

Abstract
In this paper we describe an approach that combines stable isotope labeling of amino acids in cells culture, high mass accuracy liquid chromatography tandem mass spectrometry and a novel data analysis approach to accurately determine relative peptide post-translational modification levels. This paper describes the application of this approach to the discovery of novel histone modification crosstalk networks in Saccharomyces cerevisiae. Yeast histone mutants were generated to mimic the presence/absence of 44 well-known modifications on core histones H2A, H2B, H3, and H4. In each mutant strain the relative change in H3 K79 methylation and H3 K56 acetylation were determined using stable isotope labeling of amino acids in cells culture. This approach showed relative changes in H3 K79 methylation and H3 K56 acetylation that are consistent with known histone crosstalk networks. More importantly, this study revealed additional histone modification sites that affect H3 K79 methylation and H3 K56 acetylation.
Histone post-translational modifications play a crucial role in stabilizing chromosomal structure and regulating gene transcription (1–3). There is mounting evidence that histone modifications communicate via crosstalk (4–11). Crosstalk can occur between modifications that are present on the same histone molecule (cis) or between modifications on different histones (trans). For example, H2B K123 monoubiquitination regulates H3 K4 and K79 methylation via a direct trans crosstalk interaction that involves the ubiquitin-conjugating enzyme, Rad6, and the methyltransferases, Set1 and Dot1 (4, 12–24). The methylation of histone H3 can also be regulated in trans by elements on the NH2-terminal tail of histone H4 and histone H2A (23, 25, 26). Another example of histone crosstalk is the influence of histone H3 S10 phosphorylation on the acetylation of histone H4 K16, via a trans interaction, and H3 K14, via a cis interaction (27, 28). Histone crosstalk is not strictly limited to post-translational modifications. For example, the isomerization of histone H3 P30, catalyzed by the proline isomerase Fpr4, inhibits the ability of Set2 to methylate H3 K36 (29).
In this paper we examined histone crosstalk networks for H3 K79 methylation and H3 K56 acetylation, which are among the most intensively studied histone modifications (14, 30–36). Van Leeuwen and colleagues first reported that in the budding yeast Saccharomyces cerevisiae lysine 79 of histone H3, located at the surface of the nucleosome, is methylated by the protein Dot1p. H3 K79 methylation is an abundant modification that is found in euchromatic regions of the genome and plays an important role in the discrimination between euchromatin and heterochromatin. The abundance of the mono-, di- and tri-methylation states was quantitated by liquid chromatography (LC)1-MS using the doubly charged peptides with different methylation levels whose identity was verified by tandem MS (MS/MS) (35). Using LC-MS, the Henikoff laboratory also quantified the relative abundance of doubly charged ions of unmethylated, monomethylated, and dimethylated forms of H3 K79 peptide in a Drosophila cell line system (33). Later, to prove the effect of H2B ubiquitylation in regulating the ability of Dot1 to perform successive rounds of methylation on H3 K79, the Grunstein laboratory measured the methylation profile of H3 K79 in the presence or absence of H2B ubiquitylation in yeast by chromatin immunoprecipitation and mass spectrometry (14).
The acetylation of H3 K56 by Rtt109 in S. cerevisiae occurs in S phase of the cell cycle and, therefore, serves as a mark of newly synthesized histone (32, 34, 37–41). Acetylated H3 K56 is very abundant in the yeast being found on ∼28% of the total H3 (36). H3 K56 is located near the DNA entry and exit point of the nucleosome and, as such, can influence the wrapping of DNA on the nucleosome (42, 43). The level of H3 K56 acetylation has been measured by various labs in biological systems from yeast to human (34, 36, 44–48). ChIP has been widely used to determine the abundance of H3 K56 acetylation to study its function in transcriptional regulation (34, 45). In addition, mass spectrometry has also been used to determine the relative abundance of K56 acetylation in yeast by dividing the amount of acetylated peptide by the sum of acetylated and unacetylated peptides detected by MALDI-TOF mass spectrometry (36).
Stable isotope labeling of amino acids in cell culture (SILAC) is a multiplex quantitative proteomic technique that uses metabolic labeling of proteins with isotopically heavy amino acids to determine changes in protein relative abundance (49, 50). By comparing abundances for the light and heavy labeled proteolytic peptides, the relative amount of proteins can be inferred across different experimental conditions. The challenge associated with SILAC quantitation of histone crosstalk is that the majority of data analysis software applications are designed to measure protein abundance differences rather than changes in peptides. Thus it is nontrivial to identify changes in histone PTMs. MSQuant and MaxQuant are two examples of widely used applications that support relative protein quantitation. MSQuant uses precursor ion intensities to perform quantitation (51). MaxQuant uses high-resolution mass spectrometric data to automatically detect and cluster peaks for more reliable quantitation (52). MaxQuant detects features (including peaks, isotope clusters and SILAC-labeled peptide pairs), calculates the peptide abundance ratio, and generates the intermediate files that can be automatically uploaded to Mascot for peptide identification by Mascot Daemon. Using this tool, Cuomo et al. determined methylation and acetylation profiles for peptides derived from histones H3 and H4 isolated from a set of breast cancer cell lines (53).
The purpose of this work is to discover novel histone crosstalk networks. An approach that combines yeast genetics, SILAC and LC-MS/MS has the potential to achieve this goal. In this paper we used a strategy that measures the SILAC ratios for peptides that contain H3 K79 and H3 K56 and their modified isoforms. This approach combines peptide mass accuracy, peptide retention time and the peptide's light:heavy labeling to eliminate noise for more accurate ratio calculation. Forty-four yeast mutants of modification sites on H2A, H2B, H3, and H4 were examined to determine their effect on H3 K79 methylation and H3 K56 acetylation. The data recapitulate reported crosstalk networks between histone H2B K123 and histone H3 K79 methylation. More importantly, the data identify other histone modification sites on histones H2A, H2B, H3, and H4 that affect H3 K79 methylation and H3 K56 acetylation.
EXPERIMENTAL PROCEDURES
Preparation of Histone Mutants
Lysine residues in H3 or H4 that are subject to post-translational modification were mutated to glutamine or arginine. The mutations were constructed by site-directed mutagenesis from plasmid pMP3 that carries both wild type histone H3 and H4 genes (HHT2-HHF2)(54). Plasmids containing histone mutations were shuffled into yeast strain UCC1111 (S. cerevisiae), which is deleted for the endogenous histone H3 and H4 genes. Modified residues in histones H2A and H2B were altered by site directed mutagenesis of plasmid pQQ18. These plasmids were then shuffled into strain JHY205(55).
Cell Preparation
For each wild type and histone mutant pair to be analyzed, the strains were grown separately in duplicate cultures-one containing normal l-lysine and the other containing 13C6,15N2-lysine. Cells were harvested by centrifugation in mid-log phase A600 = 0.6 to 0.8. Equal weights of cells from each culture were mixed and histones were subsequently isolated.
Histone Extraction and In-solution Digestion
After the cells were harvested, histones were purified as described previously(56). Each sample was digested with Endoproteinase Arg-C (Sequencing grade from Clostridium histolyticum, Roche Applied Science) at a 1:50 enzyme : substrate ratio in 100 mm Tris-HCl (pH 7.6). The digest was incubated at 37 °C for 8 h. The resulting peptides were desalted with a C18 Zip-tip and eluted with 0.1% trifluoroacetic acid (TFA)/50% acetonitrile (ACN).
Protein Identification
Eluting peptides were separated by reversed-phase HPLC (Dionex Ultimate 3000 capillary/nano HPLC system, Dionex, Sunnyvale, CA) and mass analyzed with a Thermo Fisher LTQ Orbitrap XL (Thermo Finnigan, San Jose, CA). Briefly, the precursor ion was first detected in the Orbitrap, and the CID product ions were generated and measured in the linear ion trap. Histones were separated on a 0.2 mm x 150 mm C18 column (3 μm, 200Å, Michrom Bioresources Inc., Auburn, CA) at a flow rate of 2 μl/min with mobile phase A containing H2O with 0.1% Formic acid (FA) and mobile phase B containing ACN with 0.1% FA. Using a 120min gradient beginning with 2% mobile phase B, the phase B linearly increased to 5% in 10min, from 5% to 15% in 20min, from 15% to 30% in 45 min, from 30% to 50% in 15 min, and from 50% to 90% in 5 min. After washing at 90% B for 1 min, the column was equilibrated at 2% B for 24 min. A blank was run between each sample injection and a bovine histone standard was run every 10 runs for quality control.
Proteins were identified and SILAC protein ratios were determined by the MassMatrix database search engine (MassMatrix 2.4.2, released on 2/22/2012). Database search parameters were set as follows: enzyme = Arg-C; maximum missed cleavages = 3, peptide mass tolerance = ±20.00 ppm, fragment mass tolerance = ±0.80 Da (CID), maximum length of peptides = 40 and minimum length of peptides = 6. Variable modifications included ubiquitination of K, acetylation of N-term and K, methylation of K (mono, di and tri) and R (mono, and di), oxidation of M and phosphorylation of STY. Peptide and protein score thresholds were set as follows: peptide score ≥ 10(CID), peptide pp value ≥ 5.0, peptide pp2 value ≥ 5.0, peptide pptag ≥ 1.3 and protein score ≥ 5.0. A maximum of two modifications per peptide were allowed for each peptide spectral match. The maximum number of matches reported per spectrum was one and the maximum number of modification combinations per peptide was three. The false discovery rate was evaluated as described by Elias (57).
MS/MS spectra were searched against the yeast histone database available from the National Human Genome Research Institute (NHGRI) histone sequence database (http://genome.nhgri.nih.gov/histones/). The search database contained 10 yeast histone sequences plus the mutant histone and a concatenated reversed decoy database. All peptide assignments were subsequently manually validated. SILAC peptide ratios were determined as described below for the H3 K79 peptide, EIAQDF79K(Me0–3)TDLR, and the H3 K56 peptide, FQ56K(Ace0–1)STELLIR (MS/MS and search results provided as supplemental Fig. S1).
Data Analysis
LC-MS/MS data files were converted to the mzXML format and then parsed by an in-house developed python script (provided in Supplemental Material). The mzXML filename, the zero-charge monoisotopic mass of the peptide, the mass of the label, the mass accuracy tolerance and signal rejection threshold were provided as arguments to python script, which was used to cluster peptide signals and calculate the global normalization factor for each sample and SILAC ratio(s) for the target peptide(s). To compensate for the systematic mixing error of different mutants, it is crucial to calculate the normalization factor for the comparison of peptide levels between mutants.
Determination of SILAC Normalization Factor
Normalization factors for each sample were determined as follows. First the mzXML data file was parsed by an in-house developed python script that identifies all co-eluting pairs of signals (i.e. “matched pairs”) that are separated by the m/z ratio for a given SILAC label. A single charge state or range of charge states (z = 1 - N) can be used to identify all matched pairs. For these data sets we only determined the normalization factor using the +3 charge state of our 8 Da SILAC label. For this SILAC label, the use of the +3 charge state produced fewer random matches. The resulting “matched pairs” separated in m/z by the triply charged SILAC label were then used to estimate the normalization factors by the following: 1) Robust linear regression (RLR) slope of light versus heavy abundance, 2) Linear regression (LR) slope, 3) Median SILAC ratio and 4) Abundance weighted median ratio. To further eliminate noise from random “matched pairs”, the normalization factors were also calculated using only the ratios with a ±log2 fold change. Median ratios, linear regression slopes, intercepts and standard deviations were calculated as described previously (58). The abundance weighted median is a heuristic method to add additional weight to ratios generated from highly abundant peptides. The Kendall-Theil method was used for robust linear regression analysis. This robust regression does not require normality of residuals and is not strongly affected by outliers (59, 60). This method first calculates all possible slopes from every pairwise combination of (x,y) data points (58). The median slope from this set is taken as the estimate for the regression slope. For large data sets, a more efficient version of the regression, called Theil's method, is used to generate a slope from fewer pairwise combinations of data points (60). Normalization factors generated by all four approaches with and without the ±log2 filter are provided in supplemental Data S1. A boxplot summarizing the effect of these normalization strategies on all data sets is provided in supplemental Fig. S2.
Quantitation of Silac Peptide Ratio
SILAC ratios for target peptides was determined from light versus heavy abundances of a given target peptide. The mzXML file was parsed using an in-house developed python script. This script iterated through every MS1 scan to find signals that correspond to the target peptide's light and heavy m/z signals for isotopes (0–4) and charge states (1 - N, where n = number of basic side chains + 1). Sets of matched signals were clustered based on similarity in retention time. The SILAC H/L ratios for a cluster signal was calculated by linear regression and robust linear regression (see normalization for description of these approaches). For these data we used a retention time window of 0.5 min to cluster peptide signals. A plot of all the light and heavy abundances pairs for each signal cluster was output along with the ratios calculated by LR and RLR. The algorithm also takes mass accuracy and signal rejection threshold as arguments. These two arguments should be set based on each instrument's mass precision and background signal abundance. Higher mass accuracy and an appropriate signal rejection threshold are important in order to decrease random signal matches. For these data sets a mass accuracy at 0.01 m/z and a signal rejection threshold at 100,000 (arb units) was used. All peptide SILAC ratios were then normalized using each experiments respective normalization factor. Because we are reporting relative ratios, normalization across samples and MS runs was not performed.
Statistical Analysis of SILAC Ratios
The mean SILAC ratio and variance of three replicates for each mutant strain were calculated using a sample-based statistics approach. The SILAC ratio of the modified peptide was first normalized to the ratio of the unmodified peptide. The ratio for a given modification state in all mutants were compared with the wild-type using a two tailed unpaired student's t test assuming unequal variance. The overall error for a set of measurements was determined by combining the sample based statistics for each measurement assuming non-overlapping sub samples. The normalized ratios and their calculated sample-based statistics are provided in supplemental Data S1 (H3 K79Me0–3) and supplemental Data S1 (H3 K56Ace0–1). A worked example of the sample-based statistics calculation is also provided in supplemental Data S1. Final ratios are reported in all tables and figures with error bars equivalent to twofold of the sample-based standard deviation.
RESULTS
In this paper we applied a novel peptide-level SILAC quantitative approach for the measurement of post-translational modifications for the study of histone crosstalk. Our initial focus was to comprehensively identify all of the sites of histone modification that influence the abundance and distribution of the methylated forms of histone H3 lysine 79 and the acetylated form of H3 lysine 56. Therefore, a collection of yeast strains was constructed that contain mutations in most of the known sites of modification on the four core histones. In general, modified lysine residues were changed to arginine and glutamine to mimic the constitutively deacetylated and acetylated states, respectively, and phosphorylated residues were changed to glutamic acid or alanine to mimic the constitutively phosphorylated and dephosphorylated states, respectively. Cultures containing wild type or mutant histone alleles were grown to mid-log phase in pairs, with one culture containing normal synthetic media and the other containing synthetic media in which the lysine had been replaced with 13C6,15N2-lysine. Equal amounts of wild type and histone mutant cells were combined and histones were purified for mass spectrometry analysis. Following proteolytic cleavage and LC-MS/MS analysis, comparison of peptide pairs that do not contain post-translational modifications allowed for a determination of the effect of the histone mutations on the total abundance of each core histone. In general, there was little or no effect on histone levels (supplemental Fig. S3).
SILAC allows for the quantitative analysis of changes in post-translational modifications by monitoring the abundance of the light versus heavy form of a target peptide. We used our informatics approach to extract all the heavy and light abundances for the target peptides in order to calculate their SILAC ratios. One way to eliminate random matches and improve precision is to use only the peptide signals that have both a light and heavy form, referred to as “matched pairs.” Normalization factors can be estimated from the ratio of matched pairs for all peptide signals in a data set. In a well-normalized data set we can assume that the total abundance for all target isoforms is conserved. Multiple strategies (described under “Experimental Procedures”) were used to normalize the H3 K79 (isoforms = 4) and H3 K56 (isoforms = 2) data obtained from all mutants. A boxplot summarizing the effect of these normalization strategies is provided as supplemental Fig. S2. We conclude that the ±log2 filtered robust linear regression slope, abundance-weighted-median and filtered abundance-weighted-median best estimate the global normalization factor of these data sets. All data are reported using the normalization factor estimated by ±log2 filtered robust linear regression slope.
Fig. 1 shows a general example of how SILAC peptide ratios were determined. In this example we compared the light and heavy SILAC labeling of the H3 K79 peptide, EIAQDF79K(Me0–3)TDLR, and the H3 K56 peptide, FQ56K(Ace0–1)STELLIR from a light wild-type:heavy wild-type mixture. These histone modifications participate in several important crosstalk pathways. These peptides also represent two cases where modified and unmodified peptides have different elution characteristics. The unmodified and methylated forms of H3 K79 peptide typically coelute under reversed-phase separation conditions whereas the acetylated form of H3 K56 peptide elutes later than the unmodified H3 K56 peptide (Fig. 1) (61). As seen in Fig. 1, the heavy/light ratios determined by either are consistent for both the H3 K56 and H3 K79 isoforms regardless of their elution pattern. The peptide ratios were calculated by robust linear regression (RLR) and linear regression (LR). In supplemental Fig. S4, the residual plots for the H3 K79Me0 show that the ratios deviation from the slope is biased by abundance. The absolute deviation for ratios at high abundance is large, however, they have relatively less relative deviation compared with ratios at low abundance. The robust regression is less sensitive to larger deviations, which is why it is useful for noisy data. These larger relative deviations at low signal abundance are caused by the fact that the variability in signal has greater impact on their ratio variability. When using the robust linear regression it is possible that an “outlier” at high abundance may be falsely rejected. In some cases the ratio may be better estimated using the median or abundance-weighted median. For this reason, the algorithm outputs all calculated ratios and a scatter plot of the light versus heavy abundance to facilitate validation of the calculated ratios.
Fig. 1.

General example of peptide-level SILAC ratio determination for the H3 K79 peptide EIAQDF79K(Me0–3)TDLR and H3 K56 peptide FQ56K(Ace0–1)STELLIR from a light wild-type:heavy wild-type mixture. A, The LC-MS/MS base peak plot. Unmodified and methylated H3 K79 peptides eluted at about the same retention time, whereas the unmodified and acetylated H3 K56 peptides were separated by 12 min. B, The average precursor (MS1) mass spectra for H3 K79 and H3 K56 isoforms. C, Scatter plots generated by the peptide-level SILAC quantitation. Peptide ratios were determined by robust linear regression and linear regression of the light versus heavy abundance. Each point in the scatter plot is a single light:heavy signal. The robust linear regression line is denoted as the solid line and the linear regression as the dashed line. The red dashed line, y = x is provided as a reference for a unity ratio.
We validated the peptide level SILAC approach by examining H3 K79 methylation and H3 K56 acetylation with mutants that are known crosstalk effectors. For example, it is well established that the ubiquitylation of histone H2B is required for the di-and tri-methylation of H3 lysine 79 (62, 63). Mutating H2B K123 to either arginine or glutamine blocks the access of Rad6-Bre1, which is responsible for ubiquitylation at lysine 123. Therefore, we performed SILAC analysis with a light WT: heavy mutant H2B K123R mixture to validate the approach. As seen in Fig. 2, there is a complete loss of peptides containing the di- and tri-methylated form of H3 lysine 79 from the H2B K123R cells and a roughly 2-fold decrease in the level of H3 K79 mono-methylation, which is in agreement with previous results (14). In addition, as shown in Fig. 2 there is a negligible amount of di- or tri-methylation of H3 lysine 79 in the H2B K123Q cells. Both mutations on H2B K123 show significant ablation of H3 K79 trimethylation. These data show that the trimethylated versions have practically disappeared in the heavy samples, but the unmodified, methylated and dimethylated are all the same or lower ratio. Perfect mass balance would assume that there are only four possible isoforms for this peptide. However this may not be the case. The deviation from a ratio sum equal to four is more likely because of another modification on this peptide that is yet to be discovered. When examining the data from this perspective, the approach can aid in discovery of other interesting peptide isoforms. Another important consideration is the reproducibility of the SILAC method. Supplemental Fig. S5 shows three replicates for the light wild type: heavy mutant H4 K8R. It is clear that the results were highly repeatable. MS/MS spectra are also provided in supplemental Fig. S1 for the H3 K79 and H3 K56 peptides used in this analysis.
Fig. 2.
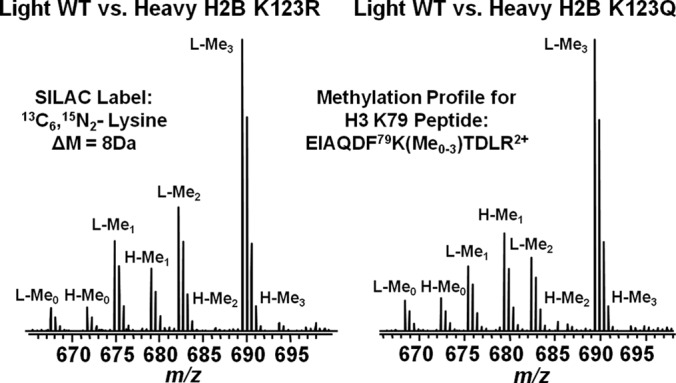
Mass spectrometry methylation profiles for the H3 K79 peptide fragment EIAQDF79K(Me0–3)TDLR2+ from SILAC light wild type:heavy mutant H2B K123R (left) and light wild type:heavy mutant H2B K123Q (right). Light and heavy peaks are labeled with L and H respectively. Peaks representing the unmethylated, mono-methylated, di-methylated or tri-methylated form of H3 K79 peptides are annotated by Me0, Me1, Me2, or Me3.
Given that the peptide level SILAC analysis recapitulates known crosstalk pathways, we used this approach to examine H3 K79 methylation and H3 K56 acetylation for our collection of 44 yeast histone modification mimetic mutants. To compare the peptide level between mutants, raw H/L ratios of each peptide were normalized using the filtered robust linear regression determined normalization factor. The normalized SILAC peptide ratios for H3 K79 are plotted in Fig. 3A and listed in Table I. Similarly, the normalized SILAC peptide ratios for H3 K56 are plotted in Fig. 3B and listed in Table II. Error bars correspond to twofold of the sample-based standard deviation based on the size and mean of each sample as described in the Experimental Procedures (an example calculation is provided in the supplemental data). These data are presented utilizing a novel plot that shows both the ratio (Fig. 4A) and significance (Fig. 4B) for H3 K79 methylation and those (Fig. 4C and 4D) for H3 K56 acetylation. This novel “Saturn plot” was developed to simplify interpretation of cross-talk data by highlighting statistically significant effects of mutations on H3 K79 methylation and H3 K56 acetylation.
Fig. 3.

A, SILAC ratios for the H3 K79 peptide EIAQDF79K(Me0–3)TDLR across all 44 yeast mutants. B, SILAC ratios for the H3 K56 peptide FQ56K(Ace0–1)STELLIR across all 44 yeast mutants. WT1 is from plasmid pMP3 that carries both wild type histone H3 and H4 genes (HHT2-HHF2). WT2 is from plasmid pQQ18 that carries both wild type histone H2A and H2B. To compare the peptide level between mutants, raw H/L ratios of each peptide are normalized using the filtered robust linear regression normalization factor. Error bars correspond to two-fold of the sample based standard deviation (see Experimental Procedures and Supplemental data for description and example).
Table I. SILAC ratios for the H3 K79 peptide EIAQDF79K(Me0–3)TDLR across all 44 yeast mutants. Each ratio is the average of three replicates. A t-test was performed between the wild type and each mutant. Peptide ratios with a p value ≤ 0.05 are reported as significant.
| H3 K79Me0 |
H3 K79Me1 |
H3 K79Me2 |
H3 K79Me3 |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Average | Std. Dev | Average | Std. Dev | p value | Average | Std. Dev | p value | Average | Std. Dev | p value | |
| WT2 | 1.15 | 0.14 | 1.12 | 0.22 | 1.0E+00 | 0.98 | 0.16 | 1.0E+00 | 0.90 | 0.03 | 1.0E+00 |
| H2A K4R | 1.17 | 0.22 | 1.42 | 0.05 | 4.3E-01 | 1.22 | 0.09 | 6.3E-01 | 0.80 | 0.09 | 2.8E-01 |
| H2A K4Q | 0.60 | 0.07 | 0.88 | 0.09 | 5.2E-03 | 1.05 | 0.13 | 1.0E-02 | 1.16 | 0.06 | 1.0E-02 |
| H2A K7R | 0.64 | 0.02 | 0.91 | 0.09 | 1.3E-02 | 1.13 | 0.05 | 9.8E-06 | 1.14 | 0.02 | 1.4E-03 |
| H2A K7Q | 0.89 | 0.07 | 1.44 | 0.01 | 3.7E-03 | 1.33 | 0.05 | 2.2E-02 | 0.74 | 0.01 | 6.9E-01 |
| H2A K21R | 0.93 | 0.08 | 0.97 | 0.03 | 2.2E-01 | 1.07 | 0.06 | 2.0E-02 | 0.89 | 0.05 | 8.4E-02 |
| H2B K6R | 0.83 | 0.09 | 1.05 | 0.11 | 4.9E-02 | 1.03 | 0.11 | 7.8E-02 | 1.16 | 0.13 | 2.8E-02 |
| H2B K6Q | 1.00 | 0.25 | 0.97 | 0.05 | 8.5E-01 | 1.03 | 0.03 | 3.5E-01 | 1.09 | 0.09 | 2.7E-01 |
| H2B S10E | 1.42 | 0.44 | 1.28 | 0.39 | 9.9E-01 | 1.25 | 0.33 | 1.0E-01 | 1.25 | 0.29 | 2.3E-01 |
| H2B S10A | 1.05 | 0.09 | 1.00 | 0.06 | 9.5E-01 | 0.94 | 0.03 | 5.5E-01 | 1.09 | 0.02 | 4.8E-02 |
| H2B K11R | 1.03 | 0.10 | 1.09 | 0.13 | 2.4E-01 | 1.02 | 0.04 | 8.9E-02 | 0.79 | 0.06 | 9.0E-01 |
| H2B K11Q | 1.13 | 0.11 | 1.19 | 0.16 | 7.9E-01 | 0.92 | 0.15 | 5.9E-01 | 1.06 | 0.13 | 4.6E-01 |
| H2B K16R | 0.87 | 0.12 | 1.10 | 0.02 | 1.2E-01 | 1.18 | 0.08 | 1.3E-01 | 1.04 | 0.07 | 7.5E-02 |
| H2B K16Q | 0.42 | 0.06 | 0.59 | 0.11 | 2.7E-02 | 0.67 | 0.17 | 2.9E-02 | 0.52 | 0.13 | 3.4E-02 |
| H2B K17R | 1.02 | 0.11 | 1.64 | 0.22 | 4.5E-03 | 1.51 | 0.09 | 2.9E-03 | 1.08 | 0.07 | 3.7E-02 |
| H2B K17Q | 0.82 | 0.04 | 0.95 | 0.06 | 8.9E-02 | 1.07 | 0.05 | 2.1E-02 | 0.93 | 0.09 | 1.5E-02 |
| H2B K21R | 0.57 | 0.21 | 1.13 | 0.05 | 1.9E-01 | 1.29 | 0.03 | 1.7E-01 | 0.96 | 0.06 | 1.8E-01 |
| H2B K21Q | 0.65 | 0.08 | 1.09 | 0.09 | 1.6E-03 | 1.25 | 0.10 | 9.5E-04 | 0.96 | 0.05 | 2.2E-03 |
| H2B K22R | 0.71 | 0.02 | 1.31 | 0.07 | 4.6E-04 | 1.33 | 0.03 | 7.2E-06 | 0.82 | 0.01 | 1.6E-02 |
| H2B K22Q | 0.96 | 0.48 | 0.77 | 0.15 | 6.3E-01 | 0.70 | 0.14 | 5.6E-01 | 0.55 | 0.13 | 8.3E-01 |
| H2B K123R | 1.39 | 0.12 | 0.69 | 0.05 | 1.5E-02 | 0.02 | 0.00 | 7.9E-04 | 0.01 | 0.00 | 6.5E-03 |
| H2B K123Q | 1.87 | 0.43 | 2.98 | 1.07 | 7.7E-02 | 0.35 | 0.10 | 1.6E-04 | 0.01 | 0.00 | 6.4E-03 |
| WT1 | 0.89 | 0.09 | 1.00 | 0.06 | 1.0E+00 | 0.98 | 0.12 | 1.0E+00 | 0.91 | 0.13 | 1.0E+00 |
| H3 K4A | 0.83 | 0.07 | 1.22 | 0.26 | 1.4E-01 | 1.42 | 0.36 | 1.2E-01 | 1.50 | 0.28 | 1.4E-02 |
| H3 K9R | 0.85 | 0.07 | 0.88 | 0.12 | 3.3E-01 | 0.79 | 0.12 | 2.0E-01 | 0.92 | 0.20 | 5.8E-01 |
| H3 T11A | 1.03 | 0.03 | 1.02 | 0.05 | 2.1E-01 | 1.00 | 0.08 | 2.9E-01 | 1.20 | 0.09 | 1.4E-01 |
| H3 K14R | 1.06 | 0.18 | 1.03 | 0.21 | 1.4E-01 | 0.97 | 0.19 | 1.8E-01 | 1.06 | 0.04 | 9.2E-01 |
| H3 K18R | 1.10 | 0.20 | 1.08 | 0.18 | 2.3E-01 | 0.96 | 0.17 | 1.3E-01 | 1.02 | 0.11 | 8.0E-01 |
| H3 K23R | 1.00 | 0.09 | 1.06 | 0.08 | 5.9E-01 | 0.98 | 0.05 | 3.4E-01 | 1.06 | 0.05 | 5.1E-01 |
| H3 K36R | 0.85 | 0.10 | 0.88 | 0.02 | 4.3E-01 | 1.09 | 0.09 | 1.8E-01 | 1.18 | 0.03 | 8.9E-02 |
| H3 K36A | 0.90 | 0.08 | 0.77 | 0.02 | 5.6E-02 | 0.81 | 0.03 | 1.4E-01 | 1.09 | 0.02 | 8.1E-02 |
| H3 K56R | 0.84 | 0.04 | 0.99 | 0.11 | 8.7E-01 | 0.96 | 0.02 | 5.8E-01 | 1.15 | 0.06 | 2.8E-02 |
| H4 K5R | 0.91 | 0.12 | 1.28 | 0.25 | 1.2E-01 | 1.09 | 0.24 | 9.2E-01 | 0.92 | 0.17 | 5.9E-01 |
| H4 K5Q | 0.91 | 0.14 | 0.95 | 0.08 | 5.0E-01 | 0.90 | 0.09 | 4.5E-01 | 0.90 | 0.11 | 8.8E-01 |
| H4 K8R | 0.95 | 0.08 | 1.21 | 0.28 | 4.0E-01 | 1.17 | 0.32 | 4.6E-01 | 0.98 | 0.14 | 9.4E-01 |
| H4 K8Q | 1.17 | 0.20 | 1.43 | 0.09 | 6.1E-01 | 1.25 | 0.11 | 8.6E-01 | 0.87 | 0.13 | 4.3E-01 |
| H4 K12R | 1.33 | 0.21 | 1.37 | 0.35 | 2.3E-01 | 1.29 | 0.23 | 5.1E-01 | 0.88 | 0.23 | 3.6E-01 |
| H4 K12Q | 0.94 | 0.14 | 1.36 | 0.15 | 3.7E-02 | 1.32 | 0.14 | 5.1E-02 | 0.55 | 0.07 | 1.2E-02 |
| H4 K16R | 1.08 | 0.11 | 1.37 | 0.12 | 5.1E-01 | 1.30 | 0.39 | 4.2E-01 | 0.94 | 0.09 | 3.9E-01 |
| H4 K16Q | 1.23 | 0.11 | 2.46 | 0.17 | 7.5E-03 | 1.59 | 0.07 | 1.2E-01 | 0.44 | 0.06 | 2.6E-03 |
| H4 K31R | 0.62 | 0.07 | 1.03 | 0.10 | 6.5E-02 | 1.12 | 0.12 | 5.9E-02 | 0.94 | 0.08 | 1.4E-02 |
| H4 K31Q | 0.77 | 0.14 | 0.74 | 0.10 | 1.3E-01 | 0.83 | 0.11 | 9.9E-01 | 1.01 | 0.15 | 3.5E-02 |
| H4 K79R | 0.95 | 0.08 | 0.98 | 0.07 | 7.1E-01 | 0.99 | 0.07 | 9.5E-01 | 1.10 | 0.07 | 1.8E-01 |
| H4 K79A | 0.81 | 0.02 | 0.94 | 0.06 | 8.7E-01 | 1.10 | 0.02 | 6.4E-02 | 1.05 | 0.02 | 3.7E-02 |
| H4 K91Q | 0.46 | 0.17 | 0.53 | 0.16 | 4.4E-01 | 0.61 | 0.19 | 3.9E-01 | 0.62 | 0.16 | 9.8E-02 |
Table II. SILAC ratios for the H3 K56 peptide FQ56K(Ace0–1)STELLIR across all 44 yeast mutants. Each ratio is the average of three replicates. A t-test was performed between the wild type and each mutant. Peptide ratios with a p value ≤ 0.05 are reported as significant.
| H3 K56Ace0 |
H3 K56Ace1 |
||||
|---|---|---|---|---|---|
| Average | Std. Dev | Average | Std. Dev | p value | |
| WT2 | 0.95 | 0.06 | 0.91 | 0.16 | 1.0E+00 |
| H2A K4R | 0.94 | 0.05 | 1.57 | 0.25 | 8.7E-03 |
| H2A K4Q | 1.05 | 0.07 | 0.89 | 0.05 | 3.7E-01 |
| H2A K7R | 1.05 | 0.03 | 0.95 | 0.14 | 6.8E-01 |
| H2A K7Q | 0.90 | 0.02 | 1.45 | 0.07 | 3.5E-03 |
| H2A K21R | 0.90 | 0.04 | 1.40 | 0.19 | 3.1E-02 |
| H2B K6R | 1.12 | 0.08 | 0.70 | 0.10 | 6.0E-02 |
| H2B K6Q | 1.06 | 0.05 | 0.91 | 0.14 | 4.7E-01 |
| H2B S10E | 1.30 | 0.46 | 1.45 | 0.45 | 1.3E-01 |
| H2B S10A | 1.05 | 0.03 | 0.97 | 0.15 | 9.3E-01 |
| H2B K11R | 0.92 | 0.04 | 1.32 | 0.10 | 1.6E-02 |
| H2B K11Q | 1.02 | 0.07 | 1.29 | 0.31 | 1.7E-01 |
| H2B K16R | 1.11 | 0.07 | 0.87 | 0.10 | 1.7E-01 |
| H2B K16Q | 0.60 | 0.15 | 0.54 | 0.13 | 7.7E-01 |
| H2B K17R | 1.19 | 0.10 | 1.68 | 0.08 | 1.4E-02 |
| H2B K17Q | 1.08 | 0.04 | 0.82 | 0.02 | 1.4E-01 |
| H2B K21R | 1.12 | 0.04 | 1.13 | 0.07 | 5.2E-01 |
| H2B K21Q | 1.03 | 0.05 | 0.87 | 0.08 | 3.8E-01 |
| H2B K22R | 1.01 | 0.03 | 0.90 | 0.05 | 6.8E-01 |
| H2B K22Q | 0.60 | 0.12 | 0.81 | 0.15 | 2.4E-01 |
| H2B K123R | 0.29 | 0.02 | 0.46 | 0.02 | 3.7E-03 |
| H2B K123Q | 0.77 | 0.20 | 1.48 | 0.45 | 4.8E-04 |
| WT1 | 0.91 | 0.16 | 0.95 | 0.16 | 1.0E+00 |
| H3 K4A | 1.10 | 0.10 | 0.52 | 0.06 | 1.9E-03 |
| H3 K9R | 0.89 | 0.17 | 0.79 | 0.10 | 7.1E-01 |
| H3 T11A | 1.15 | 0.05 | 0.87 | 0.02 | 2.5E-02 |
| H3 K14R | 1.07 | 0.08 | 0.93 | 0.20 | 6.2E-01 |
| H3 K18R | 1.04 | 0.19 | 0.71 | 0.05 | 1.4E-01 |
| H3 K23R | 1.03 | 0.02 | 0.77 | 0.07 | 3.1E-02 |
| H3 K36R | 1.09 | 0.03 | 0.96 | 0.07 | 2.8E-01 |
| H3 K36A | 1.10 | 0.03 | 0.78 | 0.06 | 1.1E-02 |
| H3 K79R | 0.93 | 0.27 | 0.51 | 0.15 | 8.6E-03 |
| H3 K79A | 0.93 | 0.06 | 0.48 | 0.10 | 1.0E-02 |
| H4 K5R | 1.00 | 0.08 | 1.14 | 0.15 | 3.3E-01 |
| H4 K5Q | 0.90 | 0.06 | 1.11 | 0.12 | 5.5E-03 |
| H4 K8R | 1.03 | 0.06 | 0.98 | 0.14 | 8.0E-01 |
| H4 K8Q | 1.04 | 0.06 | 0.82 | 0.05 | 4.0E-02 |
| H4 K12R | 0.95 | 0.05 | 1.70 | 0.12 | 1.7E-02 |
| H4 K12Q | 0.84 | 0.09 | 1.01 | 0.20 | 1.3E-01 |
| H4 K16R | 1.01 | 0.08 | 1.11 | 0.26 | 3.8E-01 |
| H4 K16Q | 0.80 | 0.06 | 1.95 | 0.05 | 2.2E-03 |
| H4 K31R | 1.03 | 0.06 | 0.95 | 0.07 | 4.6E-01 |
| H4 K31Q | 0.92 | 0.14 | 0.84 | 0.13 | 4.2E-01 |
| H4 K79R | 1.15 | 0.03 | 0.80 | 0.11 | 5.2E-02 |
| H4 K79A | 1.15 | 0.04 | 0.79 | 0.05 | 1.2E-02 |
| H4 K91Q | 0.59 | 0.20 | 0.48 | 0.14 | 8.6E-02 |
Fig. 4.

Saturn plots summarizing the relative change in post-translational modification and the significance. A, Saturn plot summary for log2(Ratio) of H3 K79 methylation and B, its significance, reported as -log(p). C, Saturn plot summary for log2(Ratio) of H3 K56 acetylation and D, its significance, reported as -log(p). The Saturn plot reveals mutants that significantly influence H3 K79 methylation or H3 K56 acetylation. The Saturn plot starts pointing east and goes anticlockwise as the arrow shows. *Dimethylation and trimethylation was negligible for mutants H2B K123R and H2B K123Q.
This analysis identified residues on each of the four core histones that affect the pattern of methylation on histone H3 lysine 79. Interestingly, in contrast to the effect seen with the H2B K123R mutant, many of these mutations result in increased levels of H3 lysine 79 methylation. The most striking example of this was observed for several mutations in the NH2-terminal tail of histone H2B. Consecutive lysine residues at positions 21 and 22 both impacted H3 lysine 79 methylation. For H2B lysine 21, the glutamine substitution led to an increase in mono- and di-methylation of H3 lysine 79. In contrast, arginine substitution of H2B lysine 22 had opposing effects where retaining the positive charge promoted mono- and di-methylation. In addition, mutations on the adjacent lysine residues at positions 16 and 17 also impacted H3 lysine 79 methylation. The glutamine substitution of H2B lysine 16 or arginine substitution of H2B lysine 17 led to an increase in mono- and di-methylation of H3 lysine 79.
Crosstalk was also identified for the NH2-terminal tail of histone H2A. Changing lysine residue 4 to glutamine resulted in an increase in the levels of the tri-methylated states of H3 lysine 79. Mutating H2A lysine 7 to glutamine caused an increase in H3 lysine 79 di-methylation where the arginine substitution increased both H3 lysine 79 di- and tri-methylation.
The NH2-terminal tail of histone H3 appeared to play only a minor role in the regulation of H3 lysine 79 methylation. Mutating the sites of acetylation in this domain had no effect on H3 lysine 79 methylation. H3 lysine 4 and lysine 36 were both mutated to alanine to test the importance of positive charge at this position. There is no obvious change of H3 K79 methylation level in mutant strain H3 K36A, however, loss of H3 lysine 4, which shares some mechanisms of regulation with H3 lysine 79, led to enhanced levels of tri-methylated H3 lysine 79 (12, 14, 19, 22).
It has been shown that deletion NH2-terminal tail of histone H4 results in the loss of di- and tri-methylation of histone H3 lysine 79. This effect was localized to a basic patch comprised of residues 17–20. From the data shown in Fig. 3A, other residues in the NH2-terminal tail of histone H4, namely lysines 12 and 16, also influenced lysine 79 methylation. Mutating these lysines to glutamine resulted in an increase in the levels of mono- and di-methylation with a concomitant decrease in tri-methylation. Previous reports have indicated that these residues do not play a role in H3 lysine 79 methylation but these conclusions were based on Western blot analyses that may not have had the sensitivity to detect these changes in H3 lysine 79 methylation (23).
Several residues on each of the four core histones were also shown to have a possible connection with H3 K56 acetylation. Interestingly, mutating H4 K16 to glutamine led to an increased level of H3 K56 acetylation. As it has been reported that substitution of H3 lysine 56 does not influence the level of H4 K16 acetylation (47), this observation suggests the possibility that there is a single-direction interaction between H3 K56 and H4 K16. The level of H3 K56 acetylation was also increased if H4 lysine 12 was mutated to arginine. Mutations of other sites of histone H4 modification did not result in significant changes in the degree of H3 K56 acetylation. Two residues in histone H3 showed an interaction with H3 K56 acetylation. Mutating lysine 79 to arginine or alanine led to a decrease in the level of H3 K56 acetylation. Lysine 4 in the NH2-terminal tail was also linked as a mutation at this site led to a twofold decrease in H3 K56 acetylation.
Crosstalk was also observed with the NH2 terminus of histone H2A. Mutating lysine 4 to arginine led to an enhanced level of H3 K56 acetylation whereas mutating lysine 4 to glutamine had no effect. Interestingly, the opposite is observed for lysine 7 where a mutation to glutamine increased the level of H3 K56 acetylation and a change to arginine does not influence the level of H3 K56 acetylation. In addition, mutating lysine 21 to arginine led to an enhanced level of H3 K56 acetylation.
Several modification sites on H2B showed effects on H3 K56 acetylation. The most striking example was lysine 123, where both arginine and glutamine mutations resulted in a significantly increased level of H3 K56 acetylation. Both arginine mutations of H2B lysine 11 and lysine 17 led to an increased level of H3 K56 whereas glutamine substitutions on these two sites have no significant effect on the level of H3 K56 acetylation.
DISCUSSION
We have demonstrated an approach for the quantitation of peptide changes using SILAC. By combining this method with the site-directed mutagenesis of yeast core histones, we identified multiple mutations that affect the level of H3 K79 methylation and H3 K56 acetylation. This is a critical first step in identifying novel pathways of histone modification crosstalk.
With the identification of potential pathways of histone modification crosstalk, the next important question is to understand the mechanism(s) through which these sites of modification communicate. One possibility is that a site of modification may directly influence the enzyme that is responsible for a second modification. For example, modifications on the NH2-terminal tails of the core histones might regulate the binding of Dot1p, the methyltransferase responsible for the methylation of histone H3 K79, to the nucleosome. These modifications could also influence H3 K79 methylation by regulating the catalytic activity or processivity of Dot1p. The second type of mechanism would involve effects on other enzymes that modulate H3 K79 methylation and H3 K56 acetylation. Principally, this would include interactions, in trans, that would regulate the binding or catalytic activity of histone demethylases (HDMs) and histone deacetylases (HDACs) that are specific for these modifications. For H3 K56 acetylation this would include the HDACs Hst3p and Hst4p whereas the histone demethylases specific for H3 lysine 79 are yet to be identified. The third class of mechanism, applicable to H3 K79 methylation, would be effects of other histone modifications on the ubiquitylation of histone H2B. As H2B ubiquitylation directly impacts H3 K79 methylation, any histone modifications that regulate H2B ubiquitylation would also indirectly effect H3 K79 methylation. Finally, this histone crosstalk could be functioning through global alterations in chromatin, transcription or DNA replication. For example, H3 K79 methylation occurs in regions of euchromatin and altering the balance between euchromatin and heterochromatin would have the effect of changing the levels of H3 K79 methylation. For example, the acetylation of histone H4 K16, identified here as a residue that influences H3 K79 methylation, regulates the binding of SIR proteins and the formation of silent chromatin structure in yeast. Hence, mutations at this site might function by altering the distribution of heterochromatin and euchromatin, which would then indirectly change H3 K79 methylation levels.
Taken together, these results identify several novel networks of histone modification crosstalk that modulate the pattern of methylation of histone H3 K79 and acetylation of H3 K56 either via direct or indirect pathways (summarized in Fig. 5). The modification state of these residues is much more sensitive to the overall state of histone modification and the functional interactions between histone post-translational modifications are more complex than previously appreciated. The approach described here will enable a more comprehensive and quantitative analysis of these complex interactions.
Fig. 5.

A network representation of A, B, and C, H3 K79 methylation crosstalk and D, H3 K56 acetylation crosstalk. Mutated sites are highlighted in red. Mutations promoting the modification are indicated by an arrow whereas the ones with reverse effects are indicated by a solid bar.
Supplementary Material
Acknowledgments
We would like to thank M. Mitchell Smith (University of Virginia) for yeast strains and plasmids.
Footnotes
* The study was funded by grants from the National Institutes of Health (R21 DK082634 to MAF and MRP, R01 CA107106 to MAF and R01 GM62970 to MRP) and support from the Ohio State University.
 This article contains supplemental Figs. S1 to S5 and Data S1 and S2.
This article contains supplemental Figs. S1 to S5 and Data S1 and S2.
1 The abbreviations used are:
- LC
- liquid chromatography
- SILAC
- stable isotope labeling of amino acids in cells culture
- LC-MS/MS
- liquid chromatography tandem mass spectrometry
- PTM
- post-translational modifications
- FA
- formic acid
- ChIP
- chromatin immunoprecipitation
- HDM
- histone demethylase
- HDAC
- histone deacetylase.
REFERENCES
- 1. Kouzarides T. (2007) Chromatin Modifications and Their Function. Cell 128, 693–705 [DOI] [PubMed] [Google Scholar]
- 2. Bhaumik S. R., Smith E., Shilatifard A. (2007) Covalent modifications of histones during development and disease pathogenesis. Nat. Struct. Mol. Biol. 14, 1008–1016 [DOI] [PubMed] [Google Scholar]
- 3. Bannister A. J., Kouzarides T. (2011) Regulation of chromatin by histone modifications. Cell Res. 21, 381–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Henry K. W., Berger S. L. (2002) Trans-tail histone modifications: wedge or bridge? Nat. Struct. Mol. Biol. 9, 565–566 [DOI] [PubMed] [Google Scholar]
- 5. Chandrasekharan M. B., Huang F., Sun Z. W. (2010) Histone H2B ubiquitination and beyond: Regulation of nucleosome stability, chromatin dynamics and the trans-histone H3 methylation. Epigenetics 5, 460–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Latham J. A., Dent S. Y. R. (2007) Cross-regulation of histone modifications. Nat Struct. Mol. Biol. 14, 1017–1024 [DOI] [PubMed] [Google Scholar]
- 7. Nathan D., Ingvarsdottir K., Sterner D. E., Bylebyl G. R., Dokmanovic M., Dorsey J. A., Whelan K. A., Krsmanovic M., Lane W. S., Meluh P. B., Johnson E. S., Berger S. L. (2006) Histone sumoylation is a negative regulator in Saccharomyces cerevisiae and shows dynamic interplay with positive-acting histone modifications. Genes Develop. 20, 966–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Martin D. G., Grimes D. E., Baetz K., Howe L. (2006) Methylation of Histone H3 Mediates the Association of the NuA3 Histone Acetyltransferase with Chromatin. Mol. Cell. Biol. 26, 3018–3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ivanovska I., Khandan T., Ito T., Orr-Weaver T. L. (2005) A histone code in meiosis: the histone kinase, NHK-1, is required for proper chromosomal architecture in Drosophila oocytes. Genes Develop. 19, 2571–2582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fingerman I. M., Li H.-C., Briggs S. D. (2007) A charge-based interaction between histone H4 and Dot1 is required for H3K79 methylation and telomere silencing: identification of a new trans-histone pathway. Genes Develop. 21, 2018–2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fingerman I. M., Du H. N., Briggs S. D. (2008) Controlling histone methylation via trans-histone pathways. Epigenetics 3, 237–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gardner R. G., Nelson Z. W., Gottschling D. E. (2005) Ubp10/Dot4p Regulates the Persistence of Ubiquitinated Histone H2B: Distinct Roles in Telomeric Silencing and General Chromatin. Mol. Cell. Biol. 25, 6123–6139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nakanishi S., Lee J. S., Gardner K. E., Gardner J. M., Takahashi Y. H., Chandrasekharan M. B., Sun Z. W., Osley M. A., Strahl B. D., Jaspersen S. L., Shilatifard A. (2009) Histone H2BK123 monoubiquitination is the critical determinant for H3K4 and H3K79 trimethylation by COMPASS and Dot1. J. Cell Biol. 186, 371–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shahbazian M. D., Zhang K., Grunstein M. (2005) Histone H2B ubiquitylation controls processive methylation but not monomethylation by Dot1 and Set1. Mol. Cell. 19, 271–277 [DOI] [PubMed] [Google Scholar]
- 15. Ng H. H., Xu R. M., Zhang Y., Struhl K. (2002) Ubiquitination of histone H2B by Rad6 is required for efficient Dot1-mediated methylation of histone H3 lysine 79. J. Biol. Chem. 277, 34655–34657 [DOI] [PubMed] [Google Scholar]
- 16. Vitaliano-Prunier A., Menant A., Hobeika M., Géli V., Gwizdek C., Dargemont C. (2008) Ubiquitylation of the COMPASS component Swd2 links H2B ubiquitylation to H3K4 trimethylation. Nat. Cell. Biol. 10, 1365–1371 [DOI] [PubMed] [Google Scholar]
- 17. Nakanishi S., Sanderson B. W., Delventhal K. M., Bradford W. D., Staehling-Hampton K., Shilatifard A. (2008) A comprehensive library of histone mutants identifies nucleosomal residues required for H3K4 methylation. Nat. Struct. Mol. Biol. 15, 881–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee J. S., Shukla A., Schneider J., Swanson S. K., Washburn M. P., Florens L., Bhaumik S. R., Shilatifard A. (2007) Histone crosstalk between H2B monoubiquitination and H3 methylation mediated by COMPASS. Cell 131, 1084–1096 [DOI] [PubMed] [Google Scholar]
- 19. Sun Z. W., Allis C. D. (2002) Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature 418, 104–108 [DOI] [PubMed] [Google Scholar]
- 20. Leung A., Cajigas I., Jia P., Ezhkova E., Brickner J. H., Zhao Z., Geng F., Tansey W. P. Histone H2B ubiquitylation and H3 lysine 4 methylation prevent ectopic silencing of euchromatic loci important for the cellular response to heat. Mol. Biol. Cell 22, 2741–2753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chandrasekharan M. B., Huang F., Sun Z. W. (2011) Decoding the trans-histone crosstalk: Methods to analyze H2B ubiquitination, H3 methylation and their regulatory factors. Methods 54, 304–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Briggs S. D., Xiao T., Sun Z. W., Caldwell J. A., Shabanowitz J., Hunt D. F., Allis C. D., Strahl B. D. (2002) Gene silencing: trans-histone regulatory pathway in chromatin. Nature 418, 498. [DOI] [PubMed] [Google Scholar]
- 23. Fingerman I. M., Li H. C., Briggs S. D. (2007) A charge-based interaction between histone H4 and Dot1 is required for H3K79 methylation and telomere silencing: identification of a new trans-histone pathway. Genes Dev. 21, 2018–2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee J. S., Shukla A., Schneider J., Swanson S. K., Washburn M. P., Florens L., Bhaumik S. R., Shilatifard A. (2007) Histone crosstalk between H2B monoubiquitination and H3 methylation mediated by COMPASS. Cell 131, 1084–1096 [DOI] [PubMed] [Google Scholar]
- 25. Zheng S., Wyrick J. J., Reese J. C. (2010) Novel trans-Tail Regulation of H2B Ubiquitylation and H3K4 Methylation by the N Terminus of Histone H2A. Mol. Cell. Biol. 30, 3635–3645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Altaf M., Saksouk N., Côté J. (2007) Histone modifications in response to DNA damage. Mutat. Res. 618, 81–90 [DOI] [PubMed] [Google Scholar]
- 27. Lo W. S., Trievel R. C., Rojas J. R., Duggan L., Hsu J. Y., Allis C. D., Marmorstein R., Berger S. L. (2000) Phosphorylation of serine 10 in histone H3 Is functionally linked in vitro and in vivo to Gcn5-mediated acetylation at lysine 14. Mol. Cell 5, 917–926 [DOI] [PubMed] [Google Scholar]
- 28. Zippo A., Serafini R., Rocchigiani M., Pennacchini S., Krepelova A., Oliviero S. (2009) Histone crosstalk between H3S10ph and H4K16ac generates a histone code that mediates transcription elongation. Cell 138, 1122–1136 [DOI] [PubMed] [Google Scholar]
- 29. Nelson C. J., Santos-Rosa H., Kouzarides T. (2006) Proline isomerization of histone H3 regulates lysine methylation and gene expression. Cell 126, 905–916 [DOI] [PubMed] [Google Scholar]
- 30. Celic I., Masumoto H., Griffith W. P., Meluh P., Cotter R. J., Boeke J. D., Verreault A. (2006) The sirtuins hst3 and Hst4p preserve genome integrity by controlling histone h3 lysine 56 deacetylation. Curr. Biol. 16, 1280–1289 [DOI] [PubMed] [Google Scholar]
- 31. Maas N. L., Miller K. M., DeFazio L. G., Toczyski D. P. (2006) Cell cycle and checkpoint regulation of histone H3 K56 acetylation by Hst3 and Hst4. Mol Cell 23, 109–119 [DOI] [PubMed] [Google Scholar]
- 32. Masumoto H., Hawke D., Kobayashi R., Verreault A. (2005) A role for cell-cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature 436, 294–298 [DOI] [PubMed] [Google Scholar]
- 33. McKittrick E., Gafken P. R., Ahmad K., Henikoff S. (2004) Histone H3.3 is enriched in covalent modifications associated with active chromatin. Proc. Natl. Acad. Sci. U.S.A. 101, 1525–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schneider J., Bajwa P., Johnson F. C., Bhaumik S. R., Shilatifard A. (2006) Rtt109 is required for proper H3K56 acetylation: A chromatin mark associated with the elongating RNA polymerase II. J. Biol. Chem. 281, 37270–37274 [DOI] [PubMed] [Google Scholar]
- 35. van Leeuwen F., Gafken P. R., Gottschling D. E. (2002) Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell 109, 745–756 [DOI] [PubMed] [Google Scholar]
- 36. Xu F., Zhang K., Grunstein M. (2005) Acetylation in histone H3 globular domain regulates gene expression in yeast. Cell 121, 375–385 [DOI] [PubMed] [Google Scholar]
- 37. Tsubota T., Berndsen C. E., Erkmann J. A., Smith C. L., Yang L., Freitas M. A., Denu J. M., Kaufman P. D. (2007) Histone H3-K56 acetylation is catalyzed by histone chaperone-dependent complexes. Mol. Cell 25, 703–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Han J., Zhou H., Li Z., Xu R. M., Zhang Z. (2007) The Rtt109-Vps75 histone acetyltransferase complex acetylates non-nucleosomal histone H3. J. Biol. Chem. 282, 14158–14164 [DOI] [PubMed] [Google Scholar]
- 39. Han J., Zhou H., Li Z., Xu R. M., Zhang Z. (2007) Acetylation of lysine 56 of histone H3 catalyzed by RTT109 and regulated by ASF1 is required for replisome integrity. J. Biol. Chem. 282, 28587–28596 [DOI] [PubMed] [Google Scholar]
- 40. Driscoll R., Hudson A., Jackson S. P. (2007) Yeast Rtt109 promotes genome stability by acetylating histone H3 on lysine 56. Science 315, 649–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Han J., Zhou H., Horazdovsky B., Zhang K., Xu R. M., Zhang Z. (2007) Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science 315, 653–655 [DOI] [PubMed] [Google Scholar]
- 42. Neumann H., Hancock S. M., Buning R., Routh A., Chapman L., Somers J., Owen-Hughes T., van Noort J., Rhodes D., Chin J. W. (2009) A method for genetically installing site-specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Mol. Cell 36, 153–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Simon M., North J. A., Shimko J. C., Forties R. A., Ferdinand M. B., Manohar M., Zhang M., Fishel R., Ottesen J. J., Poirier M. G. (2011) Histone fold modifications control nucleosome unwrapping and disassembly. Proc. Natl. Acad. Sci. U.S.A. 108, 12711–12716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Michishita E., McCord R. A., Boxer L. D., Barber M. F., Hong T., Gozani O., Chua K. F. (2009) Cell cycle-dependent deacetylation of telomeric histone H3 lysine K56 by human SIRT6. Cell Cycle 8, 2664–2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Williams S. K., Truong D., Tyler J. K. (2008) Acetylation in the globular core of histone H3 on lysine-56 promotes chromatin disassembly during transcriptional activation. Proc. Natl. Acad. Sci. U.S.A. 105, 9000–9005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xie W., Song C., Young N. L., Sperling A. S., Xu F., Sridharan R., Conway A. E., Garcia B. A., Plath K., Clark A. T., Grunstein M. (2009) Histone h3 lysine 56 acetylation is linked to the core transcriptional network in human embryonic stem cells. Mol. Cell 33, 417–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xu F., Zhang Q., Zhang K., Xie W., Grunstein M. (2007) Sir2 deacetylates histone H3 lysine 56 to regulate telomeric heterochromatin structure in yeast. Mol. Cell 27, 890–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yang B., Miller A., Kirchmaier A. L. (2008) HST3/HST4-dependent deacetylation of lysine 56 of histone H3 in silent chromatin. Mol. Biol. Cell 19, 4993–5005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mann M. (2006) Functional and quantitative proteomics using SILAC. Nat. Rev. Mol. Cell Biol. 7, 952–958 [DOI] [PubMed] [Google Scholar]
- 50. Ong S.-E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., Mann M. (2002) Stable Isotope Labeling by Amino Acids in Cell Culture, SILAC, as a Simple and Accurate Approach to Expression Proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 51. Mortensen P., Gouw J. W., Olsen J. V., Ong S.-E., Rigbolt K. T. G., Bunkenborg J., Cox J. R., Foster L. J., Heck A. J. R., Blagoev B., Andersen J. S., Mann M. (2009) MSQuant, an Open Source Platform for Mass Spectrometry-Based Quantitative Proteomics. J. Proteome Res. 9, 393–403 [DOI] [PubMed] [Google Scholar]
- 52. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotech. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 53. Cuomo A., Moretti S., Minucci S., Bonaldi T. (2011) SILAC-based proteomic analysis to dissect the “histone modification signature” of human breast cancer cells. Amino Acids 41, 387–399 [DOI] [PubMed] [Google Scholar]
- 54. Kelly T. J., Qin S., Gottschling D. E., Parthun M. R. (2000) Type B histone acetyltransferase Hat1p participates in telomeric silencing. Mol. Cell. Biol. 20, 7051–7058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ahn S. H., Henderson K. A., Keeney S., Allis C. D. (2005) H2B (Ser10) phosphorylation is induced during apoptosis and meiosis in S. cerevisiae. Cell Cycle 4, 780–783 [DOI] [PubMed] [Google Scholar]
- 56. Knapp A. R., Ren C., Su X., Lucas D. M., Byrd J. C., Freitas M. A., Parthun M. R. (2007) Quantitative profiling of histone post-translational modifications by stable isotope labeling. Methods 41, 312–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Elias J. E., Gygi S. P. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Meth. 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 58. Helsel D. R., Hirsch R. M. (2002) USGS Statistical Methods in Water Resources. U.S. Geological Survey, Techniques of Water-Resources Investigations Book 4, Chap. A2 [Google Scholar]
- 59. Helsel D. R., Hirsch R. M. (1993) Statistical methods in water resources, Elsevier; (online) [Google Scholar]
- 60. Glaister P. (2005) Robust linear regression using Theil's method. J. Chem. Ed. 82, 1472 [Google Scholar]
- 61. Yang L., Tu S., Ren C., Bulloch E. M. M., Liao C.-L., Tsai M.-D., Freitas M. A. (2010) Unambiguous determination of isobaric histone modifications by reversed-phase retention time and high-mass accuracy. Anal. Biochem. 396, 13–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Weake V. M., Workman J. L. (2008) Histone Ubiquitination: Triggering Gene Activity. Mol. Cell 29, 653–663 [DOI] [PubMed] [Google Scholar]
- 63. Shilatifard A. (2006) Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Ann. Rev. Biochem. 75, 243–269 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.