. 5F). To better visualize nuclear localization of YAP1 and to test whether YAP1 localization is regulated by cellular density, we plated wildtype NSCs onto coverslips at low and high densities. YAP1 is nuclear, albeit at different levels, in all SOX2+ cells at low density (Fig 5G) but becomes weaker in some SOX2+ cells at high density (Fig 5H). Furthermore, western blot analysis of nuclear and cytoplasmic fractionations of E15.5 wildtype cortex shows that YAP1 is present in both the nucleus and cytoplasm (Fig. 5I). Together, these results indicate that YAP1 is expressed in the right cell type to mediate Notch function in NSC proliferation and survival.
To functionally test whether Yap1 is a significant downstream target of Notch1, we ectopically expressed Yap1 in wildtype NSCs in vitro. Treatment of wildtype NSCs with DAPT, a Notch pathway inhibitor, significantly reduced neurosphere formation (Fig 5J). In contrast, Yap1 expressing cells were relatively resistant to DAPT treatment, indicating that ectopic expression of Yap1 can rescue Notch pathway inhibition (Fig 5J). Realtime RT-PCR analysis of control and Yap1-expressing cells show that expression of human Yap1 transgene elevated endogenous mouse Yap1 but not TAZ expression, suggesting an auto-feedback loop (Fig 5K). Interestingly, Yap1 also elevated Hes5, but not Hes1 or N1ICD, expression, suggesting another point of potential cross-talk between the Notch and Hippo-Yap pathways. Together, these results validate that Yap1 is an important downstream effector of the Notch pathway in NSC self-renewal and suggest mutual cross-regulation between the Notch and Hippo pathways.
Discussion
The Notch pathway has been shown to play a critical role during normal development and homeostasis of multiple stem cell compartments 4, 46, 47. Aberrant activation of the Notch pathway is implicated in multiple types of human cancer, including brain tumors 48–50. Hence, it is imperative to understand molecular mechanisms of Notch function in order to safely manipulate normal stem cells or treat cancer.
Interestingly, the age-dependent effect of N1ICD elevation is more dramatic than the dose-dependent effect (Fig. 1,2). Specifically, single copy expression of N1ICD in Foxg1-Cre+ cells leads to a much more dramatic phenotype than two-copy expression in GFAP-Cre+ cells in older neuroepithelium. These observations suggest that young NSCs may be more competent or responsive than older NSCs to Notch1 activity. This idea is further supported by several of our observations. First, N1ICD transgenic NSCs were highly responsive to bFGF but not to EGF (Fig. 2E). Early neuroepithelial stem cells depend on bFGF for proliferation, while the more mature progenitor cells in the SVZ depend on EGF 51. Second, N1ICD directly activates expression of FGFR3 (Fig 2F), presumably enhancing responsiveness of N1ICD expressing cells to FGF signaling, consistent with a previous report that FGF and Notch signaling cooperate to promote neural precursor proliferation 11. Third, N1ICD expression enhances symmetric stem cell divisions (not shown), the dominant mode of cell division by early NSCs during the neuroepithelium expansion phase. It has been reported that stem cell numbers diminish with age due to both cell-autonomous and non-autonomous effects, and that NSCs require different molecular mechanisms for self-renewal at different ages 52–54. GFAP-cre;N1ICD adult mice have increased numbers of NSCs in the SVZ compared to littermate controls of the same age (not shown). However, we do not yet know whether this is due to maintenance of higher numbers of NSCs throughout life or whether N1ICD directly promotes adult NSC self-renewal in mature GFAP-Cre;N1ICD brains.
We determined that Notch activation is not sufficient to confer the stem cell phenotype to non-stem cells. This observation complements other studies that suggested that Notch signaling is not necessary for the generation of NSCs but is required to maintain NSCs 55. Notch1 directly activates expression of Sox2, Pax6 and many other NSC regulators but does not bind to DNA sequences near other stem cell regulators such as Nanog and Oct4/Pou5f1, suggesting that only a subset of the stem cell network genes is directly regulated by N1ICD/RBPJ. Together, these observations indicate that Notch is a permissive rather than instructive factor for the NSC fate. In the future, it will be important to identify distinct molecular events required for the acquisition vs. maintenance of stemness and the types of epigenetic changes that accompany progenitor specification and loss of competence to respond to N1ICD.
Our findings suggest that Notch activity must be repressed in maturing wildtype neural progenitor cells, and multiple molecular mechanisms are in place to ensure reduced Notch activity in neural progenitors. For example, earlier studies have shown that NUMB and NUMB-LIKE antagonizes Notch and these proteins are asymmetrically localized to differentiating daughter cells 56–58. More recently, it has been shown that degradation of NICD by FBW7, an E3-ligase that ubiquitinylates NICD, plays a critical role in NSC maturation in vivo 59. Identification of Notch targets in this study provides a potential explanation for this necessity: in addition to repressing neurogenic bHLH proteins by activating Hes gene expression, N1ICD/RBPJ actively represses the transcription of many genes associated with neural progenitor specification and differentiation (Table 1). According to the dogma in the field, N1ICD functions as a transcriptional co-activator; however, our results suggest that more than 20% (21/98) of the direct targets in vivo are repressed by Notch activation. Hence, Notch1 simultaneously promotes NSC fate and actively inhibits progenitor/neuron specification at the molecular level.
It should be noted that the genes we identified are endogenous targets of NICD/RBPJ in physiological in vivo conditions since the DNA binding occurs in wildtype cortices (Fig. 4E) and endogenous levels of all activated genes tested are decreased when RBPJ is inactivated (Fig. 5D, 6C). In addition to our own data, there are corroborating evidence in the literature that these are true targets of Notch: 1) the short list of 98 genes includes most well-established canonical targets of Notch previously reported (such as Hes1, Hes5, Hey1); 2) NOTCH1 target genes identified in ES cells 23 overlap with the genes we identified (including Pax6, Sox9, Id4); 3) previous genetic analyses of many of the newly identified target genes, including Pax6, Sox2, Tlx, and Id4, showed overlapping phenotypes with Notch pathway loss- and gain-of-function in the NSC compartment 60–64. Interestingly, we did not observe significant enrichment of previously identified “consensus RBPJ” motif, although they were often present near the peaks we identified (denoted with green dots in ChIP peak figures). There are multiple possible variables that might explain this result. First, our work indicates that cellular context has a strong influence in modulating Notch/RBPJ function. Previously identified “consensus sequences” were optimized in vitro or in cultured cells, as opposed to in vivo tissues, where cell:cell contact and microenvironmental cues may be drastically different. Second, RBPJ binding site preferences may be inherently different between NSCs and other cell types used in earlier studies. In addition, epigenetic status of NSCs and other cell types are most likely very different, altering the accessibility of RBPJ binding sites. We estimate that our analysis is very conservative and that Notch1 targets include many more genes than the 98 genes we prioritized. In the future, it will be informative to test the functions of those genes whose roles in NSC regulation are unknown, particularly many of the transcription factors, cell adhesion molecules, and kinases.
Figure 6.
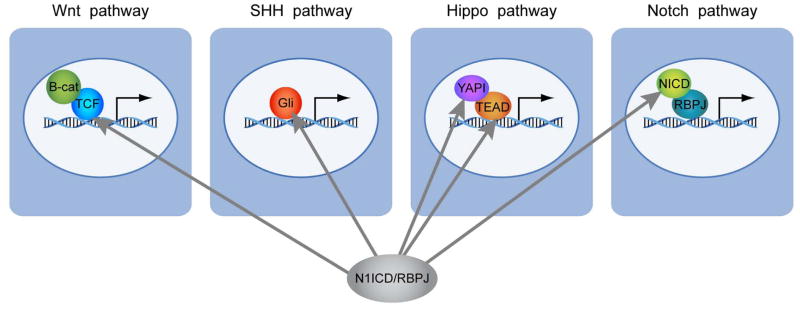
A working model based on our observation that N1ICD/RBPJ directly binds and activates expression of transcriptional effectors of the Wnt, SHH, Hippo, and Notch pathways, priming these pathways for rapid and robust responses when the canonical pathways are activated.
It is somewhat surprising that Notch activates expression of so many transcription factors (28 out of 98 genes), including key regulators of NSC proliferation and differentiation, suggesting that Notch1 is a “master regulator” of NSCs. It is even more surprising that Notch activates transcription factors that function as effectors of many other signaling pathways involved in stem cell regulation. Stem cells in vivo are under the influence of multiple signaling pathways and at each cell division, the input from all these pathways must be integrated into a single, discrete, cell fate decision. Emerging studies show that there is cross-talk among different signaling pathways via protein:protein interactions among the canonical pathway proteins or via cross-regulation of classical target genes or ligand expression 65. For example, it was recently reported that β-catenin, a transcriptional effector of the Wnt pathway, binds to N1ICD to enhance Hes1 expression 66. Others have reported that the SHH pathway activates Hes1, independent of Notch activation 67, 68. Similarly, the TGF/BMP pathway has been shown to antagonize or synergize with Notch1 activation 69. Our findings demonstrate yet another level of cross-regulation among the pathways: N1ICD regulates RNA levels of transcriptional effectors in the Wnt, SHH, and Hippo signaling pathways (Fig. 4D,E, Fig. 5A,B,C, Table 1). We propose a working model in which activation of the Notch pathway positively feeds into the activities of the other signaling pathways (Wnt, SHH and Hippo) by raising the RNA levels of their transcriptional effectors, priming them for rapid response upon their pathway activation (Fig. 6).
Another unanticipated discovery of our study is the direct crosstalk between the Hippo and Notch pathways in NSCs. The Hippo pathway responds to cellular density in vivo and in vitro by sensing cell:cell contact39. Hence, it is logical that the Notch and the Hippo pathways would be intimately interconnected; however, there is little evidence for direct interaction at the molecular level, especially in mammalian cells. In Drosophila, genetic studies have suggested that the Hippo pathway modulates Notch target gene expression in posterior follicle cells 70, and that the Hippo pathway is involved in regulating the Notch receptor level 71. Recently, Reddy et al., have shown that expression of activated Yki expands the neuroepithelial cells in Drosophila optic disc by inhibiting differentiation, and suggested that this is mediated, at least in part, through regulation of Delta1 expression. Interestingly, they also reported that the neuroblasts are refractory to the Fat-Hippo pathway 72, similar to our observation that neural progenitors are refractory to Notch activation (Fig. 2). We show that Notch1 directly activates Yap1 mRNA levels in murine NSCs and that Yap1 activates Hes5 expression, although it remains to be tested whether Hes5 is a direct target of Yap1. By combining published data by others and our observations, we propose that the coordinated activities of the Hippo and Notch pathways play a determining role in regulating the stem cell number and ultimately organ size during embryogenesis.
Conclusion
In summary, we show that Notch is prominently positioned in a transcriptional regulatory network that controls NSCs by directly regulating the expression of many stem cell factors and key components of other signaling pathways (Table 1). Clearly, N1ICD/RBPJ does not solely regulate the expression of these genes but most likely interacts with other factors that control transcription of these genes in a cooperative or competitive manner. Together with emerging studies from other laboratories, data we present here suggest a new model in which signaling pathways that are traditionally considered to act in parallel interact much more intimately than previously appreciated to control NSC fates.
Supplementary Material
Acknowledgments
We thank Ben Low, Jesse Hammer, Gene Expression Services, and Computational services at the Jackson Laboratory for their assistance. We are grateful to Dr. Igor Prudovsky at MMCRI for sharing the N1ICD expression plasmid, Dr. T. Honjo for sharing the RBPJfl/fl mice, and Tom Gridley and Christine Norton for providing us with RBPJfl/fl embryos. We also thank GENpathway Inc, and Shawn Levy at Vanderbilt Microarray Shared Resources for technical assistance. Lastly, we thank Tom Gridley, Susan Ackerman, Stewart Anderson, Marina Mione, Kevin Mills, Jeong Yoon, Ellen Burns, and Stephen Sampson for helpful comments and edits on the manuscript. The Jackson Laboratory provided funding for this work.
funding: The Jackson Laboratory provided funding for this work.
Footnotes
Disclosure of Potential Conflicts of Interest
There is no conflict of interest.
Author contributions:
Yaochen Li: collection of data, data analysis and interpretation
Matthew Hibbs: data analysis and interpretation, manuscript writing
Ashley Gard: collection of data, data analysis and interpretation
Natasha Shylo: collection of data, data analysis and interpretation
Kyuson Yun: Conception and design, financial support, provision of study material, collection of data, data analysis and interpretation, manuscript writing, final approval of manuscript
References
- 1.Gotz M, Huttner WB. The cell biology of neurogenesis. Nat Rev Mol Cell Biol. 2005;6:777–788. doi: 10.1038/nrm1739. [DOI] [PubMed] [Google Scholar]
- 2.Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 3.Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pierfelice T, Alberi L, Gaiano N. Notch in the vertebrate nervous system: an old dog with new tricks. Neuron. 2011;69:840–855. doi: 10.1016/j.neuron.2011.02.031. [DOI] [PubMed] [Google Scholar]
- 5.Ehm O, Goritz C, Covic M, et al. RBPJkappa-dependent signaling is essential for long-term maintenance of neural stem cells in the adult hippocampus. J Neurosci. 2010;30:13794–13807. doi: 10.1523/JNEUROSCI.1567-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoon K, Gaiano N. Notch signaling in the mammalian central nervous system: insights from mouse mutants. Nat Neurosci. 2005;8:709–715. doi: 10.1038/nn1475. [DOI] [PubMed] [Google Scholar]
- 7.Mason HA, Rakowiecki SM, Gridley T, et al. Loss of notch activity in the developing central nervous system leads to increased cell death. Dev Neurosci. 2006;28:49–57. doi: 10.1159/000090752. [DOI] [PubMed] [Google Scholar]
- 8.Imayoshi I, Sakamoto M, Yamaguchi M, et al. Essential roles of Notch signaling in maintenance of neural stem cells in developing and adult brains. J Neurosci. 2010;30:3489–3498. doi: 10.1523/JNEUROSCI.4987-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang X, Klein R, Tian X, et al. Notch activation induces apoptosis in neural progenitor cells through a p53-dependent pathway. Dev Biol. 2004;269:81–94. doi: 10.1016/j.ydbio.2004.01.014. [DOI] [PubMed] [Google Scholar]
- 10.Gaiano N, Nye JS, Fishell G. Radial glial identity is promoted by Notch1 signaling in the murine forebrain. Neuron. 2000;26:395–404. doi: 10.1016/s0896-6273(00)81172-1. [DOI] [PubMed] [Google Scholar]
- 11.Yoon K, Nery S, Rutlin ML, et al. Fibroblast growth factor receptor signaling promotes radial glial identity and interacts with Notch1 signaling in telencephalic progenitors. J Neurosci. 2004;24:9497–9506. doi: 10.1523/JNEUROSCI.0993-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan X, Matsui W, Khaki L, et al. Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Res. 2006;66:7445–7452. doi: 10.1158/0008-5472.CAN-06-0858. [DOI] [PubMed] [Google Scholar]
- 13.Charles N, Ozawa T, Squatrito M, et al. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell stem Cell. 2010;6:141–152. doi: 10.1016/j.stem.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu YY, Zheng MH, Cheng G, et al. Notch signaling contributes to the maintenance of both normal neural stem cells and patient-derived glioma stem cells. BMC cancer. 2011;11:82. doi: 10.1186/1471-2407-11-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iso T, Kedes L, Hamamori Y. HES and HERP families: multiple effectors of the Notch signaling pathway. J Cell Physiol. 2003;194:237–255. doi: 10.1002/jcp.10208. [DOI] [PubMed] [Google Scholar]
- 16.Anthony TE, Mason HA, Gridley T, et al. Brain lipid-binding protein is a direct target of Notch signaling in radial glial cells. Genes Dev. 2005;19:1028–1033. doi: 10.1101/gad.1302105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ge W, Martinowich K, Wu X, et al. Notch signaling promotes astrogliogenesis via direct CSL-mediated glial gene activation. J Neurosci Res. 2002;69:848–860. doi: 10.1002/jnr.10364. [DOI] [PubMed] [Google Scholar]
- 18.Purow BW, Sundaresan TK, Burdick MJ, et al. Notch-1 regulates transcription of the epidermal growth factor receptor through p53. Carcinogenesis. 2008;29:918–925. doi: 10.1093/carcin/bgn079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ronchini C, Capobianco AJ. Induction of cyclin D1 transcription and CDK2 activity by Notch(ic): implication for cell cycle disruption in transformation by Notch(ic) Mol Cell Biol. 2001;21:5925–5934. doi: 10.1128/MCB.21.17.5925-5934.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmid RS, McGrath B, Berechid BE, et al. Neuregulin 1-erbB2 signaling is required for the establishment of radial glia and their transformation into astrocytes in cerebral cortex. Proc Natl Acad Sci U S A. 2003;100:4251–4256. doi: 10.1073/pnas.0630496100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patten BA, Sardi SP, Koirala S, et al. Notch1 signaling regulates radial glia differentiation through multiple transcriptional mechanisms. J Neurosci. 2006;26:3102–3108. doi: 10.1523/JNEUROSCI.4829-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhattacharya S, Das A, Mallya K, et al. Maintenance of retinal stem cells by Abcg2 is regulated by notch signaling. J Cell Sci. 2007;120:2652–2662. doi: 10.1242/jcs.008417. [DOI] [PubMed] [Google Scholar]
- 23.Meier-Stiegen F, Schwanbeck R, Bernoth K, et al. Activated Notch1 target genes during embryonic cell differentiation depend on the cellular context and include lineage determinants and inhibitors. PLoS ONE. 2010;5:e11481. doi: 10.1371/journal.pone.0011481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cui X, Churchill GA. Statistical tests for differential expression in cDNA microarray experiments. Genome Biol. 2003;4:210. doi: 10.1186/gb-2003-4-4-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mortazavi A, Williams BA, McCue K, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 26.Mizutani K, Yoon K, Dang L, et al. Differential Notch signalling distinguishes neural stem cells from intermediate progenitors. Nature. 2007;449:351–355. doi: 10.1038/nature06090. [DOI] [PubMed] [Google Scholar]
- 27.Murtaugh LC, Stanger BZ, Kwan KM, et al. Notch signaling controls multiple steps of pancreatic differentiation. Proc Natl Acad Sci U S A. 2003;100:14920–14925. doi: 10.1073/pnas.2436557100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hébert JM, McConnell SK. Targeting of cre to the Foxg1 (BF-1) locus mediates loxP recombination in the telencephalon and other developing head structures. Developmental Biology. 2000;222:296–306. doi: 10.1006/dbio.2000.9732. [DOI] [PubMed] [Google Scholar]
- 29.Chenn A, Walsh CA. Regulation of Cerebral Cortical Size by Control of Cell Cycle Exit in Neural Precursors. Science. 2002;297:365–369. doi: 10.1126/science.1074192. [DOI] [PubMed] [Google Scholar]
- 30.Zhuo L, Theis M, Alvarez-Maya I, et al. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis. 2001;31:85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]
- 31.Monory K, Massa F, Egertova M, et al. The endocannabinoid system controls key epileptogenic circuits in the hippocampus. Neuron. 2006;51:455–466. doi: 10.1016/j.neuron.2006.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boni A, Urbanek K, Nascimbene A, et al. Notch1 regulates the fate of cardiac progenitor cells. Proc Natl Acad Sci U S A. 2008;105:15529–15534. doi: 10.1073/pnas.0808357105. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Cao Y, Yao Z, Sarkar D, et al. Genome-wide MyoD binding in skeletal muscle cells: a potential for broad cellular reprogramming. Developmental cell. 2010;18:662–674. doi: 10.1016/j.devcel.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Welboren WJ, van Driel MA, Janssen-Megens EM, et al. ChIP-Seq of ERalpha and RNA polymerase II defines genes differentially responding to ligands. The EMBO journal. 2009;28:1418–1428. doi: 10.1038/emboj.2009.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Phng LK, Gerhardt H. Angiogenesis: a team effort coordinated by notch. Dev Cell. 2009;16:196–208. doi: 10.1016/j.devcel.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 36.Gridley T. Notch signaling in vascular development and physiology. Development. 2007;134:2709–2718. doi: 10.1242/dev.004184. [DOI] [PubMed] [Google Scholar]
- 37.Han H, Tanigaki K, Yamamoto N, et al. Inducible gene knockout of transcription factor recombination signal binding protein-J reveals its essential role in T versus B lineage decision. Int Immunol. 2002;14:637–645. doi: 10.1093/intimm/dxf030. [DOI] [PubMed] [Google Scholar]
- 38.Snyder EY, Deitcher DL, Walsh C, et al. Multipotent neural cell lines can engraft and participate in development of mouse cerebellum. Cell. 1992;68:33–51. doi: 10.1016/0092-8674(92)90204-p. [DOI] [PubMed] [Google Scholar]
- 39.Zhao B, Wei X, Li W, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21:2747–2761. doi: 10.1101/gad.1602907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19:491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao B, Li L, Lei Q, et al. The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev. 2010;24:862–874. doi: 10.1101/gad.1909210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cao X, Pfaff SL, Gage FH. YAP regulates neural progenitor cell number via the TEA domain transcription factor. Genes Dev. 2008;22:3320–3334. doi: 10.1101/gad.1726608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang J, Wu S, Barrera J, et al. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell. 2005;122:421–434. doi: 10.1016/j.cell.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 44.Zhao B, Ye X, Yu J, et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008;22:1962–1971. doi: 10.1101/gad.1664408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu S, Liu Y, Zheng Y, et al. The TEAD/TEF family protein Scalloped mediates transcriptional output of the Hippo growth-regulatory pathway. Dev Cell. 2008;14:388–398. doi: 10.1016/j.devcel.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 46.Chiba S. Notch signaling in stem cell systems. Stem Cells. 2006;24:2437–2447. doi: 10.1634/stemcells.2005-0661. [DOI] [PubMed] [Google Scholar]
- 47.Androutsellis-Theotokis A, Leker RR, Soldner F, et al. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature. 2006;442:823–826. doi: 10.1038/nature04940. [DOI] [PubMed] [Google Scholar]
- 48.Pierfelice TJ, Schreck KC, Eberhart CG, et al. Notch, neural stem cells, and brain tumors. Cold Spring Harb Symp Quant Biol. 2008;73:367–375. doi: 10.1101/sqb.2008.73.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Allenspach EJ, Maillard I, Aster JC, et al. Notch signaling in cancer. Cancer biology & therapy. 2002;1:466–476. doi: 10.4161/cbt.1.5.159. [DOI] [PubMed] [Google Scholar]
- 50.Rizzo P, Osipo C, Foreman K, et al. Rational targeting of Notch signaling in cancer. Oncogene. 2008;27:5124–5131. doi: 10.1038/onc.2008.226. [DOI] [PubMed] [Google Scholar]
- 51.Tropepe V, Sibilia M, Ciruna BG, et al. Distinct neural stem cells proliferate in response to EGF and FGF in the developing mouse telencephalon. Dev Biol. 1999;208:166–188. doi: 10.1006/dbio.1998.9192. [DOI] [PubMed] [Google Scholar]
- 52.Molofsky AV, Slutsky SG, Joseph NM, et al. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature. 2006;443:448–452. doi: 10.1038/nature05091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nishino J, Kim I, Chada K, et al. Hmga2 promotes neural stem cell self-renewal in young but not old mice by reducing p16Ink4a and p19Arf Expression. Cell. 2008;135:227–239. doi: 10.1016/j.cell.2008.09.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morrison SJ, Spradling AC. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell. 2008;132:598–611. doi: 10.1016/j.cell.2008.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hitoshi S, Alexson T, Tropepe V, et al. Notch pathway molecules are essential for the maintenance, but not the generation, of mammalian neural stem cells. Genes Dev. 2002;16:846–858. doi: 10.1101/gad.975202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shen Q, Zhong W, Jan YN, et al. Asymmetric Numb distribution is critical for asymmetric cell division of mouse cerebral cortical stem cells and neuroblasts. Development. 2002;129:4843–4853. doi: 10.1242/dev.129.20.4843. [DOI] [PubMed] [Google Scholar]
- 57.Zhong W, Feder JN, Jiang MM, et al. Asymmetric localization of a mammalian numb homolog during mouse cortical neurogenesis. Neuron. 1996;17:43–53. doi: 10.1016/s0896-6273(00)80279-2. [DOI] [PubMed] [Google Scholar]
- 58.Petersen PH, Zou K, Hwang JK, et al. Progenitor cell maintenance requires numb and numblike during mouse neurogenesis. Nature. 2002;419:929–934. doi: 10.1038/nature01124. [DOI] [PubMed] [Google Scholar]
- 59.Hoeck JD, Jandke A, Blake SM, et al. Fbw7 controls neural stem cell differentiation and progenitor apoptosis via Notch and c-Jun. Nat Neurosci. 2010;13:1365–1372. doi: 10.1038/nn.2644. [DOI] [PubMed] [Google Scholar]
- 60.Estivill-Torrus G, Pearson H, van Heyningen V, et al. Pax6 is required to regulate the cell cycle and the rate of progression from symmetrical to asymmetrical division in mammalian cortical progenitors. Development. 2002;129:455–466. doi: 10.1242/dev.129.2.455. [DOI] [PubMed] [Google Scholar]
- 61.Graham V, Khudyakov J, Ellis P, et al. SOX2 functions to maintain neural progenitor identity. Neuron. 2003;39:749–765. doi: 10.1016/s0896-6273(03)00497-5. [DOI] [PubMed] [Google Scholar]
- 62.Shi Y, Chichung Lie D, Taupin P, et al. Expression and function of orphan nuclear receptor TLX in adult neural stem cells. Nature. 2004;427:78–83. doi: 10.1038/nature02211. [DOI] [PubMed] [Google Scholar]
- 63.Zhang CL, Zou Y, He W, et al. A role for adult TLX-positive neural stem cells in learning and behaviour. Nature. 2008;451:1004–1007. doi: 10.1038/nature06562. [DOI] [PubMed] [Google Scholar]
- 64.Yun K, Mantani A, Garel S, et al. Id4 regulates neural progenitor proliferation and differentiation in vivo. Development. 2004;131:5441–5448. doi: 10.1242/dev.01430. [DOI] [PubMed] [Google Scholar]
- 65.Hurlbut GD, Kankel MW, Lake RJ, et al. Crossing paths with Notch in the hyper-network. Curr Opin Cell Biol. 2007;19:166–175. doi: 10.1016/j.ceb.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 66.Shimizu T, Kagawa T, Inoue T, et al. Stabilized beta-catenin functions through TCF/LEF proteins and the Notch/RBP-Jkappa complex to promote proliferation and suppress differentiation of neural precursor cells. Mol Cell Biol. 2008;28:7427–7441. doi: 10.1128/MCB.01962-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Duncan AW, Rattis FM, DiMascio LN, et al. Integration of Notch and Wnt signaling in hematopoietic stem cell maintenance. Nat Immunol. 2005;6:314–322. doi: 10.1038/ni1164. [DOI] [PubMed] [Google Scholar]
- 68.Ingram WJ, McCue KI, Tran TH, et al. Sonic Hedgehog regulates Hes1 through a novel mechanism that is independent of canonical Notch pathway signalling. Oncogene. 2008;27:1489–1500. doi: 10.1038/sj.onc.1210767. [DOI] [PubMed] [Google Scholar]
- 69.Kluppel M, Wrana JL. Turning it up a Notch: cross-talk between TGF beta and Notch signaling. Bioessays. 2005;27:115–118. doi: 10.1002/bies.20187. [DOI] [PubMed] [Google Scholar]
- 70.Polesello C, Tapon N. Salvador-warts-hippo signaling promotes Drosophila posterior follicle cell maturation downstream of notch. Curr Biol. 2007;17:1864–1870. doi: 10.1016/j.cub.2007.09.049. [DOI] [PubMed] [Google Scholar]
- 71.Yu J, Poulton J, Huang YC, et al. The hippo pathway promotes Notch signaling in regulation of cell differentiation, proliferation, and oocyte polarity. PLoS ONE. 2008;3:e1761. doi: 10.1371/journal.pone.0001761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Reddy BV, Rauskolb C, Irvine KD. Influence of fat-hippo and notch signaling on the proliferation and differentiation of Drosophila optic neuroepithelia. Development. 2010;137:2397–2408. doi: 10.1242/dev.050013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Thomas PD, Campbell MJ, Kejariwal A, et al. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003;13:2129–2141. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.